
GeNetFormer: Transformer-Based Framework for Gene Expression Prediction in Breast Cancer



Abstract
1. Introduction
- In this work, we propose GeNetFormer, a deep learning framework for gene expression prediction tasks. We evaluate the effectiveness of transformer-based models on WSIs and ST data in breast cancer.
- Our experiments on eight benchmark transformer-based architectures provide a detailed comparison of these models. Furthermore, we analyze the influence of different loss functions and input resolutions on their performance.
- This paper shows the potential of transformer-based models to advance cancer research and set new benchmarks in the field.
2. Related Work
2.1. Approaches Using the STNet Dataset
2.2. Approaches Using HER2+ Dataset
2.3. Approaches Using Other Datasets
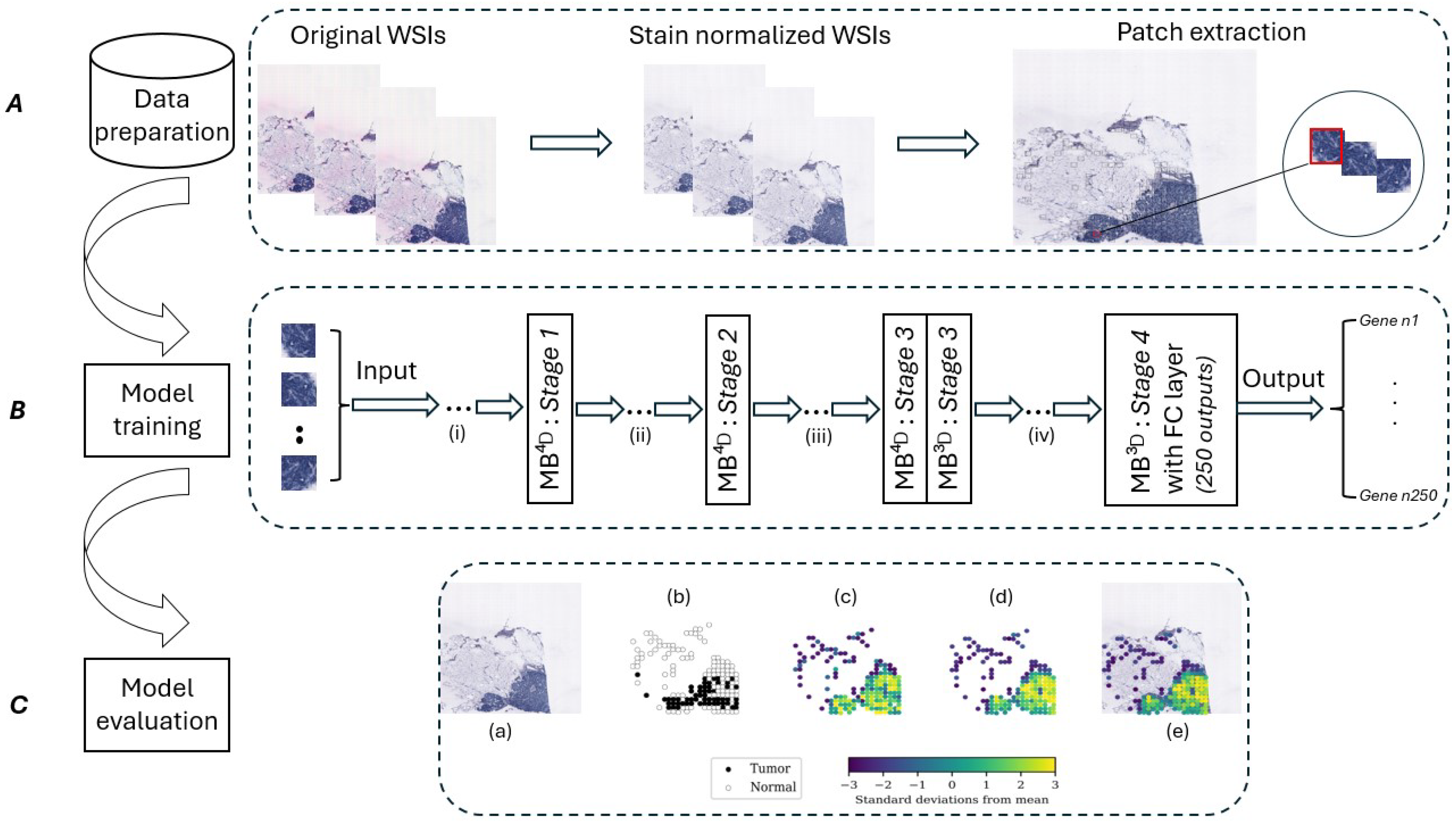
3. Materials and Methods
3.1. Dataset
3.2. Data Pre-Processing and Augmentation
3.3. Proposed Approach
- EfficientFormer is a fully transformer-based architecture designed for low-latency applications. As detailed in [20], the network starts with a patch embedding layer (PatchEmbed) followed by a series of meta transformer blocks (MB), which is expressed as follows:where represents the input image with a batch size , a channel depth of 3 (for RGB images), and spatial dimensions . denotes the final output of the network after m meta transformer blocks.Each meta transformer block (MB) consists of two components: a token mixer (TokenMixer) and a multi-layer perceptron (MLP) [20]. This relation is expressed as follows:here, represents the intermediate feature passed into the ith block, and is the output of that block [20]. The network is divided into four stages, with each stage processing features at a fixed spatial resolution [20]. Within each stage, multiple meta transformer blocks are used [20]. This design avoids the integration of MobileNet components and maintains a pure transformer-based architecture adapted for efficient computation [20]. It uses a method called latency-driven slimming to focus on the most important layers, making it faster [20]. The model also introduces a dimension-consistent design to divide the network into 4D and 3D partitions [20]. This design allows the network to benefit from the global modeling capabilities of multi-head self-attention (MHSA) [20].
