1. Introduction
Free radical polymerization (FRP) is a versatile technique, which is used with vinyl monomers, such as methacrylates, to prepare many synthetic plastics, elastomers, fibers, and oligomers [
1,
2,
3,
4,
5,
6]. Many of the material properties of polymers, such as the melt viscosity, toughness, and glass transition temperature (Tg), are dependent on the molecular weight (Mwt) [
2,
4,
7,
8]. Thus, chain transfer agents (CTAs) are often used to control the Mwt [
9,
10], where most industrially applied CTAs are non-catalytic; thus, processing requires relatively high concentrations of CTA [
11]. Furthermore, these CTAs are often volatile halogenated or sulfurous materials that produce inhalation hazards, odor emissions or generation of color [
12,
13,
14]. Moreover, as a result of the chain transfer mechanism, such CTAs often introduce an undesirable, heteroatom chain-end functionality into the final product [
13,
15,
16,
17,
18], which may significantly affect final product performance [
19,
20,
21].
Furthermore, key literature reports have highlighted the use of poly(meth)acrylate compounds as functionally active materials that can be used as CTAs and vaccine adjuvants, the latter being effective for inactivated HIV and influenza antigens [
1,
22,
23,
24,
25]. For example, poly(methyl methacrylate) (PMMA) polymers produced by γ-radiation were found to be effective due to their particle size [
22]. Meanwhile, a potential positive immunoresponse was observed from vaccine formulations containing methacrylate polymers exhibiting lower T
g values (i.e., <40 °C), an M
n in the 2000–5000 g mol
−1 range, and a >50% conversion of monomer to oligomer [
26]. Good control over the Mwt and level of conversion in the final products was shown to be important to ensure that materials could be formulated into vaccine adjuvant emulsions with the desired particle size (~200 nm or below) [
26]. However, in this study, methyl methacrylate (MMA) mixtures with oligomers of 2400 g mol
−1 and a 54% conversion did not formulate well [
26].
These lower Mwt oligomers are also more appropriate for use as CTAs when synthesized via catalytic chain transfer polymerization (CCTP) [
27], a control method which can deliver fine control over the Mwt during the FRP of (meth)acrylates and is industrially viable [
28,
29,
30]. Its mechanism generates oligomeric products that are terminated with a vinyl functionality, which (a) can act as conventional, non-catalytic CTAs via a β-scission mechanism (see Scheme S1) [
31] and do not contain any additional heteroatom functionality and (b) can also be used as the site of further post-reaction [
32,
33,
34,
35,
36].
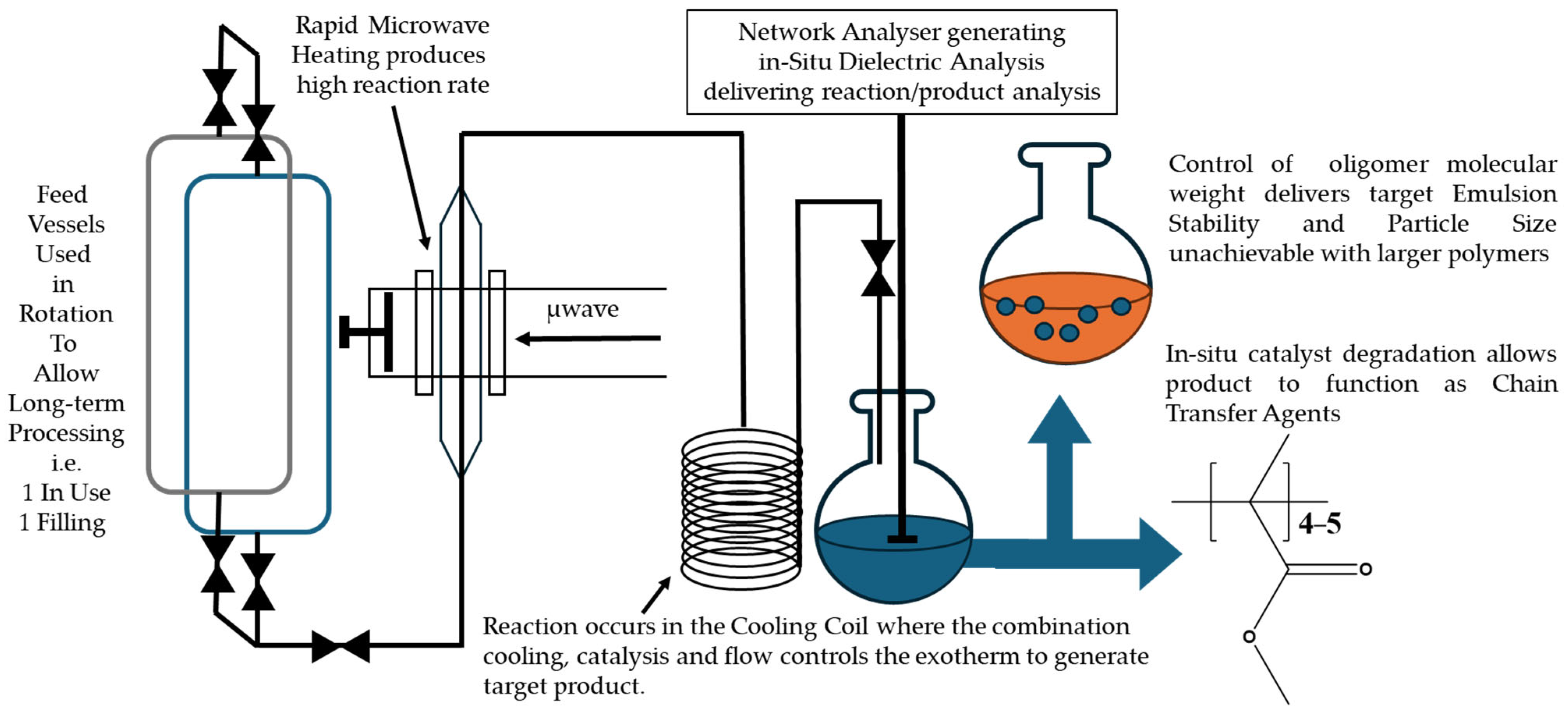
However, to realize the full potential of CCTP in small-scale, high-performance applications, development of a flow oligomerization process will be required. In this case, flow processing will significantly reduce the size of the reactor/plant required, increase process safety by reducing the potential for a Trommsdorff run away, and allow rapid grade change, thus enabling the adoption of “Just-In-Time” (JIT) methods to match batch size with demand [
37]. The former of these advantages are principally attributed to the relatively large surface area-to-volume ratio (compared to stirred tank batch reactors), resulting in efficient heat and mass transfer rates [
38]. Suddaby et al. demonstrated that continuous-flow CCTP was possible using a water-heated tubular flow reactor [
39]. However, the reactor was required to be 1.75 m long, which would hinder scale-up of this process. Outside polymerization, successful conventionally heated, continuous-flow process intensification of the synthesis of a wide range of organic systems has been reported. Examples include the solvent-free synthesis of 2-proply phenol and 2-proply hexanone [
40], the production of ortho-functionalized phenols achieved by the oxidation of aryl Grignard reagents [
41], and the in situ generation, separation, and reaction of diazomethane in a dual-channel microreactor [
42].
