
Vinyl Esters and Vinyl Sulfonates as Green Alternatives to Vinyl Bromide for the Synthesis of Monosubstituted Alkenes via Transition-Metal-Catalyzed Reactions


Abstract
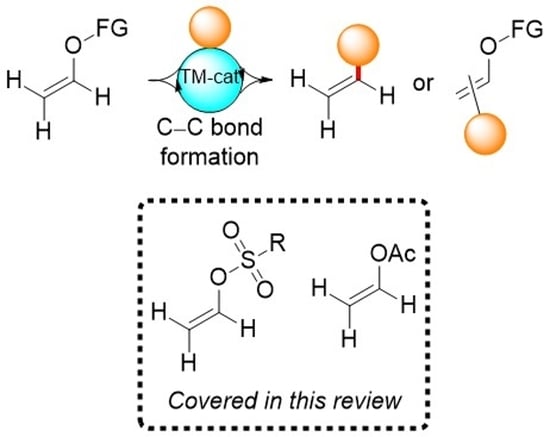
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
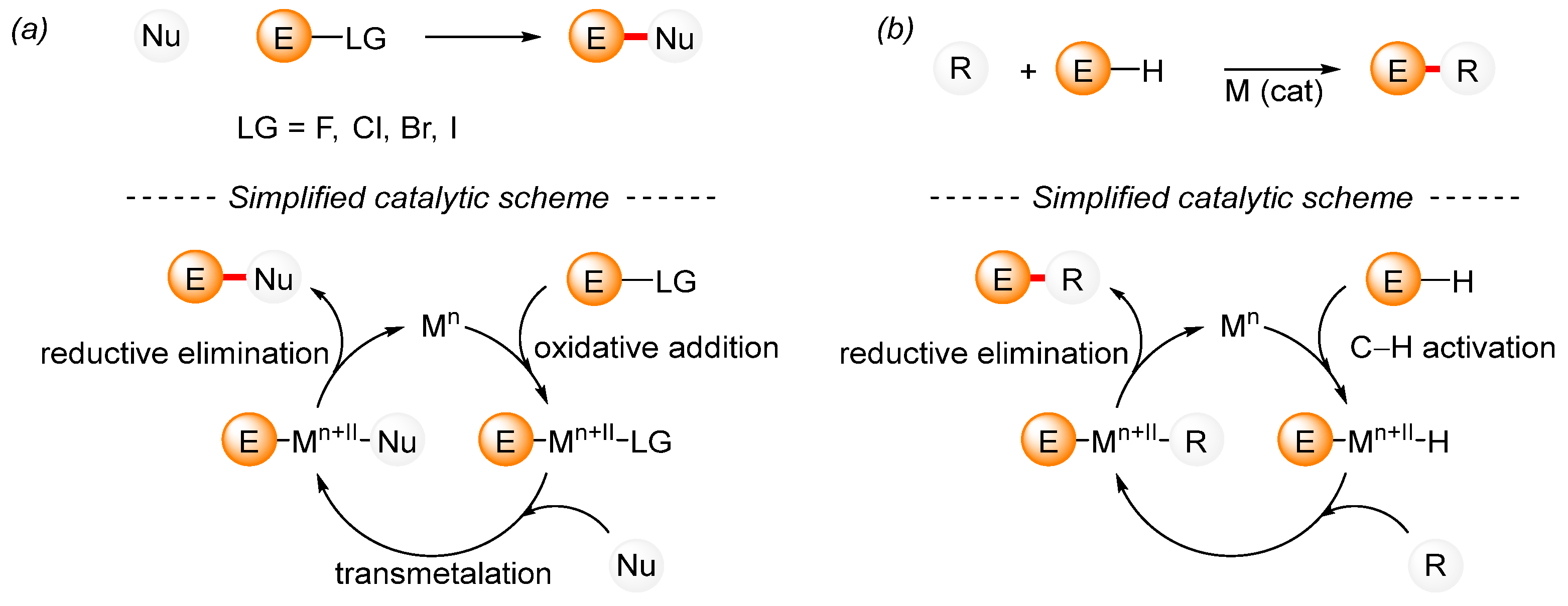
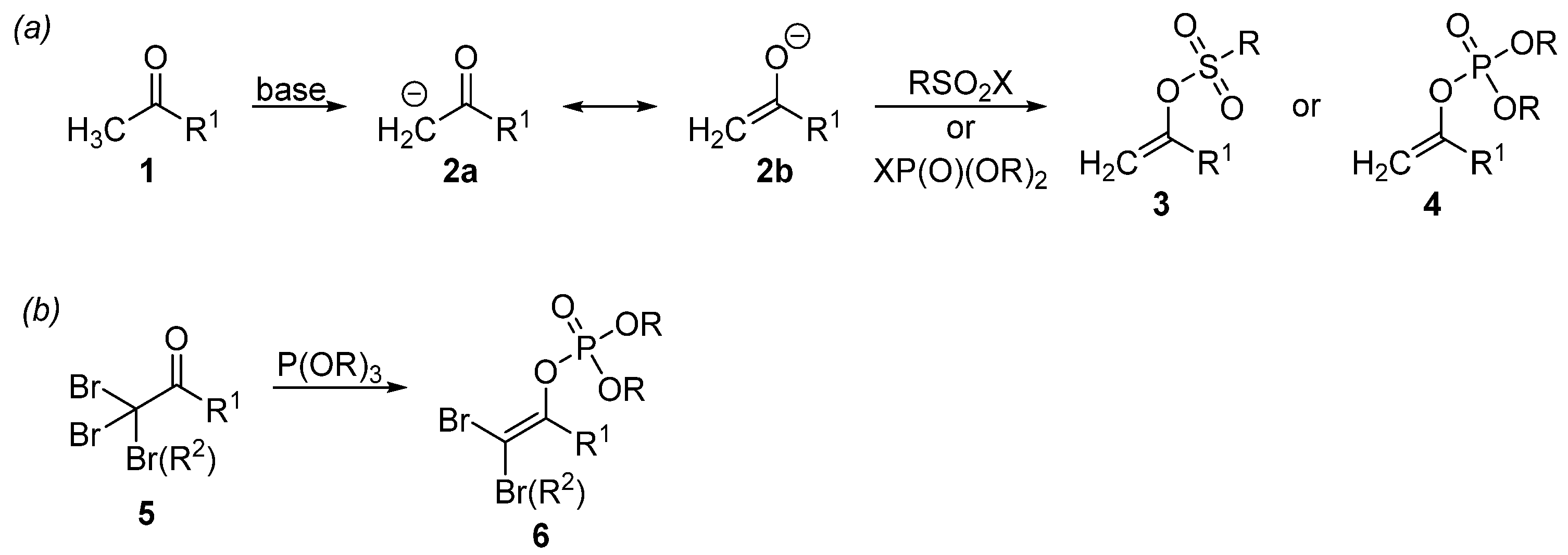
1. Introduction
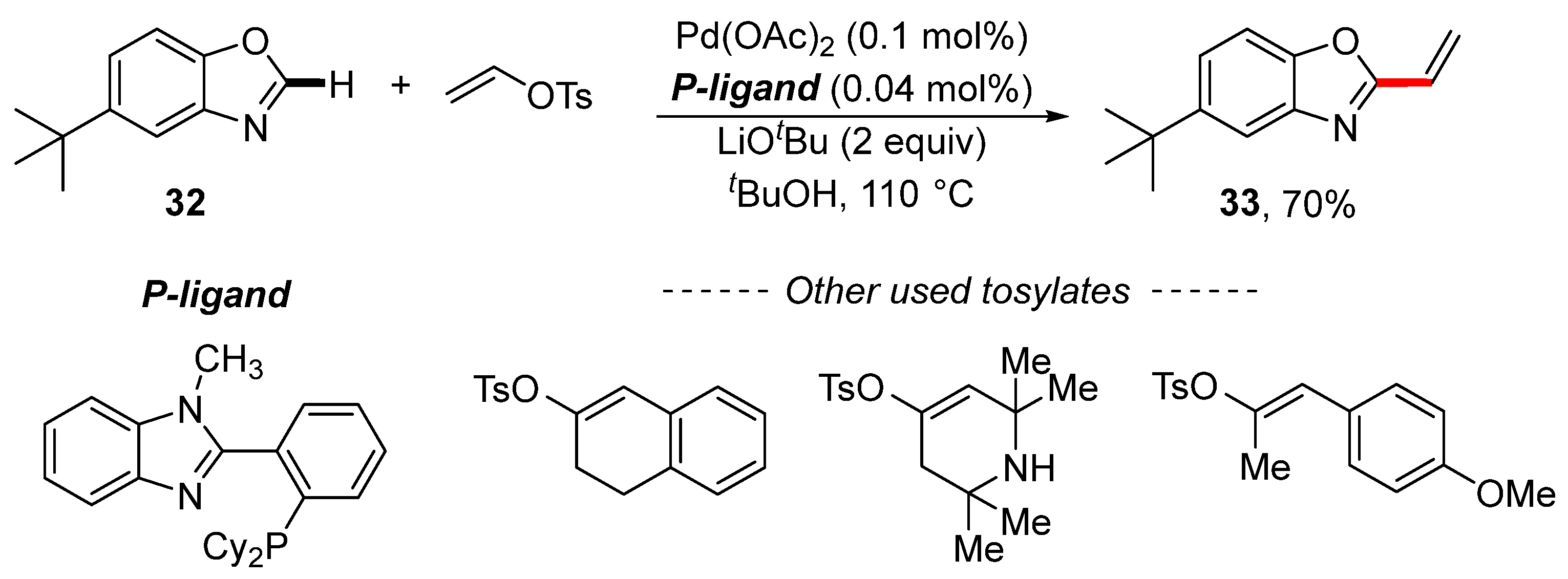
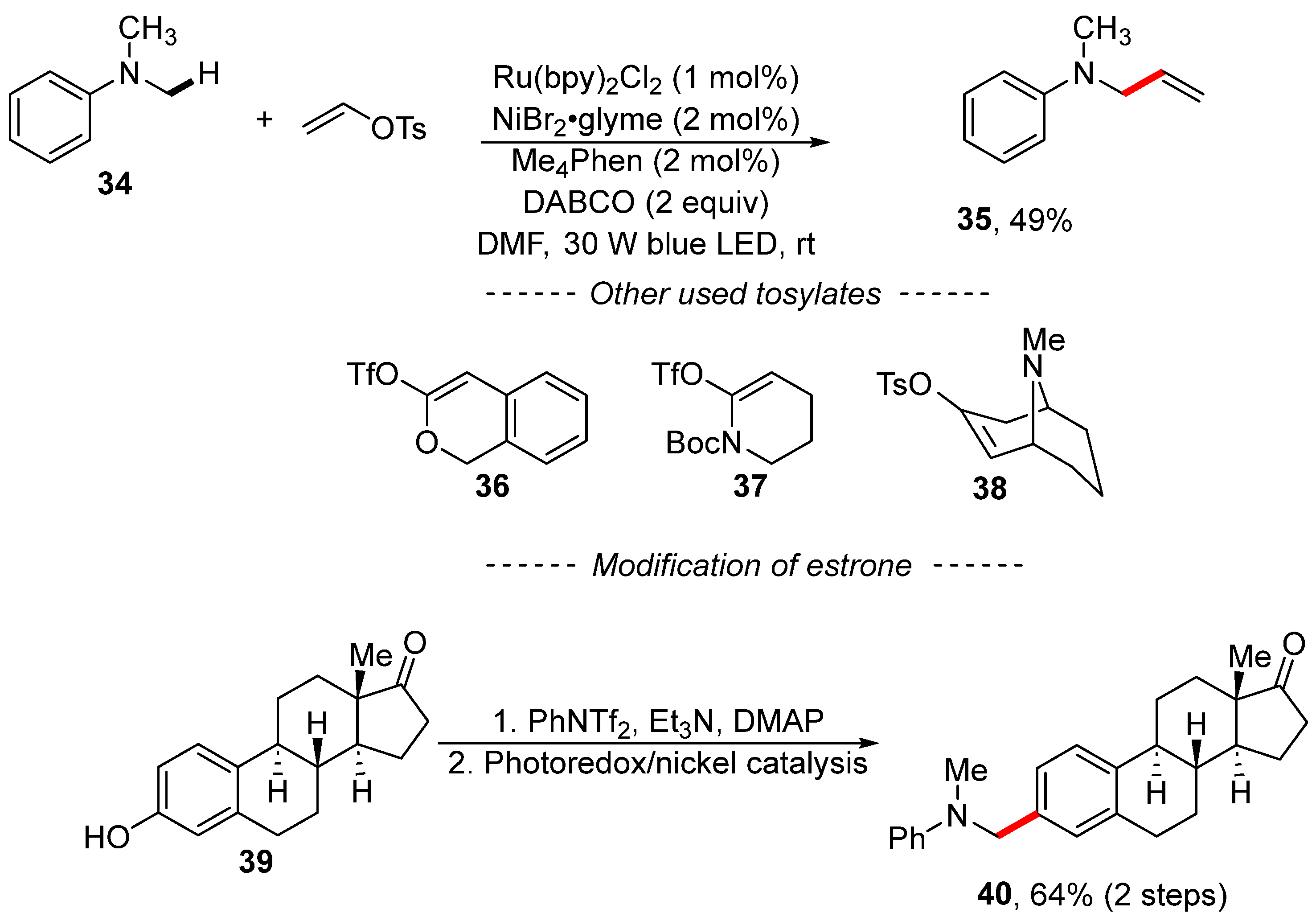
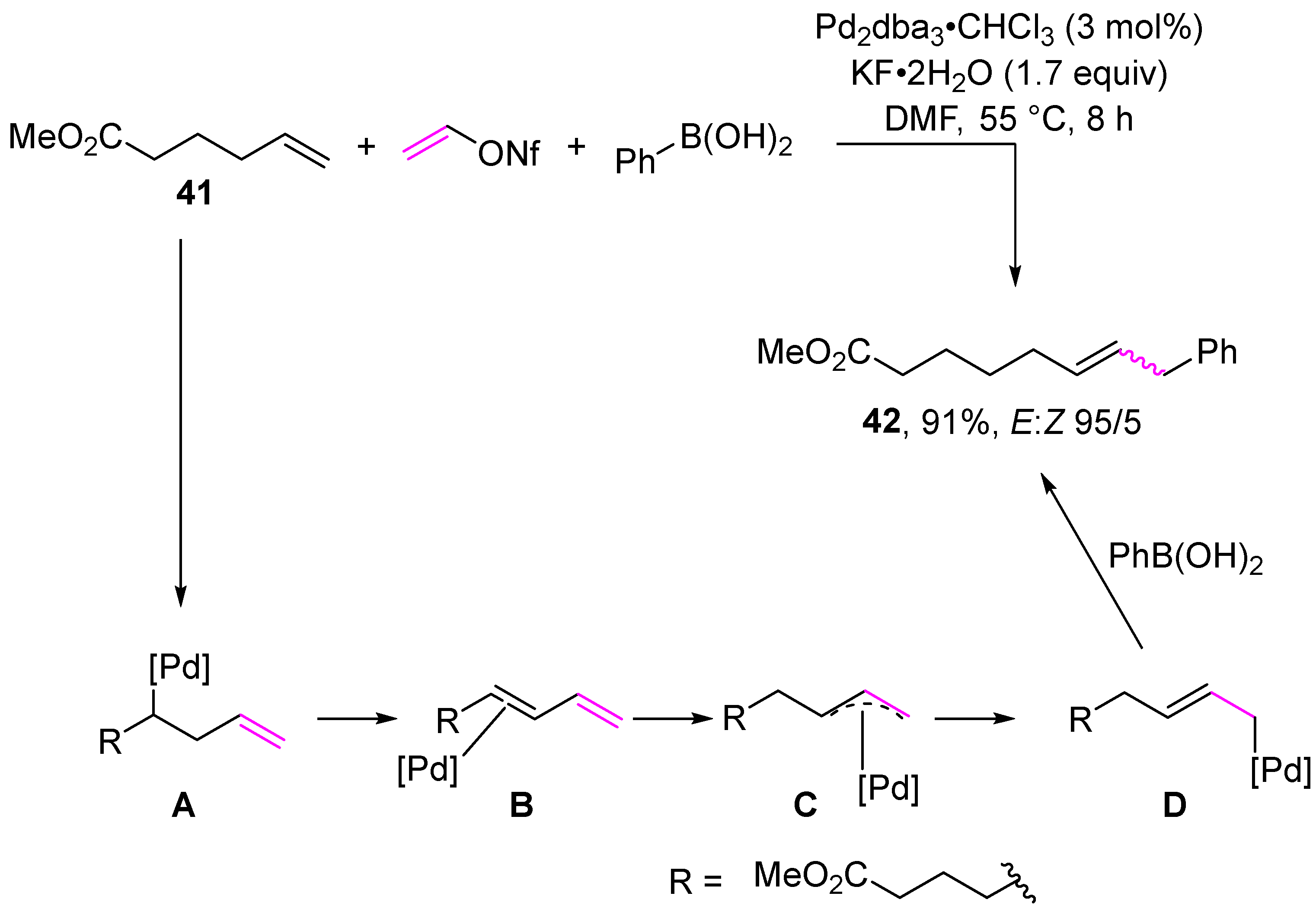
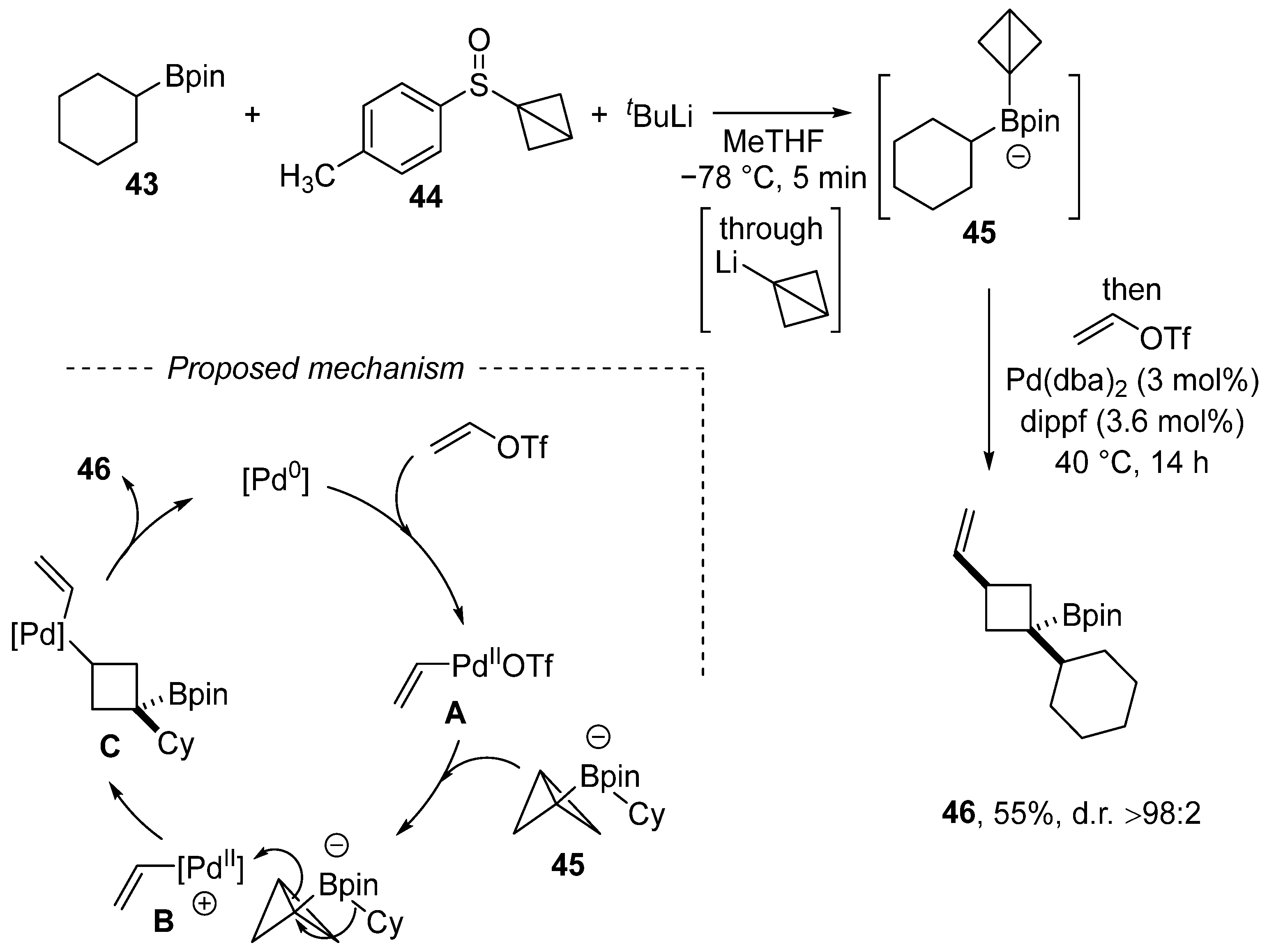
2. Cross-Coupling Reactions of Vinyl Sulfonates
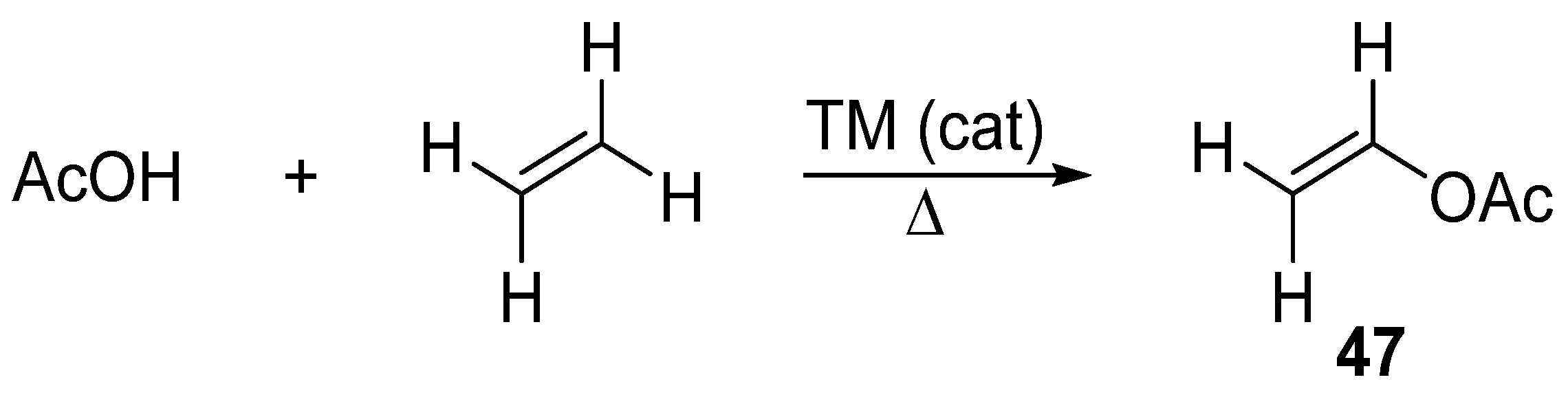
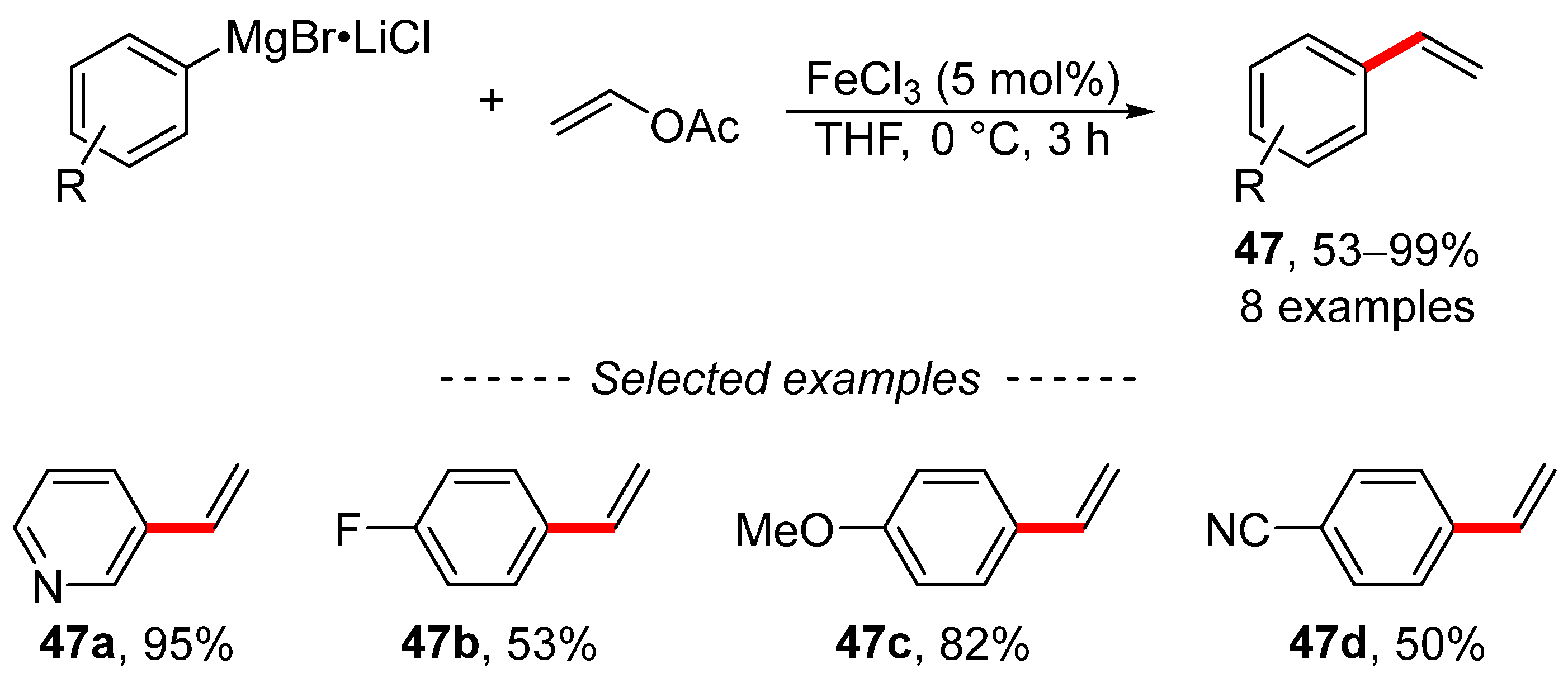
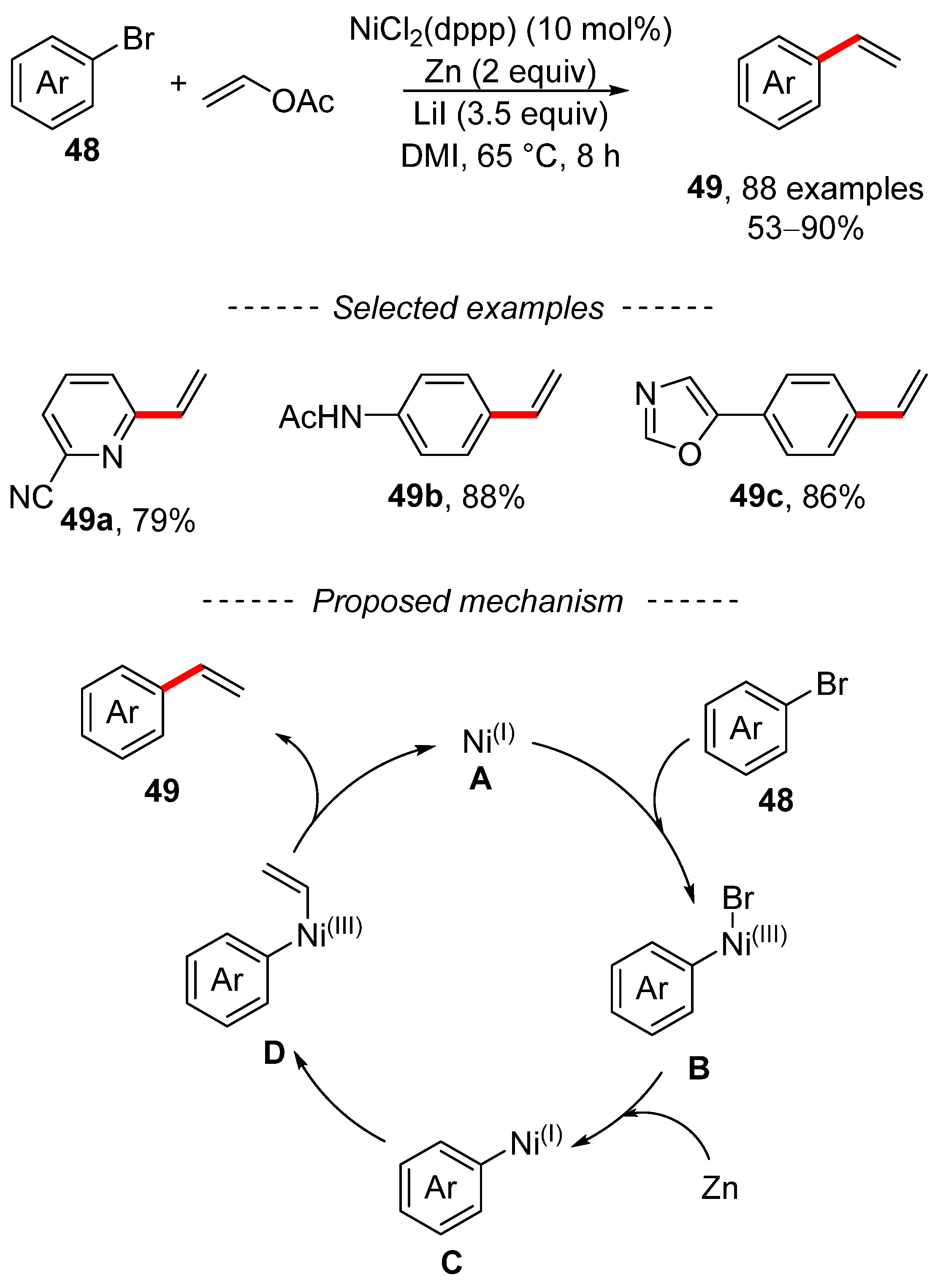
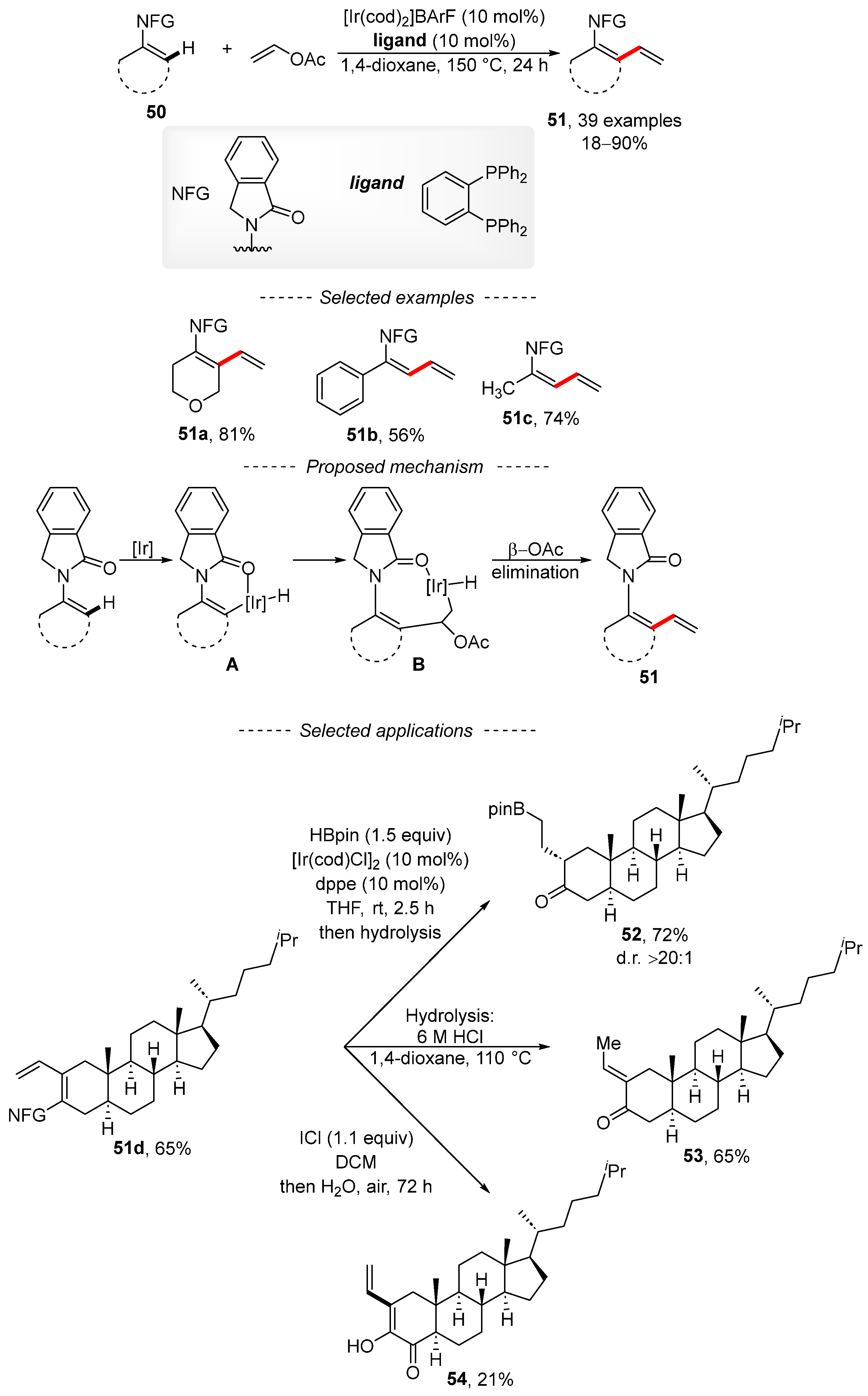
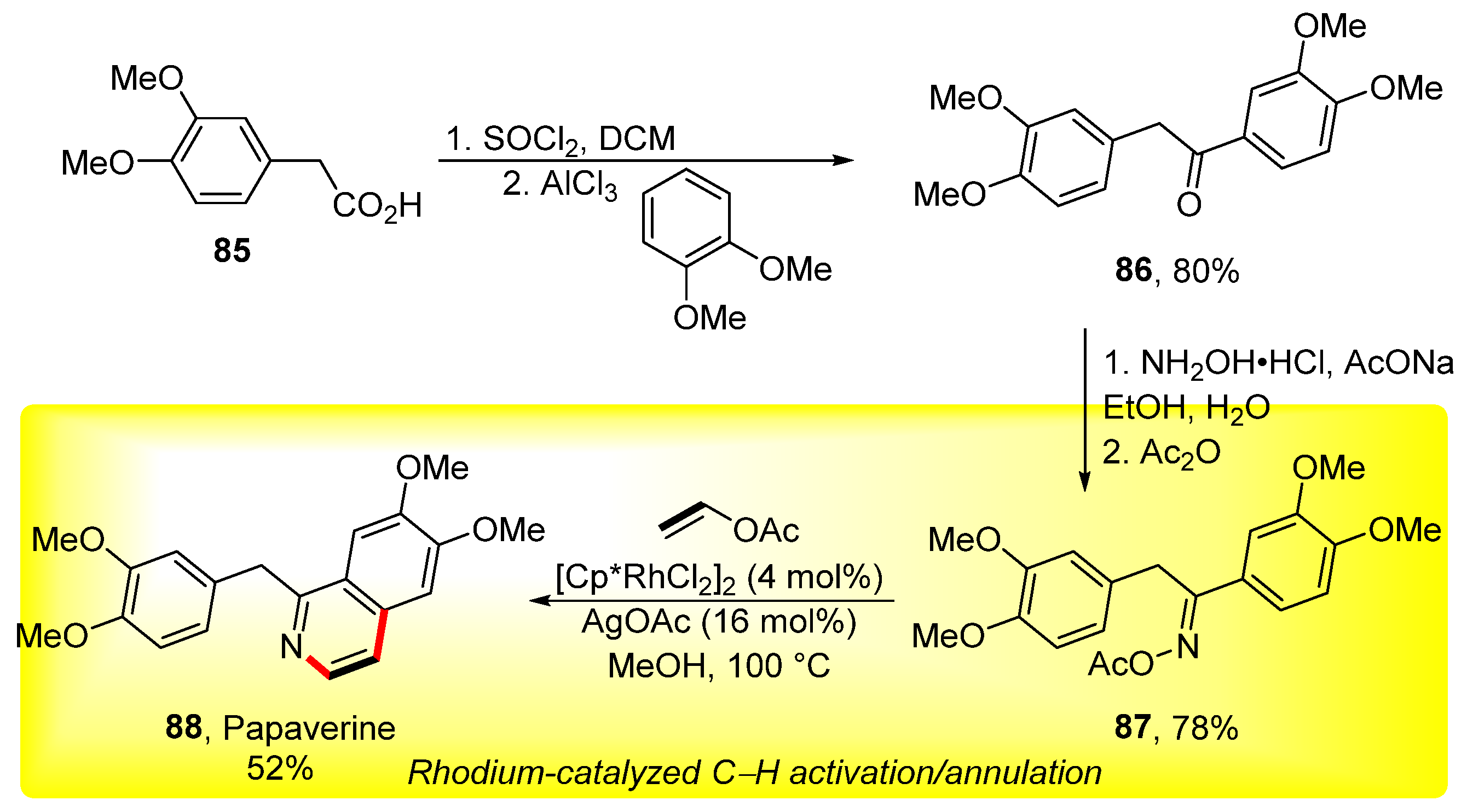
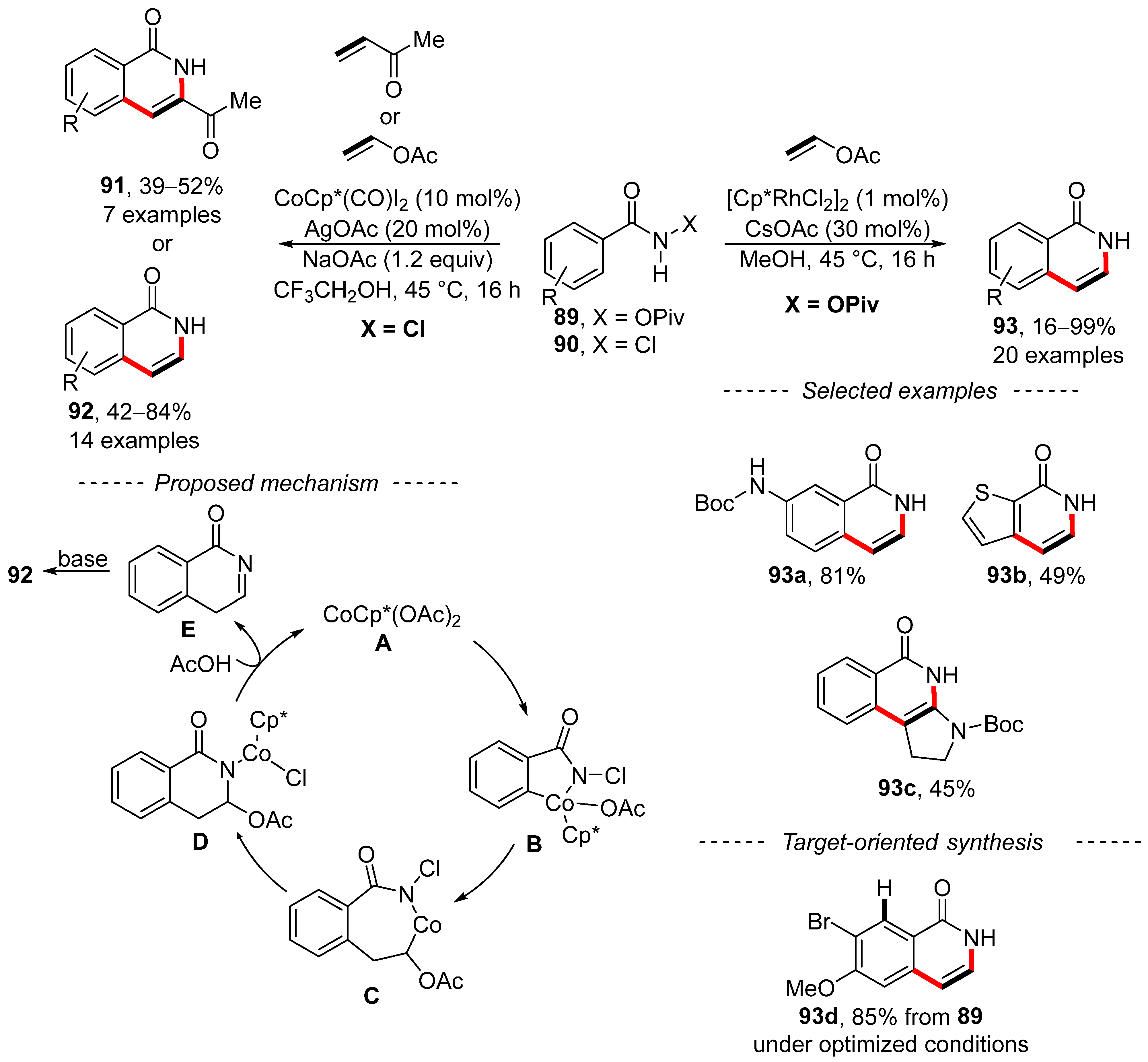
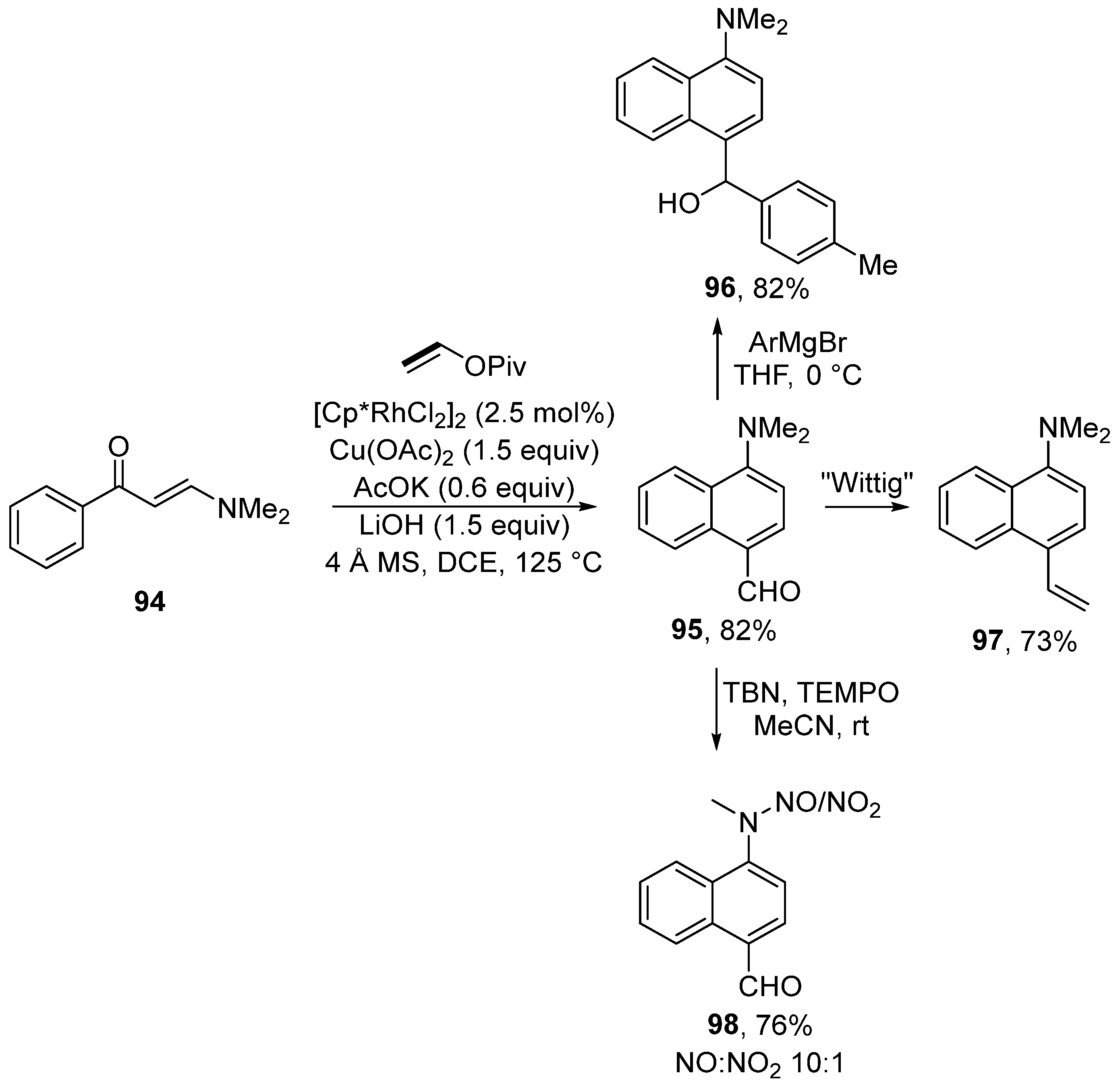
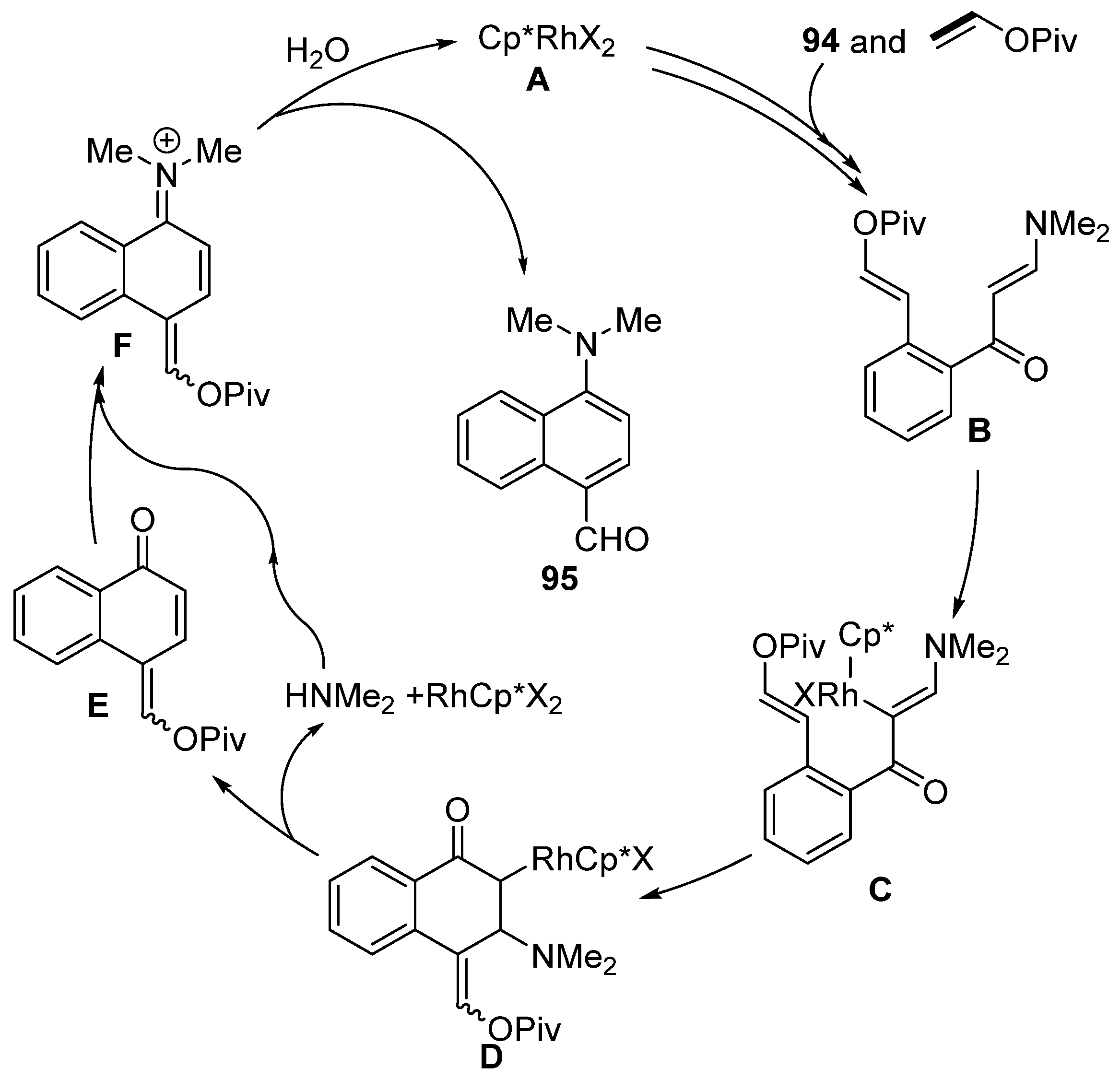
3. Cross-Coupling Reactions of Vinyl Acetates
4. Conclusions
Funding
Data Availability Statement
Conflicts of Interest
References
- Knappke, C.E.I.; Grupe, S.; Gärtner, D.; Corpet, M.; Gosmini, C.; Jacobi von Wangelin, A. Reductive Cross-Coupling Reactions between Two Electrophiles. Chem. Eur. J. 2014, 20, 6828–6842. [Google Scholar] [CrossRef]
- Lucas, E.L.; Jarvo, E.R. Stereospecific and Stereoconvergent Cross-Couplings Between Alkyl Electrophiles. Nat. Rev. Chem. 2017, 1, 0065. [Google Scholar] [CrossRef]
- Noël, T.; Buchwald, S.L. Cross-Coupling in Flow. Chem. Soc. Rev. 2011, 40, 5010–5029. [Google Scholar] [CrossRef]
- Rosen, B.M.; Quasdorf, K.W.; Wilson, D.A.; Zhang, N.; Resmerita, A.-M.; Garg, N.K.; Percec, V. Nickel-Catalyzed Cross-Couplings Involving Carbon–Oxygen Bonds. Chem. Rev. 2011, 111, 1346–1416. [Google Scholar] [CrossRef]
- Takise, R.; Muto, K.; Yamaguchi, J. Cross-Coupling of Aromatic Esters and Amides. Chem. Soc. Rev. 2017, 46, 5864–5888. [Google Scholar] [CrossRef]
- Friese, F.W.; Studer, A. New Avenues for C–B Bond Formation via Radical Intermediates. Chem. Sci. 2019, 10, 8503–8518. [Google Scholar] [CrossRef]
- Chen, K.; Wang, L.; Meng, G.; Li, P. Recent Advances in Transition-Metal-Free Aryl C–B Bond Formation. Synthesis 2017, 49, 4719–4730. [Google Scholar] [CrossRef]
- Verma, P.K.; Shegavi, M.L.; Bose, S.K.; Geetharani, K. A Nano-Catalytic Approach for C–B Bond Formation Reactions. Org. Biomol. Chem. 2018, 16, 857–873. [Google Scholar] [CrossRef]
- Yan, G.; Huang, D.; Wu, X. Recent Advances in C–B Bond Formation through a Free Radical Pathway. Adv. Synth. Catal. 2018, 360, 1040–1053. [Google Scholar] [CrossRef]
- Zhao, X.; Wang, G.; Hashmi, A.S.K. Carbene B–H Insertion Reactions for C–B Bond Formation. ChemCatChem 2021, 13, 4299–4312. [Google Scholar] [CrossRef]
- Kärkäs, M.D. Electrochemical Strategies for C–H Functionalization and C–N Bond Formation. Chem. Soc. Rev. 2018, 47, 5786–5865. [Google Scholar] [CrossRef]
- Kim, H.; Chang, S. Transition-Metal-Mediated Direct C–H Amination of Hydrocarbons with Amine Reactants: The Most Desirable but Challenging C–N Bond-Formation Approach. ACS Catal. 2016, 6, 2341–2351. [Google Scholar] [CrossRef]
- Oeser, P.; Koudelka, J.; Petrenko, A.; Tobrman, T. Recent Progress Concerning the N-Arylation of Indoles. Molecules 2021, 26, 5079. [Google Scholar] [CrossRef]
- Shin, K.; Kim, H.; Chang, S. Transition-Metal-Catalyzed C–N Bond Forming Reactions Using Organic Azides as the Nitrogen Source: A Journey for the Mild and Versatile C–H Amination. Acc. Chem. Res. 2015, 48, 1040–1052. [Google Scholar] [CrossRef]
- Song, G.; Wang, F.; Li, X. C–C, C–O and C–N Bond Formation via Rhodium(iii)-Catalyzed Oxidative C–H Activation. Chem. Soc. Rev. 2012, 41, 3651–3678. [Google Scholar] [CrossRef]
- Krylov, I.B.; Vil, V.A.; Terent’ev, A.O. Cross-Dehydrogenative Coupling for the Intermolecular C–O Bond Formation. Beilstein J. Org. Chem. 2015, 11, 92–146. [Google Scholar] [CrossRef]