- FasterViT [21] is a hybrid ViT which is designed to optimize the trade-off between accuracy and latency, focusing on high throughput for vision tasks on GPUs [21]. Its architecture combines dense convolutions in the early stages and hierarchical attention mechanisms in the later stages to balance memory-bound and compute-bound operations [21]. For different datasets and model sizes, it can scale effectively to higher-resolution input images [21]. The architecture begins with a stem that transforms the input image into overlapping patches [21]. Moreover, downsampler blocks reduce the spatial resolution between stages by a factor of 2 [21]. FasterViT introduces carrier tokens (CTs), which formulate the concept of hierarchical attention to facilitate efficient global learning [21]. The input feature map is first partitioned into local windows:where k is the window size. Carrier tokens are then initialized by pooling:where is the spatial size, is the number of carrier tokens, and includes positional encoding. The complexity of hierarchical attention is given by the following:where k is the window size, L is the number of carrier tokens, is the spatial size, and d is the feature dimension.
- BEiT v2 [22] builds on the masked image modeling (MIM) framework by using a semantic visual tokenizer to convert images into discrete visual tokens [22]. It uses ViTs as its backbone, which divide input images into patches [22]. To train the tokenizer, BEiT v2 introduces vector-quantized knowledge distillation (VQ-KD), where a transformer encoder and a quantizer map image patches to discrete codes [22]. BEiT v2 follows the MIM approach, masking 40% of the image patches and replacing them with a learnable embedding [22]. The model reconstructs the masked patches by predicting their visual tokens with a softmax classifier [22], optimizing the MIM loss:to improve the global representation, the [CLS] token is pre-trained with a bottleneck mechanism that aggregates patch-level information, and refined with a flat transformer decoder [22]. The final pre-training loss combines the masked image modeling (MIM) loss and the decoder’s masked prediction loss [22]. BEiT v2 can learn both patch-level and global representations by ensuring feature quantization and global information aggregation [22].
- Swin Transformer v2 [23] addresses important challenges in scaling model capacity and resolution for vision tasks. To improve training stability, it introduces residual post-normalization, which normalizes residual outputs before merging them into the main branch [23]. This reduces the accumulation of activation amplitudes in deeper layers [23]. It also replaces dot product attention with scaled cosine attention [23], defined as follows:where is a learnable scalar and represents the relative position bias.To handle mismatches between low-resolution pre-training and high-resolution fine-tuning, Swin Transformer v2 introduces log-spaced continuous position bias (Log-CPB) [23], which smooths the transfer of relative position biases across different window sizes [23]. The position bias is computed using a meta-network:where G is a small MLP, and coordinates are transformed into log space as
- PyramidViT v2 [24] improves on the original PyramidViT v1 by addressing three major limitations: high computational complexity, loss of local continuity, and inflexibility in handling variable input resolutions [24]. To address these issues, PyramidViT v2 introduces three innovations: a linear spatial reduction attention layer (SRA) to reduce the computational cost of attention, overlapping patch embedding instead of non-overlapping patches to model local continuity, and convolutional feed-forward networks to eliminate the need for fixed-size positional embeddings [24]. PyramidViT v2’s hierarchical architecture follows a pyramid structure, with spatial resolution decreasing and channel dimensions increasing over four stages [24]. It scales with different variants by adjusting parameters such as channel dimensions and number of layers [24]. PyramidViT v2 shows improvements in classification, detection, and segmentation tasks [24].
- MobileViT [25] is a lightweight ViT designed to combine the strengths of CNNs and transformers for mobile vision tasks [25]. It incorporates convolutional properties to improve global representation learning [25]. The core of the MobileViT architecture is the MobileViT block [25], which combines convolutions and transformers to encode both local and global information [25]. For an input tensor , an convolutional layer followed by a convolutional layer is applied to produce where the convolution encodes local spatial information and the convolution projects the tensor into a higher dimensional space [25]. To capture global information, is unfolded into non-overlapping patches [25], where P is the patch size and N is the number of patches. Transformers are then applied to learn the relationships between patches [25]:The resulting global representation is folded back to [25], projected to the original dimension C, and merged with the input X via concatenation followed by another convolution [25]. The hierarchical structure of MobileViT consists of an initial convolution layer, followed by MobileNetV2 blocks for downsampling and MobileViT blocks for feature extraction [25]. The spatial dimensions decrease progressively through the network, allowing for multi-scale representation learning [25]. MobileViT shows high performance in a variety of tasks [25].
- MobileViT v2 [26] is a hybrid network of CNNs and ViTs optimized for mobile devices that addresses the inefficiencies of multi-head self-attention (MHA) by introducing separable self-attention [26]. Multi-head self-attention (MHA) on MobileVit, with its complexity (where k is the number of tokens or patches), is replaced by a separable self-attention approach that reduces complexity to [26]. Separable self-attention computes context scores which re-weight the input tokens to produce a context vector that encodes global information [26]. This is achieved through element-wise operations such as summation and multiplication, eliminating expensive operations such as batch-wise matrix multiplication [26]. The simplicity of element-wise operations ensures faster inference and lower memory consumption compared to its previous version, MobileViT [26]. MobileViT v2 is also state of the art for tasks such as object detection and segmentation, with marked improvements in both accuracy and latency over MobileViT v1 [26].
- EfficientViT [27] is a lightweight and memory-efficient ViT designed for high-speed and resource-constrained applications. It addresses inefficiencies in memory access, computation redundancy, and parameter usage through three innovations: a sandwich layout, cascaded group attention (CGA), and parameter reallocation [27]. EfficientViT uses a single memory-bound multi-head self-attention (MHSA) layer sandwiched between feed-forward network (FFN) layers to reduce memory overhead and improve inter-channel communication [27]. To minimize redundancy in the attention heads, cascaded group attention (CGA) splits input features across the heads and refines them progressively [27]. EfficientViT reallocates parameters by increasing channel width for critical modules and decreasing redundant dimensions [27]. This ensures that parameters are used optimally without affecting performance [27]. EfficientViT achieves state-of-the-art performance in tasks such as classification and detection.
3.4. Evaluation Metrics
- The MAE measures the average difference between predicted () and actual () gene expression values. It treats all errors equally.lower MAE values mean that the model predicts closer to the actual gene expressions.
- The RMSE also measures the difference between predictions and actual gene expression values, but gives more weight to larger errors by squaring them.smaller RMSE values indicate that the model is making fewer major errors.