Meanwhile, the application of microwave heating when applied to chemical synthesis is itself a relatively mature technology and has been widely reported in the literature for more than 30 years [
43]. In more recent work related to this study, we reported the acceleration of FRP by using microwave heating [
35,
44]. However, it was proposed that microwave heating of the system led to bulk heating instead of selective heating after a short time [
45]. Thus, it was envisaged that adopting this heating method had the potential to overcome the issues with polymerization reaction rates and make them more applicable to transfer to flow processing. This has precedence in general chemistry; the utilization of microwave-induced volumetric or selective heating effects has been attributed to improved reaction rates [
43,
46,
47,
48,
49,
50]. Early investigations into integration of continuous flow with microwave heating utilized relatively crude reactor configurations [
51,
52]. While these studies were an important first step, they demonstrated poor control for the further exploitation/scaling of the technology [
53]. However, more recently, the design of microwave cavities specifically focused on integrating these flow processing and microwave technologies has yielded reliable systems that can operate up to 300 °C and at pressures up to 200 bar with excellent parameter control [
54]. For example, Koyama et al. reported the development of a single-mode resonant microwave applicator incorporating a helical flow reactor to conduct a Claisen rearrangement that delivered a yield of 91% [
55], whilst Barham et al. achieved C-alkylation of dimethylacetamide with styrene using a continuous-flow microwave reactor at a scale of ~65 g/h [
56]. Additionally, Diels–Alder synthesis using microwave-heated flow chemistry has been reported, using a resonant microwave cavity giving yields of up to 85% [
57].
In addition, we also reported a range of halogenated organometallic CCTP catalysts that were made “in situ” prior to polymerization [
58], so removing the need for a catalyst manufacturing process from the full commercial process. Linking these developments would present a significant step toward achieving a commercially viable, continuous-flow route to CCTP adjuvant oligomer mixture production. This study takes advantage of rapid (a) in situ catalyst production and (b) bulk heating of the reaction medium to develop direct, continuous-flow manufacture of model PMMA oligomeric resin products, i.e., CCTP oligomer in monomer mixtures, that can be used directly in vaccine adjuvant emulsions or as CTA mixtures. We focus on the synthesis of lower Mwt MMA oligomer mixtures that would exhibit lower T
g’s and viscosities (i.e., dimer–pentamer mixtures with a ~50% conversion) and the evaluation of their suitability for use in the emulsification/chain transfer process. This was because, in a prior paper, the authors had shown that it was not possible for MMA oligomers that were in the 1000s of g mol
−1 molecular weight range to produce emulsion micelles of the correct size due to their viscosity [
26]. Thus, as they were too large, they would not pass through the last filtering stage of the vaccine formulation process. Hence, this paper investigated if the correct emulsion particles size could be achieved using these sub-1000 g mol
−1 oligomer species. Thus, the focus of the application testing in this study was solely upon the emulsion micelle size analysis to verify this proposed explanation. Further testing of these candidates as adjuvant, which takes a considerable number of months, is an ongoing study and will be published in subsequent papers. The overall unique combination of technologies that have been brought together to enable the delivery of the novel larger-scale manufacturing process to generate these target polymer-in-monomer solutions are depicted in
Scheme 1.
2. Experimental Section
2.1. Materials
All chemicals were used as supplied without further purification. MMA was purchased from Acros Organics (Verona, Italy). Azobisisobutyronitrile (AIBN; 98%, V-65); chloroform—HPLC grade (99%); triethylamine (TEA; 99%)—lab reagent grade; methanol (99.9%); anhydrous cobalt chloride (CoCl2; 99%); dimethyl (DMG) and diphenylglyoxime (DPG) (93%); and chloroform-d (CDCl3; 99.8 atom % D) were purchased from Sigma Aldrich (Gillingham, UK). Bis[(difluoroboryl)diphenyl glyoximato]cobalt(II) (PhCoBF) catalyst was purchased from DuPont (Wilmington, DE, USA). 1,2-dimyristoyl-snglycero-3-phosphocholine (DMPC) was obtained from Lipoid (Ludwigshafen, Germany). Poloxamer 188 and glycerol were obtained from Spectrum Chemical (New Brunswick, NJ, USA). Buffer components were obtained from J.T. Baker (Phillipsburg, NJ, USA) or Fluka (Charlotte, NC, USA).
2.2. General Polymerization for PhCoBF-Mediated Reactions
A stock solution was used for all polymerizations, which consisted of the chosen monomer, cobalt catalyst (PhCoBF) (0.6 g/L), and AIBN (1 w/w %). This was prepared in the following way: The monomer was purged with nitrogen (oxygen free) for 2 h prior to the addition of the catalyst. Following this, PhCoBF (0.6 g/L) was added, and degassing continued for 30 min. The vessel was then sealed and sonicated for 30 s to aid dissolution of the catalyst/catalyst precursors. AIBN (1 w/w %) was then added, and the solution was degassed for 30 min. For subsequent polymerization, the reaction vessels/feed tanks were charged (by cannulation) with this stock solution using nitrogen pressure from the sealed mixing vessel to prevent exposure to air.
