Abstract
Scavenging of electrogenerated superoxide radical anion (O2•−) by pyrogallol (PyH3) was investigated on the basis of cyclic voltammetry and in situ electrolytic electron spin resonance spectrum in N,N-dimethylformamide with the aid of density functional theory (DFT) calculations. Quasi-reversible dioxygen/O2•− redox couple was modified by the presence of PyH3, suggesting that O2•− was scavenged by PyH3 through proton-coupled electron transfer (PCET) involving two proton transfer and one electron transfer. DFT calculation suggested that the pre-reactive formation of a hydrogen-bond (HB) complex and the subsequent concerted two-proton-coupled electron transfer characterized by catechol moiety in PyH3 is plausible mechanism that embodies the superior kinetics of the O2•− scavenging by PyH3 as shown in the electrochemical results. Furthermore, it was clarified that the three hydroxyl groups of PyH3 promote the formation of HB complex, in comparative analyses using related compounds, resulting in the promotion of the O2•− scavenging.
1. Introduction
Benzene-1,2,3-triol (pyrogallol, PyH3) is one of the most sensitive organic compounds toward oxygen and causes autoxidation [1,2,3,4,5,6]. Its characteristic structure, based on three hydroxyl (OH) groups substituted onto a benzene ring, constitutes some antioxidants, such as gallocatechin, epigallocatechin gallate, and prodelphinidin (the polymeric tannins composed of gallocatechin). It is well recognized that PyH3 is a much more efficient antioxidant than benzene-1,2-diol (catechol, CatH2). Therefore, numerous studies have investigated the antioxidant reaction mechanism with polyphenols containing PyH3 moiety, experimentally and theoretically [1,5,7,8,9,10]. Three isometric benzenetriols (PyH3, benzene-1,2,4-triol, and benzene-1,3,5-triol) are differentiated by the position of OH groups. The larger the number of OH groups on phenolic compounds is, the higher the antioxidant properties are, although these isomers show different reactivity depending on their positions of OH groups. Therefore, the antioxidant property of PyH3 is considered as due to electron donation derived from its π-conjugated quinoid structure, which directly reacts with any reactive oxygen species (ROS) such as superoxide radical anion (O2•−), precursor of other ROS, and grand state molecular oxygen (O2) in the autoxidation. The antioxidant activity of PyH3 with its autoxidation involves complicated reaction mechanisms, though the mechanism between PyH3 and O2 involves two-proton transfer (PT) and two-electron transfer (ET) forming pyrogallol-ortho-quinone (PyH) and hydroperoxide (H2O2).
Simultaneously, it is well recognized that the autoxidation is promoted in an alkali solution, showing that the primary deprotonation occurs forming corresponding anion (PyH2−) followed by the autoxidation reaction [2,3]. Furthermore, it is considered that forming intermediate peroxiradical (ROS) such as hydroperoxiradical (HO2•) followed by a free radical chain reaction is a main mechanism for the net autoxidation. O2 is a moderately good electrophile that can accept electrons from PyH3, rather than O2•−. However, HO2• formed after protonation of O2•− as a Brønsted base is a strong oxidant. Thus, the deprotonation and subsequent oxidation, i.e., PT and ET between PyH3 and oxygen species involving O2 and ROS, are closely related. Most of the reported papers on the antioxidant activity of PyH3 are in aqueous solvents [2,3,6,11,12], and even under pH control, many protons are present. Among these papers, antioxidant reactions of PyH3 involving the autoxidation and ROS scavenging were detected and measured in aqueous media, by increases in O2 consumption or absorbance changes of the solution. Hence, the antioxidant reactions cannot be estimated separately, because several reactant species, i.e., PyH3, PyH2−, O2, O2•−, and HO2• coexist in the experimental solution. Thus, there are still some uncertain issues to be clarified regarding the antioxidant mechanism with the relationship between PT and ET, although the plausible main mechanism for O2•− scavenging by different reactants, PyH3/PyH2−, are simply denoted in Scheme 1, forming pyrogallol-ortho-quinone radical anion (PyH•−)/radical dianion (Py•2−) and H2O2.
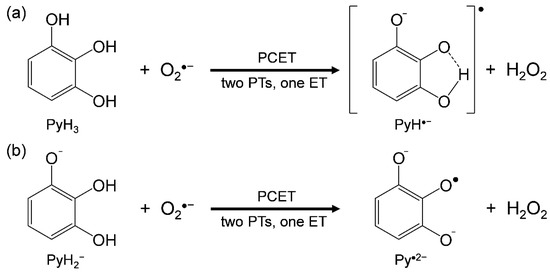
Scheme 1.
Reaction schemes of O2•− scavenging by (a) PyH3 and (b) PyH2−, through proton-coupled electron transfer (PCET) involving two PTs and one ET.
Nasr et al. reported the electrochemical oxidation of PyH3 in acidic aqueous solution [12]. In their pioneering work, the voltammetric results showed that PyH3 oxidation occurs in the same potential region as that of phenol (PhOH). Conversely, the initial deprotonation of PyH3 forming PyH2− increases electron density in the benzene ring and consequently increases its reactivity to electrophilic attack. Considering the above, the deprotonation first occurs in an aqueous buffer media, then the oxidation processes occur either directly on the electrode surface or can be mediated by O2 (autoxidation) and other oxygen species electrogenerated at the anode surface, implying that it is the main pathway for the oxidation of PyH3 by oxygen species in natural environments such as living body. Several reaction mechanisms for ROS scavenging by phenolic antioxidants such as PyH3 are known, including the superoxide-facilitated oxidation (SFO) [13,14,15], hydrogen atom transfer (HAT) involving PCET [16,17,18,19,20], and sequential proton-loss electron transfer (SPLET) [21]. In the SFO mechanism, the initial PT from the substrate to O2•− to give HO2• is followed by rapid dismutation to give H2O2 and O2. Then, the substrate anion is oxidized by the O2 formed in the dismutation process [15]. Conversely, the other two mechanisms involve direct oxidation by O2•−/HO2•.
