
Anticholinesterase Inhibition, Drug-Likeness Assessment, and Molecular Docking Evaluation of Milk Protein-Derived Opioid Peptides for the Control of Alzheimer’s Disease

 , , ,
, , ,  and
and
Abstract
:
1. Introduction
2. Materials and Methods
2.1. Preparation and Characterization of In Silico Opioid Peptides Used in This Study
2.2. In Silico Drug-Likeness Assessment of Opioid Peptides
2.3. Molecular Docking of Peptides
2.4. Peptides Synthesis, Purification, and Characterization
2.4.1. Synthesis of β-casomorphin 5
2.4.2. Synthesis of β-casomorphin 7
2.4.3. Peptide Characterization
2.5. In Vitro Anticholinesterase Inhibitory Enzyme Activity of Casomorphins
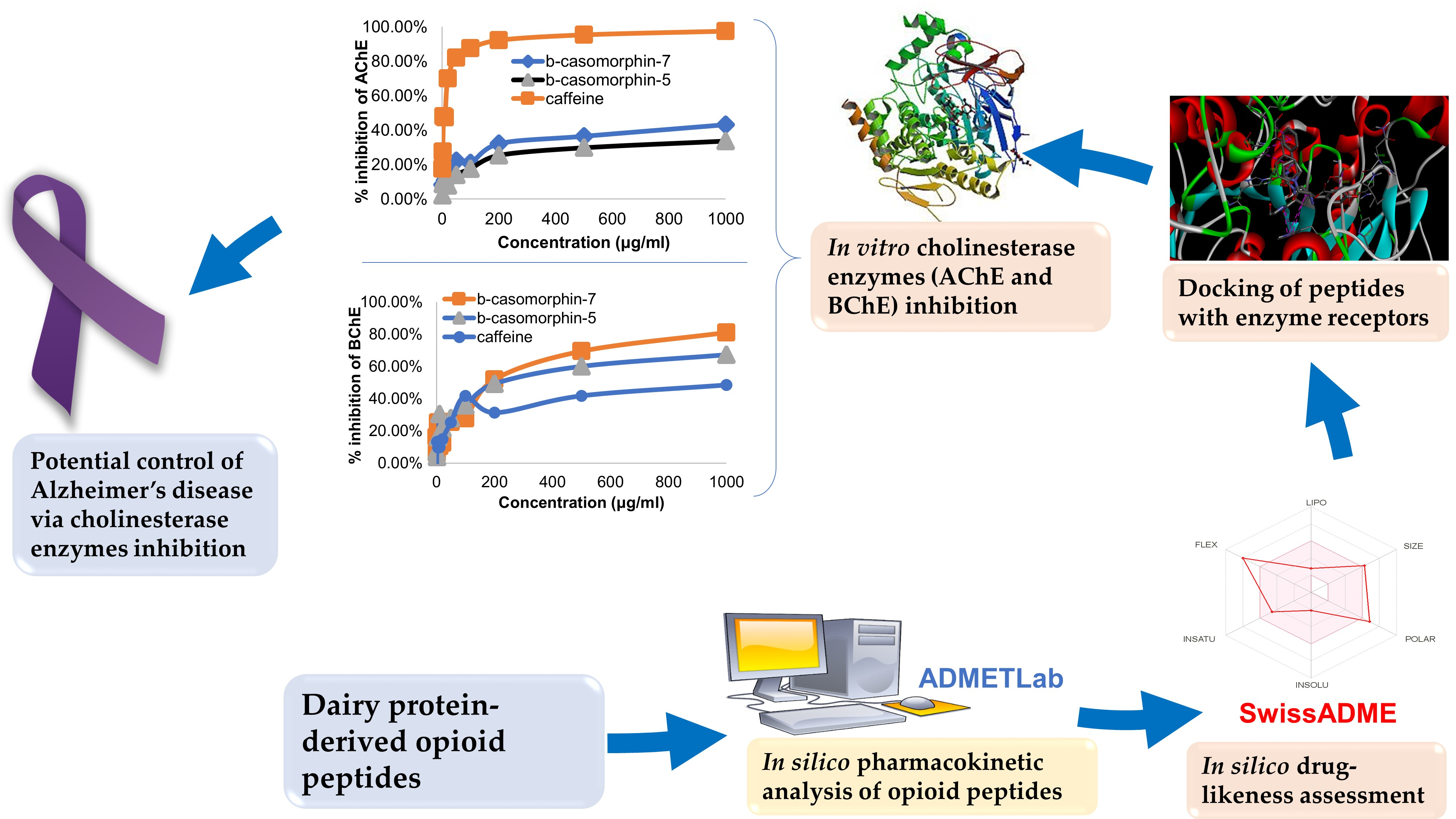
3. Results and Discussion
3.1. Hypothesis and Justification
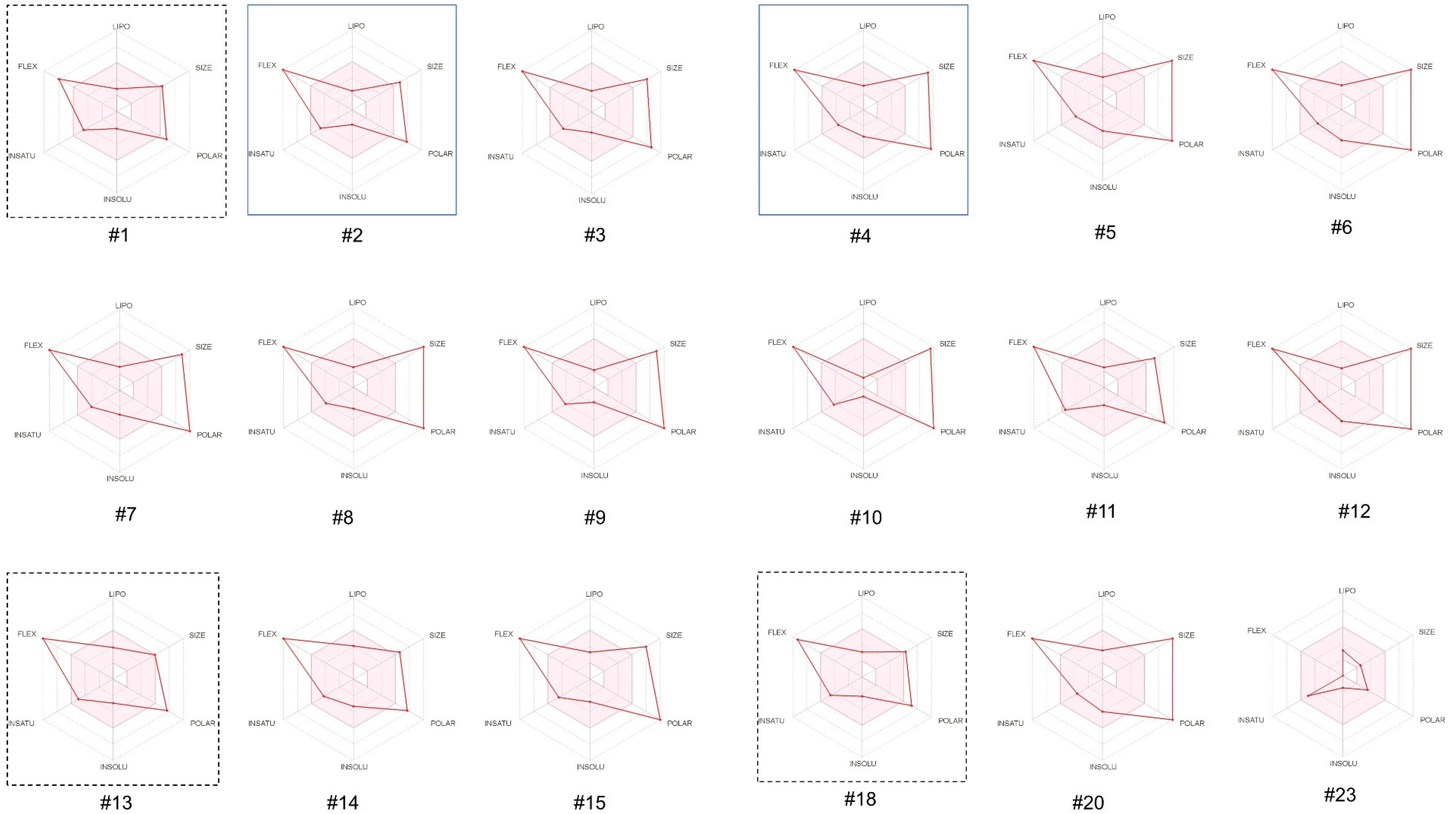
3.2. Drug-Likeness and Related Properties of Opioid Peptides
3.2.1. In Silico Evaluation of Physicochemical Properties of the Opioid Peptides
3.2.2. In Silico Evaluation of the Pharmacokinetic Properties of the Opioid Peptides
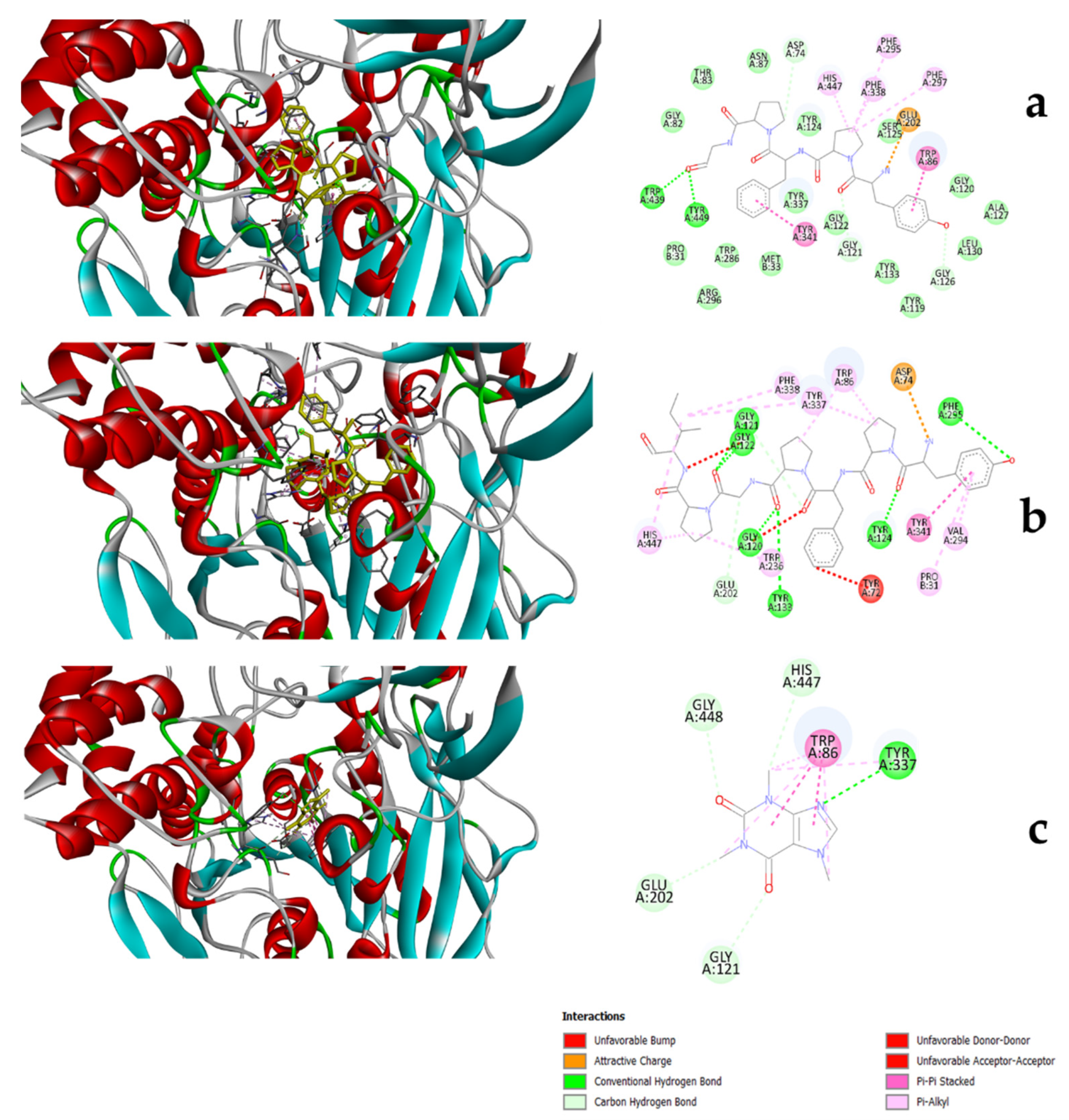
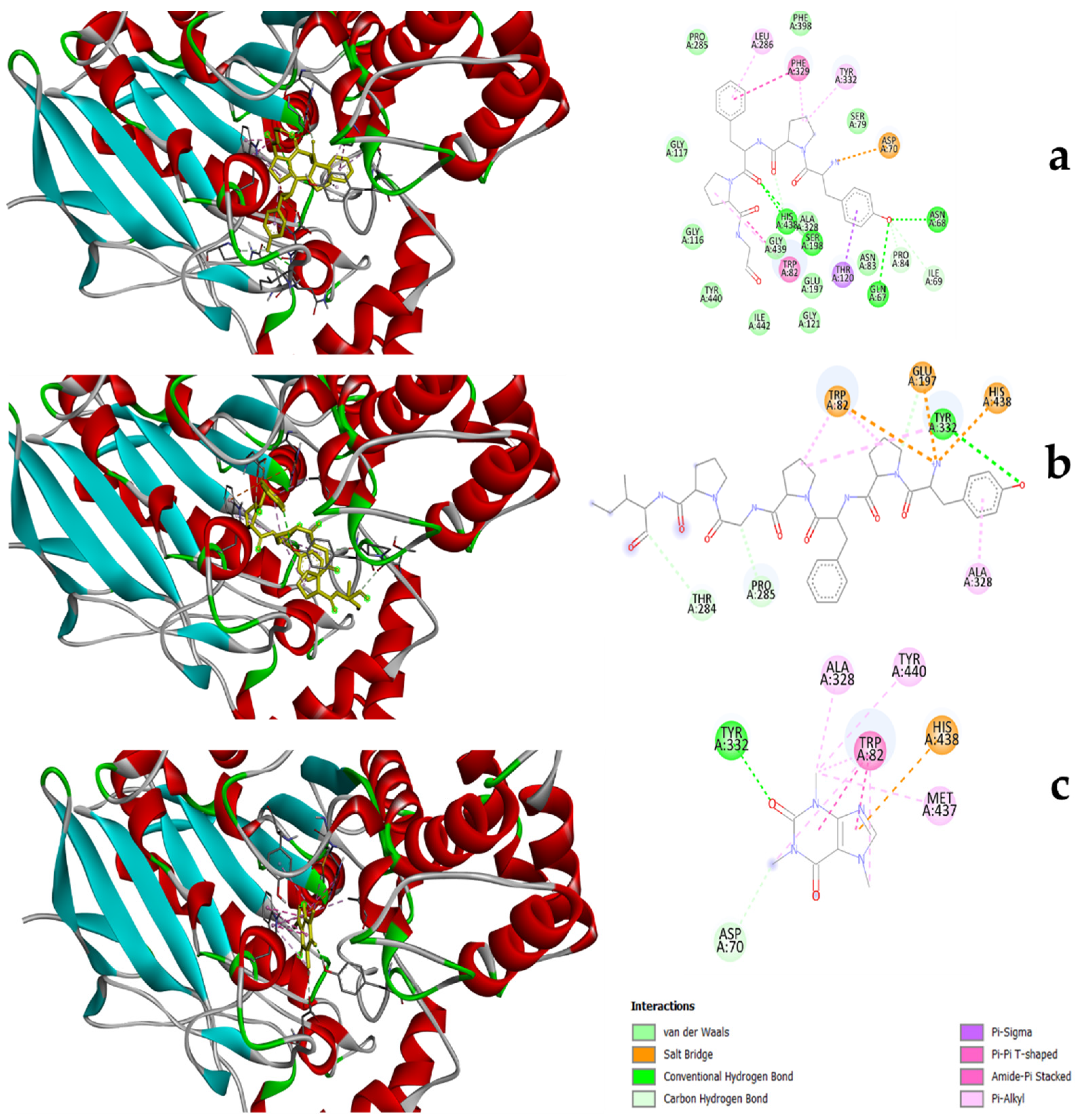
3.3. Molecular Docking of Peptides to Substrates
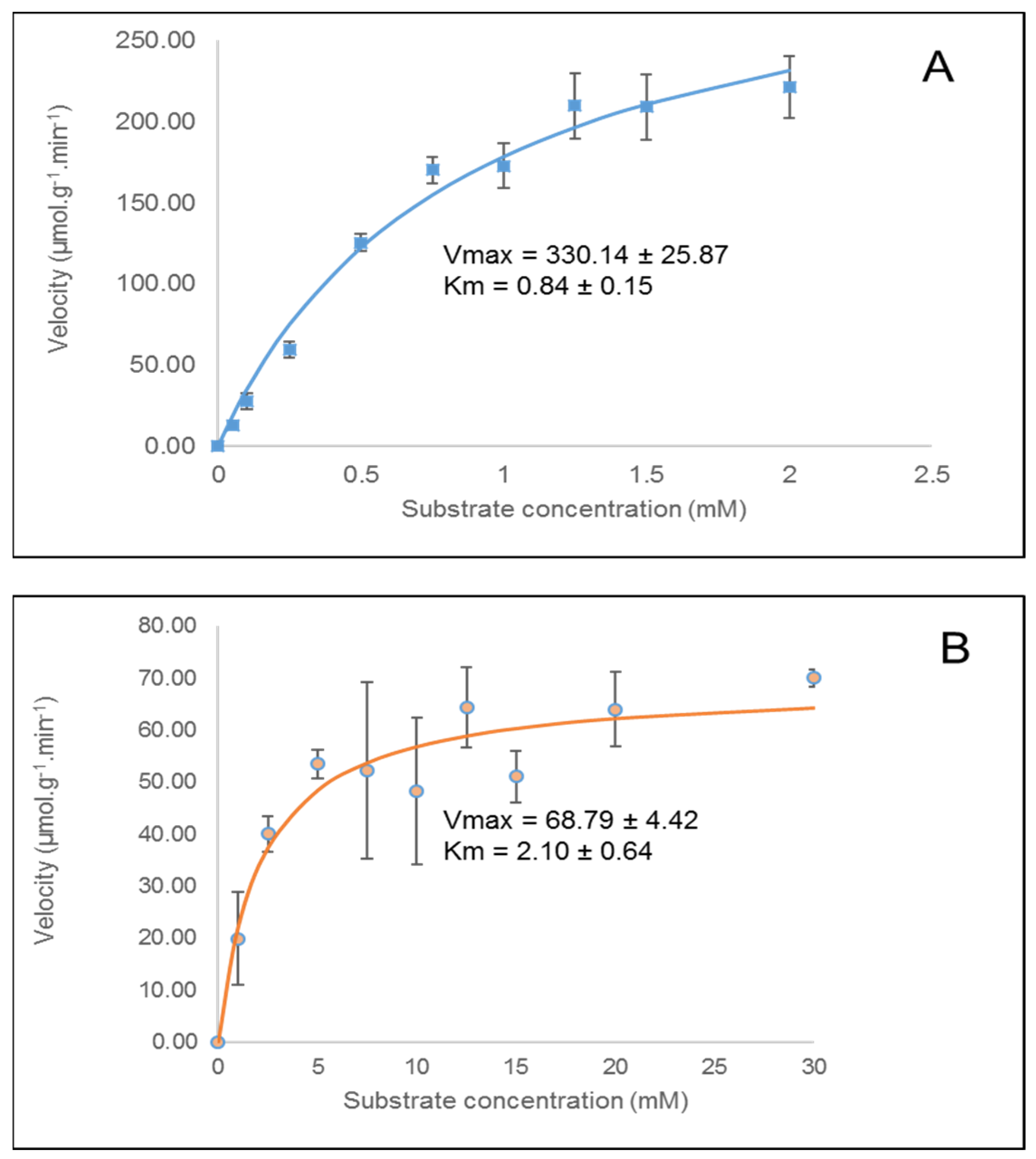
3.4. In Vitro Cholinesterase Inhibition by β-casomorphin-5 and β-casomorphin-7
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Mathys, H.; Davila-Velderrain, J.; Peng, Z.; Gao, F.; Mohammadi, S.; Young, J.Z.; Menon, M.; He, L.; Abdurrob, F.; Jiang, X. Single-cell transcriptomic analysis of Alzheimer’s disease. Nature 2019, 570, 332–337. [Google Scholar] [CrossRef] [PubMed]
- Jung, M.; Park, M. Acetylcholinesterase inhibition by flavonoids from Agrimonia pilosa. Molecules 2007, 12, 2130–2139. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grossberg, G.T. Cholinesterase Inhibitors for the Treatment of Alzheimer’s Disease: Getting on and Staying on. Curr. Ther. Res. 2003, 64, 216–235. [Google Scholar] [CrossRef] [Green Version]
- Yener, I.; Kocakaya, S.O.; Ertas, A.; Erhan, B.; Kaplaner, E.; Oral, E.V.; Yilmaz-Ozden, T.; Yilmaz, M.A.; Ozturk, M.; Kolak, U. Selective in vitro and in silico Enzymes Inhibitory Activities of Phenolic Acids and Flavonoids of Food Plants: Relations with Oxidative Stress. Food Chem. 2020, 327, 127045. [Google Scholar] [CrossRef]
- Aldrich, J.V.; McLaughlin, J.P. Opioid Peptides: Potential for Drug Development. Drug Discov. Today Technol. 2012, 9, e23–e31. [Google Scholar] [CrossRef] [Green Version]