- Lefèvre, G.; Franc, G.; Tlili, A.; Adamo, C.; Taillefer, M.; Ciofini, I.; Jutand, A. Contribution to the Mechanism of Copper-Catalyzed C–N and C–O Bond Formation. Organometallics 2012, 31, 7694–7707. [Google Scholar] [CrossRef]
- Stürmer, R. Take the Right Catalyst: Palladium-Catalyzed C–C, C–N, and C–O Bond Formation on Chloroarenes. Angew. Chem. Int. Ed. 1999, 38, 3307–3308. [Google Scholar] [CrossRef]
- Wu, X.-F.; Neumann, H. Zinc-Catalyzed Organic Synthesis: C–C, C–N, C–O Bond Formation Reactions. Adv. Synth. Catal. 2012, 354, 3141–3160. [Google Scholar] [CrossRef]
- Pan, X.-Q.; Zou, J.-P.; Yi, W.-B.; Zhang, W. Recent Advances in Sulfur- and Phosphorous-Centered Radical Reactions for the Formation of S–C and P–C Bonds. Tetrahedron 2015, 71, 7481–7529. [Google Scholar] [CrossRef]
- Tappe, F.M.J.; Trepohl, V.T.; Oestreich, M. Transition-Metal-Catalyzed C–P Cross-Coupling Reactions. Synthesis 2010, 3037–3062. [Google Scholar] [CrossRef]
- Wang, L.; Chen, H.; Duan, Z. Synthetic Applications of Transition-Metal-Catalyzed C–P Bond Cleavage. Chem. Asian J. 2018, 13, 2164–2173. [Google Scholar] [CrossRef]
- Wauters, I.; Debrouwer, W.; Stevens, C.V. Preparation of Phosphines Through C–P Bond Formation. Beilstein J. Org. Chem. 2014, 10, 1064–1096. [Google Scholar] [CrossRef]
- Zhang, H.; Zhang, X.-Y.; Dong, D.-Q.; Wang, Z.-L. Copper-Catalyzed Cross-Coupling Reactions for C–P Bond Formation. RSC Adv. 2015, 5, 52824–52831. [Google Scholar] [CrossRef]
- Bhunia, S.; Pawar, G.G.; Kumar, S.V.; Jiang, Y.; Ma, D. Selected Copper-Based Reactions for C–N, C–O, C–S, and C–C Bond Formation. Angew. Chem. Int. Ed. 2017, 56, 16136–16179. [Google Scholar] [CrossRef]
- Eichman, C.C.; Stambuli, J.P. Transition-Metal-Catalyzed Synthesis of Aryl Sulfides. Molecules 2011, 16, 590–608. [Google Scholar] [CrossRef]
- Li, J.; Yang, S.; Wu, W.; Jiang, H. Recent Developments in Palladium-Catalyzed C–S Bond Formation. Org. Chem. Front. 2020, 7, 1395–1417. [Google Scholar] [CrossRef]
- Shen, C.; Zhang, P.; Sun, Q.; Bai, S.; Hor, T.S.A.; Liu, X. Recent Advances in C–S Bond Formation via C–H Bond Functionalization and Decarboxylation. Chem. Soc. Rev. 2015, 44, 291–314. [Google Scholar] [CrossRef]
- Buttard, F.; Sharma, J.; Champagne, P.A. Recent Advances in the Stereoselective Synthesis of Acyclic All-Carbon Tetrasubstituted alkenes. Chem. Commun. 2021, 57, 4071–4088. [Google Scholar] [CrossRef]
- Edlová, T.; Čubiňák, M.; Tobrman, T. Cross-Coupling Reactions of Double or Triple Electrophilic Templates for Alkene Synthesis. Synthesis 2020, 53, 255–266. [Google Scholar]
- Flynn, A.B.; Ogilvie, W.W. Stereocontrolled Synthesis of Tetrasubstituted Olefins. Chem. Rev. 2007, 107, 4698–4745. [Google Scholar] [CrossRef]
- Mukherjee, N.; Planer, S.; Grela, K. Formation of Tetrasubstituted C–C Double Bonds via Olefin Metathesis: Challenges, Catalysts, and Applications in Natural Product Synthesis. Org. Chem. Front. 2018, 5, 494–516. [Google Scholar] [CrossRef]
- Negishi, E.-i.; Huang, Z.; Wang, G.; Mohan, S.; Wang, C.; Hattori, H. Recent Advances in Efficient and Selective Synthesis of Di-, Tri-, and Tetrasubstituted Alkenes via Pd-Catalyzed Alkenylation–Carbonyl Olefination Synergy. Acc. Chem. Res. 2008, 41, 1474–1485. [Google Scholar] [CrossRef]
- Paek, S.M. Synthesis of Tetrasubstituted Alkenes via Metathesis. Molecules 2012, 17, 3348–3358. [Google Scholar] [CrossRef]
- Polák, P.; Váňová, H.; Dvořák, D.; Tobrman, T. Recent Progress in Transition Metal-Catalyzed Stereoselective Synthesis of Acyclic All-Carbon Tetrasubstituted Alkenes. Tetrahedron Lett. 2016, 57, 3684–3693. [Google Scholar] [CrossRef]
- Reiser, O. Palladium-Catalyzed Coupling Reactions for the Stereoselective Synthesis of Tri- and Tetrasubstituted Alkenes. Angew. Chem. Int. Ed. 2006, 45, 2838–2840. [Google Scholar] [CrossRef]
- Tobrman, T.; Mrkobrada, S. Palladium-Catalyzed Cross-Coupling Reactions of Borylated Alkenes for the Stereoselective Synthesis of Tetrasubstituted Double Bond. Organics 2022, 3, 210–239. [Google Scholar] [CrossRef]