Considering the relationship between the structure of PyH3 and the mechanism of O2•−/HO2• scavenging, quinone–hydroquinone π-conjugation is inferred to play a role in a PCET mechanism. In our previous studies, it has been reported that O2•−/HO2• is scavenged by polyphenols [20], diphenols (hydroquinone [22] and CatH2 [23]), and monophenols [24,25], through a PCET mechanism. In these studies, a concerted two-proton-coupled electron transfer (2PCET) involving two PTs and one ET is a plausible reaction pathway for CatH2 moiety based on the energetics and kinetics for successful O2•− scavenging. Therefore, CatH2 moiety comprised in PyH3 is expected to play through the concerted 2PCET, although the third OH group (3OH) gives a different chemical mechanism of PyH3 from CatH2.
In this study, we analyzed the reaction between PyH3 and electrogenerated O2•− comparatively using some related compounds (Figure 1) in dehydrated N,N-dimethylformamide (DMF) by using electrochemistry and density functional theory (DFT) calculation. Accordingly, herein we present a mechanistic insight into PCET for the O2•− scavenging reaction by PyH3 that constitute some highly reactive antioxidants.
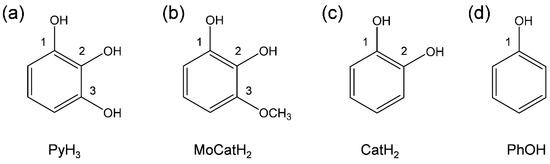
Figure 1.
Structures of PyH3 and the related compounds considered in this study. (a) PyH3, (b) 3-methoxybenzene-1,2-diol (MoCatH2), (c) CatH2, and (d) PhOH.
2. Materials and Methods
2.1. Chemicals
We obtained PyH3 (98.0%), MoCatH2 (99.0%), and CatH2 (99.0%), from Sigma-Aldrich Inc. (Tokyo, Japan), and purified by repeated sublimation under reduced pressure immediately before use. PhOH (99.5%) was purchased from Tokyo Chemical Industry Co., Ltd. (Tokyo, Japan), at the best available grade and was used as received. Dinitrogen (N2) gas (99.0%) and O2 gas (99.0%) were purchased from Medical Sakai Co., Ltd. (Gifu, Japan), and were used as received. The solvent for electrochemical and electron spin resonance (ESR)/ultraviolet-visible (UV-vis) spectral-measurements was spectrograde purity DMF (99.7%) available from Nacalai Tesque Inc. (Kyoto, Japan) and used as received. Tetrapropylammonium perchlorate (TPAP) was prepared as described previously [26] and used as a supporting electrolyte for DMF. Ferrocene (Fc), used as a potential reference compound, was commercially available from Nacalai Tesque Inc. and used as received.
2.2. Electrochemical and In Situ Electrolytic ESR/UV-vis Spectrum Measurements
Cyclic voltammetry was performed using a three-electrode system comprising a 1.0 mm-diameter glassy carbon (GC) working electrode, a coiled platinum (Pt) counter electrode, and a silver/silver nitrate (Ag/AgNO3) reference electrode (containing acetonitrile solution of 0.1 mol dm−3 tetrabutylammonium perchlorate and 0.01 mol dm−3 AgNO3; BAS RE-5) at 25 °C using BAS 100B electrochemical workstation, coupled to BAS electrochemical software to record data (Supplementary Materials, Table S1). In situ electrolytic ESR/UV–vis spectra were measured using a JEOL JES-FA200 X-band spectrometer/an OCEAN HDX spectrometer (OptoSirius Co., Ltd.). The controlled-potential electrolysis was performed at room temperature in an electrochemical ESR cell using a 0.5 mm-diameter straight Pt wire sealed in a glass capillary as a working electrode/an optically transparent thin-layer electrochemical (OTTLE) cell (path length: 1.0 mm) using a Pt mesh working electrode (Supplementary Materials, Figure S1). Samples were prepared in a glove box completely filled with N2 gas to prevent contamination by moisture. The DMF solution containing 0.1 mol dm−3 TPAP as a supporting electrolyte was saturated with O2 by air-bubbling the gas for ca. 2–3 min and the gas was passed over the solutions during the electrochemical and spectral measurements to maintain the concentration of O2 at a constant level. The equilibrium concentration of O2 was calculated as 4.8 × 10−3 mol dm−3.
2.3. Calculation
All solution phase calculations were performed at the DFT level with the Becke three-parameter Lee–Yang–Parr (B3LYP) hybrid functional as implemented in Gaussian 16 Program package [27]. This functional was chosen because it has been shown to give good geometries of the reactants, products and transition states (TS) in PCET reactions between phenolic compounds and free radicals [28]. Geometry optimization, vibrational frequency calculations, the intrinsic reaction coordinate (IRC) calculations, and population analysis of each compound was performed by employing the standard split-valence triple ζ basis sets augmented by the polarization 3df,2p and diffusion orbitals 6-311+G(3df,2p). The solvent contribution of DMF to the standard Gibbs free energies was computed employing the polarized continuum model (PCM) at the default settings of the Gaussian 16, which is widely employed in the description of the thermodynamic characteristics of solvation. The zero-point energies and thermal correction, together with entropy, were used to convert the internal energies to standard Gibbs energy at 298.15 K. The natural bond orbital (NBO) technique was used for electron and spin calculations in population analysis [29].
