Abstract
Using a very recently proposed theoretical model, electron transfer rates in solution are calculated from first principles for different donor-acceptor pairs in tetrahydrofuran. We show that this approach, which integrates tunneling effects into a classical treatment of solvent motion, is able to provide reliable rate constants and their temperature dependence, even in the case of highly exergonic reactions, where Marcus’ theory usually fails.
1. Introduction
Electron transfer (ET) reactions are at foundation of modern chemistry, being exploited in numerous applications (e.g., photovoltaics, biosensing, molecular electronics), and are still the subject of active and vibrant research. The seminal work of Marcus paved the way to the development of this vast field [1,2,3]. Marcus’ theory relates the kinetics of ET process to two well-defined physical quantities: (i) the of the whole ET reaction and (ii) the reorganization energy associated with the motions of both solvent and reactants . While this elegant theory is still successfully used to address ET phenomena, as for instance the rational design of organic molecules to be used in technological devices, its classical treatment of nuclear quantum motion does not allow to explore regimes in which tunneling is crucial [4,5,6], and drammatically fails in reproducing ET rates of strongly exothermic reactions [7].
Alternative theories have been formulated [8], and several papers have focused on implementing quantum effects arising from high frequency vibrational modes into Marcus’ theory [9,10,11,12,13,14,15,16], but only recently the task of including the whole heat bath provided by intramolecular coordinates of the redox pair—which represented the real breakthrough for understanding the unusual temperature dependence of an early ET step occurring in bacterial photosynthetic reaction center [17,18,19]—has been undertaken [20]. That objective has been achieved by introducing a multistep kinetic model of ET reactions, described in refs. [21,22], in which the motion of the solvent is separated from that of the redox pair [23,24]. That separation of motion makes it possible the employment of an effective treatment of tunnelling effects, which incorporates the whole set of nuclear coordinates of the redox pair and takes into account changes of both the equilibrium nuclear positions and of vibrational frequencies upon electronic transition, allowing to include effects due to normal mode mixing too [17,25,26]. The approach has been tested on different D-A pairs in both polar and non-polar solvents: it proved to provide ET rates in excellent agreement with measurements [27,28] and to reproduce the expected temperature dependence [20]. The achievement of such a satisfactory result was however subdued to the use of solvent reorganization energy as extrapolated from available experimental data. Nevertheless, to make the model predictive, an ab initio determination of such a quantity would be highly desirable. The evaluation of the solvent reorganization energy based on molecular dynamics with an explicit treatment of solvent molecules [29,30,31,32] is certainly a possible choice, but it might be unaffordable for high-throughput screening procedures. Therefore, the use of implicit solvent methods would be desirable, provided that their accuracy is properly tested. Herein, we show that the simple non-equilibrium Marcus’ approach for the evaluation of solvent reorganization energy is extremely effective and leads to calculated ET rates which are in excellent agreement with the observed ones, yielding also the correct temperature dependence.
2. Theory
2.1. The Kinetic Model
For a given donor-acceptor system, the multistep mechanism reads as Scheme 1:
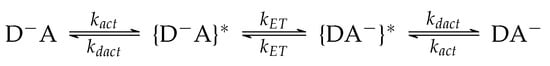
Scheme 1.
The multistep kinetic mechanism of ET.
Where and represent the initial and the final states, respectively, and and indicate the ensembles of transient structures in vibronic resonance with each other.
First, we consider the activation step bringing the donor and the acceptor species into electronic degeneracy, which is mandatory when initial and final states are not in vibronic resonance at frozen solvent coordinates. This may typically occur in polar solvents for which . The rate constant of this step is hence estimated assuming the sole involvement of the solvent coordinate and a typical Arrhenius dependence on temperature:
where is a transmission coefficient and is the standard activation free energy, i.e. the free energy difference of with respect to . Applying Marcus’ reasoning to frozen intramolecular coordinates [3], is defined as [1,3]:
In Equation (2), is the free energy difference between initial and final states, is the total reorganization energy, composed by the sum of the solvent and reactants contributions. These in turn may be splitted in the individual contributions pertaining to the donor and the acceptor, e.g., for the solvent . Following Ref. [20], appearing in Equation (1), is defined as:
where and are the total degeneracies of the activated and the equilibrium states and is the rate constant for deactivation, available from experiments, in consistence with the principle of microscopic reversibility [33].