- Bhaskaran, S.; Padusha, M.S.A.; Sajith, A.M. Application of Palladium Based Precatalytic Systems in the Suzuki-Miyaura Cross-Coupling Reactions of Chloro- Heterocycles. ChemistrySelect 2020, 5, 9005–9016. [Google Scholar] [CrossRef]
- Fairlamb, I.J.S. Regioselective (Site-Selective) Functionalisation of Unsaturated Halogenated Nitrogen, Oxygen and Sulfur Heterocycles by Pd-Catalysed Cross-Couplings and Direct Arylation Processes. Chem. Soc. Rev. 2007, 36, 1036–1045. [Google Scholar] [CrossRef]
- Gandhi, S. Catalytic Enantioselective Cross Dehydrogenative Coupling of sp3 C–H of Heterocycles. Org. Biomol. Chem. 2019, 17, 9683–9692. [Google Scholar] [CrossRef]
- Heravi, M.M.; Hashemi, E. Recent Advances in Application of Intramolecular Suzuki Cross-Coupling in Cyclization and Heterocyclization. Monatsh. Chem. 2012, 143, 861–880. [Google Scholar] [CrossRef]
- Langer, P. Cross-Coupling Reactions of Polyhalogenated Heterocycles. Synlett 2022, 33, 1029–1051. [Google Scholar] [CrossRef]
- Liu, Y.; Wan, J.-P. Tandem Reactions Initiated by Copper-Catalyzed Cross-Coupling: A New Strategy Towards Heterocycle Synthesis. Org. Biomol. Chem. 2011, 9, 6873–6894. [Google Scholar] [CrossRef]
- Hopkinson, M.N.; Gee, A.D.; Gouverneur, V. AuI/AuIII Catalysis: An Alternative Approach for C–C Oxidative Coupling. Chem. Eur. J. 2011, 17, 8248–8262. [Google Scholar] [CrossRef]
- Jones, K.M.; Klussmann, M. Oxidative Coupling of Tertiary Amines: Scope, Mechanism and Challenges. Synlett 2012, 159–162. [Google Scholar] [CrossRef]
- Kozlowski, M.C. Oxidative Coupling in Complexity Building Transforms. Acc. Chem. Res. 2017, 50, 638–643. [Google Scholar] [CrossRef]
- Liu, J.; Ye, Y.; Sessler, J.L.; Gong, H. Cross-Electrophile Couplings of Activated and Sterically Hindered Halides and Alcohol Derivatives. Acc. Chem. Res. 2020, 53, 1833–1845. [Google Scholar] [CrossRef]
- Pang, X.; Peng, X.; Shu, X.-Z. Reductive Cross-Coupling of Vinyl Electrophiles. Synthesis 2020, 52, 3751–3763. [Google Scholar] [CrossRef]
- Pang, X.; Su, P.-F.; Shu, X.-Z. Reductive Cross-Coupling of Unreactive Electrophiles. Acc. Chem. Res. 2022, 55, 2491–2509. [Google Scholar] [CrossRef]
- Meng, G.; Shi, S.; Szostak, M. Cross-Coupling of Amides by N–C Bond Activation. Synlett 2016, 27, 2530–2540. [Google Scholar] [CrossRef]
- Pound, S.M.; Watson, M.P. Asymmetric Synthesis via Stereospecific C–N and C–O Bond Activation of Alkyl Amine and Alcohol Derivatives. Chem. Commun. 2018, 54, 12286–12301. [Google Scholar] [CrossRef]
- Wang, H.; Zhang, S.-Q.; Hong, X. Computational Studies on Ni-Catalyzed Amide C–N Bond Activation. Chem. Commun. 2019, 55, 11330–11341. [Google Scholar] [CrossRef]
- Wang, Q.; Su, Y.; Li, L.; Huang, H. Transition-Metal Catalysed C–N Bond Activation. Chem. Soc. Rev. 2016, 45, 1257–1272. [Google Scholar] [CrossRef]
- Cheng, H.-G.; Chen, H.; Liu, Y.; Zhou, Q. The Liebeskind–Srogl Cross-Coupling Reaction and its Synthetic Applications. Asian J. Org. Chem. 2018, 7, 490–508. [Google Scholar] [CrossRef]
- Otsuka, S.; Nogi, K.; Yorimitsu, H. C–S Bond Activation. Top. Curr. Chem. 2018, 376, 13. [Google Scholar] [CrossRef]
- Pan, F.; Shi, Z.-J. Recent Advances in Transition-Metal-Catalyzed C–S Activation: From Thioester to (Hetero)aryl Thioether. ACS Catal. 2014, 4, 280–288. [Google Scholar] [CrossRef]
- Bisz, E.; Szostak, M. Iron-Catalyzed C–O Bond Activation: Opportunity for Sustainable Catalysis. ChemSusChem 2017, 10, 3964–3981. [Google Scholar] [CrossRef]
- Sellars, J.D.; Steel, P.G. Transition Metal-Catalysed Cross-Coupling Reactions of P-activated Enols. Chem. Soc. Rev. 2011, 40, 5170–5180. [Google Scholar] [CrossRef]
- Tobisu, M.; Chatani, N. Cross-Couplings Using Aryl Ethers via C–O Bond Activation Enabled by Nickel Catalysts. Acc. Chem. Res. 2015, 48, 1717–1726. [Google Scholar] [CrossRef]
- Tobisu, M.; Chatani, N. Metal-Catalyzed Aromatic C–O Bond Activation/Transformation. In Organometallics for Green Catalysis; Dixneuf, P.H., Soulé, J.-F., Eds.; Springer International Publishing: Cham, Switzerland, 2019; pp. 103–140. [Google Scholar]