3. Results
3.1. Cyclic Voltammetry and ESR Analysis of O2/O2•− in the Presence of PyH3
In Figure 2, cyclic voltammograms (CV) of saturated O2 (4.8 × 10−3 mol dm−3) in the presence of PyH3 and related compounds (Figure 1a–d) in DMF, and ESR spectra of the CV solutions (b) obtained via in situ electrolytic ESR system are demonstrated. CV and ESR in the presence of (c) CatH2 and CV in the presence of (d) PhOH, were already reported in our previous paper, though are shown here for comparison [22,24]. In aprotic solvents such as DMF, O2 shows quasi-reversible redox at −1.284 V vs. the ferrocenium ion/ferrocene (Fc+/Fc) couple (Equation (1)) corresponding to generation of O2•− in the initial cathodic scan and reoxidation to the starting materials (O2), in the returned anodic scan (1c/1a, bold lines in Figure 2), where O2•− is not particularly reactive toward aprotic DMF. The reversible CVs investigated here were all modified to irreversible one by the presence of any compounds (a–d) with concentration dependency (0 to 3.0, 5.0 × 10−3 mol dm−3), supported that CVs of bubbled N2 showed no peak over the potential range. The reactivity of PyH3 estimated from a loss of reversibility of the CV is higher than the others. Thus, the loss of reversibility in the CVs of O2/O2•− is caused by the acid–base reaction; the initial PT from the compounds to O2•− as a Brønsted base forming HO2• (Equation (2)).
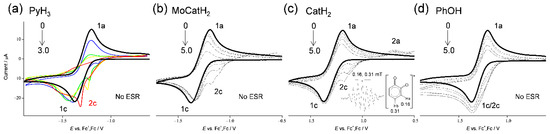
Figure 2.
CVs of 4.8 × 10−3 mol dm−3 O2 in the presence of (a) PyH3, (b) MoCatH2, (c) CatH2, and (d) PhOH, in DMF containing 0.1 mol dm−3 TPAP recorded with a GC electrode (1.0 mm) at a scan rate of 0.1 V s−1. Concentrations (×10−3 mol dm−3) are (a) 0 (black), 1.0 (blue), 2.0 (green), and 3.0 (red), and (b–d) 0, 1.0, 2.0, 3.0, and 5.0, (the concentration changes are shown by arrows). ESR spectra obtained via in situ controlled-potential electrolysis (at −1.3 V vs. V vs. Fc+/Fc) of the CV solutions (c).
With the generation of HO2•, bielectronic CVs were observed derives from the reduction of HO2• to hydroperoxyl anion (HO2−) (Equation (3)) as shown in Figure 2d, cathodic current 2c. Conversely, in the presence of (a) PyH3, (b) MoCatH2, and (c) MoCatH2, the bielectronic CVs do not appear due to the scavenging of HO2• by the subsequent ET (Equation (4)) from the deprotonated anion forming its radical (PyH2−/PyH2•, MoCatH−/MoCatH•, CatH−/CatH•), where a cathodic prepeak appeared. In our previous study, the ET involved in the PCET mechanism for successful O2•− scavenging required two structural characteristics; (1) the quinone–hydroquinone π-conjugated structure characterized by ortho/para-diphenol, and (2) the OH proton for the second PT [20,22,23,24,30].
O2 + e− ↔ O2•− (E° = −1.284 V vs. Fc+/Fc)
O2•− + PyH3 → HO2• + PyH2− (the initial PT)
HO2• + e− → HO2− (E° = −0.4 to −0.2 V vs. Fc+/Fc)
HO2• + PyH2− → HO2− + PyH2• (ET)
HO2− + PyH2• → H2O2 + PyH•− (the second PT)
Considering these results, we rationalized that O2•− formation after the primary electrode process associated with PT from the OH group leads to the irreversible overall reduction of O2 to H2O2, which is driven by the exergonic reduction of the resulting HO2•/HO2−. Therefore, the CV traces for O2/O2•− in the presence of phenolic compounds are divided into two typical curves: type A, an irreversible two-electron process observed in electro–chemical–electro reactions (Equations (1)–(3)), and type B, an irreversible one-electron process (Equations (1), (2), (4), and (5)) leading to O2•− scavenging. Figure 3 shows the plausible electrochemical mechanism for O2/O2•− in the presence of (a) PyH3 and (b) PhOH, summarizing Equations (1)–(5).
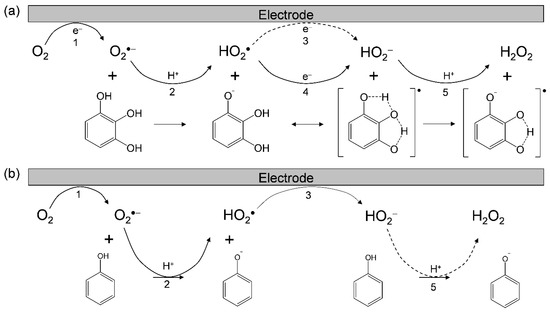
Figure 3.
Plausible electrochemical mechanisms of O2/O2•− in the presence of (a) PyH3 and (b) PhOH in DMF. 1 one-electron reduction of O2/O2•−, 2 the initial PT from acidic substrate to O2•−, 3 one-electron reduction of HO2•/HO2−, 4 ET from substrate anion to HO2•, 5 the second PT to HO2−. The net PCET reaction between PyH3 and O2•− forming PyH•− and H2O2 involves two PTs and one ET.