The second step is the elementary ET process in resonant conditions, for which the rate can be calculated within the framework of the Fermi Golden Rule, considering a non-radiative transition between two electronic states and :
where is:
where is the electronic coupling element for ET reaction, assumed to be independent of the vibrational coordinates, and indicate the vectors of the vibrational quantum states of the and , respectively, and are the vibronic energies of and , respectively, and is the equilibrium (Boltzmann) population of . Overall, consists of an electronic coupling term and of , the Franck-Condon weighted density of states of the elementary → transition at the of the reaction, averaged over a thermal equilibrium distribution of initial vibrational states. For weakly interacting molecules, the vibrational motions of each molecular site upon electron transfer can be assumed to be independent from each other. It follows that:
where and are the spectral distributions of the donor photoelectronic and of the acceptor electron attachment spectra [34,35], which can be measured [36,37,38] and reliably estimated from ab initio calculations [17,35,39,40,41,42,43,44,45].
From the description of the kinetic scheme, it is clear that the determination of the solvent reorganization energy is apical in the model. In fact, while ET reactions are exothermic when both solvent (Q) and molecular () nuclear coordinates are in their equilibrium conditions, we here assume the elementary ET step to occur at fixed Q. In particular, we define:
and consider two cases: (i) , for which ET occurs at , the equilibrium solvent coordinate of the initial state, without thus requiring any solvent activation; (ii) , for which ET occurs at , the point at which the potential energy surfaces of the initial and final states cross each other when intramolecular coordinates are kept fixed at their initial equilibrium value. From Equation (7), it follows that determines the occurrence and the rate of the activation step. Furthermore, even in cases when ET is exothermic and activation is not required, i.e. the kinetic model is not applied, still influences the ET rate, inasmuch as the Franck-Condon weighted density of states is evaluated from Equation (4) at , which in turns depend on , Equation (7).
2.2. Solvent Reorganization Energy
In a previous study [20], in tetrahydrofuran (THF) has been directly estimated from the experimental rate constants of the extremely exothermic BIP-BQO or BIP-NQO pairs, see Figure 1, and from the electronic coupling elements reported in ref. [29]. However, this approach provides values of independent of the chemical nature of the acceptor, an approximation which works well for the A/D pairs of Figure 1 [29], but likely impracticable in other cases, and, more importantly, restricts the feasibility of the model to the study of cases in which can be extrapolated from measurements, thus limiting its predictive power.
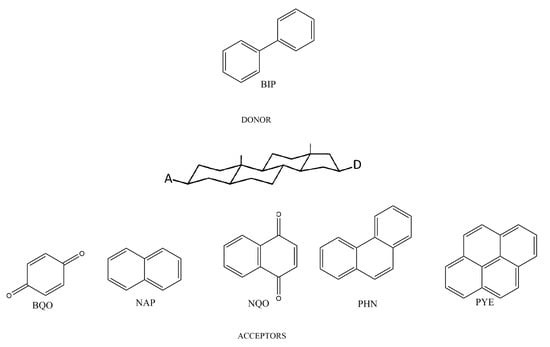
Figure 1.
Chemical structures of the donor and the acceptors considered in this study. BIP = 1,1-biphenyl, BQO = 2-benzoquinonyl, NAP = 2-naphthyl, NQO = 2-naphtoquinonyl, PHN = 9-phenanthryl, PYE = 1-pyrenyl.
Here, we calculate in THF using Marcus’ non-equilibrium approach, according to which [1,46]:
where n is the number of exchanged electrons, e is the electron charge, and are the radii of cavity accommodating the donor and acceptor, respectively, R the intermolecular distance, and the high-frequency and static dielectric constants of the solvent, respectively.
Since solvent reorganization energies are sensitive to the chosen radii of the dielectric cavities, only for comparison purposes, we have also employed a nonlocal response function theory, formulated in the inverted-space representation of the electrostatic fields, in which the cavity is determined from the atomic radii of A/D pair and solvent [47]. Within this theory, the solvent reorganization is given by two contributions [47]:
where is the usual orientational reorganization energy, associated with the energetic strength of anisotropic fluctuations of the solvent polarization, and the density reorganization energy, which describes how the translational motion of the solvent alters its response by locally changing solvent density around the solute. We will not consider here the latter effect, which is not included in Marcus’ theory.