- Cornella, J.; Zarate, C.; Martin, R. Metal-Catalyzed Activation of Ethers via C–O Bond Cleavage: A New Strategy for Molecular Diversity. Chem. Soc. Rev. 2014, 43, 8081–8097. [Google Scholar] [CrossRef]
- Gandeepans, P.; Müller, T.; Zell, D.; Cera, G.; Warratz, S.; Ackermann, L. 3d Transition Metals for C–H Activation. Chem. Rev. 2019, 119, 2192–2452. [Google Scholar] [CrossRef]
- Boutadla, Y.; Davies, D.L.; Macgregor, S.A.; Poblador-Bahamonde, A.I. Mechanisms of C–H bond activation: Rich synergy between computation and experiment. Dalton Trans. 2009, 30, 5820–5831. [Google Scholar] [CrossRef]
- Kotek, V.; Polák, P.; Tobrman, T. Efficient and Simple Preparation of Functionalized 1,1-Dibromoenol Phosphates. Monat. Chem. 2016, 147, 405–412. [Google Scholar] [CrossRef]
- Ashida, Y.; Tanabe, Y. Cover Picture: Stereocomplementary and Parallel Syntheses of Multi-Substituted (E)-, (Z)-Stereodefined α,β-Unsaturated Esters: Application to Drug Syntheses. Chem. Rec. 2020, 20, 1409. [Google Scholar] [CrossRef]
- Chassaing, S.; Specklin, S.; Weibel, J.-M.; Pale, P. Vinyl and Aryl Sulfonates: Preparations and Applications in Total Synthesis. Curr. Org. Synth. 2012, 9, 806–827. [Google Scholar] [CrossRef]
- Li, Z.; Ning, Z.; Yu, H.; Jiao, L. Research Progress in the Preparation of Aryl and Alkyl Mixed Phosphates. Chin. J. Org. Chem. 2021, 41, 4180–4191. [Google Scholar] [CrossRef]
- Perkow, W.; Ullerich, K.; Meyer, F. Neue Phosphorsäureester mit pupillenverengender Wirkung. Naturwissenschaften 1952, 39, 353. [Google Scholar] [CrossRef]
- Kotek, V.; Polák, P.; Dvořáková, H.; Tobrman, T. Aluminum Chloride Promoted Cross-Coupling of Trisubstituted Enol Phosphates with Organozinc Reagents En Route to the Stereoselective Synthesis of Tamoxifen and Its Analogues. Eur. J. Org. Chem. 2016, 5037–5044. [Google Scholar] [CrossRef]
- Kotek, V.; Dvořáková, H.; Tobrman, T. Modular and Highly Stereoselective Approach to All-Carbon Tetrasubstituted Alkenes. Org. Lett. 2015, 17, 608–611. [Google Scholar] [CrossRef]
- Koudelka, J.; Tobrman, T. Synthesis of 2-Substituted Cyclobutanones by a Suzuki Reaction and Dephosphorylation Sequence. Eur. J. Org. Chem. 2021, 3260–3269. [Google Scholar] [CrossRef]
- Edlová, T.; Dvořáková, H.; Eigner, V.; Tobrman, T. Substrate-Controlled Regioselective Bromination of 1,2-Disubstituted Cyclobutenes: An Application in the Synthesis of 2,3-Disubstituted Cyclobutenones. J. Org. Chem. 2021, 86, 5820–5831. [Google Scholar] [CrossRef]
- Čubiňák, M.; Bigeon, J.; Galář, P.; Ondič, L.; Tobrman, T. The Synthesis of Tetrasubstituted Cycloalkenes Bearing π-Conjugated Substituents and Their Optical Properties. ChemistrySelect 2021, 6, 9904–9910. [Google Scholar] [CrossRef]
- Čubiňák, M.; Tobrman, T. Room-Temperature Negishi Reaction of Trisubstituted Vinyl Phosphates for the Synthesis of Tetrasubstituted Alkenes. J. Org. Chem. 2020, 85, 10728–10739. [Google Scholar] [CrossRef]
- Polák, P.; Tobrman, T. Novel Selective Approach to Terminally Substituted [n]Dendralenes. Eur. J. Org. Chem. 2019, 957–968. [Google Scholar] [CrossRef]
- Polák, P.; Tobrman, T. The synthesis of polysubstituted indoles from 3-bromo-2-indolyl phosphates. Org. Biomol. Chem. 2017, 15, 6233–6241. [Google Scholar] [CrossRef]
- Barkley, L.B.; Knowles, W.S.; Raffelson, H.; Thompson, Q.E. Studies in Steroid Total Synthesis. IV. A Stereoselective Ring A Synthesis1. J. Am. Chem. Soc. 1956, 78, 4111–4116. [Google Scholar] [CrossRef]
- Kumar, S.; Khatri, V.; Mangla, P.; Chhatwal, R.J.; Parmar, V.S.; Prasad, A.K. C-Glycopyranosyl Aldehydes: Emerging Chiral Synthons in Organic Synthesis. RSC Adv. 2023, 13, 19898–19954. [Google Scholar] [CrossRef]
- Ilia, G.; Simulescu, V.; Plesu, N.; Chiriac, V.; Merghes, P. Wittig and Wittig–Horner Reactions under Sonication Conditions. Molecules 2023, 28, 1958. [Google Scholar] [CrossRef]