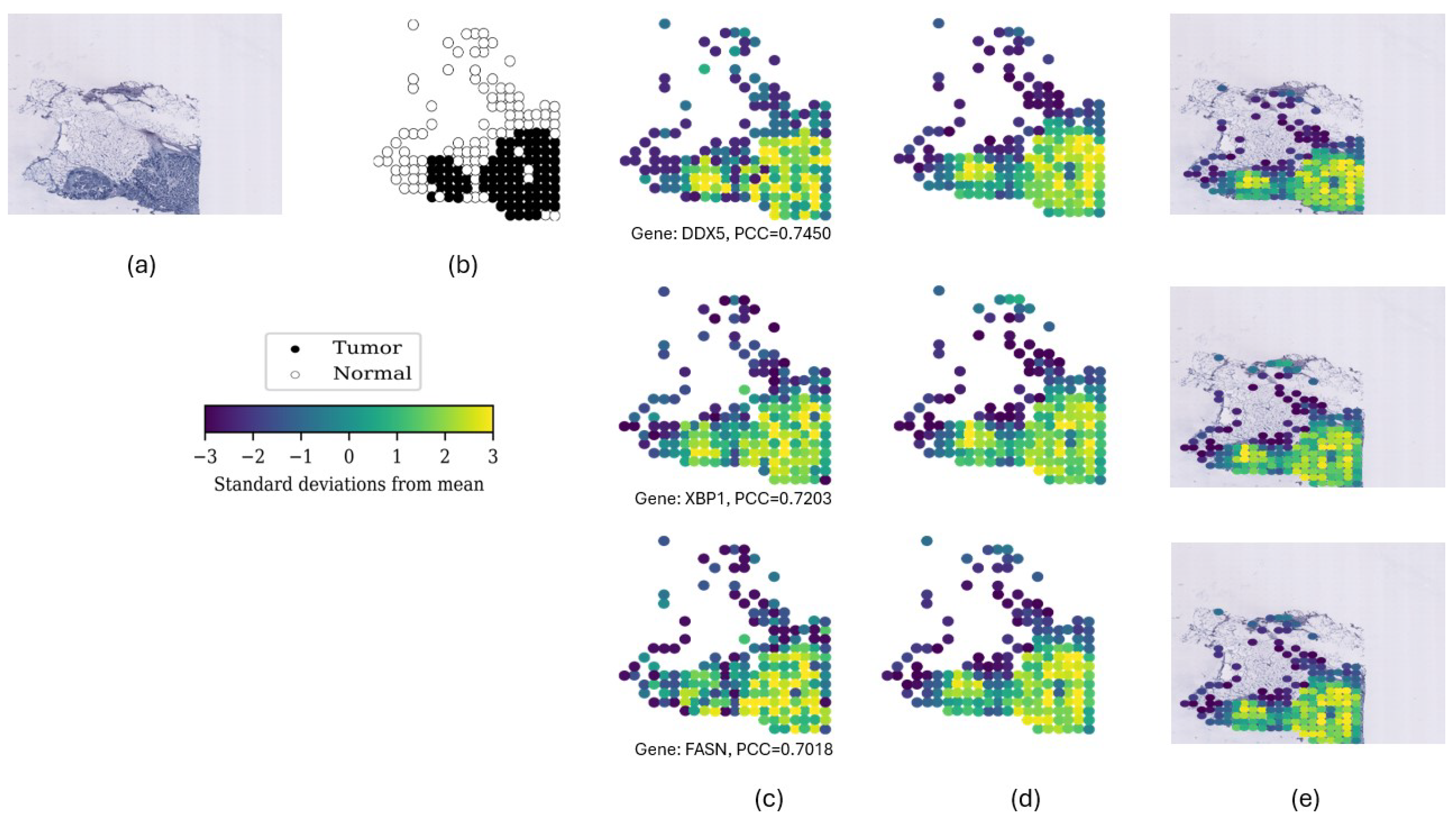
- The PCC indicates the strength of the relationship between predicted and actual gene expression values.PCC values close to 1 indicate a strong positive correlation.
3.5. Loss Functions
- The MSELoss function calculates the average squared difference between predicted and actual gene expression values during training. Larger errors are penalized more heavily than smaller errors.
- The SL1Loss function combines the advantages of the MAE and MSE. It treats small errors like an MAE and large errors like an MSE. This balance makes the model more robust, especially in datasets with outliers or noise.
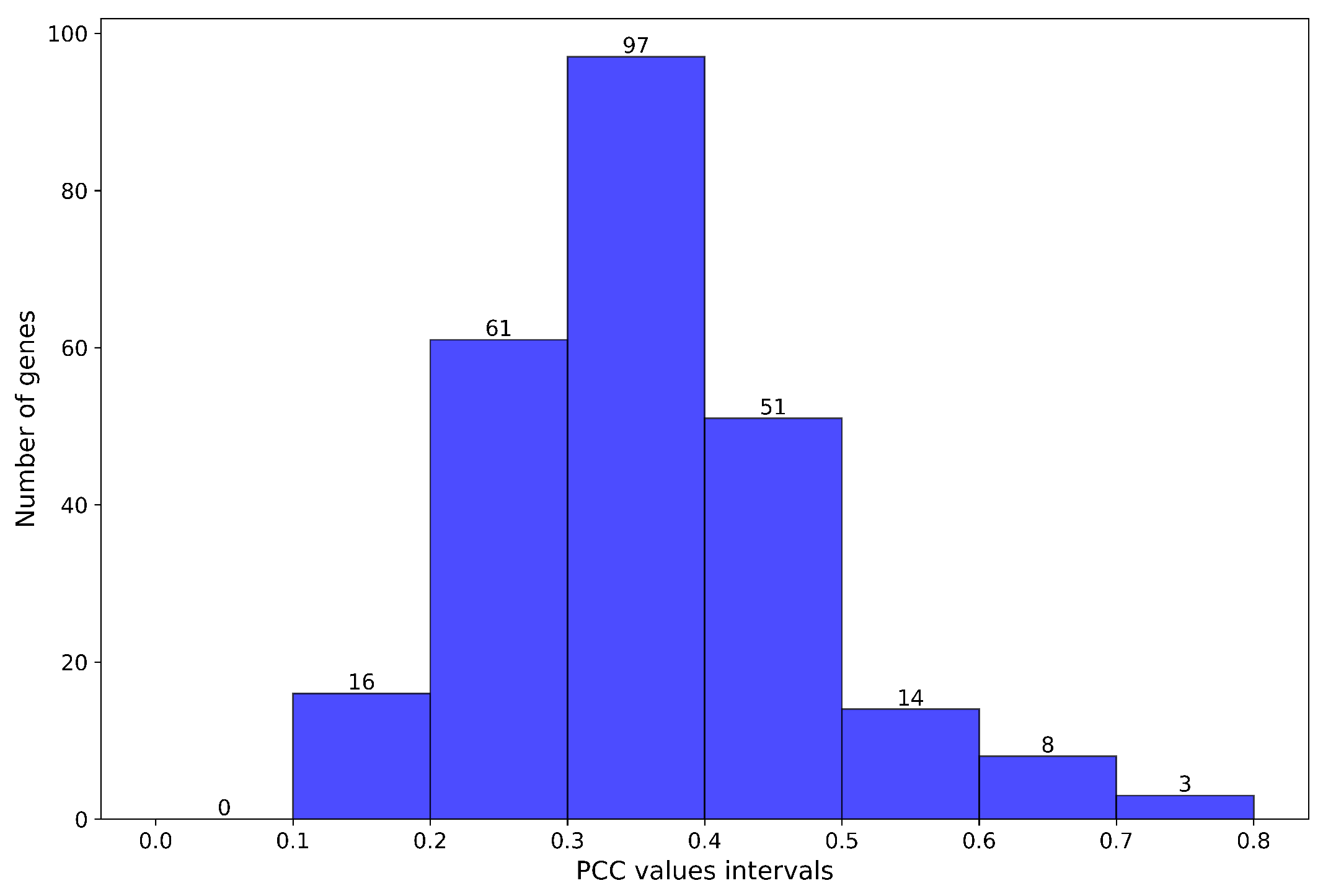
4. Results
4.1. Experiments
4.2. MSELoss Function
4.2.1. Resolution 224 × 224 Pixels
4.2.2. Resolution 256 × 256 Pixels
4.3. Sl1loss Function
4.3.1. Resolution 224 × 224 Pixels
4.3.2. Resolution 256 × 256 Pixels
4.4. Comparison with ST-Net Performance
5. Discussion
Role of the Predicted Biomarkers in BC Progression
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| H&E | Hematoxylin and eosin |
| ST | Spatial transcriptomics |
| WSIs | Whole-slide images |
| CNN | Convolutional neural network |
| GNN | Graph neural network |
| ViT | Vision transformer |
| MAE | Mean Absolute Error |
| RMSE | Root Mean Squared Error |
| PCC | Pearson Correlation Coefficient |
| NLP | Natural language processing |
References
- Obeagu, E.I.; Obeagu, G.U. Breast cancer: A review of risk factors and diagnosis. Medicine 2024, 103, e36905. [Google Scholar] [CrossRef] [PubMed]
- Jiang, B.; Bao, L.; He, S.; Chen, X.; Jin, Z.; Ye, Y. Deep learning applications in breast cancer histopathological imaging: Diagnosis, treatment, and prognosis. Breast Cancer Res. 2024, 26, 137. [Google Scholar] [CrossRef] [PubMed]
- Thaalbi, O.; Akhloufi, M.A. Deep learning for breast cancer diagnosis from histopathological images: Classification and gene expression: Review. Netw. Model. Anal. Health Inform. Bioinform. 2024, 13, 52. [Google Scholar] [CrossRef]
- He, B.; Bergenstråhle, L.; Stenbeck, L.; Abid, A.; Andersson, A.; Borg, Å.; Maaskola, J.; Lundeberg, J.; Zou, J. Integrating spatial gene expression and breast tumour morphology via deep learning. Nat. Biomed. Eng. 2020, 4, 827–834. [Google Scholar] [CrossRef]
- Rahaman, M.M.; Millar, E.K.; Meijering, E. Breast cancer histopathology image-based gene expression prediction using spatial transcriptomics data and deep learning. Sci. Rep. 2023, 13, 13604. [Google Scholar] [CrossRef]
- Mejia, G.; Cárdenas, P.; Ruiz, D.; Castillo, A.; Arbeláez, P. SEPAL: Spatial Gene Expression Prediction from Local Graphs. In Proceedings of the IEEE/CVF International Conference on Computer Vision, Paris, France, 1–6 October 2023; pp. 2294–2303. [Google Scholar] [CrossRef]
- Liu, Z.; Qian, S.; Xia, C.; Wang, C. Are transformer-based models more robust than CNN-based models? Neural Netw. 2024, 172, 106091. [Google Scholar] [CrossRef]
- Yang, Y.; Hossain, M.Z.; Stone, E.A.; Rahman, S. Exemplar guided deep neural network for spatial transcriptomics analysis of gene expression prediction. In Proceedings of the IEEE/CVF Winter Conference on Applications of Computer Vision, Waikoloa, HI, USA, 2–7 January 2023; pp. 5039–5048. [Google Scholar] [CrossRef]