3. Computational Details
Equilibrium geometries, normal coordinates and vibrational frequencies of the neutral and anionic species are calculated at the density functional theory (DFT) using Gaussian 16 [48]. We employ the B3LYP (Becke [49], 3-parameter [50], Lee–Yang–Parr) [51] functional with the 6-31+G(d,p) basis set. In all the calculations, solvent effect was included by using the continuum polarizable medium (PCM) approach [52,53]. To determine in Equation (8), the radii of cavities have been obtained from the computed PCM cavity volumes, assuming spherical shapes, whereas R has been evaluated as the sum of , and the androstane spacer length, the latter taken from geometry optimization of the whole molecules.
Solvent reorganization energies from nonlocal response theory are evaluated employing the freely available SOLVMOL package [54]; for each redox state, ESP charges evaluated by Gaussian 16 at the same level of the theory cited above have been employed [55].
Franck-Condon weighted density of states (FCWDs) are calculated using a development version of the MolFC package, available on request. The internal (curvilinear) representation of normal coordinates have been adopted in all the cases [56]. The electronic coupling elements reported in ref. [29] have been employed throughout. When activation step is not necessary, ET rates are calculated by Equation (4) with the FCWD evaluated at . In the other case, the kinetic scheme is used and the set of coupled ordinary differential equations (ODEs) is solved by using the Dormand-Prince method of order 4, a member of the Runge-Kutta family of ODE solvers, as implemented in MATLAB package [57] with from Equation (1) and from Equation (4) but with the FCWD evaluated at .
4. Results and Discussion
Calculated values of s and s for each A/D pair in THF are listed in Table 1, along with values of s, the standard activation free energy (c.f. Equation (2)), and the calculated and the observed rate constants. Solvent reorganization energies calculated by Marcus’ solvent treatment and by nonlocal response theory function are very similar each other, when only solvent polarization effects are considered; we have thus considered only the former ones for computing ET rates. Herein, assuming that the entropy of reaction due to intramolecular vibrations is negligible, we consider the calculated energy differences as free energies. The extremely exergonic ET reactions involving BIP-BQO and BIP-NQO pairs do not require an activation step because they retain exergonicity also at frozen solvent coordinates (see Table 1), so that their rate constants can be directly evaluated by Equations (4)–(6), using . Vice versa, all the other A/D pairs require a solvent activation step, so that their rates have been obtained by numerically solving the systems of ordinary differential equations associated to the multistep kinetic ET mechanism, c.f. Scheme 1. The rate of the solvent response to a nonequilibrium charge distribution of the solute () has been taken from time dependent spectroscopic measurements (Stokes shifts) [58], and the transmission coefficient has been set to 5 s for all species, as in previous work [20]. Table 1 shows that for all systems considered here the calculated rates differ from the experimental ones by a factor 2–3, the order of magnitude being well reproduced in all the cases.

Table 1.
Calculated equilibrium energy changes (), reorganization energies (), activation free energy (), and rate constants (k) for ET of each BIP-bridge-A pair. All energies are in eV. Experimental rate constants from ref. [27].
In Figure 2, the temperature dependence of ET for two acceptors, BQO and NAP, the only two for which experimental results are available [59], is reported. ET from BIP to NAP needs activation by solvent motion and therefore the T dependence of ET rate constant is dominated by the Arrenhius exponential factor, and thus ET rates change by over 4 orders of magnitude. Conversely, ET from BIP to BQO occurs by tunneling and it is almost temperature independent. In this case, we have considered that the dielectric constant of THF exhibits a significant temperature dependence, which, in the temperature range to 30 °C, can be expressed by the following relation: [60]. It is interesting to remark as increases with T, so does solvent reorganization energy. However, since ET rates occurs by tunneling, the larger values of do not produce an exponentially decrease of the rate. In fact, we observed an increase of the Franck-Condon weighted density of states with a mild effect on ET rates, c.f. Figure 2.