- Morodo, R.; Bianchi, P.; Monbaliu, J.-C.M. Continuous Flow Organophosphorus Chemistry. Eur. J. Org. Chem. 2020, 5236–5277. [Google Scholar] [CrossRef]
- Heravi, M.M.; Zadsirjan, V.; Daraie, M.; Ghanbarian, M. Applications of Wittig Reaction in the Total Synthesis of Natural Macrolides. ChemistrySelect 2020, 5, 9654–9690. [Google Scholar] [CrossRef]
- Heravi, M.M.; Ghanbarian, M.; Zadsirjan, V.; Alimadadi Jani, B. Recent Advances in the Applications of Wittig Reaction in the Total Synthesis of Natural Products Containing Lactone, Pyrone, and Lactam as a Scaffold. Monatsh. Chem. 2019, 150, 1365–1407. [Google Scholar] [CrossRef]
- Ager, D.J. Peterson Alkenation. Sci. Synth. 2010, 47, 85. [Google Scholar] [CrossRef]
- Frances van Staden, L.; Gravestock, D.; Ager, D.J. New developments in the Peterson olefination reaction. Chem. Soc. Rev. 2002, 31, 195–200. [Google Scholar] [CrossRef]
- Wang, Y.; Wu, Y.; Lia, Y.; Tang, Y. Denitrogenative Suzuki and carbonylative Suzuki coupling reactions of benzotriazoles with boronic acids. Chem. Sci. 2017, 8, 3852–3857. [Google Scholar] [CrossRef]
- Kálai, T.; Jekő, J.; Hideg, K. Synthesis of Isoindoline Nitroxides by Electrocyclic Reactions. Synthesis 2009, 2591–2595. [Google Scholar] [CrossRef]
- Peyroux, E.; Berthiol, F.; Doucet, H.; Santelli, M. Suzuki Cross-Coupling Reactions between Alkenylboronic Acids and Aryl Bromides Catalysed by a Tetraphosphane-Palladium Catalyst. Eur. J. Org. Chem. 2004, 1075–1082. [Google Scholar] [CrossRef]
- Okazaki, R.; Ooka, M.; Tokitoh, N.; Inamoto, N. Synthesis and Reactions of 1,6-Dithiocyanato- and 1,6-Diiodo-1,3,5-cycloheptatrienes. J. Org. Chem. 1985, 50, 180–185. [Google Scholar] [CrossRef]
- McLaughlin, M.L.; McKinney, J.A.; Paquette, L.A. Efficient Preparation of Homochiral Bicyclo-Annulated Cyclopentadienes via the Skattebøl Rearrangement. Avoidance of limitations due to angle strain. Tetrahedron Lett. 1986, 27, 5595–5598. [Google Scholar] [CrossRef]
- Labadie, J.W.; Stille, J.K. Mechanisms of the Palladium-Catalyzed Couplings of Acid Chlorides with Organotin Reagents. J. Am. Chem. Soc. 1983, 105, 6129–6137. [Google Scholar] [CrossRef]
- Adamson, N.J.; Jeddi, H.; Malcolmson, S.J. Preparation of Chiral Allenes through Pd-Catalyzed Intermolecular Hydroamination of Conjugated Enynes: Enantioselective Synthesis Enabled by Catalyst Design. J. Am. Chem. Soc. 2019, 141, 8574–8583. [Google Scholar] [CrossRef]
- Du, F.; Yin, L.; Ning, Y.; Peng, Y. Catalytic Asymmetric Synthesis of Phosphoryl-1,4-dihydropyridazines via an Enantioselective Allylic Alkylation/1,3-Dipolar Cycloaddition/Rearrangement Reaction Sequence. Adv. Synth. Catal. 2016, 358, 2280–2285. [Google Scholar] [CrossRef]
- Sirotkina, J.; Grigorjeva, L.; Jirgensons, A. Synthesis of Alkynyl-Glycinols by Lewis Acid Catalyzed Propargylic Substitution of Bis-Imidates. Eur. J. Org. Chem. 2015, 6900–6908. [Google Scholar] [CrossRef]
- Cheng, J.-K.; Loh, T.-P. Copper- and Cobalt-Catalyzed Direct Coupling of sp3 α-Carbon of Alcohols with Alkenes and Hydroperoxides. J. Am. Chem. Soc. 2015, 137, 42–45. [Google Scholar] [CrossRef]
- Seath, C.P.; Vogt, D.B.; Xu, Z.; Boyington, A.J.; Jui, N.T. Radical Hydroarylation of Functionalized Olefins and Mechanistic Investigation of Photocatalytic Pyridyl Radical Reactions. J. Am. Chem. Soc. 2018, 140, 15525–15534. [Google Scholar] [CrossRef]
- Groves, A.; Martínez, J.I.; Smith, J.J.; Lam, H.W. Remote Nucleophilic Allylation by Allylrhodium Chain Walking. Chem. Eur. J. 2018, 24, 13432–13436. [Google Scholar] [CrossRef]
- Le Fèvre, R.J.W.; Sundaram, A. 787. Molecular polarisability: The conformations of ten cyclic dibasic acid anhydrides indicated by their dipole moments, molar Kerr constants, etc. J. Chem. Soc. (Resumed) 1962, 4009–4019. [Google Scholar] [CrossRef]
- Agents Classified by the IARC Monographs, Volumes 1–134. Available online: https://monographs.iarc.who.int/list-of-classifications (accessed on 20 August 2023).