In this scenario, the CV results recorded in the presence of (d) PhOH demonstrate type A (O2•− is not scavenged) showing the appearance of a cathodic current ascribed HO2•. Conversely, each of the CV results in the presence of (a) PyH3, (b) MoCatH2, and (c) CatH2 demonstrate type B (scavenging of O2•−/HO2•). Then, the O2•−/HO2• scavenging by (c) CatH2 was confirmed via in situ electrolytic ESR measurements of the CV solutions at an applied potential of −1.3 V corresponding to the O2 reduction (Equation (1)) with ESR scanning during 4 min. With reference to (a) PyH3 and (b) MoCatH2, ESR shows no signal and the CV shows type B with a large reactivity. The cathodic prepeaks (2c) appearing in Figure 2a–c are inferred to be assigned to reduction of the product radical/dianion (PyH•−/PyH2−, MoCat•−/MoCat2−, Cat•−/Cat2−), although the corresponding anodic peaks are observed only for (c). It is presumed that the ESR spectra for (a, b) were undetectable because the generated PyH•− and MoCat•− were further reduced to dianions (PyH2− and MoCat−) at the applied potential (−1.3 V) for the electrogeneration of O2•−.
Next, in situ electrolytic UV–vis spectra for the CV solution containing PyH3 (1.0 × 10−3 mol dm−3) using the OTTLE cell (Supporting Information, Figure S2) were measured in the absence of O2 under purging N2 and in the presence of O2 (Figure 4). The spectrum of PyH3 alone has a characteristic absorption band centered at 272 nm. Under the applied potential at 1.0 to −2.0 V vs. Fc+/Fc without O2, the spectrum did not change where any potential was applied (data not shown), demonstrating that PyH3 is not electrolyzed without deprotonation. Conversely, the spectrum has changed in the presence of CH3ONa (5.0 × 10−3 mol dm−3) without applying a potential (red line). Since PyH3 is deprotonated by CH3ONa as a Brønsted base, the spectrum will be attributed to PyH2− or PyH2−. On the other side, the spectrum of PyH3 in the presence of saturated O2 (4.8 × 10−3 mol dm−3) has changed, showing the appearance of an absorption band centered at 292 nm at applied cathodic potentials over −1.3 V corresponding electrogeneration of O2•−.
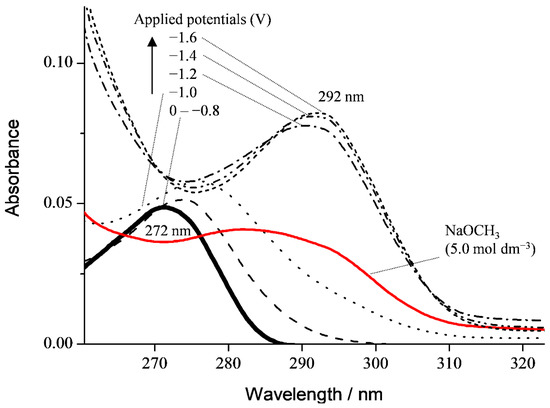
Figure 4.
UV–vis spectral changes in PyH3 (1.0 × 10−3 mol dm−3) solutions in the absence (black bold) and presence (red bold) of CH3ONa (5.0 × 10−3 mol dm−3) and in situ electrolytic UV–vis spectra of PyH3 in the presence of saturated O2. The controlled potential at −1.0, −1.2, −1.4, and −1.6 V vs. Fc+/Fc was applied to the solutions.
These spectral changes have demonstrated that the product of homogeneous reaction between PyH3 and O2•− is PyH•−, and the observed spectrum derives from PyH2− via further 1-electron reduction of PyH•− at the electrode. By analogy with the CV results, the initial PT (Equation (1)) and the following reactions including ET between HO2• and PyH2− (Equation (4)) rapidly undergo base-catalyzed oxidation. Since PyH•− would be electrolyzed to PyH2− at the electrode, the radical product was undetectable using the in situ electrolytic ESR system.
Notably, the CV and spectral results demonstrated that (a) PyH3 with its two OH groups can scavenge O2•− through the PCET involving two PTs and one ET, whereas the role of the 3OH group is unclear. These results imply that the reaction mechanism of (a) PyH3 is similar to that of (c) CatH2 (Scheme 1), however their reactivities are different.
3.2. Change in HOMO–LUMO Energies upon PCET between PyH3 and O2•− in DFT Analyses
DFT calculations with the frontier molecular orbital analysis were performed to aid the mechanistic analysis of O2•− scavenging by PyH3 in DMF. Figure 5 shows HOMO–LUMO changes upon PCET between PyH3/PyH2− and O2•−. After the initial PT, some reactant species, i.e., PyH3, PyH2−, O2•−, and HO2•, coexist in the solution. The singly occupied molecular orbital (SOMO) energy (Hartree) for HO2• (−0.3142) is much lower than HOMO energies of PyH3 and PyH2−. Thus, the electron acceptor will be HO2•, not O2•−. Considering that CV in DMF revealed that HO2• formed after the initial PT is scavenged (Figure 2a), the electron donor will be PyH2−, for which the downhill energy relationship is indicated by the bold red line. Thus, this change in HOMO–LUMO (SOMO) energies upon PT between PyH3 and O2•− forming PyH2− and HO2• is reasonable for subsequent ET. Next, the HOMO–LUMO relationship between the products after ET (i.e., PyH2•, and HO2−) is reversed, which is rational for orbital energies in the reverse ET (red dotted line). However, the HOMO (−0.2754) of the PT-forming H2O2 is lower than HOMO (−0.1648) of HO2−, making the reverse ET improbably. Thus, the subsequent PT is dominant in determining the ET direction.
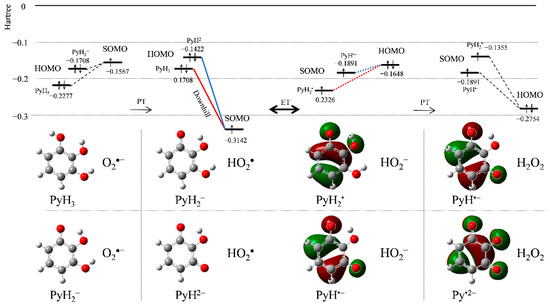
Figure 5.