2.3. General Polymerization for CoDPGCl2-Mediated (In Situ) Reactions
The procedures were the same as those for the PhCoBF reaction, except that the required quantities of CoCl2 and DPG ligand were added instead of PhCoBF under an inert atmosphere to the degassed monomer solution.
With both catalyst systems, the catalyst was not removed from the products in this current version of the process, and so they will remain in the product, whether as the catalyst in the case of PhCoBF or the deactivated form with DeCoDPGCl2.
2.4. Microwave-Heated (MWH) Batch Polymerization
First, 30 mL of stock solution was transferred into the reaction vessel, containing a magnetic stirrer, and sealed with a septum such that the internal pressure of the monomer could be maintained above its boiling point. The solution was then introduced to one of two microwave reactors that operated at a frequency of 2450 MHz: (a) a CEM Discover SP reactor (maximum output power of 300 W), which could not be electromagnetically tuned, or (b) a Sairem Miniflow reactor (200 W maximum power), incorporating an adjustable short-circuit plate terminating the transmission line, which enabled the formation of a standing wave within the cavity. A single manually adjustable tuning stub was also fixed in the waveguide transmission line enabling impedance matching to the load (reaction mixture and vessel held in the applicator section). Direct temperature measurements of the reaction mixture were made using a glass-sheathed optical fiber (OF) probe (FISO FTO-10, −10–300 °C) inserted directly into the reaction mixture, and the data were continuously recorded on a data-logger. Microwave energy was then supplied to the samples to allow the bulk mixture to rapidly reach three target reaction temperatures: 80, 90, and 100 °C. Afterwards, the application of microwaves ceased, and the flask was removed from the cavity. The exothermic energy of the polymerization was used to continue the reaction for varying periods of time before cooling. This method was adopted to confirm that after initial catalyst formation, the rapid production of initiated oligomers in an MWH process would lead to bulk heating [
45]. Therefore, the further application of microwave energy beyond this initial rapid heating stage was unnecessary. This strategy enabled the use of internal exothermic energy to achieve the desired products in a controlled, sustainable way. After cooling to room temperature, the products were analyzed by gel permeation chromatography (GPC) and
1H nuclear magnetic resonance (NMR) to determine monomer conversion and product quality.
2.5. Conventionally Heated (CH) Batch Polymerization
CH experiments were performed utilizing a thermostatically controlled, stirred oil bath pre-heated to 100 °C. The reaction was prepared following the method detailed in the microwave procedure, and then immersed in the pre-heated oil bath. Once the solution reached the desired reaction temperature, determined using a glass-sheathed OF probe (FISO FTO-10, −10–300 °C) inserted into the bulk, the data were continuously recorded on a data-logger. The vessel was removed from the oil bath and allowed to react using the exotherm for varying time periods prior to cooling. The reaction products were then isolated and analyzed by GPC and
1H NMR. An example NMR spectrum of such conventionally produced oligomers has been included in the ESI document as
Figure S1.
2.6. MWH Flow Polymerization
The flow system consisted of twin 3750 cm
3 feed vessels, which allowed both simultaneous use and charging of individual vessels. Flow within the system was produced via direct application of a nitrogen head pressure (maximum 10 bar) to ensure non-pulsed flow. It was monitored via a load cell accurate to two decimal places, recording the mass entering the receiver vessel, and controlled via a manually operated 12.7 mm needle valve. The medium flowed against gravity through the reactor cavity, which had microwave energy applied from a Sairem 2000 W maximum power generator operating at 2450 MHz. The microwave cavity and cylindrical choke section, included for safety purposes to prevent microwave leakage, were contained in a microwave-transparent 40 cm polytetrafluoroethylene (PTFE) tube capable of withstanding pressures up to 40 bar. Direct product stream temperature measurements were made using a stainless steel-sheathed K-type thermocouple inserted into the flow of the reagents prior to them entering the cavity and into the product after it had exited the cavity (see Item 5 in
Figure S2 for placement). In this way, a direct measurement of the bulk could be safely made without interference from the input microwave energy. The medium was cooled using a cooling coil held within an active air stream to below MMA’s atmospheric boiling point prior to exiting the apparatus into the receiver vessel.
2.7. CH Flow Polymerization
A lagged stainless-steel tube with similar dimensions to the PTFE tube used in the microwave experiments was heated using electrically operated heating tape (Omegalux SRT102-200, (Omega, Manchester, UK)) with a maximum operating power of 1050 W. The rest of the apparatus was as described in the microwave flow experiments.
2.8. Post-Polymerization with Oligomer Mixtures
Secondary stage polymer products were produced using the CCTP-mediated reaction products from the continuous manufacturing procedure as CTAs. The Mwt achieved allowed assessment of any residual, active CCTP catalyst. These post-polymerizations used MMA monomer as the monomeric reagent, the polymerization of which was mediated by either the (a) molecular distilled MMA2 oligomers, (b) continuous CCTP oligomer mixture containing residual CoDPGCl2, or (c) continuous CCTP oligomer mixture containing residual PhCoBF. In the case of the continuous CCTP oligomer mixtures, a second set of experiments was conducted where the mixture had been sparged with air.
A typical polymerization mediated by MMA2 is as follows: The desired quantity of MMA monomer (20.0 mL, 17.52 g, 175.2 mmol) and cyclohexanone solvent (10 mL, 9.48 g, 96.6 mmol) were introduced into a Schlenk flask equipped with a magnetic stir bar under inert atmosphere. These reagents were then degassed for either 30 or 120 min using a flow of nitrogen gas (for the benchmark reaction comparisons) or air (for the air-sparged equivalents). The desired quantities of mediating agent (e.g., MMA2 (7 g, 35 mmol)) and AIBN initiator (1 w/w %) were then added, and the system was stirred until it became homogeneous. The calculation of the amount of oligomer-containing mixture used in the post-reaction was based on the molar concentration of the oligomers present in the solution used. For example, in the case of the molecularly distilled MMA2 reactions, this was 100%. The flask was then submerged into an oil bath pre-heated to 80 °C. The reaction mixture was then stirred at this reaction temperature for the desired reaction time (between 3 and 5 h), after which it was removed from the oil and quenched to room temperature. Once cooled, the solution was added dropwise to cold (0 °C) heptane, and the resulting precipitate was collected via decanting the solvent (lowest Mwt oligomers) or filtration (higher Mwt polymers), which was then dried to constant mass under vacuum. Reaction products were analyzed by GPC to determine the Mwt and polydispersity index (PDI) values.