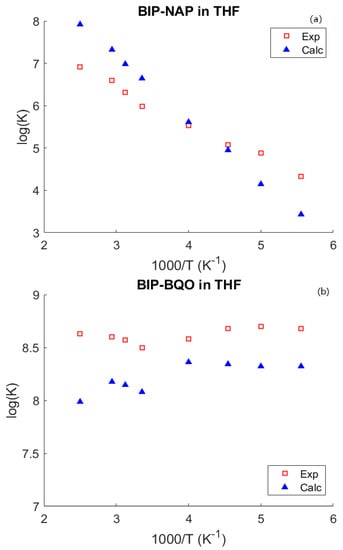
Figure 2.
Experimental (red squares) and calculated (blu triangles) temperature dependence of rate constat for BIP-NAP (panel (a)) and BIP-BQO (panel (b)) pairs in THF.
5. Conclusions
In our approach, we have assumed that elementary ET reactions always occurs by tunneling and that solvent motion modulates the energy condition under which this phenomenon occurs. Herein, at variance with a previous study, we calculate solvent reorganization energy using Marcus’ original expression, showing that it yields satisfying results for the systems considered here. That is a significant step toward the development of a predictive first-principles approach for the calculation of ET rates, which ultimately tends to avoid the long and laborious calculations of molecular dynamics, routinely used for determining ET rates in solution. However, the approach still makes use of an adjustable parameter to be set from experimental data, i.e. the transmission coefficient , and this parameter has to be related to solvent properties; work is in progress along this line.
Author Contributions
Conceptualization, A.P.; funding acquisition, A.P.; investigation, A.L. (Anna Leo) and A.L. (Alessandro Landi) and F.A. and A.P.; methodology, A.L. (Anna Leo) and A.L. (Alessandro Landi) and A.P.; project administration and resources, A.L. (Anna Leo) and A.L. (Alessandro Landi) and A.P.; software, A.L. (Anna Leo) and A.L. (Alessandro Landi) and F.A. and A.P.; writing, A.L. (Anna Leo), A.L. (Alessandro Landi), F.A. and A.P. All authors have read and agreed to the published version of the manuscript.
Funding
Financial support of University of Salerno is gratefully acknowledged.
Acknowledgments
We thank D Mathyushov for sharing the SOLVMOL package.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Marcus, R.A. On the Theory of Oxidation-Reduction Reactions Involving Electron Transfer. I. J. Chem. Phys. 1956, 24, 966–978. [Google Scholar] [CrossRef]
- Marcus, R.A. Chemical and Electrochemical Electron-Transfer Theory. Annu. Rev. Phys. Chem. 1964, 15, 155–196. [Google Scholar] [CrossRef]
- Sumi, H.; Marcus, R.A. Dinamical Effects in Electron Transfer Reactions. J. Chem. Phys. 1986, 84, 4894. [Google Scholar] [CrossRef]
- Landi, A.; Borrelli, R.; Capobianco, A.; Velardo, A.; Peluso, A. Hole Hopping Rates in Organic Semiconductors: A Second-Order Cumulant Approach. J. Chem. Theory Comput. 2018, 14, 1594–1601. [Google Scholar] [CrossRef] [PubMed]
- Landi, A. Charge Mobility Prediction in Organic Semiconductors: Comparison of Second-Order Cumulant Approximation and Transient Localization Theory. J. Phys. Chem. C 2019, 123, 18804–18812. [Google Scholar] [CrossRef]
- Landi, A.; Borrelli, R.; Capobianco, A.; Velardo, A.; Peluso, A. Second-Order Cumulant Approach for the Evaluation of Anisotropic Hole Mobility in Organic Semiconductors. J. Phys. Chem. C 2018, 122, 25849–25857. [Google Scholar] [CrossRef]
- Akesson, E.; Walker, G.C.; Barbara, P.F. Dinamical Solvent Effects on Electron Transfer Rates in the Inverted Regime: Ultrafast Studies on the Betaines. J. Chem. Phys. 1991, 95, 4188. [Google Scholar] [CrossRef]
- Formosinho, S.J.; Arnaut, L.G.; Fausto, R. A Critical Assessment of Classical and Semi-classical Models for Electron Transfer Reactions in Solution. Prog. React. Kinet 1998, 23, 1–90. [Google Scholar]