- Andersson, A.; Larsson, L.; Stenbeck, L.; Salmén, F.; Ehinger, A.; Wu, S.Z.; Al-Eryani, G.; Roden, D.; Swarbrick, A.; Borg, Å.; et al. Spatial deconvolution of HER2-positive breast cancer delineates tumor-associated cell type interactions. Nat. Commun. 2021, 12, 6012. [Google Scholar] [CrossRef]
- Pang, M.; Su, K.; Li, M. Leveraging information in spatial transcriptomics to predict super-resolution gene expression from histology images in tumors. bioRxiv 2021. [Google Scholar] [CrossRef]
- Gao, R.; Yuan, X.; Ma, Y.; Wei, T.; Johnston, L.; Shao, Y.; Lv, W.; Zhu, T.; Zhang, Y.; Zheng, J.; et al. Predicting Gene Spatial Expression and Cancer Prognosis: An Integrated Graph and Image Deep Learning Approach Based on HE Slides. bioRxiv 2023. [Google Scholar] [CrossRef]
- Zeng, Y.; Wei, Z.; Yu, W.; Yin, R.; Yuan, Y.; Li, B.; Tang, Z.; Lu, Y.; Yang, Y. Spatial transcriptomics prediction from histology jointly through transformer and graph neural networks. Briefings Bioinform. 2022, 23, bbac297. [Google Scholar] [CrossRef]
- Mondol, R.K.; Millar, E.K.; Graham, P.H.; Browne, L.; Sowmya, A.; Meijering, E. hist2RNA: An efficient deep learning architecture to predict gene expression from breast cancer histopathology images. Cancers 2023, 15, 2569. [Google Scholar] [CrossRef] [PubMed]
- Qu, H.; Zhou, M.; Yan, Z.; Wang, H.; Rustgi, V.K.; Zhang, S.; Gevaert, O.; Metaxas, D.N. Genetic mutation and biological pathway prediction based on whole slide images in breast carcinoma using deep learning. NPJ Precis. Oncol. 2021, 5, 87. [Google Scholar] [CrossRef] [PubMed]
- Vahadane, A.; Peng, T.; Sethi, A.; Albarqouni, S.; Wang, L.; Baust, M.; Steiger, K.; Schlitter, A.M.; Esposito, I.; Navab, N. Structure-preserving color normalization and sparse stain separation for histological images. IEEE Trans. Med. Imaging 2016, 35, 1962–1971. [Google Scholar] [CrossRef] [PubMed]
- Rehman, Z.; Wan Ahmad, W.; Ahmad Fauzi, M.; Abas, F.S.; Cheah, P.L.; Looi, L.; Toh, Y. Comprehensive analysis of color normalization methods for HER2-SISH histopathology images. J. Eng. Sci. Technol 2024, 19, 146–159. [Google Scholar]
- Vaswani, A. Attention is all you need. Adv. Neural Inf. Process. Syst. 2017. [Google Scholar] [CrossRef]
- Dosovitskiy, A. An image is worth 16x16 words: Transformers for image recognition at scale. arXiv 2020, arXiv:2010.11929. [Google Scholar] [CrossRef]
- Deng, J.; Dong, W.; Socher, R.; Li, L.J.; Li, K.; Fei-Fei, L. ImageNet: A large-scale hierarchical image database. In Proceedings of the 2009 IEEE Conference on Computer Vision and Pattern Recognition, Miami, FL, USA, 20–25 June 2009; pp. 248–255. [Google Scholar] [CrossRef]
- Li, Y.; Yuan, G.; Wen, Y.; Hu, J.; Evangelidis, G.; Tulyakov, S.; Wang, Y.; Ren, J. Efficientformer: Vision transformers at mobilenet speed. Adv. Neural Inf. Process. Syst. 2022, 35, 12934–12949. [Google Scholar] [CrossRef]
- Hatamizadeh, A.; Heinrich, G.; Yin, H.; Tao, A.; Alvarez, J.M.; Kautz, J.; Molchanov, P. Fastervit: Fast vision transformers with hierarchical attention. arXiv 2023, arXiv:2306.06189. [Google Scholar] [CrossRef]
- Peng, Z.; Dong, L.; Bao, H.; Ye, Q.; Wei, F. Beit v2: Masked image modeling with vector-quantized visual tokenizers. arXiv 2022, arXiv:2208.06366. [Google Scholar] [CrossRef]
- Liu, Z.; Hu, H.; Lin, Y.; Yao, Z.; Xie, Z.; Wei, Y.; Ning, J.; Cao, Y.; Zhang, Z.; Dong, L.; et al. Swin transformer v2: Scaling up capacity and resolution. In Proceedings of the IEEE/CVF Conference on Computer Vision and Pattern Recognition, New Orleans, LA, USA, 18–24 June 2022; pp. 12009–12019. [Google Scholar] [CrossRef]