- Mykura, R.C.; Songara, P.; Luc, E.; Rogers, J.; Stammers, E.; Aggarwal, V.K. Studies on the Lithiation, Borylation, and 1,2-Metalate Rearrangement of O-Cycloalkyl 2,4,6-Triisopropylbenzoates. Angew. Chem. Int. Ed. 2021, 60, 11436–11441. [Google Scholar] [CrossRef]
- Wang, Y.; Noble, A.; Myers, E.L.; Aggarwal, V.K. Enantiospecific Alkynylation of Alkylboronic Esters. Angew. Chem. Int. Ed. 2016, 55, 4270–4274. [Google Scholar] [CrossRef]
- Kumpulainen, H.; Järvinen, T.; Saari, R.; Lehtonen, M.; Vepsäläinen, J. An Efficient Strategy for the Synthesis of 1-Chloroethyl Phosphates and Phosphoramidates. J. Org. Chem. 2005, 70, 9056–9058. [Google Scholar] [CrossRef]
- Zhao, S.; Cai, X.; Lu, Y.; Hu, J.; Xiong, Z.; Jin, J.; Li, Y.; Wang, H.; Wu, J.-Q. Cp*Ir(III) and Cp*Rh(III)-Catalyzed Annulation of Salicylaldehydes with Fluorinated Vinyl Tosylates. Chem. Commun. 2022, 58, 8966–8969. [Google Scholar] [CrossRef]
- Gøgsig, T.M.; Søbjerg, L.S.; Lindhardt, A.T.; Jensen, K.L.; Skrydstrup, T. Direct Vinylation and Difluorovinylation of Arylboronic Acids Using Vinyl- and 2,2-Difluorovinyl Tosylates via the Suzuki–Miyaura Cross Coupling. J. Org. Chem. 2008, 73, 3404–3410. [Google Scholar] [CrossRef]
- McCammant, M.S.; Shigeta, T.; Sigman, M.S. Palladium-Catalyzed 1,3-Difunctionalization Using Terminal Alkenes with Alkenyl Nonaflates and Aryl Boronic Acids. Org. Lett. 2016, 18, 1792–1795. [Google Scholar] [CrossRef]
- Lyapkalo, I.M.; Webel, M.; Reißig, H.-U. Synthesis and Heck Reactions of Ethenyl- and (Z)-Butadien-1-yl Nonaflate Obtained by the Fragmentation of Furan Derivatives. Eur. J. Org. Chem. 2001, 4189–4194. [Google Scholar] [CrossRef]
- Song, B.; Knauber, T.; Gooßen, L.J. Decarboxylative Cross-Coupling of Mesylates Catalyzed by Copper/Palladium Systems with Customized Imidazolyl Phosphine Ligands. Angew. Chem. Int. Ed. 2013, 52, 2954–2958. [Google Scholar] [CrossRef]
- Fawcett, A.; Biberger, T.; Aggarwal, V.K. Carbopalladation of C–C σ-Bonds Enabled by Strained Boronate Complexes. Nat. Chem. 2019, 11, 117–122. [Google Scholar] [CrossRef]
- Shirakawa, E.; Sato, T.; Imazaki, Y.; Kimura, T.; Hayashi, T. Cobalt-Catalyzed Cross-Coupling of Alkynyl Grignard Reagents with Alkenyl Triflates. Chem. Commun. 2007, 43, 4513–4515. [Google Scholar] [CrossRef]
- Harkness, G.J.; Clarke, M.L. A Highly Enantioselective Alkene Methoxycarbonylation Enables a Concise Synthesis of (S)-Flurbiprofen. Eur. J. Org. Chem. 2017, 4859–4863. [Google Scholar] [CrossRef]
- Dunet, G.; Knochel, P. Iron-Catalyzed Cross-Coupling between Alkenyl and Dienyl Sulfonates and Functionalized Arylcopper Reagents. Synlett 2006, 3, 0407–0410. [Google Scholar] [CrossRef]
- Li-Yuan Bao, R.; Zhao, R.; Shi, L. Progress and Developments in the Turbo Grignard Reagent i-PrMgCl·LiCl: A Ten-Year Journey. Chem. Commun. 2015, 51, 6884–6900. [Google Scholar] [CrossRef]
- Adrio, J.; Carretero, J.C. Functionalized Grignard Reagents in Kumada Cross-Coupling Reactions. ChemCatChem 2010, 2, 1384–1386. [Google Scholar] [CrossRef]
- Ila, H.; Baron, O.; Wagner, A.J.; Knochel, P. Functionalized Magnesium Organometallics as Versatile Intermediates for the Synthesis of Polyfunctional Heterocycles. Chem. Commun. 2006, 6, 583–593. [Google Scholar] [CrossRef]
- Knochel, P.; Dohle, W.; Gommermann, N.; Kneisel, F.F.; Kopp, F.; Korn, T.; Sapountzis, I.; Vu, V.A. Highly Functionalized Organomagnesium Reagents Prepared through Halogen–Metal Exchange. Angew. Chem. Int. Ed. 2003, 42, 4302–4320. [Google Scholar] [CrossRef]
- Tobrman, T.; Dvořák, D. Heck reactions of 6- and 2-halopurines. Eur. J. Org. Chem. 2008, 2923–2928. [Google Scholar] [CrossRef]
- Khan, R.I.; Pitchumani, K. A Pyridinium Modified β-Cyclodextrin: An Ionic Supramolecular Ligand for Palladium Acetate in C–C Coupling Reactions in Water. Green Chem. 2016, 18, 5518–5528. [Google Scholar] [CrossRef]
- Berthiol, F.; Doucet, H.; Santelli, M. Heck Reaction of Vinyl Bromides with Alkenes in the Presence of a Tetra phosphine/Palladium Catalyst. Synlett 2003, 0841–0844. [Google Scholar] [CrossRef]
- Hansen, A.L.; Ebran, J.-P.; Ahlquist, M.; Norrby, P.-O.; Skrydstrup, T. Heck Coupling with Nonactivated Alkenyl Tosylates and Phosphates: Examples of Effective 1,2-Migrations of the Alkenyl Palladium(II) Intermediates. Angew. Chem. Int. Ed. 2006, 45, 3349–3353. [Google Scholar] [CrossRef]
- Fu, W.C.; Wu, Y.; So, C.M.; Wong, S.M.; Lei, A.; Kwong, F.Y. Catalytic Direct C2-Alkenylation of Oxazoles at Parts per Million Levels of Palladium/PhMezole-Phos Complex. Org. Lett. 2016, 18, 5300–5303. [Google Scholar] [CrossRef]
- Gui, Y.-Y.; Liao, L.-L.; Sun, L.; Zhang, Z.; Ye, J.-H.; Shen, G.; Lu, Z.-P.; Zhou, W.-J.; Yu, D.-G. Coupling of C(sp3)–H bonds with C(sp2)–O Electrophiles: Mild, General and Selective. Chem. Commun. 2017, 53, 1192–1195. [Google Scholar] [CrossRef]
- Kumar, D.; Chen, M.S.; Goodman, D.W. Synthesis of Vinyl Acetate on Pd-Based Catalysts. Catal. Today 2007, 123, 77–85. [Google Scholar] [CrossRef]
- Gärtner, D.; Stein, A.L.; Grupe, S.; Arp, J.; Jacobi von Wangelin, A. Iron-Catalyzed Cross-Coupling of Alkenyl Acetates. Angew. Chem. Int. Ed. 2015, 54, 10545–10549. [Google Scholar] [CrossRef]
- Su, M.; Huang, X.; Lei, C.; Jin, J. Nickel-Catalyzed Reductive Cross-Coupling of Aryl Bromides with Vinyl Acetate in Dimethyl Isosorbide as a Sustainable Solvent. Org. Lett. 2022, 24, 354–358. [Google Scholar] [CrossRef]
- Zhou, B.; Qi, X.; Liu, P.; Dong, G. Development and Mechanistic Studies of the Iridium-Catalyzed C–H Alkenylation of Enamides with Vinyl Acetates: A Versatile Approach for Ketone Functionalization. Angew. Chem. Int. Ed. 2021, 60, 20926–20934. [Google Scholar] [CrossRef]
- Sk, M.R.; Maji, M.S. Cobalt(III)-Catalyzed Ketone-Directed C–H Vinylation Using Vinyl Acetate. Org. Chem. Front. 2020, 7, 19–24. [Google Scholar] [CrossRef]
- Sarkar, D.; Venkateswaran, R.V. Synthesis of Bruguierol A Employing Ring-Closing Metathesis. Tetrahedron Lett. 2011, 52, 3232–3233. [Google Scholar] [CrossRef]
- Otley, K.D.; Ellman, J.A. An Efficient Method for the Preparation of Styrene Derivatives via Rh(III)-Catalyzed Direct C–H Vinylation. Org. Lett. 2015, 17, 1332–1335. [Google Scholar] [CrossRef]
- Mei, S.-T.; Jiang, K.; Wang, N.-J.; Shuai, L.; Yuan, Y.; Wei, Y. Rhodium-Catalyzed Direct C–H Vinylation of Arenes To Access Styrenes with Vinyl Acetate as a Vinyl Source. Eur. J. Org. Chem. 2015, 6135–6140. [Google Scholar] [CrossRef]
- Gophane, D.B.; Endeward, B.; Prisner, T.F.; Sigurdsson, S.T. A Semi-Rigid Isoindoline-Derived Nitroxide Spin Label for RNA. Org. Biomol. Chem. 2018, 16, 816–824. [Google Scholar] [CrossRef]