Change in HOMO−LUMO (SOMO) energies (Hartree) upon PCET between PyH3 and O2•− with its corresponding chemical species in DMF, calculated using the DFT-(U)B3LYP/PCM/6-311+G(3df,2p).
Alternatively, the HOMO–LUMO relationship for O2•− scavenging by PyH2− preformed by the initial deprotonation indicates that the similar PCET involving two PTs and one ET occurs: the downhill ET (blue bold line) and reverse ET (blue dotted line). As reported previously, 2PCET occurs between CatH2 moiety and O2•− after the formation of the pre-reactive hydrogen-bond (HB) complex from the free reactants (FR), and the proton and electron are concertedly transferred in one kinetic step via a TS forming the product complex (PC) [20,23]. Therefore, it is expected that the 2PCET between PyH3 and O2•− occurs in a similar concerted manner through the HB formed between two OH groups of PyH3 and O2•−.
3.3. Free Energy Calculations of PCET between PyH3 and O2•−
For a mechanistic analysis of the O2•− scavenging by PyH3 in DMF, DFT calculations were performed at the (U)B3LYP/PCM/6-311+G(3df,2p) level. In Figure 6, the equilibrium schemes and standard Gibbs free energy changes (ΔG°/kJ mol−1, 298.15 K) of the six diabatic electronic states for the PCET involving two PTs and one ET between (a) PyH3 and O2•−, and between (b) O2•− and PyH2− formed after the initial deprotonation are shown. The important factors in determining the sequential processes shown in this scheme are the ΔG°s for the individual reactions; the acid–base interaction and redox potentials of the components. In Figure 6a, ET1 (ΔG° = 405.3 kJ mol−1) is strongly endergonic, thus, PT1 (17.9) forming PyH2− and HO2• must primarily occur, as shown in the CV result. In the following pathway shown in the lower rectangle, both PT3 (357.9) and ET2 (28.2) are uphill endergonic, suggesting that the sequential PCET does not proceed but the concerted PCET (−39.5) is a thermodynamically feasible pathway. Alternatively, one-step one-electron transfer concerted with sequential two-proton transfer after initial formation of the HB complexes between PyH3 and O2•− without generating high-energy intermediates, which we refer to as concerted 2PCET reactions, is another feasible pathway [23,28]. For a successful O2•− scavenging in either PCET pathway, the second PT coupled to ET is necessary, as reported in our previous studies [25,31]. On the other side, a PCET reaction between PyH2− and O2•− shown in Figure 6b is also plausible, in case that the initial deprotonation of PyH3 will partially occur in an aprotic DMF solution. Although, since both PT1 (88.5) and ET1 (253.5) are uphill, the only feasible pathway is 2PCET forming quinone-radical-dianion (Py•2−) as a product of the net reaction involving three PTs and one ET from PyH3.
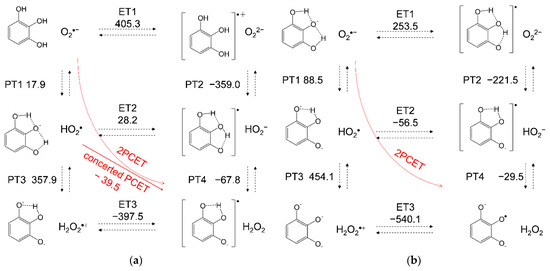
Figure 6.
Six diabatic electronic states and the ΔG° values for PCET between (a) PyH3 and O2•−, and (b) PyH2− and O2•−, involving two PTs and one ET in DMF. The ΔG° (kJ mol−1, 298.15 K) for the (PT1-PT4) and ET (ET1-ET3) were calculated using DFT-(U)B3LYP/PCM/6-311+G(3df,2p) method.
For a comparative study, the ΔG° values of the PCET pathways for CatH2 and MoCatH2 were calculated (Table 1). From a thermodynamic viewpoint, the total values of ΔG° for the net PCET were obtained from the sum of the values for the two PTs and one ET. If the PCET occurs along a pathway involving the unfeasible single PT/ET (PT1, PT3, ET1, and ET2), the total values cannot embody the energetic driving force because the ΔG° for the unfeasible PT/ET has been summed in it. However, since the concerted PCET (ET2–PT4/PT3–ET3) after the initial PT1 is endergonic for both CatH2 (−55.7) and MoCatH2 (−79.1), the total values (−36.2 and −32.9) can embody the exergonic driving force through PT1–concerted PCET pathway, similar to PyH3 (concerted: −39.6, total: −21.6). Notably, both the ΔG° values (concerted and total) for PyH3 are larger than those for CatH2 and MoCatH2, showing a lower reactivity of PyH3. The effect of the substituted group of MoCatH2, PyH3, and CatH2, on the O2•− scavenging through the PCET is primarily considered to be due to the electron-donating ability (-OCH3 > -OH > -H) increasing electron density in the benzene ring, known as the Hammett equation [32]. Additionally, the intramolecular HB formed at the 3OH group strongly stabilizes the negatively charged deprotonated species along the PCET; PyH2−, PyH2−, and its trianion (Py3−), consequently suppressing their reactivities to electrophilic attack. Thus, these ΔG° values confirm that the PCET mechanism in Figure 6a alone cannot explain the reason for the higher reactivity of PyH3 than the others toward electrogenerated O2•− shown in the CVs (Figure 2). As a result of the comparative analyses of the ΔG° values, the involvement of three reaction pathways is plausible for efficient O2•− scavenging by PyH3; PT–concerted PCET and 2PCET between PyH3 and O2•−, and 2PCET between PyH2− and O2•−.

Table 1.