2.9. Gel Permeation Chromatography (GPC)
GPC was conducted on a Varian GPC50 system at 40 °C. The system used a PLgel 3-μm guard column and two PolarGel 5 μm Mixed-D columns coupled to a refractive index detector. The mobile phase consisted of an HPLC-grade 95:5 mixture of chloroform (95%) to TEA (99%) at a flow rate of 1.0 cm3/min. Analysis was carried out using ASTRA 6.1.7.17 (Wyatt Technology Corporation, Goleta, CA, USA) software. The number- and weight-average molecular weight (Mn and Mw, respectively) and polydispersity (Ð) were calculated using narrow standards of PMMA for the calibration curve. The GPC standards used were purchased from Agilent (Santa Clara, CA, USA) as the InfinityLab EasiVial PMMA pre-weighed calibration kit with samples covering the range from Mn ~600 to 2,000,000.
2.10. Thermogravimetric Analysis (TGA)
Measurements were performed in triplicate using a Q5000 (TA instruments, Cheadle, UK) TGA apparatus. Sample masses were typically 10–15 mg and were measured using a stepwise temperature ramp from ambient to 300 °C, holding the temperature 30–90 min under a flowing nitrogen carrier gas (25 mL/min).
2.11. Nuclear Magnetic Resonance (NMR) Spectroscopy
1H spectra were recorded at 25 °C using a Bruker DPX-300 (Bruker Inc., Karlsruhe, Germany), operating at 300 MHz. All samples were analyzed in a CDCl3 solution that acted as a standard reference peak at 7.26 ppm. Spectral analysis was performed using the ACDLABS 12 software.
2.12. Coupled Gas Chromatography–Electron Ionization Mass Spectrometry (GC/MS)
Products were analyzed via GC/MS on a Waters Autospec Micromass VG (Manchester, UK). The instrument was fitted with a BP1 GC column (15 m, 0.25 mm, 0.25 µm), using helium as the carrier gas (100 KPa). The gradient was set to 50 °C (3 min) to 300 °C (20 min) at 6 °C/min.
2.13. Dielectric Coaxial Probe Measurements
The technique used a 7 mm coaxial probe coupled with a flexible coaxial cable connected to an Agilent 8753 ES VNA (30–6000 MHz) network analyzer. The probe itself consisted of a 7 mm coaxial line with a 2 mm inner conductor, the tip of which was terminated with a radiating aperture surrounded by a circular metallic flange. During measurements, the head of the probe was directly inserted into the sample while the network analyzer transmitted a range of signals at frequencies of 100–3000 MHz and recorded the reflection coefficient (Γ) of the sample. The amplitude and phase of Γ were used to calculate the dielectric properties of the solution, as detailed previously [
44,
59]. Calibration of the equipment prior to collection of the data was performed to account for systematic errors/experimental variation due to the presence of internal imperfections in the transmission line resulting in spurious reflections of the swept signal. This calibration involved the measurement of three different standards: open line (air), short-circuited line (aluminum foil), and a reference liquid of known dielectric response (methanol), as previously described [
60,
61].
2.14. Emulsion Formulation and Characterization
The synthesized PMMA oligomer–monomer solution was formulated with a mixture of phospholipid and poloxamer emulsifiers, glycerol, and a 25 mM ammonium phosphate buffer (pH 5.8), as previously described [
26,
62,
63]. The crude formulations were then processed by high-shear mixing and high-pressure homogenization to produce oil-in-water nano-emulsions containing 4%
v/
v PMMA. Emulsion droplet diameter and droplet polydispersity index (DPDI) were determined by dynamic light scattering using a Zetasizer-S, -ZS, or -APS (Malvern), as previously described [
63].
3. Results and Discussion
The aim of this study was to achieve the continuous manufacture of adjuvant/CTA mixture products that contain a mixture of oligomeric methacrylate species and monomer using an in situ CCTP catalyst. These mixtures represent the feedstock for subsequent vaccine adjuvant emulsion or polymerization processes. The key to success was the synthesis of these end-use oligomers with the target Mwt and percent conversion [
33], so that the T
g and viscosity will enable the manufacture of bio-instructive emulsion droplets of the correct size (i.e., ~200 nm or below) or maximize their efficiency as CTAs. Droplet size is a critical value as it allows for sterilization of the formulation via filtration. By following this strategy, the overall manufacturing chain will be reduced from a multi-step process (i.e., CCTP catalyst synthesis, crude oligomer CTA resin manufacture, catalyst deactivation, and final product isolation) to a single stage. These initial end-use targets were selected to potentially allow the entire product stream to be utilized in the application. This was a strategy deliberately chosen to reduce the environmental impact of these early-stage process investigations. This is because purification usually involves the use of a high-energy tariff for distillation and/or generates considerable amounts of waste (e.g., solvents and antisolvents). However, it is expected that in subsequent studies targeting alternative applications, purification of the mixtures will be required. Therefore, when designing these subsequent processes, minimizing the level of purification to match the needs of the specific end-use application will be a major consideration.