- Witkin, J.M.; Statnick, M.A.; Rorick-Kehn, L.M.; Pintar, J.E.; Ansonoff, M.; Chen, Y.; Tucker, R.C.; Ciccocioppo, R. The biology of Nociceptin/Orphanin FQ (N/OFQ) related to obesity, stress, anxiety, mood, and drug dependence. Pharmacol. Ther. 2014, 141, 283–299. [Google Scholar] [CrossRef] [Green Version]
- Cai, Z.; Ratka, A. Opioid system and Alzheimer’s disease. Neuromol. Med. 2012, 14, 91–111. [Google Scholar] [CrossRef]
- Crichton, G.E.; Murphy, K.J.; Bryan, J. Dairy intake and cognitive health in middle-aged South Australians. Asia Pac. J. Clin. Nutr. 2010, 19, 161–171. [Google Scholar]
- Camfield, D.A.; Owen, L.; Scholey, A.B.; Pipingas, A.; Stough, C. Dairy constituents and neurocognitive health in ageing. Br. J. Nutr. 2011, 106, 159–174. [Google Scholar] [CrossRef] [Green Version]
- Ano, Y.; Ozawa, M.; Kutsukake, T.; Sugiyama, S.; Uchida, K.; Yoshida, A.; Nakayama, H. Preventive Effects of a Fermented Dairy Product against Alzheimer’s Disease and Identification of a Novel Oleamide with Enhanced Microglial Phagocytosis and Anti-Inflammatory Activity. PLoS ONE 2015, 10, e0118512. [Google Scholar] [CrossRef] [Green Version]
- Garg, S.; Nurgali, K.; Kumar Mishra, V. Food proteins as source of opioid peptides—A review. Curr. Med. Chem. 2016, 23, 893–910. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Udenigwe, C.C. Role of food-derived opioid peptides in the central nervous and gastrointestinal systems. J. Food Biochem. 2019, 43, e12629. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Trivedi, M.; Zhang, Y.; Lopez-Toledano, M.; Clarke, A.; Deth, R. Differential neurogenic effects of casein-derived opioid peptides on neuronal stem cells: Implications for redox-based epigenetic changes. J. Nutr. Biochem. 2016, 37, 39–46. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brantl, V.; Teschemacher, H.; Bläsig, J.; Henschen, A.; Lottspeich, F. Opioid activities of β-casomorphins. Life Sci. 1981, 28, 1903–1909. [Google Scholar] [CrossRef]
- Han, D.-N.; Zhang, D.-H.; Wang, L.-P.; Zhang, Y.-S. Protective effect of β-casomorphin-7 on cardiomyopathy of streptozotocin-induced diabetic rats via inhibition of hyperglycemia and oxidative stress. Peptides 2013, 44, 120–126. [Google Scholar] [CrossRef]
- Lister, J.; Fletcher, P.J.; Nobrega, J.N.; Remington, G. Behavioral effects of food-derived opioid-like peptides in rodents: Implications for schizophrenia? Pharmacol. Biochem. Behav. 2015, 134, 70–78. [Google Scholar] [CrossRef]
- Froehlich, J.C. Opioid peptides. Alcohol Health Res. World 1997, 21, 132–136. [Google Scholar]