ΔG° values (kJ mol−1, 298.15 K) for PCET between O2•− and phenolic compounds (PyH3, PyH2−, CatH2, and MoCatH2) in DMF, calculated using DFT at the (U)B3LYP/PCM/6-311+G(3df,2p) level.
3.4. Potential Energy Surfaces of the PCET between PyH3 and O2•−
For gaining deeper insight into the PCET mechanism for the O2•− scavenging by PyH3 in DMF, potential energy surfaces were investigated at the (U)B3LYP/PCM/6-311+G(3df,2p) level of theory. It is assumed that the reaction involves three elementary steps: (i) formation of the prereactive HB complex (PRC) from the FRs, (ii) reaction to the PC via a TS, and (iii) dissociation of the PC yielding free products (FP). Furthermore, the structural and electronic changes during the reaction were analyzed with the NBO calculations. First, we started with an analysis of potential energy scanning for the stable HB complexes (PRC, intermediate HB complex, and PC) along the PCET reaction (Figure 7a). Then, optimized structures of plausible PRC (PyH3−O2•−) formed from the FRs via two HBs (step i) were obtained, resulting in a lower ΔG° by 81.2 kJ mol−1 (set as zero). Next, an energy profile (ΔG°, kJ mol−1) along the IRC for the 2PCET involving concerted two PTs and one ET forming the PC (PyH•−–H2O2) was obtained (step ii). The IRC shows that a 2PCET occurs between PyH3 and O2•− in one kinetic process via the TS of a low activation energy (Ea) at 53.9 kJ mol−1, without generating any intermediates such as HO2•, HO2−, PyH2−, and PyH2•.
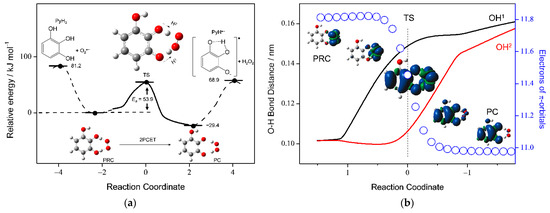
Figure 7.
(a) Energy profile (kJ mol−1) along the IRC of the 2PCET between PyH3 and O2•− in DMF with structures of FRs (PyH3, O2•−), PRC (PyH3−O2•−), TS, PC (PyH•−−H2O2), and FPs (PyH•−, H2O2). (b) Changes in O−H bond distances (OH1: black line, OH2: red line/nm) corresponding to the labels of the left vertical axes, and the number of electrons (open circles) on the π-orbitals in the PyH3 moiety (corresponding to the labels of the right vertical axes). The calculations were performed using the DFT-(U)B3LYP/PCM/6-311+G(3df,2p) method. The NBO analyses obtained π-electrons and spin density distributions localizing the atoms constituting the radicals along the PCET.
Figure 7b shows changes in O−H bond distances (OH1: black line, and OH2: red line) with the number of electrons on the π-orbital of PyH3 moiety along the IRC (blue circle). Then, spin density distributions localizing the atoms consisting of the radical before and after the TS along the 2PCET are demonstrated, showing that the radical localized on O2•− in the initial PRC was transferred to PyH•− in the resulting PC. Changes in the spins on the electron donor side (PyH3) and acceptor side (O2•−) are in good correlation with the changes in the π-electron of the PyH3. Furthermore, careful observation of changes in structures and OH1/OH2 in the IRC indicates that the π-electron transfer occurs simultaneously with sequential lengthening of the two O−H bond distances of PyH3. The first step of the reaction is the attraction of one phenolic proton (H1) by O2•−. This attraction results in nearly complete deprotonation of PyH3 and transfer of one-half of the π-electron from PyH3 to O2•− in the TS. Movement of the second proton (H2) accelerates the ET from the TS forward and leads to formation of the PC as the resulting reaction system. The results also demonstrate that the electronic state of the TS is characterized by the delocalization of the radical anion over the HB complex of the components.
For a comparative study, potential energy surfaces for the PCET with IRC and TS between O2•− and the other compounds—CatH2, MoCatH2, and the anion PyH2−—were investigated. Similar 2PCET mechanisms for CatH2 and MoCatH2 were obtained without generating intermediates (Supplementary Data, Figures S2 and S3), but was not obtained for PyH2−. Then, the ΔG° values of the complexes (FR, PC, and FP) and the Ea values, for PyH3, CatH2, and MoCatH2 (Geometries of the TS are shown in Supplementary Materials, Tables S3–S5), are listed in Table 2. Notably, there is almost no difference in each Ea value (PyH3: 53.9, CatH2: 52.5, and MoCatH2: 50.7), and either of those is as low as the hydrogen-bonding energy. Conversely, there are about 10–20 kJ mol−1 differences between the ΔG° values for forming each of the PRC from the FRs (PyH3: 81.2, CatH2: 71.6, and MoCatH2: 61.4). Thus, the ΔG° values for the formation of the PRC from the FRs (step i), rather than the kinetics of the 2PCET reaction to the PC via a TS (step ii), embody the superior O2•−-scavenging ability of PyH3 with a good correlation with the CV results in DMF. Considering the IRC results together with the CV (Figure 2) and the ΔG° results (Figure 6), the 2PCET between O2•− and PyH2− formed after the deprotonation of PyH3 is not feasible. In an aprotic DMF solution, the initial reaction between PyH3 and O2•− will be the formation of the PRC stabilized at −81.2 kJ mol−1 via two HBs, rather than the initial PT (PT1 in Figure 6a: 17.9 kJ mol−1) or a deprotonation (proton loss in the solution).
Table 2.
ΔG° and Ea values (kJ mol−1, 298.15 K) for 2PCET between O2•− and the compounds (PyH3, CatH2, and MoCatH2) in DMF, calculated using DFT at the (U)B3LYP/PCM/6-311+G(3df,2p) level.