- Levich, V.G. Present State of the Theory of Oxidation-Reduction in Solution; Interscience: New York, NY, USA, 1965; Volume 4. [Google Scholar]
- Dogonadze, R.; Kuznetsov, A.; Margishvili, T. The Present State of the Theory of Charge Transfer Processes in Condensed Phase. Electrochim. Acta 1980, 25, 1–28. [Google Scholar] [CrossRef]
- Kestner, N.; Logan, J.; Jortner, J. Thermal Electron Transfer Reactions in Polar Solvents. J. Phys. Chem. 1974, 78, 2148. [Google Scholar] [CrossRef]
- Jortner, J.; Bixon, M. Intramolecular Vibrational Excitations Accompanying Solvent-Controlled Electron Transfer Reactions. J. Chem. Phys. 1988, 88, 167. [Google Scholar] [CrossRef]
- Bixon, M.; Jortner, J. Electron Transfer—From Isolated Molecules to Biomolecules, Part One; John Wiley & Sons: New York, NY, USA, 1999; Volume 106. [Google Scholar]
- Zusman, L.D. Outer-Sphere Electron Transfer in Polar Solvents. Chem. Phys. 1980, 49, 295. [Google Scholar] [CrossRef]
- Sparpaglione, M.; Mukamel, S. Adiabatic vs. Nonadiabatic Electron Transfer and Longitudinal Solvent Dielectric Relaxation: Beyond the Debye Model. J. Phys. Chem. 1987, 91, 15. [Google Scholar] [CrossRef]
- Ovchinnikova, M.Y. Teor. Eksp. Kim. Theor. Exper. Chem. 1982, 17, 507. [Google Scholar] [CrossRef]
- Borrelli, R.; Peluso, A. The Temperature Dependence of Radiationless Transition Rates from Ab Initio Computations. Phys. Chem. Chem. Phys. 2011, 13, 4420–4426. [Google Scholar] [CrossRef]
- Schenck, C.; Parson, W.; Holten, D.; Windsor, M.W.; Sarai, A. Temperature Dependence of Electron Transfer Between Bacteriopheophytin and Ubiquinone in Protonated and Deuterated Reaction Centers of Rhodopseudomonas shaeroides. Biophys. J. 1981, 36, 479–489. [Google Scholar] [CrossRef]
- Kirmaier, C.; Holten, D.; Parson, W.W. Temperature and Detection-Wevelenght Dependence of the Picosecond Electron-Transfer Kinetics Mesured in Rhodopseudomonas shaeroides Reaction Centers. Resolution of New Spectral and Kinetic Components in the Primary Charge-Separation Process. Biochim. Biophys. Acta Bioenerg. 1985, 810, 33–48. [Google Scholar] [CrossRef]
- Leo, A.; Peluso, A. Electron Transfer Rate in Polar and Non-Polar Environments: A Generalization of Marcus’ Theory to Include an Effective Treatment of Tunneling Effects. J. Phys. Chem. Lett. 2022, 13, 9148–9155. [Google Scholar] [CrossRef]
- Landi, A.; Borrelli, R.; Capobianco, A.; Peluso, A. Transient and Enduring Electronic Resonances Drive Coherent Long Distance Charge Transport in Molecular Wires. J. Phys. Chem. Lett. 2019, 10, 1845–1851. [Google Scholar] [CrossRef]
- Landi, A.; Capobianco, A.; Peluso, A. Coherent Effects in Charge Transport in Molecular Wires: Toward a Unifying Picture of Long-Range Hole Transfer in DNA. J. Phys. Chem. Lett. 2020, 11, 7769–7775. [Google Scholar] [CrossRef]
- Peluso, A.; Caruso, T.; Landi, A.; Capobianco, A. The Dynamics of Hole Transfer in DNA. Molecules 2019, 24, 4044. [Google Scholar] [CrossRef]
- Landi, A.; Capobianco, A.; Peluso, A. The Time Scale of Electronic Resonance in Oxidized DNA as Modulated by Solvent Response: An MD/QM-MM Study. Molecules 2021, 26, 5497. [Google Scholar] [CrossRef]
- Kubo, R.; Toyozawa, Y. Application of the Method of Generating Function to Radiative and Non-Radiative Transitions of a Trapped Electron in a Crystal. Prog. Theor. Phys. 1955, 13, 160–182. [Google Scholar] [CrossRef]
- Lax, M. The Franck-Condon Principle and Its Application to Crystals. J. Chem. Phys. 1952, 20, 1752–1760. [Google Scholar] [CrossRef]