- Minkiewicz, P.; Iwaniak, A.; Darewicz, M. BIOPEP-UWM database of bioactive peptides: Current opportunities. Int. J. Mol. Sci. 2019, 20, 5978. [Google Scholar] [CrossRef] [Green Version]
- Daina, A.; Michielin, O.; Zoete, V. SwissADME: A free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Sci. Rep. 2017, 7, 42717. [Google Scholar] [CrossRef] [Green Version]
- Dong, J.; Wang, N.-N.; Yao, Z.-J.; Zhang, L.; Cheng, Y.; Ouyang, D.; Lu, A.-P.; Cao, D.-S. ADMETlab: A platform for systematic ADMET evaluation based on a comprehensively collected ADMET database. J. Cheminform. 2018, 10, 29. [Google Scholar] [CrossRef]
- Lipinski, C.A.; Lombardo, F.; Dominy, B.W.; Feeney, P.J. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv. Drug Deliv. Rev. 1997, 23, 3–25. [Google Scholar] [CrossRef]
- Wang, N.-N.; Dong, J.; Deng, Y.-H.; Zhu, M.-F.; Wen, M.; Yao, Z.-J.; Lu, A.-P.; Wang, J.-B.; Cao, D.-S. ADME properties evaluation in drug discovery: Prediction of Caco-2 cell permeability using a combination of NSGA-II and boosting. J. Chem. Inf. Model. 2016, 56, 763–773. [Google Scholar] [CrossRef] [PubMed]
- Didziapetris, R.; Japertas, P.; Avdeef, A.; Petrauskas, A. Classification analysis of P-glycoprotein substrate specificity. J. Drug Target. 2003, 11, 391–406. [Google Scholar] [CrossRef] [PubMed]
- Ji, D.; Xu, M.; Udenigwe, C.C.; Agyei, D. Physicochemical characterisation, molecular docking, and drug-likeness evaluation of hypotensive peptides encrypted in flaxseed proteome. Curr. Res. Food Sci. 2020, 3, 41–50. [Google Scholar] [CrossRef] [PubMed]
- Ellman, G.L.; Courtney, K.D.; Andres, V., Jr.; Featherstone, R.M. A new and rapid colorimetric determination of acetylcholinesterase activity. Biochem. Pharmacol. 1961, 7, 88–95. [Google Scholar] [CrossRef]
- Cer, R.Z.; Mudunuri, U.; Stephens, R.; Lebeda, F.J. IC 50-to-K i: A web-based tool for converting IC 50 to K i values for inhibitors of enzyme activity and ligand binding. Nucleic Acids Res. 2009, 37, W441–W445. [Google Scholar] [CrossRef] [Green Version]
- Mondal, P.; Gupta, V.; Das, G.; Pradhan, K.; Khan, J.; Gharai, P.K.; Ghosh, S. Peptide-Based Acetylcholinesterase Inhibitor Crosses the Blood-Brain Barrier and Promotes Neuroprotection. ACS Chem. Neurosci. 2018, 9, 2838–2848. [Google Scholar] [CrossRef]
- Trivedi, M.S.; Shah, J.S.; Al-Mughairy, S.; Hodgson, N.W.; Simms, B.; Trooskens, G.A.; Van Criekinge, W.; Deth, R.C. Food-derived opioid peptides inhibit cysteine uptake with redox and epigenetic consequences. J. Nutr. Biochem. 2014, 25, 1011–1018. [Google Scholar] [CrossRef] [Green Version]
- Holzer, P. Opioid receptors in the gastrointestinal tract. Regul. Pept. 2009, 155, 11–17. [Google Scholar] [CrossRef] [Green Version]