- Wang, W.; Xie, E.; Li, X.; Fan, D.P.; Song, K.; Liang, D.; Lu, T.; Luo, P.; Shao, L. Pvt v2: Improved baselines with pyramid vision transformer. Comput. Vis. Media 2022, 8, 415–424. [Google Scholar] [CrossRef]
- Mehta, S.; Rastegari, M. Mobilevit: Light-weight, general-purpose, and mobile-friendly vision transformer. arXiv 2021, arXiv:2110.02178. [Google Scholar] [CrossRef]
- Mehta, S.; Rastegari, M. Separable self-attention for mobile vision transformers. arXiv 2022, arXiv:2206.02680. [Google Scholar] [CrossRef]
- Liu, X.; Peng, H.; Zheng, N.; Yang, Y.; Hu, H.; Yuan, Y. Efficientvit: Memory efficient vision transformer with cascaded group attention. In Proceedings of the IEEE/CVF Conference on Computer Vision and Pattern Recognition, Vancouver, BC, Canada, 17–24 June 2023; pp. 14420–14430. [Google Scholar] [CrossRef]
- Mazurek, A.; Luo, W.; Krasnitz, A.; Hicks, J.; Powers, R.S.; Stillman, B. DDX5 regulates DNA replication and is required for cell proliferation in a subset of breast cancer cells. Cancer Discov. 2012, 2, 812–825. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Iliopoulos, D.; Zhang, Q.; Tang, Q.; Greenblatt, M.B.; Hatziapostolou, M.; Lim, E.; Tam, W.L.; Ni, M.; Chen, Y.; et al. XBP1 promotes triple-negative breast cancer by controlling the HIF1α pathway. Nature 2014, 508, 103–107. [Google Scholar] [CrossRef]
- Chaturvedi, S.; Biswas, M.; Sadhukhan, S.; Sonawane, A. Role of EGFR and FASN in breast cancer progression. J. Cell Commun. Signal. 2023, 17, 1249–1282. [Google Scholar] [CrossRef]
- Suresh, R.; Diaz, R.J. The remodelling of actin composition as a hallmark of cancer. Transl. Oncol. 2021, 14, 101051. [Google Scholar] [CrossRef]
- Liu, H.; Zhang, Z.; Huang, Y.; Wei, W.; Ning, S.; Li, J.; Liang, X.; Liu, K.; Zhang, L. Plasma HSP90AA1 predicts the risk of breast cancer onset and distant metastasis. Front. Cell Dev. Biol. 2021, 9, 639596. [Google Scholar] [CrossRef]
- de Oliveira, R.C.; Dos Reis, S.P.; Cavalcante, G.C. Mutations in Structural Genes of the Mitochondrial Complex IV May Influence Breast Cancer. Genes 2023, 14, 1465. [Google Scholar] [CrossRef]
- Kumar, A.; Kumar, V.; Arora, M.; Kumar, M.; Ammalli, P.; Thakur, B.; Prasad, J.; Kumari, S.; Sharma, M.C.; Kale, S.S.; et al. Overexpression of prothymosin-α in glioma is associated with tumor aggressiveness and poor prognosis. Biosci. Rep. 2022, 42, BSR20212685. [Google Scholar] [CrossRef]
- Huo, Q.; Wang, J.; Xie, N. High HSPB1 expression predicts poor clinical outcomes and correlates with breast cancer metastasis. BMC Cancer 2023, 23, 501. [Google Scholar] [CrossRef]
- Moutafi, M.; Robbins, C.J.; Yaghoobi, V.; Fernandez, A.I.; Martinez-Morilla, S.; Xirou, V.; Bai, Y.; Song, Y.; Gaule, P.; Krueger, J.; et al. Quantitative measurement of HER2 expression to subclassify ERBB2 unamplified breast cancer. Lab. Investig. 2022, 102, 1101–1108. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Model | Innovations | Processing Time | Advantages in State of the Art |
|---|---|---|---|
| EfficientFormer | MetaBlock + Latency slimming | Fast | High accuracy with efficiency, real-time tasks. |
| FasterViT | Hierarchical attention mechanisms | Moderate | High-resolution data processing efficiency. |