- Beng, T.K.; Wilkerson-Hill, S.M.; Sarpong, R. Direct Access to Functionalized Azepanes by Cross-Coupling with α-Halo Eneformamides. Org. Lett. 2014, 16, 916–919. [Google Scholar] [CrossRef]
- Liu, W.; Li, Y.; Xu, B.; Kuang, C. Palladium-Catalyzed Olefination and Arylation of 2-Substituted 1,2,3-Triazole N-Oxides. Org. Lett. 2013, 15, 2342–2345. [Google Scholar] [CrossRef]
- Yang, Y.; Kuang, C. Room-Temperature Direct Alkenylation of 3-Arylsydnones. Eur. J. Org. Chem. 2014, 7810–7813. [Google Scholar] [CrossRef]
- Li, S.-S.; Wang, C.-Q.; Lin, H.; Zhang, X.-M.; Dong, L. Rhodium(III)-Catalyzed C–C Coupling of 7-Azaindoles with Vinyl Acetates and Allyl Acetates. Org. Biomol. Chem. 2016, 14, 229–237. [Google Scholar] [CrossRef]
- Lin, W.; Li, W.; Lu, D.; Su, F.; Wen, T.-B.; Zhang, H.-J. Dual Effects of Cyclopentadienyl Ligands on Rh(III)-Catalyzed Dehydrogenative Arylation of Electron-Rich Alkenes. ACS Catal. 2018, 8, 8070–8076. [Google Scholar] [CrossRef]
- Qin, W.-B.; Li, W.-W.; Zhu, P.-F.; Mo, X.-G.; Zhang, H.-J.; Wen, T.-B. Rh(III)-Catalyzed Regio- and Stereoselective Bisindolylation of Vinyl Acetate: An Efficient Approach toward (E)-1,2-Bis(2-indolyl)ethenes. Org. Chem. Front. 2018, 5, 1096–1100. [Google Scholar] [CrossRef]
- Zhang, H.-J.; Lin, W.; Su, F.; Wen, T.-B. Rhodium-Catalyzed β-Selective Oxidative Heck-Type Coupling of Vinyl Acetate via C–H Activation. Org. Lett. 2016, 18, 6356–6359. [Google Scholar] [CrossRef]
- Moghaddam, F.M.; Pourkaveh, R.; Karimi, A. Oxidative Heck Reaction as a Tool for Para-selective Olefination of Aniline: A DFT Supported Mechanism. J. Org. Chem. 2017, 82, 10635–10640. [Google Scholar] [CrossRef]
- Wu, J.; Cao, K.; Xu, T.-T.; Zhang, X.-J.; Jiang, L.; Yang, J.; Huang, Y. Palladium-Catalyzed Regioselective Mono-Alkenylation of o-Carboranes via Heck Type Coupling Reaction of a Cage B–H Bond. RSC Adv. 2015, 5, 91683–91685. [Google Scholar] [CrossRef]
- Karimi, B.; Behzadnia, H.; Elhamifar, D.; Akhavan, P.F.; Esfahani, F.K.; Zamani, A. Transition-Metal-Catalyzed Oxidative Heck Reactions. Synthesis 2010, 1399–1427. [Google Scholar] [CrossRef]
- Meng, L.; Liu, C.; Zhang, W.; Zhou, C.; Lei, A. Palladium-Catalysed β-Selective Oxidative Heck Reaction of an Electron-Rich Olefin. Chem. Commun. 2014, 50, 1110–1112. [Google Scholar] [CrossRef]
- Odell, L.R.; Lindh, J.; Gustafsson, T.; Larhed, M. Continuous Flow Palladium(II)-Catalyzed Oxidative Heck Reactions with Arylboronic Acids. Eur. J. Org. Chem. 2010, 2270–2274. [Google Scholar] [CrossRef]
- Öhrngren, P.; Fardost, A.; Russo, F.; Schanche, J.-S.; Fagrell, M.; Larhed, M. Evaluation of a Nonresonant Microwave Applicator for Continuous-Flow Chemistry Applications. Org. Process Res. Dev. 2012, 16, 1053–1063. [Google Scholar] [CrossRef]
- Chu, H.; Sun, S.; Yu, J.-T.; Cheng, J. Rh-Catalyzed Sequential Oxidative C–H Activation/Annulation with Geminal-Substituted Vinyl Acetates to Access Isoquinolines. Chem. Commun. 2015, 51, 13327–13329. [Google Scholar] [CrossRef]
- Webb, N.J.; Raw, S.A.; Marsden, S.P. Isoquinoline Synthesis by C–H Activation/Annulation Using Vinyl Acetate as an Acetylene Equivalent. Tetrahedron 2018, 74, 5200–5205. [Google Scholar] [CrossRef]
- Jothi Murugan, S.; Jeganmohan, M. Cp*Co(III)-Catalyzed Regioselective [4 + 2]-Annulation of N-Chlorobenzamides with Vinyl Acetate/Vinyl Ketones. J. Org. Chem. 2023, 88, 1578–1589. [Google Scholar] [CrossRef]
- Webb, N.J.; Marsden, S.P.; Raw, S.A. Rhodium(III)-Catalyzed C–H Activation/Annulation with Vinyl Esters as an Acetylene Equivalent. Org. Lett. 2014, 16, 4718–4721. [Google Scholar] [CrossRef]
- Liang, G.; Rong, J.; Sun, W.; Chen, G.; Jiang, Y.; Loh, T.-P. Synthesis of Polyaromatic Rings: Rh(III)-Catalyzed [5 + 1] Annulation of Enaminones with Vinyl Esters through C–H Bond Functionalization. Org. Lett. 2018, 20, 7326–7331. [Google Scholar] [CrossRef]
- Zhang, M.; Zhang, H.-J.; Han, T.; Ruan, W.; Wen, T.-B. Rh(III)-Catalyzed Oxidative Coupling of Benzoic Acids with Geminal-Substituted Vinyl Acetates: Synthesis of 3-Substituted Isocoumarins. J. Org. Chem. 2015, 80, 620–627. [Google Scholar] [CrossRef]
- Lou, M.; Deng, Z.; Mao, X.; Fu, Y.; Yang, Q.; Peng, Y. Rhodium-catalyzed C–H bond activation alkylation and cyclization of 2-arylquinazolin-4-ones. Org. Biomol. Chem. 2018, 16, 1851–1859. [Google Scholar] [CrossRef]
- Li, S.-S.; Liu, C.-F.; Xia, Y.-Q.; Li, W.-H.; Zhang, G.-T.; Zhang, X.-M.; Dong, L. A Unique Annulation of 7-Azaindoles with Alkenyl Esters to Produce π-Conjugated 7-Azaindole Derivatives. Org. Biomol. Chem. 2016, 14, 5214–5218. [Google Scholar] [CrossRef]
- Pinna, C.; Martino, P.A.; Meroni, G.; Sora, V.M.; Tamborini, L.; Dallavalle, S.; Contente, M.L.; Pinto, A. Biocatalyzed Synthesis of Vanillamides and Evaluation of Their Antimicrobial Activity. J. Agric. Food Chem. 2022, 70, 223–228. [Google Scholar] [CrossRef]
- Cavallari, E.; Carrera, C.; Aime, S.; Reineri, F. 13C MR Hyperpolarization of Lactate by Using ParaHydrogen and Metabolic Transformation in Vitro. Chem. Eur. J. 2017, 23, 1200–1204. [Google Scholar] [CrossRef]
- Gregorić, T.; Makarević, J.; Štefanić, Z.; Žinić, M.; Frkanec, L. Gamma Radiation- and Ultraviolet-Induced Polymerization of Bis(amino acid)fumaramide Gel Assemblies. Polymers 2022, 14, 214. [Google Scholar] [CrossRef]
- Deng, W.; Hu, Y.; Hu, J.; Li, X.; Li, Y.; Huang, Y. Electrochemically induced Markovnikov-type selective hydro/deuterophosphonylation of electron-rich alkenes. Chem. Commun. 2022, 58, 12094–12097. [Google Scholar] [CrossRef]
- Zeng, T.; You, W.; Chen, G.; Nie, X.; Zhang, Z.; Xia, L.; Hong, C.; Chen, C.; You, Y. Degradable PE-Based Copolymer with Controlled Ester Structure Incorporation by Cobalt-Mediated Radical Copolymerization under Mild Condition. iScience 2020, 23, 100904. [Google Scholar] [CrossRef]
- Young, C.M.; Taylor, J.E.; Smith, A.D. Evaluating Aryl Esters as Bench-Stable C(1)-Ammonium Enolate Precursors in Catalytic, Enantioselective Michael Addition–Lactonisations. Org. Biomol. Chem. 2019, 17, 4747–4752. [Google Scholar] [CrossRef]
- Kadidae, L.O.; Usami, A.; Honda, M. Palladium(II) Acetate as Catalyst in Transvinylation Reactions of Hydroxycinnamic Acid and Its Derivatives. Asian J. Chem. 2018, 30, 589–593. [Google Scholar] [CrossRef]
- Sato, K.; Kobayashi, S.; Sekishita, A.; Wakui, M.; Tanaka, M. Synthesis and Thrombogenicity Evaluation of Poly(3-methoxypropionic acid vinyl ester): A Candidate for Blood-Compatible Polymers. Biomacromolecules 2017, 18, 1609–1616. [Google Scholar] [CrossRef]