- Korhonen, H.; Pihlanto, A. Bioactive Peptides from Food Proteins. In Handbook of Food Products Manufacturing; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2006; pp. 1–37. [Google Scholar] [CrossRef]
- Tyagi, A.; Daliri, E.B.-M.; Kwami Ofosu, F.; Yeon, S.-J.; Oh, D.-H. Food-Derived Opioid Peptides in Human Health: A Review. Int. J. Mol. Sci. 2020, 21, 8825. [Google Scholar] [CrossRef]
- Di, L.; Fish, P.V.; Mano, T. Bridging solubility between drug discovery and development. Drug Discov. Today 2012, 17, 486–495. [Google Scholar] [CrossRef] [PubMed]
- Lipinski, C.A. Drug-like properties and the causes of poor solubility and poor permeability. J. Pharmacol. Toxicol. Methods 2000, 44, 235–249. [Google Scholar] [CrossRef]
- Wang, N.-N.; Huang, C.; Dong, J.; Yao, Z.-J.; Zhu, M.-F.; Deng, Z.-K.; Lv, B.; Lu, A.-P.; Chen, A.F.; Cao, D.-S. Predicting human intestinal absorption with modified random forest approach: A comprehensive evaluation of molecular representation, unbalanced data, and applicability domain issues. RSC Adv. 2017, 7, 19007–19018. [Google Scholar] [CrossRef] [Green Version]
- Wang, Z.; Yan, A.; Yuan, Q. Classification of Blood-Brain Barrier Permeation by Kohonen’s Self-Organizing Neural Network (KohNN) and Support Vector Machine (SVM). QSAR Comb. Sci. 2009, 28, 989–994. [Google Scholar] [CrossRef]
- Wolking, S.; Schaeffeler, E.; Lerche, H.; Schwab, M.; Nies, A.T. Impact of Genetic Polymorphisms of ABCB1 (MDR1, P-Glycoprotein) on Drug Disposition and Potential Clinical Implications: Update of the Literature. Clin. Pharmacokinet. 2015, 54, 709–735. [Google Scholar] [CrossRef]
- Rostkowski, M.; Spjuth, O.; Rydberg, P. WhichCyp: Prediction of cytochromes P450 inhibition. Bioinformatics 2013, 29, 2051–2052. [Google Scholar] [CrossRef] [Green Version]
- Veith, H.; Southall, N.; Huang, R.; James, T.; Fayne, D.; Artemenko, N.; Shen, M.; Inglese, J.; Austin, C.P.; Lloyd, D.G. Comprehensive characterization of cytochrome P450 isozyme selectivity across chemical libraries. Nat. Biotechnol. 2009, 27, 1050–1055. [Google Scholar] [CrossRef] [Green Version]
- van Waterschoot, R.A.; Schinkel, A.H. A critical analysis of the interplay between cytochrome P450 3A and P-glycoprotein: Recent insights from knockout and transgenic mice. Pharmacol. Rev. 2011, 63, 390–410. [Google Scholar] [CrossRef] [Green Version]
- Simeon, S.; Anuwongcharoen, N.; Shoombuatong, W.; Malik, A.A.; Prachayasittikul, V.; Wikberg, J.E.; Nantasenamat, C. Probing the origins of human acetylcholinesterase inhibition via QSAR modeling and molecular docking. PeerJ 2016, 4, e2322. [Google Scholar] [CrossRef] [Green Version]