| BEiT v2 | Semantic visual tokenizer | Slow | High global feature comprehension. |
| Swin Transformer v2 | Scaled cosine attention + Log-spaced continuous position bias | Moderate | High resolution and large data processing efficiency. |
| PyramidViT v2 | Linear spatial reduction attention + Overlapping patch embedding + Convolutional feed-forward networks | Moderate | High efficiency of large image processing. |
| MobileViT v2 | Separable self-attention | Very Fast | Lightweight, ideal for mobile devices. |
| MobileViT | MobileViT blocks | Very Fast | Lightweight, ideal for mobile devices. |
| EfficientViT | Sandwich layout + Cascaded group attention + Parameter reallocation | Fast | Balancing memory efficiency and accuracy. |
| Gene | GeNetFormer-Eff | GeNetFormer-FVT | GeNetFormer-BE2 | GeNetFormer-MVT2 | GeNetFormer-ST2 | GeNetFormer-PVT2 | GeNetFormer-MVTS | GeNetFormer-EffV | ST-Net |
|---|---|---|---|---|---|---|---|---|---|
| DDX5 | 0.7411 | 0.7155 | 0.7091 | 0.7088 | 0.6940 | 0.6888 | 0.6511 | 0.6186 | 0.6738 |
| XBP1 | 0.7101 | 0.7040 | 0.7124 | 0.6953 | 0.6947 | 0.6417 | 0.6933 | 0.5899 | 0.7273 |
| FASN | 0.6946 | 0.6589 | 0.6774 | 0.6771 | 0.6134 | 0.6035 | 0.5902 | 0.5983 | 0.6996 |
| ACTG1 | 0.6737 | 0.6827 | 0.6885 | 0.6375 | 0.6355 | 0.6620 | 0.6545 | 0.5637 | 0.5921 |
| HSP90AB1 | 0.6645 | 0.6327 | 0.6714 | 0.6408 | 0.6275 | 0.6227 | 0.5839 | 0.5804 | 0.6294 |
| COX6C | 0.6527 | 0.4918 | 0.6331 | 0.5159 | 0.6390 | 0.5564 | 0.6172 | 0.5210 | 0.6390 |
| PTMA | 0.6412 | 0.6265 | 0.4942 | 0.6306 | 0.5283 | 0.5341 | 0.5810 | 0.5671 | 0.6356 |
| HSPB1 | 0.6332 | 0.4969 | 0.5976 | 0.5058 | 0.4641 | 0.4516 | 0.4955 | 0.4281 | 0.5630 |
| ERBB2 | 0.6323 | 0.5694 | 0.5071 | 0.5771 | 0.5209 | 0.5110 | 0.4346 | 0.4993 | 0.6072 |
| ENSG00000269028 | 0.6258 | 0.6078 | 0.6331 | 0.6441 | 0.6158 | 0.5492 | 0.4174 | 0.5574 | 0.5847 |
| Gene | GeNetFormer-Eff | GeNetFormer-FVT | GeNetFormer-BE2 | GeNetFormer-MVT2 | GeNetFormer-ST2 | GeNetFormer-PVT2 | GeNetFormer-MVTS | GeNetFormer-EffV | ST-Net |
|---|---|---|---|---|---|---|---|---|---|
| DDX5 | 0.7450 | 0.7068 | 0.6556 | 0.7222 | 0.7101 | 0.7064 | 0.6666 | 0.5210 | 0.6713 |
| XBP1 | 0.7203 | 0.7270 | 0.6862 | 0.7128 | 0.7160 | 0.6601 | 0.7040 | 0.5693 | 0.7320 |
| FASN | 0.7018 | 0.6483 | 0.6535 | 0.6900 | 0.6461 | 0.6197 | 0.6225 | 0.5457 | 0.6968 |
| ACTG1 | 0.6761 | 0.6771 | 0.6362 | 0.6494 | 0.6615 | 0.6693 | 0.6728 | 0.4435 | 0.5886 |
| HSP90AB1 | 0.6726 | 0.6396 | 0.6145 | 0.6462 | 0.6556 | 0.6456 | 0.5964 | 0.5077 | 0.6204 |
| COX6C | 0.6548 | 0.5068 | 0.5784 | 0.5460 | 0.5444 | 0.5721 | 0.6456 | 0.3430 | 0.6306 |
| PTMA | 0.6486 | 0.6072 | 0.5639 | 0.6447 | 0.5154 | 0.5333 | 0.5859 | 0.3475 | 0.6345 |
| HSPB1 | 0.6391 | 0.5379 | 0.5606 | 0.5809 | 0.5330 | 0.4458 | 0.5003 | 0.3496 | 0.5581 |
| ERBB2 | 0.6241 | 0.5894 | 0.6233 | 0.5797 | 0.5073 | 0.5042 | 0.4870 | 0.4838 | 0.6211 |
| ENSG00000269028 | 0.6269 | 0.6392 | 0.5742 | 0.6390 | 0.5872 | 0.5566 | 0.4422 | 0.5175 | 0.5812 |
| Gene | GeNetFormer-Eff | GeNetFormer-FVT | GeNetFormer-BE2 | GeNetFormer-MVT2 | GeNetFormer-ST2 | GeNetFormer-PVT2 | GeNetFormer-MVTS | GeNetFormer-EffV | ST-Net |
|---|---|---|---|---|---|---|---|---|---|
| DDX5 | 0.7437 | 0.7211 | 0.7077 | 0.7152 | 0.7044 | 0.7163 | 0.6773 | 0.6529 | 0.6712 |
| XBP1 | 0.7085 | 0.7122 | 0.6223 | 0.6797 | 0.6951 | 0.6852 | 0.7084 | 0.5523 | 0.7273 |