3.1. Batch Production for Optimization of Reaction and Stoichiometry Parameters
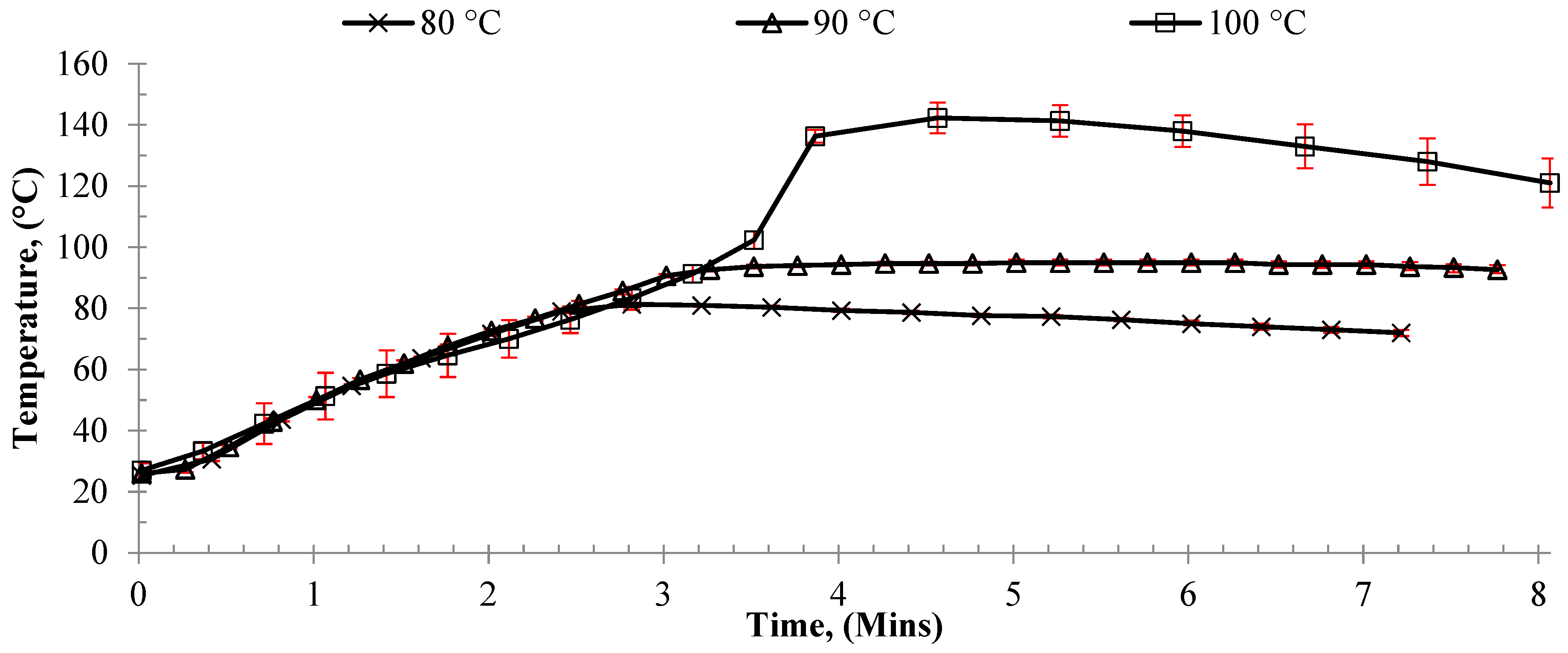
Initially, CH and MWH bulk polymerization were conducted via batch processing in an oil bath and a microwave reactor (CEM with maximum power 300 W), respectively. Initially, we investigated the effect of target reaction temperature (80, 90, or 100 °C) on monomer conversion. These experiments used a benchmark CCTP agent (PhCoBF) as a pre-prepared catalyst to ensure that the expected control over the reaction was obtained whilst the reaction conditions were optimized to achieve the reaction rates necessary for continuous flow. Sealed flasks were utilized to prevent loss of the monomer (MMA boiling point = 100 °C), and the power input was monitored to maintain the set point by using a direct OF measurement of the reaction medium. Monomer conversion was observed to increase in both the CH and MWH reactions when the temperature increased from 80 to 90 °C (
Table 1).
This enhancement was attributed to an increase in radicals released as the initiator’s half-life temperature decreased, leading to more initiation per unit time. However, when the temperature increased to 100 °C, a difference in heating methods was observed. The CH conversion remained similar to that of the 90 °C experiment, whilst that of the MWH increased to 65%. This difference was previously observed when investigating the influence of MWH on the efficiency of chain transfer in methacrylate systems [
35]. This observation was attributed to the MWH selectively heating the radicals, and thus increasing their diffusivity and reactivity. In so doing, MWH promoted propagation over termination, whilst CH bulk heating of the medium led to “initiator burn-out”, i.e., too many radicals were released per unit time leading to direct radical–radical termination. However, the conversion data from the current study showed higher conversions for both heating methods compared to the previous report.
Examination of the reaction thermal profiles showed that these reactions were not comparable to previously reported data. Rather, the reaction exotherm in both CH and MWH experiments was observed to have overcome the thermal equilibrium of the system causing the bulk temperature to rapidly rise to 140 °C (
Figure 1 and see CH:MWH comparison in
Figure S1).
It was proposed that the rapid reaction temperature increase was due to auto-initiation and resulted in increased reactions rates and percentage conversions. The onset temperature for MMA auto-initiation is reported to be around 120 °C [
64], and both CH and MWH experiments were observed to produce very similar heating profiles. Thus, the difference in conversion between the current study and prior study was attributed to the increased heating rate achieved.
When applying MWH, 140 °C was reached after approximately 3 min, whilst the CH experiments took over 10 min, confirming that more efficient heating was achieved with microwave energy. Thus, in these experiments, the CH initiator burnout that was previously reported when the experiment’s exotherm was well controlled such that the reaction medium remained at 100 °C was counteracted by the onset of auto-initiation. Therefore, these batch experiments suggested that, by enabling the reaction to undergo an exotherm, auto-initiation could be used to rapidly achieve a rapid polymerization within a stable flow process using MWH, as flow processes typically utilize a much-reduced reactor volume than batch systems of similar-sized output, so heat removal can be achieved much more efficiently. Thus, a rapid heating MWH process design was developed to exploit the two process benefits obtained from an exothermic flow reaction: (a) once the set point was reached, additional energy was not required to maintain the temperature, and (b) the initial small-scale batch experiments showed that conversions above the target 50% yield were possible in only 8 min whilst achieving the target Mwt of oligomer.
Prior to performing reactions in continuous flow, we investigated the effect of increasing microwave power on oligomer production. To achieve this, batch experiments were repeated using a Sairem microwave generator with an operating frequency of 2450 MHz and maximum power of 2000 W, connected to a single mode cavity. Power levels of 500 and 1000 W were utilized, and the amount of absorbed energy required to reach the temperature set-point was measured.