- Lu, S.-H.; Wu, J.W.; Liu, H.-L.; Zhao, J.-H.; Liu, K.-T.; Chuang, C.-K.; Lin, H.-Y.; Tsai, W.-B.; Ho, Y. The discovery of potential acetylcholinesterase inhibitors: A combination of pharmacophore modeling, virtual screening, and molecular docking studies. J. Biomed. Sci. 2011, 18, 8. [Google Scholar] [CrossRef] [Green Version]
- Santoni, G.; de Sousa, J.; de la Mora, E.; Dias, J.; Jean, L.; Sussman, J.L.; Silman, I.; Renard, P.-Y.; Brown, R.C.; Weik, M. Structure-based optimization of nonquaternary reactivators of acetylcholinesterase inhibited by organophosphorus nerve agents. J. Med. Chem. 2018, 61, 7630–7639. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wajid, S.; Khatoon, A.; Khan, M.A.; Zafar, H.; Kanwal, S.; Choudhary, M.I.; Basha, F.Z. Microwave-Assisted Organic Synthesis, structure–activity relationship, kinetics and molecular docking studies of non-cytotoxic benzamide derivatives as selective butyrylcholinesterase inhibitors. Bioorg. Med. Chem. 2019, 27, 4030–4040. [Google Scholar] [CrossRef] [PubMed]
- Kobus-Cisowska, J.; Szczepaniak, O.; Szymanowska, D.; Przeor, M.; Jarzębski, M.; Ligaj, M.; Gramza-Michałowska, A.; Szwajgier, D.; Foksowicz-Flaczyk, J. Chocolate desserts with ricotta hydrolysates: In vitro study of inhibitory activity against angiotensin-converting enzyme and cholinesterase. J. Food Sci. 2020, 85, 3003–3011. [Google Scholar] [CrossRef] [PubMed]
- Naik, A.S.; Mora, L.; Hayes, M. Characterisation of Seasonal Mytilus edulis By-Products and Generation of Bioactive Hydrolysates. Appl. Sci. 2020, 10, 6892. [Google Scholar] [CrossRef]
- Su, G.; Zhao, T.; Zhao, Y.; Sun-Waterhouse, D.; Qiu, C.; Huang, P.; Zhao, M. Effect of anchovy (Coilia mystus) protein hydrolysate and its Maillard reaction product on combating memory-impairment in mice. Food Res. Int. 2016, 82, 112–120. [Google Scholar] [CrossRef]
- Moreira, T.F.M.; Pessoa, L.G.A.; Seixas, F.A.V.; Ineu, R.P.; Gonçalves, O.H.; Leimann, F.V.; Ribeiro, R.P. Chemometric evaluation of enzymatic hydrolysis in the production of fish protein hydrolysates with acetylcholinesterase inhibitory activity. Food Chem. 2022, 367, 130728. [Google Scholar] [CrossRef]
- Li, S.; Li, A.J.; Travers, J.; Xu, T.; Sakamuru, S.; Klumpp-Thomas, C.; Huang, R.; Xia, M. Identification of Compounds for Butyrylcholinesterase Inhibition. SLAS Discov. Adv. Sci. Drug Discov. 2021, 26, 1355–1364. [Google Scholar] [CrossRef]