- Closs, G.L.; Calcaterra, L.T.; Green, N.J.; Penfield, K.W.; Miller, J.R. Distance, Stereoelectronic Effects, and the Marcus Inverted Region in Intramolecular Electron Transfer in Organic Radical Anions. J. Chem. Phys. 1989, 90, 3673–3683. [Google Scholar] [CrossRef]
- Liang, N.; Miller, J.R.; Closs, G.L. Correlating Temperature Dependence to Free Energy Dependence of Intramolecular Long-Range Electron Transfer. J. Am. Chem. Soc. 1989, 11, 8740–8741. [Google Scholar] [CrossRef]
- Parson, W.W. Effects of Free Energy and Solvent on Rates of Intramolecular Electron Transfer in Organic Radical Anions. J. Phys. Chem. A 2017, 121, 7297–7306. [Google Scholar] [CrossRef]
- Parson, W.W. Vibrational Relaxations and Dephasing in Electron-Transfer Reactions. J. Phys. Chem. 2016, 120, 11412–11418. [Google Scholar] [CrossRef]
- Parson, W.W. Electron-Transfer Dynamics in a Zn-Porphyrin-Quinone Cyclophane: Effects of Solvent, Vibrational Relaxations, and Conical Intersections. J. Phys. Chem. B 2018, 122, 3854–3863. [Google Scholar] [CrossRef]
- Parson, W.W. Temperature Dependence of the Rate of Intramolecular Electron Transfer. J. Phys. Chem. B 2018, 122, 8824–8833. [Google Scholar] [CrossRef]
- Mahan, B.H. Microscopic Reversibility and Detailed Balance. J. Chem. Educ. 1975, 52, 299–302. [Google Scholar] [CrossRef]
- Hopfield, J.J. Electron transfer between biological molecules by thermal activated tunneling. Proc. Natl. Acad. Sci. USA 1974, 71, 3640–3644. [Google Scholar] [CrossRef] [PubMed]
- Borrelli, R.; Peluso, A. Elementary Electron Transfer Reactions: From Basic Concepts to Recent Computational Advances. WIREs: Comput. Mol. Sci. 2013, 3, 542–559. [Google Scholar] [CrossRef]
- Rabalais, J.W.; Karlsson, L.; Werme, L.O.; Bergmark, T.; Siegbahn, K. Analysis of Vibrational Structure and Jahn-Teller Effects in the Electron Spectrum of Ammonia. J. Chem. Phys. 1973, 58, 3370–3372. [Google Scholar] [CrossRef]
- Wang, X.B.; Woo, H.K.; Wang, L.S. Vibrational Cooling in a Cold Ion Trap: Vibrationally Resolved Photoelectron Spectroscopy of Cold C60− Anions. J. Chem. Phys. 2005, 123, 051106. [Google Scholar] [CrossRef]
- Capobianco, A.; Carotenuto, M.; Caruso, T.; Peluso, A. The Charge-Transfer Band of an Oxidized Watson-Crick Guanosine-Cytidine Complex. Angew. Chem. Int. Ed. 2009, 48, 9526–9528. [Google Scholar] [CrossRef]
- Niu, Y.; Peng, Q.; Deng, C.; Gao, X.; Shuai, Z. Theory of Excited State Decays and Optical Spectra: Application to Polyatomic Molecules. J. Phys. Chem. A 2010, 114, 7817–7831. [Google Scholar] [CrossRef] [PubMed]
- Santoro, F.; Lami, A.; Improta, R.; Bloino, J.; Barone, V. Effective Method for the Computation of Optical Spectra of Large Molecules at Finite Temperature Including the Duschinsky and Herzberg-Teller Effect: The Qx Band of Porphyrin as A Case Study. J. Chem. Phys. 2008, 128, 224311. [Google Scholar] [CrossRef]
- Capobianco, A.; Borrelli, R.; Landi, A.; Velardo, A.; Peluso, A. Absorption Band Shapes of a Push-Pull Dye Approaching the Cyanine Limit: A Challenging Case for First Principle Calculations. J. Phys. Chem. A 2016, 120, 5581–5589. [Google Scholar] [CrossRef]
- Peluso, A.; Borrelli, R.; Capobianco, A. Photoelectron Spectrum of Ammonia, a Test Case for the Calculation of Franck–Condon Factors in Molecules Undergoing Large Geometrical Displacements upon Photoionization. J. Phys. Chem. A 2009, 113, 14831–14837. [Google Scholar] [CrossRef]