As in the 300 W MWH experiments, temperatures achieved were significantly above 100 °C due to the self-generated exotherm (
Figure S3 and
Table 2).
At 500 and 1000 W, the maximum temperature was in the same range as at 300 W (~140 °C), but this temperature was now reached in less than 2 min. Further analysis of the absorbed power data showed that the microwave energy was only applied at a significant level (i.e., ≥500 W) for a short period because after the set point had been reached the input power was automatically reduced. Thus, the continued increase in the observed reaction temperature was attributed to the internal exotherm produced by the auto-initiation of the polymerization and was largely independent from the microwave energy. Hence, the similarity between the 500 and 1000 W reaction profiles was due to the length of time that the microwave power was applied by the control system and indicated no evidence of selective heating as a direct result of increasing power. Additionally, these results suggested that, under these batch conditions, 500 W was sufficient to achieve the processing goals. Comparison of the 500 and 1000 W data (
Table 2 and
Figure S4) showed no negative effect on the reaction yield and reaction times. Reactions were then investigated in flow because these batch reactions were self-limiting due to the vapor pressure achieved within the reactor.
3.2. Comparison of MWH and CH Flow CCTP Synthesis of Methacrylate Oligomers
An initial series of flow experiments were conducted using the designs in
Figure S2 and 500 W of input power to determine if a stable, high-yielding polymerization could be achieved in this reactor configuration that yielded the target product. However, to achieve the same results in the CH experiment, an extended heat zone, as shown in
Figure S2, and a reduction in the mass flow rate of 120 g/min was required to achieve comparable energy transfer to the MWH experiment. The results shown in
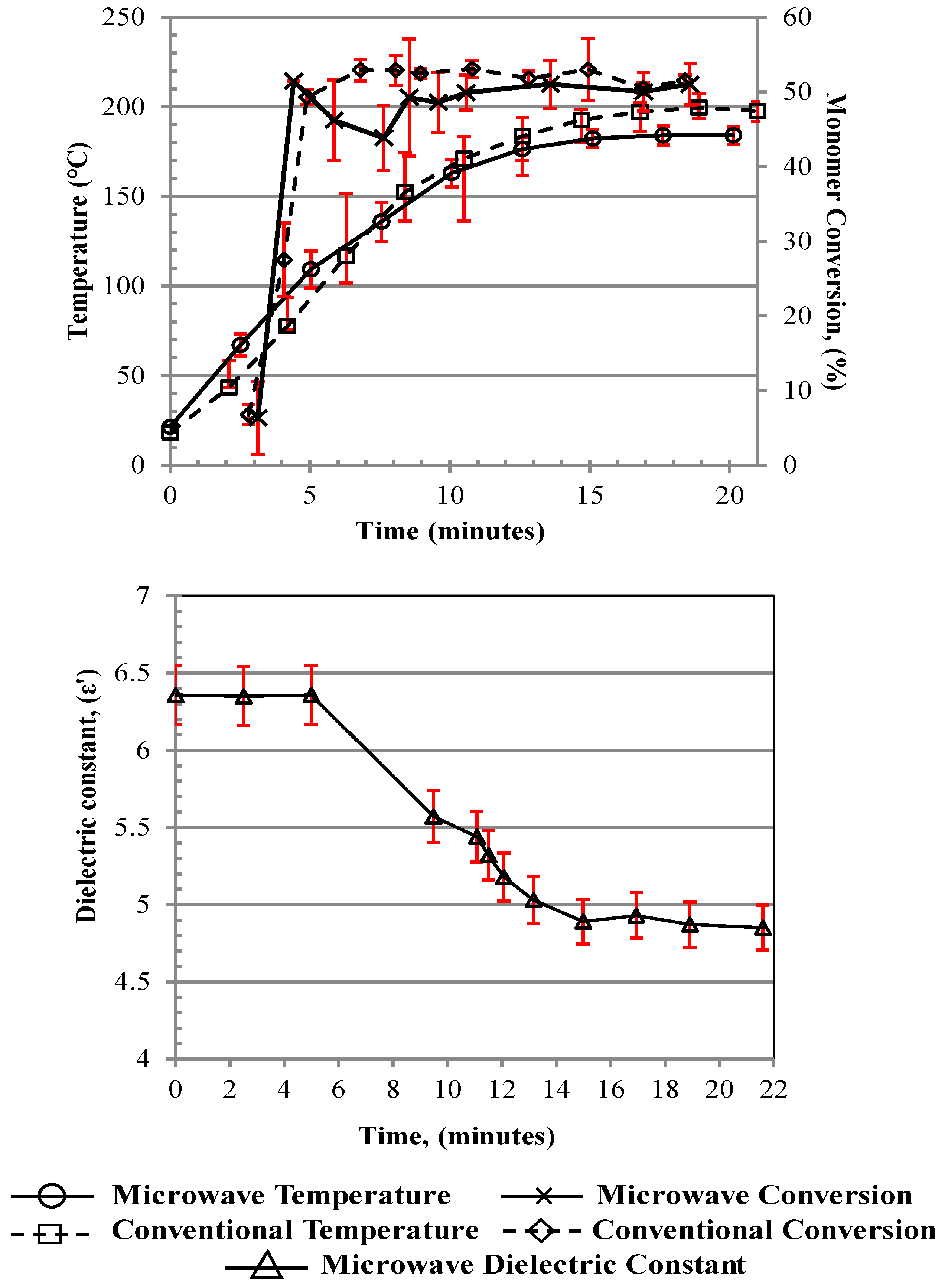
Figure 2 indicate that both the dielectric experiments and the conversion results may be subject to a potential time lag of ~1–2 min because they are measured offline by sampling from the receiver vessel, as compared to the temperature measurements (see discussion on this below), which are made directly as the medium leaves the “heated zone”.