Taken together, these findings indicate that the O2•− scavenging by PyH3 in DMF is governed by the concerted 2PCET after forming PRC via two HBs, which corresponds to a moving along the red diagonal line of the two rectangles shown in Figure 6a. In Figure 8, the net mechanism of the O2•− scavenging by PyH3 in DMF is shown. In the 2PCET mechanism, ET occurs between oxygen-π-orbitals orthogonal to the molecular framework, then, PT occurs between oxygen-σ-orbitals along the HBs [23]. It is presumed that the higher reactivity of PyH3 with O2•− than that for CatH2, is due to the sequential reactions; the initial formation of the PRC, followed by the 2PCET.
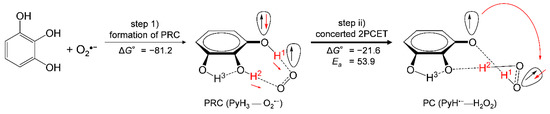
Figure 8.
Plausible mechanism and the ΔG° and Ea values (kJ mol−1, 298.15 K) for the PCET pathways between PyH3 and O2•− involving the initial formation of PRC followed by concerted 2PCET in DMF. The ΔG° and Ea values were calculated using DFT-(U)B3LYP/PCM/6-311+G(3df,2p) method.
4. Conclusions
In conclusion, we have investigated the O2•− scavenging by PyH3 through the PCET in DMF. As a result, we have clarified;
- PyH3 scavenges O2•− through the 2PCET involving concerted two PTs and one ET, in a similar mechanism for CatH2;
- the net mechanism involves the initial formation of PRC followed by concerted 2PCET;
- the 3OH group thermodynamically promotes the formation of PRC via two HBs but does not promote the latter 2PCET, resulting in an effective O2•− scavenging ability of PyH3.
Although the results presented in this manuscript are for a chemical reaction in aprotic DMF solvent, the PCET theory is adaptable to biological processes in such as a lipid bilayer. Therefore, we hope that the findings obtained in this study will provide evidence for the mechanistic actions of O2•− scavenging by various antioxidants involving PyH3 moiety.
Supplementary Materials
The following is available online at https://www.mdpi.com/article/10.3390/electrochem3010008/s1, Table S1: CV parameters, Tables S2–S5: ΔG° values for the PCET in various solvents, Table S6: Calculated geometry of complexes, Figure S1: In situ electrolytic ESR/UV-vis system, Figure S2: Energy profiles along IRC of 2PCET between CatH2 and O2•−, Figure S3: Energy profiles along IRC of 2PCET between MoCatH2 and O2•−.
Author Contributions
Conceptualization, T.N.; data curation, R.H.; methodology, B.U.; resources, K.K., S.U.; writing—original draft preparation, T.N.; writing—review and editing, B.U. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by Grant-in-Aid for Scientific Research, grant number 19K16338 from Japan Society for the Promotion of Science (JSPS).
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Data available in a publicly accessible repository.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Furuno, K.; Akasako, T.; Sugihara, N. The contribution of the pyrogallol moiety to the superoxide radical scavenging activity of flavonoids. Biol. Pharm. Bull. 2002, 25, 19–23. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, X. Improved pyrogallol autoxidation method: A reliable and cheap superoxide-scavenging assay suitable for all antioxidants. J. Agric. Food Chem. 2012, 60, 6418–6424. [Google Scholar] [CrossRef] [PubMed]
- Ramasarma, T.; Rao, A.V.S.; Devi, M.M.; Omkumar, R.V.; Bhagyashree, K.S.; Bhat, S.V. New insights of superoxide dismutase inhibition of pyrogallol autoxidation. Mol. Cell. Biochem. 2015, 400, 277–285. [Google Scholar] [CrossRef] [PubMed]
- Doona, C.J.; Kustin, K. Kinetics and mechanism of pyrogallol autoxidation: Calibration of the dynamic response of an oxygen electrode. Int. J. Chem. Kinet. 1993, 25, 239–247. [Google Scholar] [CrossRef]
- Wright, J.S.; Johnson, E.R.; DiLabio, G.A. Predicting the activity of phenolic antioxidants: Theoretical method, analysis of substituent effects, and application to major families of antioxidants. J. Am. Chem. Soc. 2001, 123, 1173–1183. [Google Scholar] [CrossRef]
- Marklund, S.; Marklund, G. Involvement of the Superoxide Anion Radical in the Autoxidation of Pyrogallol and a Convenient Assay for Superoxide Dismutase. Eur. J. Biochem. 1974, 47, 469–474. [Google Scholar] [CrossRef] [PubMed]
- Alavi Rafiee, S.; Farhoosh, R.; Sharif, A. Antioxidant Activity of Gallic Acid as Affected by an Extra Carboxyl Group than Pyrogallol in Various Oxidative Environments. Eur. J. Lipid Sci. Technol. 2018, 120, 1800319. [Google Scholar] [CrossRef]
- Sutanto, H.; Susanto, B.H.; Nasikin, M. Solubility and antioxidant potential of a pyrogallol derivative for biodiesel additive. Molecules 2019, 24, 2439. [Google Scholar] [CrossRef] [Green Version]
- Romero, M.; Rojano, B.; Mella-Raipán, J.; Pessoa-Mahana, C.D.; Lissi, E.; López-Alarcón, C. Antioxidant Capacity of pure compounds and complex mixtures evaluated by the ORAC-pyrogallol red assay in the presence of Triton X-100 micelles. Molecules 2010, 15, 6152–6167. [Google Scholar] [CrossRef] [Green Version]