- Udenigwe, C.C. Bioinformatics approaches, prospects and challenges of food bioactive peptide research. Trends Food Sci. Technol. 2014, 36, 137–143. [Google Scholar] [CrossRef]
- Rahman, M.; Browne, J.J.; Van Crugten, J.; Hasan, M.F.; Liu, L.; Barkla, B.J. In Silico, Molecular Docking and In Vitro Antimicrobial Activity of the Major Rapeseed Seed Storage Proteins. Front. Pharmacol. 2020, 11. [Google Scholar] [CrossRef]
- Ramírez, D.; Caballero, J. Is It Reliable to Use Common Molecular Docking Methods for Comparing the Binding Affinities of Enantiomer Pairs for Their Protein Target? Int. J. Mol. Sci. 2016, 17, 525. [Google Scholar] [CrossRef] [Green Version]
- Phillips, M.A.; Stewart, M.A.; Woodling, D.L.; Xie, Z.-R. Has Molecular Docking Ever Brought us a Medicine? In Molecular Docking; IntechOpen: London, UK, 2018. [Google Scholar] [CrossRef] [Green Version]
- Cava, C.; Castiglioni, I. Integration of Molecular Docking and In Vitro Studies: A Powerful Approach for Drug Discovery in Breast Cancer. Appl. Sci. 2020, 10, 6981. [Google Scholar] [CrossRef]
- Wu, J.; Aluko, R.E.; Nakai, S. Structural Requirements of Angiotensin I-Converting Enzyme Inhibitory Peptides: Quantitative Structure−Activity Relationship Study of Di- and Tripeptides. J. Agric. Food Chem. 2006, 54, 732–738. [Google Scholar] [CrossRef] [PubMed]
- Politi, A.; Durdagi, S.; Moutevelis-Minakakis, P.; Kokotos, G.; Papadopoulos, M.G.; Mavromoustakos, T. Application of 3D QSAR CoMFA/CoMSIA and in silico docking studies on novel renin inhibitors against cardiovascular diseases. Eur. J. Med. Chem. 2009, 44, 3703–3711. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Number | Opioid Peptide | Source | Sequence |
|---|---|---|---|
| 1 | βb-casomorphin-4 | bovine milk β-casein | YPFP |
| 2 | βb-casomorphin-5 | bovine milk β-casein | YPFPG |
| 3 | βb-casomorphin-6 | bovine milk β-casein | YPFPGP |
| 4 | βb-casomorphin-7 | bovine milk β-casein | YPFPGPI |
| 5 | βb-casomorphin-8 (A1) | bovine milk A1 β-casein | YPFPGPIH |
| 6 | βb-casomorphin-8 (A2) | bovine milk A2 β-casein | YPFPGPIP |
| 7 | neocasomorphin-6 | bovine milk β-casein | YPVEPF |
| 8 | αb-casein exorphin (1-7) | bovine milk α-casein | RYLGYLE |
| 9 | αb-casein exorphin (2-7) | bovine milk α-casein | YLGYLE |
| 10 | casoxin A | bovine milk κ-casein | YPSYGLN |
| 11 | casoxin B | bovine milk κ-casein | YPYY |
| 12 | casoxin C | bovine milk κ-casein | YIPIQYVLSR |
| 13 | α-lactorphin | bovine/human milk α-lactalbumin | YGLF-NH2 |
| 14 | βb-lactorphin | bovine milk β-lactoglobulin | YLLF-NH2 |