| FASN | 0.6935 | 0.6663 | 0.5975 | 0.6777 | 0.6129 | 0.6151 | 0.5938 | 0.6100 | 0.6948 |
| ACTG1 | 0.6645 | 0.6859 | 0.6127 | 0.6450 | 0.6501 | 0.6881 | 0.6596 | 0.5475 | 0.5961 |
| HSP90AB1 | 0.6608 | 0.6450 | 0.5830 | 0.6446 | 0.6478 | 0.6357 | 0.6053 | 0.6005 | 0.6304 |
| COX6C | 0.6455 | 0.5324 | 0.5353 | 0.5042 | 0.5185 | 0.5609 | 0.6352 | 0.6192 | 0.6399 |
| PTMA | 0.6418 | 0.6273 | 0.5277 | 0.6296 | 0.5551 | 0.5477 | 0.5789 | 0.5651 | 0.6352 |
| HSPB1 | 0.6318 | 0.5169 | 0.4639 | 0.4812 | 0.4793 | 0.4713 | 0.5033 | 0.4215 | 0.5740 |
| ENSG00000269028 | 0.6314 | 0.6095 | 0.5467 | 0.6436 | 0.5872 | 0.5710 | 0.3759 | 0.5069 | 0.5693 |
| ENSG00000255823 | 0.6260 | 0.6047 | 0.5252 | 0.6252 | 0.5872 | 0.5519 | 0.3458 | 0.5131 | 0.5615 |
| Gene | GeNetFormer-Eff | GeNetFormer-FVT | GeNetFormer-BE2 | GeNetFormer-MVT2 | GeNetFormer-ST2 | GeNetFormer-PVT2 | GeNetFormer-MVTS | GeNetFormer-EffV | ST-Net |
|---|---|---|---|---|---|---|---|---|---|
| DDX5 | 0.7525 | 0.7185 | 0.6707 | 0.7319 | 0.7174 | 0.7272 | 0.6560 | 0.5698 | 0.6907 |
| XBP1 | 0.6930 | 0.7250 | 0.6953 | 0.7023 | 0.7168 | 0.6966 | 0.7138 | 0.6422 | 0.7320 |
| FASN | 0.6954 | 0.6661 | 0.6498 | 0.6839 | 0.6483 | 0.6255 | 0.6006 | 0.6457 | 0.6991 |
| ACTG1 | 0.6903 | 0.6847 | 0.6441 | 0.6556 | 0.6677 | 0.6912 | 0.6504 | 0.5699 | 0.6037 |
| HSP90AB1 | 0.6666 | 0.6544 | 0.6338 | 0.6576 | 0.6718 | 0.6572 | 0.5788 | 0.6105 | 0.6474 |
| COX6C | 0.6282 | 0.5449 | 0.5819 | 0.5222 | 0.5600 | 0.5851 | 0.6330 | 0.3488 | 0.6439 |
| PTMA | 0.6492 | 0.6198 | 0.5441 | 0.6432 | 0.5469 | 0.5427 | 0.5852 | 0.5188 | 0.6379 |
| HSPB1 | 0.6294 | 0.5645 | 0.5872 | 0.5139 | 0.5421 | 0.5083 | 0.4979 | 0.2902 | 0.5969 |
| ERBB2 | 0.6272 | 0.5939 | 0.6183 | 0.5898 | 0.4508 | 0.5288 | 0.5082 | 0.4880 | 0.6224 |
| KRT19 | 0.6173 | 0.5438 | 0.6483 | 0.4220 | 0.5801 | 0.3554 | 0.2608 | 0.2339 | 0.5980 |
| Gene | GeNetFormer-Eff | GeNetFormer-FVT | GeNetFormer-BE2 | GeNetFormer-MVT2 | GeNetFormer-ST2 | GeNetFormer-PVT2 | GeNetFormer-MVTS | GeNetFormer-EffV | ST-Net |
|---|---|---|---|---|---|---|---|---|---|
| MSELoss—224 × 224 resolution | |||||||||
| DDX5 | 0.7411 | 0.7155 | 0.7091 | 0.7088 | 0.6940 | 0.6888 | 0.6511 | 0.6186 | 0.6738 |
| XBP1 | 0.7101 | 0.7040 | 0.7124 | 0.6953 | 0.6947 | 0.6417 | 0.6933 | 0.5899 | 0.7273 |
| FASN | 0.6946 | 0.6589 | 0.6774 | 0.6771 | 0.6134 | 0.6035 | 0.5902 | 0.5983 | 0.6996 |
| ACTG1 | 0.6737 | 0.6827 | 0.6885 | 0.6375 | 0.6355 | 0.6620 | 0.6545 | 0.5637 | 0.5921 |
| HSP90AB1 | 0.6645 | 0.6327 | 0.6714 | 0.6408 | 0.6275 | 0.6227 | 0.5839 | 0.5804 | 0.6294 |
| COX6C | 0.6527 | 0.4918 | 0.6331 | 0.5159 | 0.6390 | 0.5564 | 0.6172 | 0.5210 | 0.6390 |
| PTMA | 0.6412 | 0.6265 | 0.4942 | 0.6306 | 0.5283 | 0.5341 | 0.5810 | 0.5671 | 0.6356 |
| HSPB1 | 0.6332 | 0.4969 | 0.5976 | 0.5058 | 0.4641 | 0.4516 | 0.4955 | 0.4281 | 0.5630 |
| ERBB2 | 0.6323 | 0.5694 | 0.5071 | 0.5771 | 0.5209 | 0.5110 | 0.4346 | 0.4993 | 0.6072 |
| ENSG00000269028 | 0.6258 | 0.6078 | 0.6331 | 0.6441 | 0.6158 | 0.5492 | 0.4174 | 0.5574 | 0.5847 |
| MSELoss—256 × 256 resolution | |||||||||
| DDX5 | 0.7450 | 0.7068 | 0.6556 | 0.7222 | 0.7101 | 0.7064 | 0.6666 | 0.5210 | 0.6713 |
| XBP1 | 0.7203 | 0.7270 | 0.6862 | 0.7128 | 0.7160 | 0.6601 | 0.7040 | 0.5693 | 0.7320 |
| FASN | 0.7018 | 0.6483 | 0.6535 | 0.6900 | 0.6461 | 0.6197 | 0.6225 | 0.5457 | 0.6968 |