- Hibert, G.; Grau, E.; Pintori, D.; Lecommandoux, S.; Cramail, H. ADMET Polymerization of α,ω-Unsaturated Glycolipids: Synthesis and Physico-Chemical Properties of the Resulting Polymers. Polym. Chem. 2017, 8, 3731–3739. [Google Scholar] [CrossRef]
- Hedir, G.G.; Arno, M.C.; Langlais, M.; Husband, J.T.; O’Reilly, R.K.; Dove, A.P. Poly(oligo(ethylene glycol) vinyl acetate)s: A Versatile Class of Thermoresponsive and Biocompatible Polymers. Angew. Chem. Int. Ed. 2017, 56, 9178–9182. [Google Scholar] [CrossRef]
- Krejzová, J.; Šimon, P.; Vavříková, E.; Slámová, K.; Pelantová, H.; Riva, S.; Spiwok, V.; Křen, V. Enzymatic Synthesis of New C-6-acylated Derivatives of NAG-thiazoline and Evaluation of their Inhibitor Activities Towards Fungal β-N-acetylhexosaminidase. J. Mol. Catal. B Enzym. 2013, 87, 128–134. [Google Scholar] [CrossRef]
- Martínez-Montero, S.; Fernández, S.; Sanghvi, Y.S.; Gotor, V.; Ferrero, M. An Expedient Biocatalytic Procedure for Abasic Site Precursors Useful in Oligonucleotide Synthesis. Org. Biomol. Chem. 2011, 9, 5960–5966. [Google Scholar] [CrossRef]
- Padrosa, D.R.; Contente, M.L. Multi-Gram Preparation of Cinnamoyl Tryptamines as Skin Whitening Agents Through a Chemo-Enzymatic Flow Process. Tetrahedron Lett. 2021, 86, 153453. [Google Scholar] [CrossRef]
- Jebrane, M.; Terziev, N.; Heinmaa, I. Biobased and Sustainable Alternative Route to Long-Chain Cellulose Esters. Biomacromolecules 2017, 18, 498–504. [Google Scholar] [CrossRef]
- Vilela, C.; Rua, R.; Silvestre, A.J.D.; Gandini, A. Polymers and Copolymers from Fatty Acid-Based Monomers. Ind. Crops Prod. 2010, 32, 97–104. [Google Scholar] [CrossRef]
- Lou, S.; Gao, S.; Wang, W.; Zhang, M.; Zhang, J.; Wang, C.; Li, C.; Kong, D.; Zhao, Q. Galactose-Functionalized Multi-Responsive Nanogels for Hepatoma-Targeted Drug Delivery. Nanoscale 2015, 7, 3137–3146. [Google Scholar] [CrossRef]
- Shen, F.-W.; Zhou, K.-C.; Cai, H.; Zhang, Y.-N.; Zheng, Y.-L.; Quan, J. One-Pot Synthesis of Thermosensitive Glycopolymers Grafted Gold Nanoparticles and Their Lectin Recognition. Colloids Surf. B 2019, 173, 504–511. [Google Scholar] [CrossRef]
- Adelman, R.L. The Interchange Reaction of Vinyl Acetate with Organic Acids. J. Org. Chem. 1949, 14, 1057–1077. [Google Scholar] [CrossRef]
- Jiang, R.; Chen, Z.; Zhan, K.; Liu, L.; Zhou, J.; Ai, Y.; Li, S.; Bao, H.; Hu, Z.n.; Qi, L.; et al. Reusable Rhodium Catalyst for the Selective Transvinylation of sp2-C Linked Carboxylic Acid. Tetrahedron Lett. 2018, 59, 3279–3282. [Google Scholar] [CrossRef]
- Ketterling, A.A.; Lisitsyn, A.S.; Nosov, A.V.; Likholobov, V.A. Carboxylic Acid Transvinylation as Catalysed by Complexes of Palladium Acetate with Phenanthroline-Like Ligands. Appl. Catal. 1990, 66, 123–131. [Google Scholar] [CrossRef]
- Henry, P.M. Palladium(II)-Catalyzed Exchange and Isomerization Reactions. I. Exchange of Enol Acetates with Acetic Acid Catalyzed by Palladium(II) Chloride. J. Am. Chem. Soc. 1971, 93, 3853–3859. [Google Scholar] [CrossRef]
- Ziriakus, J.; Zimmermann, T.K.; Pöthig, A.; Drees, M.; Haslinger, S.; Jantke, D.; Kühn, F.E. Ruthenium-Catalyzed Transvinylation—New Insights. Adv. Synth. Catal. 2013, 355, 2845–2859. [Google Scholar] [CrossRef]
- Winternheimer, D.J.; Shade, R.E.; Merlic, C.A. Methods for Vinyl Ether Synthesis. Synthesis 2010, 2497–2511. [Google Scholar] [CrossRef]
- Ober, M.S.; Romer, D.R.; Etienne, J.; Thomas, P.J.; Jain, V.; Cameron, J.F.; Thackeray, J.W. Backbone Degradable Poly(aryl acetal) Photoresist Polymers: Synthesis, Acid Sensitivity, and Extreme Ultraviolet Lithography Performance. Macromolecules 2019, 52, 886–895. [Google Scholar] [CrossRef]
- Cluzeau, J.; Nettekoven, U.; Kovačevič, M.P.; Časar, Z. Concise Six-Step Asymmetric Approach to Ramelteon from an Acetophenone Derivative Using Ir, Rh, Cu, and Ni Catalysis. J. Org. Chem. 2022, 87, 2129–2135. [Google Scholar] [CrossRef]
- Alavez-Rosas, D.; Maldonado-Domínguez, M.; González-Antonio, O.; Romero-Ávila, M.; Méndez-Stivalet, J.; Flores-Pérez, B. Synthesis of 1,3- and 1,2,3-Functionalized Pyrroles via Ir(I)-Catalyzed Vinylation of Allyl Alcohols. Chem. Heterocycl. Compd. 2017, 53, 526–531. [Google Scholar] [CrossRef]
- Spiegelberg, B.; Jiao, H.; Grauke, R.; Kubis, C.; Spannenberg, A.; Brandt, A.; Taden, A.; Beck, H.; Tin, S.; de Vries, J.G. Use of Iridium-Catalyzed Transfer Vinylation for the Synthesis of Bio-Based (bis)-Vinyl Ethers. Adv. Synth. Catal. 2022, 364, 1251–1263. [Google Scholar] [CrossRef]
- Jena, S.; Chanda, K. Copper Catalyzed Synthesis of Heterocyclic Molecules via C–N and C–O Bond Formation under Microwaves: A Mini-Review. ACS Omega 2023, 8, 23240–23256. [Google Scholar] [CrossRef]
- Goldberg, I. Ueber Phenylirungen bei Gegenwart von Kupfer als Katalysator. Ber. Dtsch. Chem. Ges. 1906, 39, 1691–1692. [Google Scholar] [CrossRef]
- Serrano, E.; de Nanteuil, F.; Waser, J. Diester-Substituted Aminocyclopropanes: Synthesis and Use in [3+2]-Annulation Reactions. Synlett 2014, 25, 2285–2288. [Google Scholar] [CrossRef]
- Kimura, J.; Nakamichi, S.; Ogawa, S.; Obora, Y. Iridium-Catalyzed Vinylation of Carbazole Derivatives with Vinyl Acetate. Synlett 2017, 28, 719–723. [Google Scholar] [CrossRef]


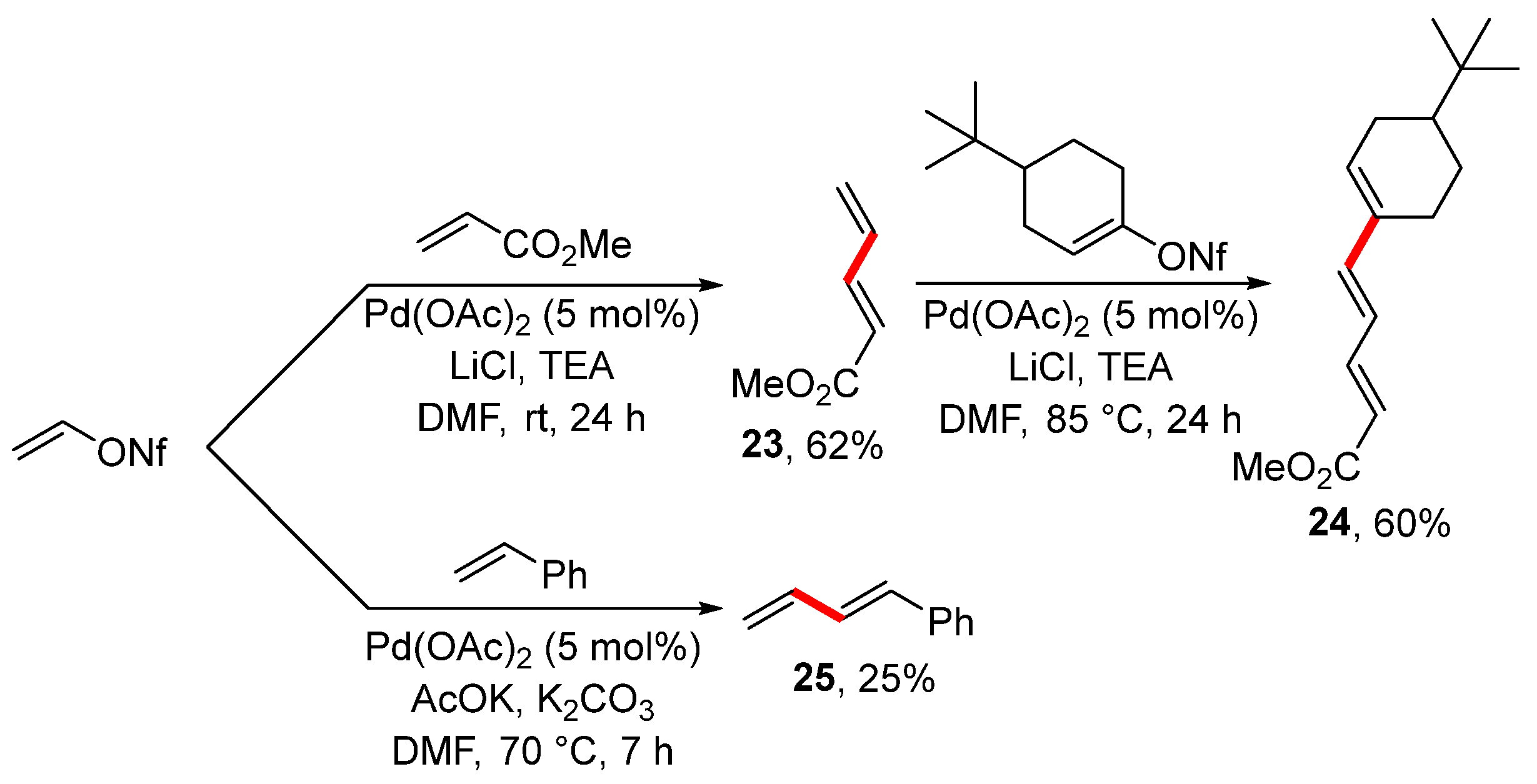


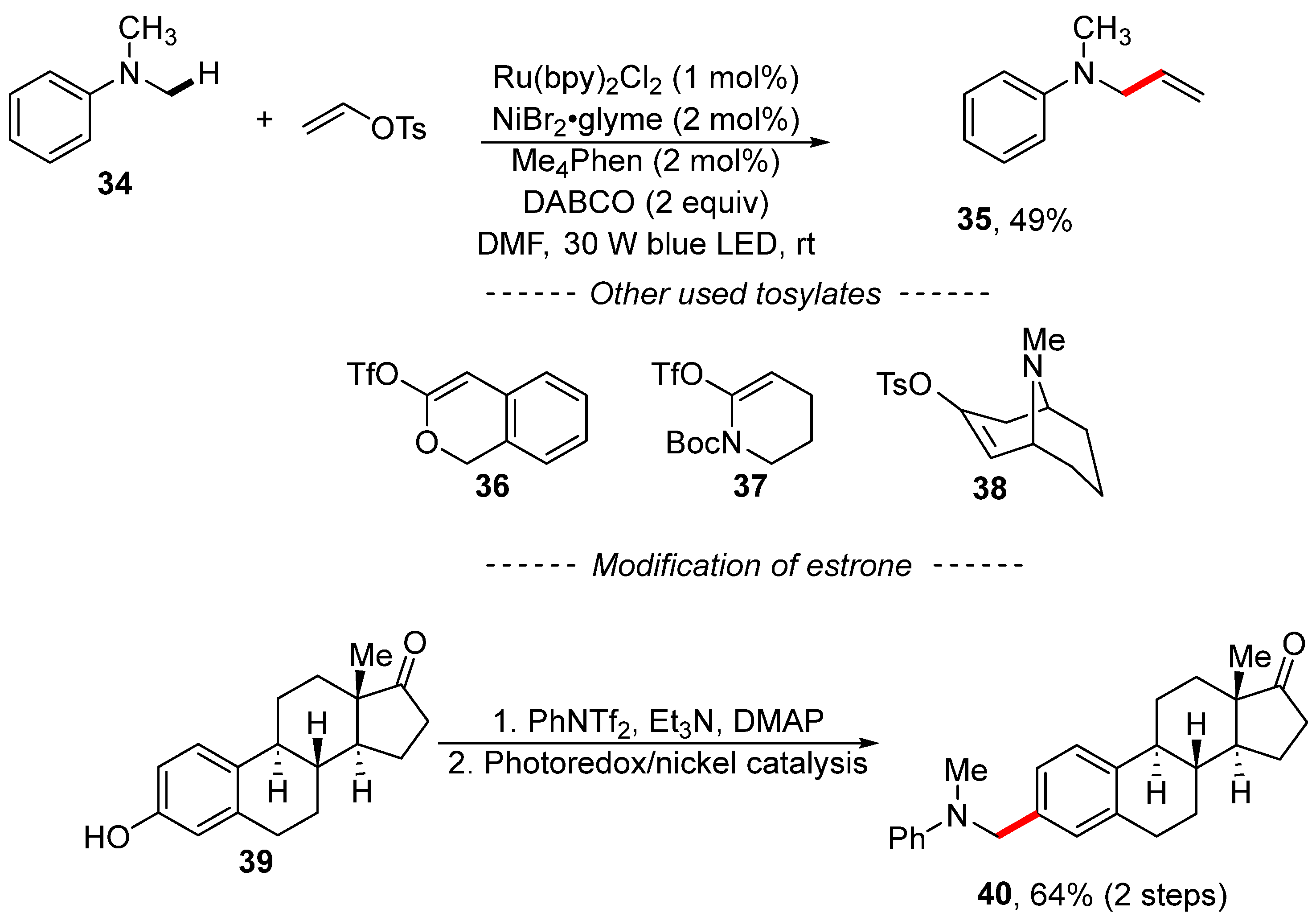
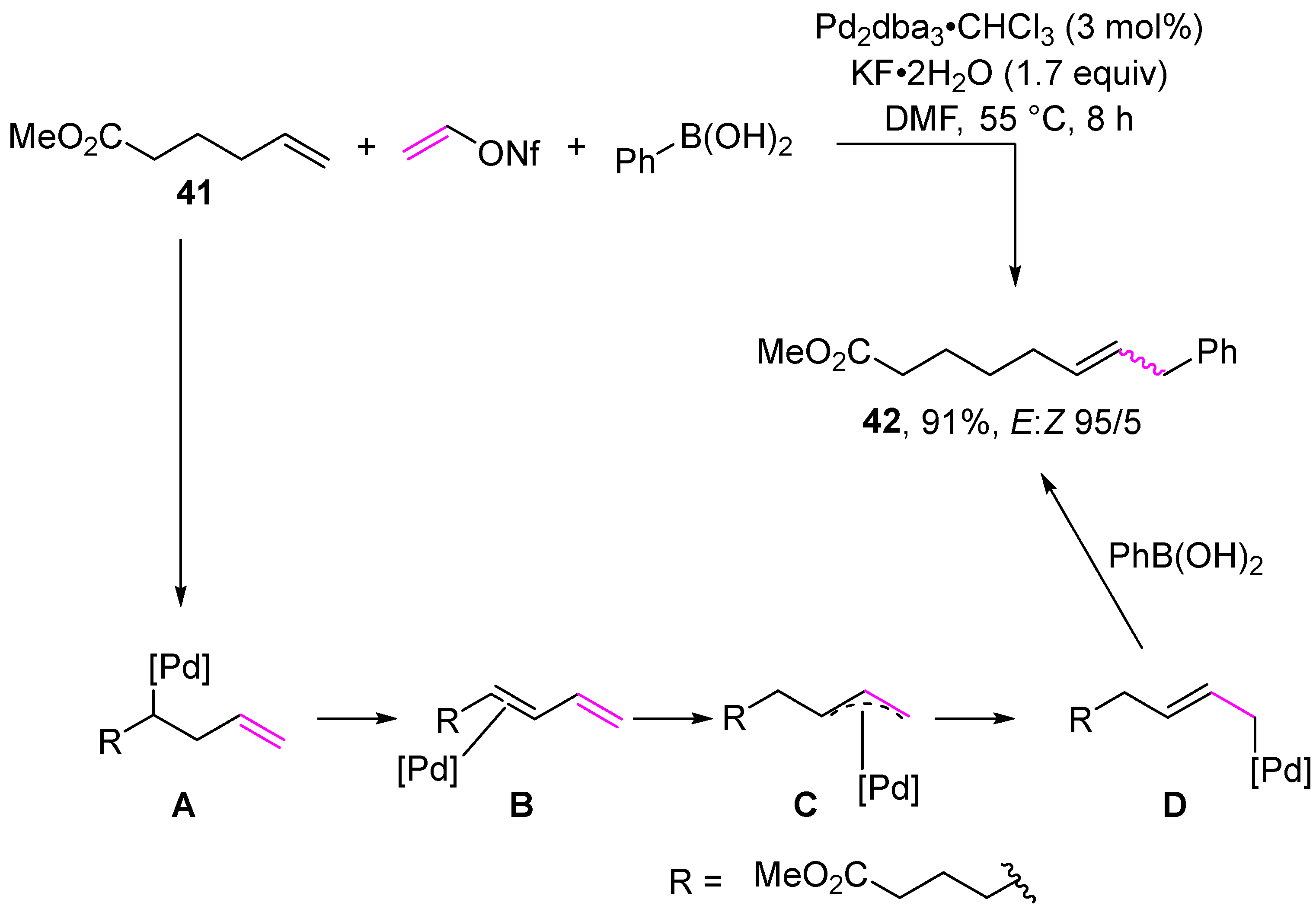
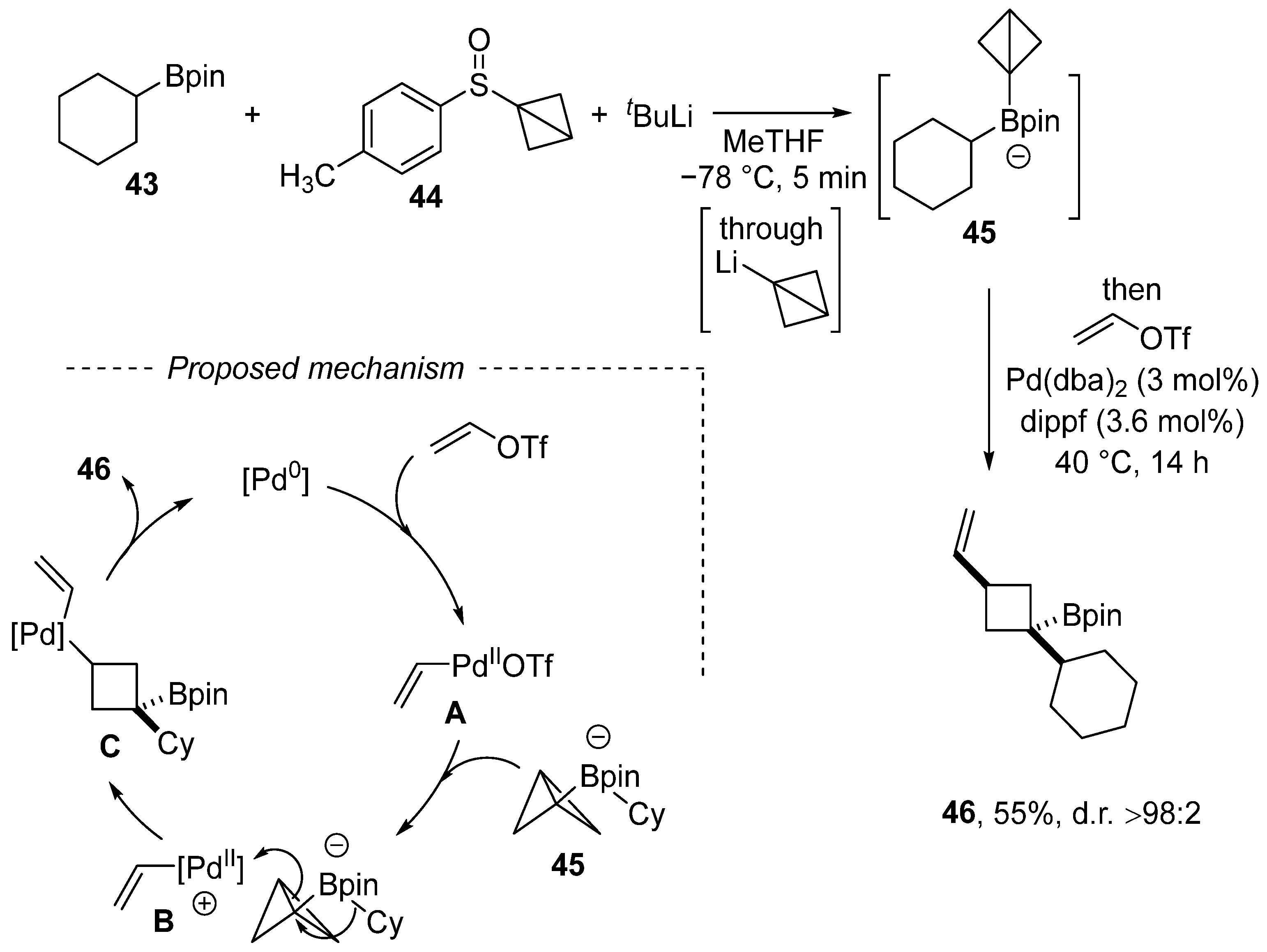











Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tobrman, T. Vinyl Esters and Vinyl Sulfonates as Green Alternatives to Vinyl Bromide for the Synthesis of Monosubstituted Alkenes via Transition-Metal-Catalyzed Reactions. Chemistry 2023, 5, 2288-2321. https://doi.org/10.3390/chemistry5040153
Tobrman T. Vinyl Esters and Vinyl Sulfonates as Green Alternatives to Vinyl Bromide for the Synthesis of Monosubstituted Alkenes via Transition-Metal-Catalyzed Reactions. Chemistry. 2023; 5(4):2288-2321. https://doi.org/10.3390/chemistry5040153
Chicago/Turabian StyleTobrman, Tomáš. 2023. "Vinyl Esters and Vinyl Sulfonates as Green Alternatives to Vinyl Bromide for the Synthesis of Monosubstituted Alkenes via Transition-Metal-Catalyzed Reactions" Chemistry 5, no. 4: 2288-2321. https://doi.org/10.3390/chemistry5040153
APA StyleTobrman, T. (2023). Vinyl Esters and Vinyl Sulfonates as Green Alternatives to Vinyl Bromide for the Synthesis of Monosubstituted Alkenes via Transition-Metal-Catalyzed Reactions. Chemistry, 5(4), 2288-2321. https://doi.org/10.3390/chemistry5040153