| 15 | lactoferroxin A | bovine/human milk lactoferrin | YLGSGY-OCH3 |
| 16 | lactoferroxin B | bovine/human milk lactoferrin | RYYGY-OCH3 |
| 17 | lactoferroxin C | bovine/human milk lactoferrin | KYLGPQY-OCH3 |
| 18 | βh-casomorphin-4 | human milk β-casein | YPFV |
| 19 | βh-casomorphin-5 | human milk β-casein | YPFVE |
| 20 | βh-casomorphin-7 | human milk β-casein | YPFVEPI |
| 21 | βh-casomorphin-8 | human milk β-casein | YPFVEPIP |
| 22 | casoxin D | human milk α-casein | YVPFPPF |
| 23 | αh-casomorphin | human milk α-casein | YVPFP |
| Rule-of-5 | HIA (+/−) | Pgp-Substrate (Y/N) | BBB (+/−) | CYP3A4-Inhibitor (Y/N) | ||||||
|---|---|---|---|---|---|---|---|---|---|---|
| No. | Molecular weight | HBD | HBA | LogP | Solubility (µg/mL) | |||||
| Optimal range | ≤500 | ≤5 | ≤10 | ≤5 | >10 | |||||
| 1 | βb-casomorphin-4 | 523 | 4 | 6 | 1.056 | 158 | (−) | N | (−) | N |
| 2 | βb-casomorphin-5 | 580 | 5 | 7 | 0.172 | 341 | (−) | Y | (−) | Y |
| 3 | βb-casomorphin-6 | 677 | 5 | 8 | 0.163 | 300 | (−) | Y | (−) | Y |
| 4 | βb-casomorphin-7 | 790 | 6 | 9 | 0.694 | 217 | (−) | Y | (−) | Y |
| 5 | βb-casomorphin-8 (A1) | 927 | 8 | 11 | 0.145 | 272 | (−) | Y | (−) | Y |
| 6 | βb-casomorphin-8 (A2) | 887 | 6 | 10 | 0.685 | 216 | (−) | Y | (−) | Y |
| 7 | neocasomorphin-6 | 751 | 7 | 9 | 0.547 | 245 | (−) | Y | (−) | Y |
| 8 | αb-casein exorphin (1-7) | 913 | 14 | 12 | −0.945 | 438 | (−) | Y | (+) | Y |
| 9 | αb-casein exorphin (2-7) | 757 | 10 | 10 | 0.307 | 374 | (−) | Y | (+) | N |
| 10 | casoxin A | 813 | 11 | 12 | −2.745 | 415 | (−) | Y | (−) | Y |
| 11 | casoxin B | 605 | 7 | 8 | 1.204 | 159 | (−) | N | (−) | N |
| 12 | casoxin C | 1251 | 17 | 16 | −1.52 | 331 | (−) | Y | (−) | Y |
| 13 | α-lactorphin | 498 | 6 | 6 | 0.122 | 255 | (−) | Y | (+) | Y |
| 14 | βb-lactorphin | 554 | 6 | 6 | 1.537 | 123 | (+) | Y | (+) | N |
| 15 | lactoferroxin A | 673 | 9 | 11 | −1.891 | 430 | (−) | Y | (−) | N |
| 16 | lactoferroxin B | 735 | 10 | 11 | −1.044 | 257 | (−) | Y | (+) | Y |
| 17 | lactoferroxin C | 882 | 10 | 13 | −1.13 | 380 | (−) | Y | (−) | Y |
| 18 | βh-casomorphin-4 | 525 | 5 | 6 | 1.206 | 162 | (−) | Y | (−) | Y |
| 19 | βh-casomorphin-5 | 654 | 7 | 8 | 0.556 | 237 | (−) | Y | (−) | Y |
| 20 | βh-casomorphin-7 | 864 | 8 | 10 | 1.077 | 210 | (−) | Y | (−) | Y |
| 21 | βh-casomorphin-8 | 961 | 8 | 11 | 1.068 | 207 | (−) | Y | (−) | Y |
| 22 | casoxin D | 866 | 6 | 19 | 1.915 | 125 | (−) | Y | (−) | Y |
| 23 | αh-casomorphin | 622 | 5 | 7 | 1.197 | 199 | (−) | Y | (−) | Y |
| 24 | Caffeine | 194 | 6 | 0 | −1.029 | 11,435 | (+) | N | (+) | N |