| ACTG1 | 0.6761 | 0.6771 | 0.6362 | 0.6494 | 0.6615 | 0.6693 | 0.6728 | 0.4435 | 0.5886 |
| HSP90AB1 | 0.6726 | 0.6396 | 0.6145 | 0.6462 | 0.6556 | 0.6456 | 0.5964 | 0.5077 | 0.6204 |
| COX6C | 0.6548 | 0.5068 | 0.5784 | 0.5460 | 0.5444 | 0.5721 | 0.6456 | 0.3430 | 0.6306 |
| PTMA | 0.6486 | 0.6072 | 0.5639 | 0.6447 | 0.5154 | 0.5333 | 0.5859 | 0.3475 | 0.6345 |
| HSPB1 | 0.6391 | 0.5379 | 0.5606 | 0.5809 | 0.5330 | 0.4458 | 0.5003 | 0.3496 | 0.5581 |
| ERBB2 | 0.6241 | 0.5894 | 0.6233 | 0.5797 | 0.5073 | 0.5042 | 0.4870 | 0.4838 | 0.6211 |
| ENSG00000269028 | 0.6269 | 0.6392 | 0.5742 | 0.6390 | 0.5872 | 0.5566 | 0.4422 | 0.5175 | 0.5812 |
| SL1Loss—224 × 224 resolution | |||||||||
| DDX5 | 0.7437 | 0.7211 | 0.7077 | 0.7152 | 0.7044 | 0.7163 | 0.6773 | 0.6529 | 0.6712 |
| XBP1 | 0.7085 | 0.7122 | 0.6223 | 0.6797 | 0.6951 | 0.6852 | 0.7084 | 0.5523 | 0.7273 |
| FASN | 0.6935 | 0.6663 | 0.5975 | 0.6777 | 0.6129 | 0.6151 | 0.5938 | 0.6100 | 0.6948 |
| ACTG1 | 0.6645 | 0.6859 | 0.6127 | 0.6450 | 0.6501 | 0.6881 | 0.6596 | 0.5475 | 0.5961 |
| HSP90AB1 | 0.6608 | 0.6450 | 0.5830 | 0.6446 | 0.6478 | 0.6357 | 0.6053 | 0.6005 | 0.6304 |
| COX6C | 0.6455 | 0.5324 | 0.5353 | 0.5042 | 0.5185 | 0.5609 | 0.6352 | 0.6192 | 0.6399 |
| PTMA | 0.6418 | 0.6273 | 0.5277 | 0.6296 | 0.5551 | 0.5477 | 0.5789 | 0.5651 | 0.6352 |
| HSPB1 | 0.6318 | 0.5169 | 0.4639 | 0.4812 | 0.4793 | 0.4713 | 0.5033 | 0.4215 | 0.5740 |
| ENSG00000269028 | 0.6314 | 0.6095 | 0.5467 | 0.6436 | 0.5872 | 0.5710 | 0.3759 | 0.5069 | 0.5693 |
| ENSG00000255823 | 0.6260 | 0.6047 | 0.5252 | 0.6252 | 0.5872 | 0.5519 | 0.3458 | 0.5131 | 0.5615 |
| SL1Loss—256 × 256 resolution | |||||||||
| DDX5 | 0.7525 | 0.7185 | 0.6707 | 0.7319 | 0.7174 | 0.7272 | 0.6560 | 0.5698 | 0.6907 |
| XBP1 | 0.6930 | 0.7250 | 0.6953 | 0.7023 | 0.7168 | 0.6966 | 0.7138 | 0.6422 | 0.7320 |
| FASN | 0.6954 | 0.6661 | 0.6498 | 0.6839 | 0.6483 | 0.6255 | 0.6006 | 0.6457 | 0.6991 |
| ACTG1 | 0.6903 | 0.6847 | 0.6441 | 0.6556 | 0.6677 | 0.6912 | 0.6504 | 0.5699 | 0.6037 |
| HSP90AB1 | 0.6666 | 0.6544 | 0.6338 | 0.6576 | 0.6718 | 0.6572 | 0.5788 | 0.6105 | 0.6474 |
| COX6C | 0.6282 | 0.5449 | 0.5819 | 0.5222 | 0.5600 | 0.5851 | 0.6330 | 0.3488 | 0.6439 |
| PTMA | 0.6492 | 0.6198 | 0.5441 | 0.6432 | 0.5469 | 0.5427 | 0.5852 | 0.5188 | 0.6379 |
| HSPB1 | 0.6294 | 0.5645 | 0.5872 | 0.5139 | 0.5421 | 0.5083 | 0.4979 | 0.2902 | 0.5969 |
| ERBB2 | 0.6272 | 0.5939 | 0.6183 | 0.5898 | 0.4508 | 0.5288 | 0.5082 | 0.4880 | 0.6224 |
| KRT19 | 0.6173 | 0.5438 | 0.6483 | 0.4220 | 0.5801 | 0.3554 | 0.2608 | 0.2339 | 0.5980 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Thaalbi, O.; Akhloufi, M.A. GeNetFormer: Transformer-Based Framework for Gene Expression Prediction in Breast Cancer. AI 2025, 6, 43. https://doi.org/10.3390/ai6030043
Thaalbi O, Akhloufi MA. GeNetFormer: Transformer-Based Framework for Gene Expression Prediction in Breast Cancer. AI. 2025; 6(3):43. https://doi.org/10.3390/ai6030043
Chicago/Turabian StyleThaalbi, Oumeima, and Moulay A. Akhloufi. 2025. "GeNetFormer: Transformer-Based Framework for Gene Expression Prediction in Breast Cancer" AI 6, no. 3: 43. https://doi.org/10.3390/ai6030043
APA StyleThaalbi, O., & Akhloufi, M. A. (2025). GeNetFormer: Transformer-Based Framework for Gene Expression Prediction in Breast Cancer. AI, 6(3), 43. https://doi.org/10.3390/ai6030043