- Mu, S.; Chen, C. Electrochemical Oxidation of Pyrogallol: Formation and Characterization of Long-Lived Oxygen Radicals and Application To Assess the Radical Scavenging Abilities of Antioxidants. J. Phys. Chem. B 2012, 116, 12567–12573. [Google Scholar] [CrossRef]
- Zhang, Q.A.; Wang, X.; Song, Y.; Fan, X.H.; García-Martín, J.F. Optimization of pyrogallol autoxidation conditions and its application in evaluation of superoxide anion radical scavenging capacity for four antioxidants. J. AOAC Int. 2016, 99, 504–511. [Google Scholar] [CrossRef] [PubMed]
- Nasr, B.; Hsen, T.; Abdellatif, G. Electrochemical treatment of aqueous wastes containing pyrogallol by BDD-anodic oxidation. J. Environ. Manag. 2009, 90, 523–530. [Google Scholar] [CrossRef]
- Das, K.; Roychoudhury, A. Reactive oxygen species (ROS) and response of antioxidants as ROS-scavengers during environmental stress in plants. Front. Environ. Sci. 2014, 2, 53. [Google Scholar] [CrossRef] [Green Version]
- Dikalov, S.I.; Harrison, D.G. Methods for detection of mitochondrial and cellular reactive oxygen species. Antioxid. Redox Signal. 2014, 20, 372–382. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fridovich, I. Superoxide dismutase. In Encyclopedia of Biological Chemistry, 2nd ed.; Elsevier Inc.: Amsterdam, The Netherlands, 2013; pp. 352–354. ISBN 9780123786319. [Google Scholar]
- Costentin, C.; Robert, M.; Savéant, J.M. Concerted proton-electron transfers: Electrochemical and related approaches. Acc. Chem. Res. 2010, 43, 1019–1029. [Google Scholar] [CrossRef]
- Wenger, O.S. Proton-coupled electron transfer with photoexcited metal complexes. Acc. Chem. Res. 2013, 46, 1517–1526. [Google Scholar] [CrossRef] [Green Version]
- Hammes-Schiffer, S. Theory of proton-coupled electron transfer in energy conversion processes. Acc. Chem. Res. 2009, 42, 1881–1889. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Singh, P.S.; Evans, D.H. Study of the electrochemical reduction of dioxygen in acetonitrile in the presence of weak acids. J. Phys. Chem. B 2006, 110, 637–644. [Google Scholar] [CrossRef]
- Nakayama, T.; Uno, B. Importance of proton-coupled electron transfer from natural phenolic compounds in superoxide scavenging. Chem. Pharm. Bull. Tokyo 2015, 63, 967–973. [Google Scholar] [CrossRef] [Green Version]
- Biela, M.; Rimarčík, J.; Senajová, E.; Kleinová, A.; Klein, E. Antioxidant action of deprotonated flavonoids: Thermodynamics of sequential proton-loss electron-transfer. Phytochemistry 2020, 180, 112528. [Google Scholar] [CrossRef]
- Nakayama, T.; Uno, B. Quinone-hydroquinone π-conjugated redox reaction involving proton-coupled electron transfer plays an important role in scavenging superoxide by polyphenolic antioxidants. Chem. Lett. 2010, 39, 162–164. [Google Scholar] [CrossRef]
- Nakayama, T.; Uno, B. Concerted two-proton-coupled electron transfer from catechols to superoxide via hydrogen bonds. Electrochim. Acta 2016, 208, 304–309. [Google Scholar] [CrossRef]
- Nakayama, T.; Uno, B. Structural properties of 4-substituted phenols capable of proton-coupled electron transfer to superoxide. Int. J. Adv. Res. Chem. Sci. 2016, 3, 11–19. [Google Scholar] [CrossRef]
- Nakayama, T.; Honda, R.; Kuwata, K.; Usui, S.; Uno, B. Electrochemical and Mechanistic Study of Reactivities of α-, β-, γ-, and δ-Tocopherol toward Electrogenerated Superoxide in N, N-Dimethylformamide through Proton-Coupled Electron Transfer. Antioxidants 2021, 11, 9. [Google Scholar] [CrossRef] [PubMed]
- Okumura, N.; Uno, B. Electronic spectra of the electrogenerated 1,4-benzoquinone π-dianion and the strongly hydrogen-bonded charge-transfer complex with methanol. Bull. Chem. Soc. Jpn. 1999, 72, 1213–1217. [Google Scholar] [CrossRef]
- Frisch, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; Li, X.; et al. Gaussian 16, Rev. B.01; Gaussian, Inc.: Wallingford, CT, USA, 2016; ISBN 9781935522027. [Google Scholar]
- Quintero-Saumeth, J.; Rincón, D.A.; Doerr, M.; Daza, M.C. Concerted double proton-transfer electron-transfer between catechol and superoxide radical anion. Phys. Chem. Chem. Phys. 2017, 19, 26179–26190. [Google Scholar] [CrossRef]
- Reed, A.E.; Weinstock, R.B.; Weinhold, F. Natural population analysis. J. Chem. Phys. 1985, 83, 735–746. [Google Scholar] [CrossRef]
- Nakayama, T.; Honda, R. Electrochemical and mechanistic study of superoxide elimination by mesalazine through proton-coupled electron transfer. Pharmaceuticals 2021, 14, 120. [Google Scholar] [CrossRef]
- Nakayama, T.; Honda, R. Electrochemical and Mechanistic Study of Oxidative Degradation of Favipiravir by Electrogenerated Superoxide through Proton-Coupled Electron Transfer. ACS Omega 2021, 6, 21730–21740. [Google Scholar] [CrossRef]
- Hammett, L.P. The Effect of Structure upon the Reactions of Organic Compounds. Benzene Derivatives. J. Am. Chem. Soc. 1937, 59, 96–103. [Google Scholar] [CrossRef]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).