| AChE | BChE | |||
|---|---|---|---|---|
| Ligand | Binding free energy (kcal/mol) | Estimated Ki (μM) | Binding free energy (kJ/mol) | Estimated Ki (μM) |
| βb-casomorphin-5 | −14.6 | −14.8 | ||
| βb-casomorphin-7 | −10.1 | 0.04 | −14.4 | |
| Caffeine | −6.7 | 12.12 | −6.5 | 16.99 |
| βb-casomorphin-5 | βb-casomorphin-7 | Caffeine | |
|---|---|---|---|
| PRO31 | ☐ | ||
| TYR72 | ∗ | ||
| ASP74 | △ | ● | |
| TRP86 | ☐ | ☐ | ☐ |
| GLY120 | △ | ||
| GLY121 | △ | △ | △ |
| GLY122 | △ ∗ | ||
| TYR124 | △ | ||
| GLY126 | △ | ||
| GLU202 | △ ● | △ | △ |
| TRP236 | ☐ | ||
| VAL294 | ☐ | ||
| PHE295 | ☐ | △ | |
| PHE297 | ☐ | ||
| TYR337 | ☐ | ☐ | |
| PHE338 | ☐ | ☐ | |
| TYR341 | ☐ | ☐ | |
| TRP439 | △ | ||
| HIS447 | ☐ | ☐ | ☐ |
| GLY448 | △ | ||
| TYR449 | △ |
| βb-casomorphin-5 | βb-casomorphin-7 | Caffeine | |
|---|---|---|---|
| GLN67 | △ | ||
| ASN68 | △ | ||
| ILE69 | △ | ||
| ASP70 | △ | △ | |
| TRP82 | ☐ | ☐ ● | ☐ |
| PRO84 | △ | ||
| THR120 | ☐ | ||
| GLU197 | △ ● | ||
| SER198 | △ | ||
| THR284 | △ | ||
| PRO285 | △ | ||
| LEU286 | ☐ | ||
| ALA328 | ☐ | ☐ | |
| PHE329 | ☐ | ||
| TYR332 | ☐ | △ ☐ | △ |
| TRP430 | |||
| MET437 | ☐ | ||
| HIS438 | △ | △ ● | ☐ ● |
| TYR440 | ☐ |
| Ligand | Experimental Ki (μM) | Inhibition (%) at 1000 μg/mL | IC50 (μg/mL) | |||
|---|---|---|---|---|---|---|
| AChE | BChE | AChE | BChE | AChE | BChE | |
| β-casomorphin-5 | 3814.6 | 127.2 | 34% | 67% | 7151 | 238.5 |
| β-casomorphin-7 | 1666.4 | 76.3 | 43% | 81% | 4255 | 194.7 |
| Caffeine | 11.8 | 1514.0 | 97% | 48% | 7.4 | 949.7 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ji, D.; Ma, J.; Dai, J.; Xu, M.; Harris, P.W.R.; Brimble, M.A.; Agyei, D. Anticholinesterase Inhibition, Drug-Likeness Assessment, and Molecular Docking Evaluation of Milk Protein-Derived Opioid Peptides for the Control of Alzheimer’s Disease. Dairy 2022, 3, 422-437. https://doi.org/10.3390/dairy3030032
Ji D, Ma J, Dai J, Xu M, Harris PWR, Brimble MA, Agyei D. Anticholinesterase Inhibition, Drug-Likeness Assessment, and Molecular Docking Evaluation of Milk Protein-Derived Opioid Peptides for the Control of Alzheimer’s Disease. Dairy. 2022; 3(3):422-437. https://doi.org/10.3390/dairy3030032
Chicago/Turabian StyleJi, Dawei, Jingying Ma, Junyi Dai, Min Xu, Paul W. R. Harris, Margaret A. Brimble, and Dominic Agyei. 2022. "Anticholinesterase Inhibition, Drug-Likeness Assessment, and Molecular Docking Evaluation of Milk Protein-Derived Opioid Peptides for the Control of Alzheimer’s Disease" Dairy 3, no. 3: 422-437. https://doi.org/10.3390/dairy3030032