- Landi, A.; Landi, A.; Velardo, A.; Peluso, A. Efficient Charge Dissociation of Triplet Excitons in Bulk Heterojunction Solar Cells. ACS Appl. Energy Mater. 2022, 5, 10815–10824. [Google Scholar] [CrossRef]
- Landi, A.; Padula, D. Multiple Charge Separation Pathways in New-Generation Non-Fullerene Acceptors: A Computational Study. J. Mater. Chem. A 2021, 9, 24849–24856. [Google Scholar] [CrossRef]
- Velardo, A.; Borrelli, R.; Capobianco, A.; Landi, A.; Peluso, A. Disentangling Electronic and Vibrational Effects in the Prediction of Band Shapes for Singlet-Triplet Transitions. J. Phys. Chem. C 2019, 123, 14173–14179. [Google Scholar] [CrossRef]
- Marcus, R.A. Electrostatic Free Energy and Other Properties of States Having Nonequilibrium Polarization. II. J. Chem. Phys. 1956, 24, 979–989. [Google Scholar] [CrossRef]
- Matyushov, V.D. Solvent Reorganization Energy of Electron-Transfer Reactions in Polar Solvent. J. Chem. Phys. 2004, 120, 7532–7556. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 16 Revision C.01; Gaussian Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
- Kim, K.; Jordan, K.D. Comparison of Density Functional and MP2 Calculations on the Water Monomer and Dimer. J. Phys. Chem. 1994, 98, 10089–10094. [Google Scholar] [CrossRef]
- Stephens, P.; Devlin, F.J.; Chabalowski, C.F.; Frisch, M.J. Ab Initio Calculation of Vibrational Absorption and Circular Dichroism Spectra Using Density Functional Force Fields. J. Phys. Chem. 1994, 98, 11623–11627. [Google Scholar] [CrossRef]
- Cramer, C.J. Essentials of Computational Chemistry: Theories and Models, 2nd ed.; John Wiley & Sons: New York, NY, USA, 2004. [Google Scholar]
- Miertiuš, S.; Scrocco, E.; Tomasi, J. Electrostatic Interaction of a Solute with a Continuum. A Direct Utilization of Ab Initio Molecular Potentials for the Prevision of Solvent effects. Chem. Phys. 1981, 55, 117–129. [Google Scholar] [CrossRef]
- Tomasi, J.; Mennucci, B.; Cammi, R. Quantum Mechanical Continuum Solvation Models. Chem. Rev. 2005, 105, 2999–3094. [Google Scholar] [CrossRef]
- Matyushov, D. Software Package for the Solvation Thermodynamics of Biomolecules. 2007. Available online: https://skysonginnovations.com/technology/software-package-for-the-solvation-thermodynamics-of-biomolecules/ (accessed on 16 December 2022).
- Singh, U.C.; Kollman, P.A. An Approach to Computing Electrostatic Charges for Molecules. J. Comput. Chem. 1984, 5, 129–145. [Google Scholar] [CrossRef]
- Capobianco, A.; Borrelli, R.; Noce, C.; Peluso, A. Franck-Condon Factors in Curvilinear Coordinates: The Photoelectron Spectrum of Ammonia. Theor. Chem. Acc. 2012, 131, 1181. [Google Scholar] [CrossRef]
- Dormand, J.R.; Prince, P.J. A Family of Embedded Runge-Kutta Formulae. J. Comput. Appl. Math. 1980, 6, 19–26. [Google Scholar] [CrossRef]
- Simon, J.D. Time-Resolved Studies of Solvation in Polar Media. Res. Chem. Acc. 1988, 21, 128–134. [Google Scholar] [CrossRef]
- Liang, N.; Miller, J.R.; Closs, G.L. Temperature-Independent Long-Range Electron Transfer Reactions in the Marcus Inverted Region. J. Am. Chem. Soc. 1990, 112, 5353–5354. [Google Scholar] [CrossRef]
- Metz, D.J.; Glines, A. Density, Viscosity and Dielectric Constant of Tetrahydrofuran Between -78 and 30. J. Chem. Phys. 1966, 11973. [Google Scholar] [CrossRef]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).