Prior to 65 °C, the initiator’s 10 h half-life temperature, there was negligible monomer conversion. Therefore, this was defined as the first stage of the start-up phase of the process, where the system overcame the thermal inertia of the reactor surfaces. Once this temperature was reached, the rate of monomer conversion sharply rose from 6% to ~50% in ~1 min with MWH and ~1.5 min with CH, during which time the reaction bulk temperature rose from 65 °C to ~95 °C with MWH and ~75 °C with CH. Up to this point, the heating profiles were very similar between MWH and CH, with the slower increase in the CH temperature attributed to the need for a slower flow rate and an extended heating zone in the design, as previously indicated. However, after this point, the monomer conversion profiles began to differ, with the MWH dropping and the CH continuing to increase. At the same time, the rate of the increase in temperature for the MWH was reduced compared to the CH until the temperature profiles crossed at ~130 °C. This behavior was linked to the different heating modes and was defined as the second stage of the start-up phase. In the MWH experiment, having reached and surpassed the set-point, the control system removed the energy input, and conversion was reduced. Meanwhile in the CH experiment, the instantaneous removal of all input energy was not possible due to thermal lag in the thermo-fluid, causing conversion to continue to rise, but at a much-reduced rate. After 130 °C, the MWH conversion level started to rise again and reached similar levels to the CH conversion. This was attributed to the onset of auto-initiation and the reaction being driven by the internal exotherm rather than the external heating source. This explained why, after ~8 to 9 min, both the conversion (50–55%) and bulk temperature ranges (180–200 °C) were steady, as the reaction was defined as having reached steady state once auto-initiation was fully established. The recorded conversion levels were slightly lower than those from the batch polymerization, which was attributed to the longer residence time in the batch processing regime (300 s) compared to flow processes (90 s, i.e., the time it takes for the material to flow through the heated zone). During the steady-state operation, very little microwave energy was drawn for the generator, making the process very efficient, as it primarily used the exotherm to drive the operation. Comparison between heating methods under flowing conditions is difficult, as it is complicated to estimate the energy drawn from the thermofluidic during steady-state operation. However, the higher flow rate allowed by the microwave system indicated that a 20% higher production rate was achieved with MWH. Furthermore, the MWH processes could achieve the target monomer conversion under flowing conditions at a faster production rate due to the increased efficiency of energy transfer into the bulk via the volumetric heating mechanisms involved.
Furthermore, the data in
Figure 2 demonstrated that in situ monitoring of the output stream’s ε’ value was a viable method of online process monitoring, as it could define both (a) the onset of oligomerization and (b) when the thermally dictated steady-state operation had been achieved; see Discussion S1 for further explanation.
3.3. Characterization of Target Continuous-Flow Oligomerization Products
The final product mixtures from the optimized continuous-flow CCTP were analyzed for oligomer compositions with GPC, GC-MS, and TGA data. In the case of the GPC and GC-MS data, the relative abundance of the oligomers was ascertained by ratioing the relative peak areas for the separate moieties. Meanwhile, for the TGA data, the weight loss percentages were ratioed to obtain the relative masses of the oligomers present in the sample (
Table 3 and
Figures S5–S7).
The peaks for individual oligomer species could be resolved in the GPC spectra obtained for the product mixtures but were not fully baseline separated. Meanwhile, a comparison between oligomer compositions from CH and MWH flow processing confirmed that the percentage spread of oligomers was very similar in the product mixtures and showed that it was not affected by the heating method applied (
Figure S5). Additionally,
Table 3 shows good agreement in the predicted oligomer mixture composition among the GPC, GC/MS, and TGA data. However, the TGA showed only four distinct thermal mass loss events due to the high boiling points of the penta- and hexamer. Furthermore, it showed that, within the error of the measurements, the target of 50% of dimer, trimer, and tetramer had been achieved within the overall product mixture.
3.4. Use of a Deactivatable In Situ CCTP Catalyst
A chloride variant of an in situ CCTP catalyst, defined as CoDPGCl
2 (see the molecular structure in
Figure S8), was used to control the flow chemistry within the flow apparatus. We had previously used this catalyst to control batch experiments, but it was less stable to the ingress of oxygen and water than bromide equivalents in batch experiments [
35]. CoDPGCl
2 was applied in this reactor format to determine if it would deliver both (a) sufficient control over the Mwt to produce the target dimer–pentamer oligomer mixtures and (b) a route to deactivate the catalyst post-polymerization. Because of the demonstrated capability of rapidly increasing the reaction medium’s temperature using a single degassed feed, we were able to apply this less-stable catalyst when operating in flow. Thus, the reagents required to create the in situ catalyst were added to the stock solution, replacing the PhCoBF used up to this point. The oligomer mixture product was then produced using MWH–continuous flow, which was collected and used in subsequent polymerization to determine if residual CCTP catalyst was present and active as a CTA. Any catalyst still active would influence the final Mwt of the secondary polymer. The target was for any Mwt control observed to be due to the CCTP-derived oligomer mixtures acting alone as CTAs. This evaluation was conducted via a series of solution polymerization using dimer–trimer mixtures containing (a) CoDPGCl
2 without deactivation, (b) CoDPGCl
2 with deactivation by sparging (named DeCoDPGCl
2), (c) PhCoBF without deactivation, (d) PhCoBF with deactivation by degassing (DePhCoBF), and (e) a 40% wt:wt molecularly distilled dimer (MMA
2) in monomer (see
Table 4).
The chloride catalyst could produce the target dimer/trimer/tetramer either in flow or batch (
Table 4, Entries 1 and 2). Furthermore, when the in situ catalyst was synthesized in monomer only, but subjected to being degassed/sparged prior to polymerization, it was shown to be deactivated, as it produced a final product with an M
n of 62,000 g mol
−1 (
Table 4, Entry 3). When the CoDPGCl
2-produced dimer–trimer solution was polymerized under a nitrogen atmosphere, the CCTP agent was still affecting the polymerization as a product of 1400 g mol
−1 was isolated (
Table 4, Entry 4). This M
n was also much lower than the product polymer isolated from the application of the distilled dimer benchmark (
Table 4, Entry 8). Thus, the catalyst was still influencing the post-polymerization and must still be active. However, if the solution was degassed with air prior to polymerization, the products’ Mwt was similar to that of the dimer (
Table 4, Entry 5). The slight difference between MMA
2 and DeCoDPGCl
2 was attributed to the mixture of oligomers that would be present in the DeCoDPGCl
2 case. By comparison, PhCoBF was not deactivated by the degassing treatment; the only effect was to inhibit the post-polymerization by the inclusion of additional oxygen after excessive degassing.
These data showed that it was possible to manufacture the target dimer/trimer/tetramer mixtures using the chloride-liganded CoDPGCl2 catalyst in flow, thus removing the need for an entire, multi-step, catalysis manufacturing process from the overall manufacturing process to produce these oligomers. We also demonstrated that it was possible to deactivate the catalyst so that the flow-produced resins could be used directly as CTAs or solutions containing CTA with limited additional processing (i.e., the addition of a degassing stage and, if necessary, the removal of the free monomer by distillation). Thus, these oligomers could be used directly as the feedstock for subsequent solution, emulsion, suspension, or formulation processes, without the need for additional purification stages to remove the CCTP catalyst. We also showed that use of the in situ catalyst aligned with the simplification of the flow process by continuing to allow the use of a single feedline into the reactor and simplifying process control.
3.5. Emulsion Formulation and Physical Characteristics
Finally, a batch of crude oligomer mixture with an M
n of 300 g mol
−1, PDI of 1.5, and a conversion of ~90% was formulated with a mixture of phospholipid, poloxamer emulsifiers, glycerol, and a 25 mM ammonium phosphate buffer (pH 5.8) and processed by high-pressure homogenization. The objective was to generate a homogeneous emulsion with a droplet size of ~200 nm or below to allow for filtration through a terminal sterile filter. Following high-pressure homogenization, the emulsion droplet size (Z-ave) was observed to be 88 nm, with a size polydispersity index of 0.253 (
Figure 3).
The visual appearance of the emulsion was homogeneous milky-white and the pH was in the ~5.7 region, as expected. Thus, this low-PMMA oligomer mixture was successfully formulated in an oil-in-water nano-emulsion with comparable physical characteristics to our previously reported vaccine adjuvant emulsions [
18,
26,
62]. The physical properties of these emulsions were subsequently monitored over 3 months at different storage temperatures, and stability data for the particle size, polydispersity index (PDI), visual appearance, and emulsion pH are shown in
Tables S1–S4. Emulsion pH, PDI, and appearance were shown to be stable over this time-period at different storage temperatures. The particle size data showed some small emulsion particle size growth during month one only, but all the emulsions remained under the target size of ~200 nm during the entire monitoring period, even at 40 °C.
These observations further support the conclusions made in our prior work, which demonstrated the success in nano-emulsion formulation was related to the T
g and viscosity of the oligomeric adjuvant mixture [
26]. In our prior study, PMMA polymers with an M
n of 2400 g mol
−1, a PDI of 2.4, and a conversion of 54% were shown not to formulate well [
26]. This difference in processability was attributed to the fact that T
g is a property that is Mwt-dependent when the sample of materials is below its chain entanglement length, which is in the region of 10,000 g mol
−1 for PMMA [
7]. Thus, reducing the Mwt of a mixture of oligomers to a mixture of chain lengths from dimer to pentamer has significantly reduced the overall polymer T
g. For example, dimers and trimers are known to be viscous liquids that can be molecularly distilled under reduced pressure indicating that they have a sub-room temperature T
g [
58].
Finally, to gauge the sustainability for this process, the Process Mass Intensity (PMI) (i.e., total mass of raw materials (except water)/total mass of final product) was calculated. However, there are two views that can be taken for what constitutes the full product. The first hypothesis would consider the entire mixture as the adjuvant, with the monomer playing an active role in the adjuvant efficiency as a viscosity modifier. In this case, the only “non-active” raw materials would be the CCTP catalyst, as the initiator would become part of the oligomeric product. The second hypothesis would consider that the monomeric component is only present as reaction “solvent” in the calculation. In
Calculation S1 in the Supplementary Information, the PMI calculations are detailed, which give values of 1 for the first hypothesis and 2 for the second. Thus, the first scenario exhibits high sustainability. From our previous study, we believe that viscosity is a vital characteristic if the adjuvant is to be successful; thus, the former figure is a true reflection of the PMI. However, further study will be required to define the vital nature of the monomeric component in these mixtures before the PMI can be definitively attributed to the process.
4. Conclusions
A novel reactor design and methodology for the continuous-flow production of low-Mwt MMA oligomers using both microwave and conventional heating has been demonstrated. This process was successfully performed to produce a target set of oligomers of a specific Mwt that exhibited a monomer conversion of >50%. These oligomer mixtures are models for vaccine adjuvant or CTA systems. The products were successfully trialed in both of these end uses and found to successfully deliver the target behavior sought, i.e., emulsion droplet sizes below 220 nm or Mwt reductions in post-polymerization equivalent to isolated CCTP dimers, respectively. The process design was also shown to deliver several significant sustainability benefits. These included significantly limiting the input energy required, especially in the case of the microwave experiments, where the process required significant heat energy for only ~4 min to achieve catalyst manufacture and auto-initiation. This represents the first time this in situ technology has been conducted in-flow, which represents a significant advancement, as typically CCTP catalyst have previously needed to be pre-synthesized before use. Thus, this paper demonstrated this methodology can be applied in a flow system, by successfully achieving this “fast” catalysts synthesis within the timescale of this flow process. This point then principally drove the steady-state operation utilizing the exotherm energy of the reaction. At steady state, only very low levels of microwave energy were required to maintain this operation. Direct comparison of microwave and conventionally heated processes showed no evidence of selective heating effects under the conditions investigated, although an increase in production rate (20%) for MWH was noted due to the efficient energy transfer associated with volumetric heating. Enabling rapid heating from room temperature allowed the use of a single feed into the heated zone, and the use of an in situ method to create the CTA in flow simplified the design. This removes an entire catalyst synthesis process from the overall process footprint and removes the need to store active organometallic catalysts. Furthermore, the flow system allowed the use of a less stable CTA, which could be deactivated by gas sparging, removing the need for additional post-polymerization purification of the oligomer mixtures before use. Finally, in situ monitoring of the dielectric constant showed that significant changes in this property could be seen between the feed (start-up) and product materials (steady-state production), such that its value could be utilized as a precise online process monitoring technique.


 ,
, 


{kind=link}
{kind=link}
{kind=link}
{kind=link}