

Theoretical Study of Excited-State Dynamics of Hypercoordinated Carbon Molecule


Abstract
1. Introduction
2. Computational Methodology
2.1. Static Calculations
2.2. Vibronic Hamiltonian
2.3. State Minima and Conical Intersection
2.4. MCTDH Wavepacket Dynamics
3. Results
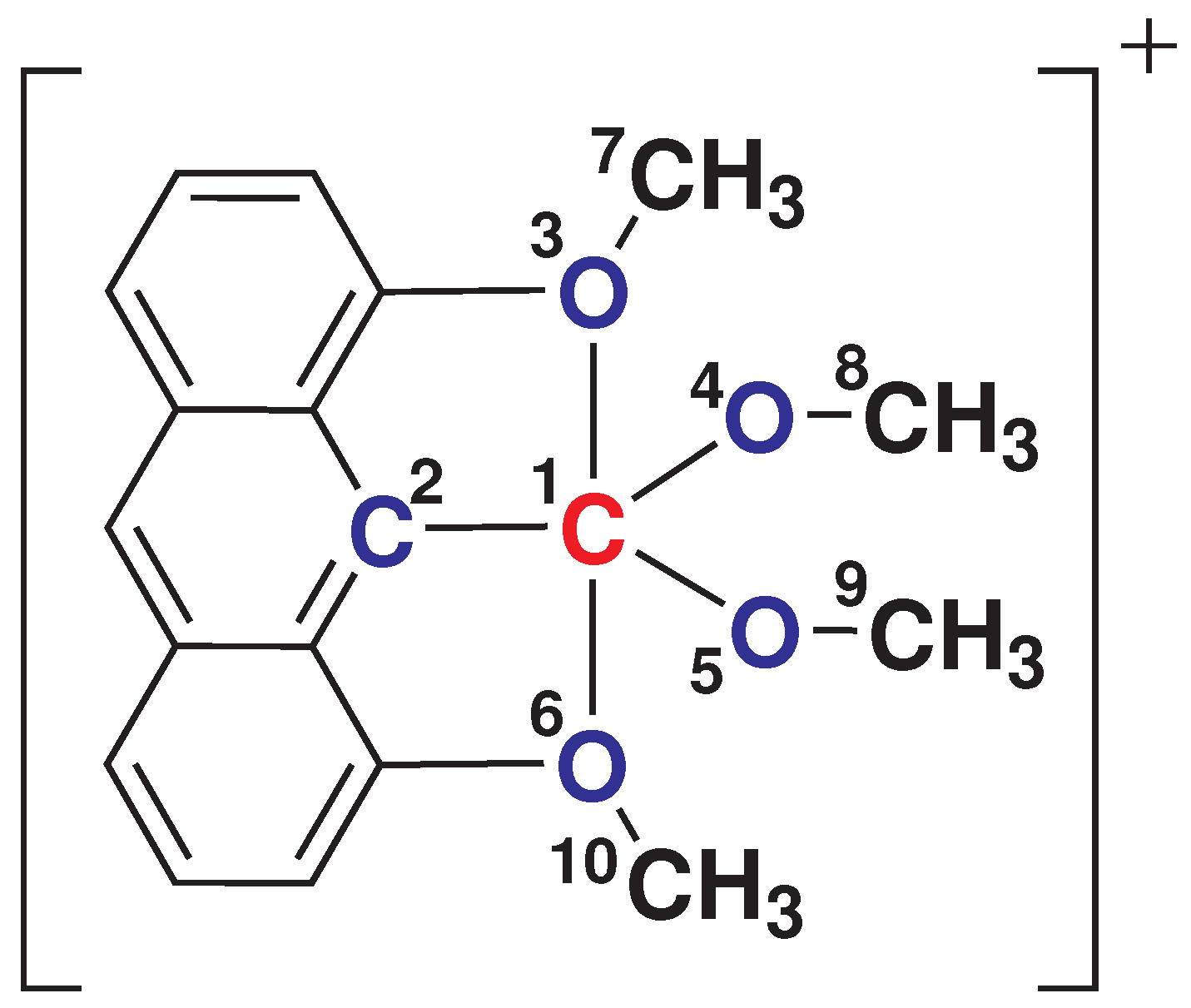
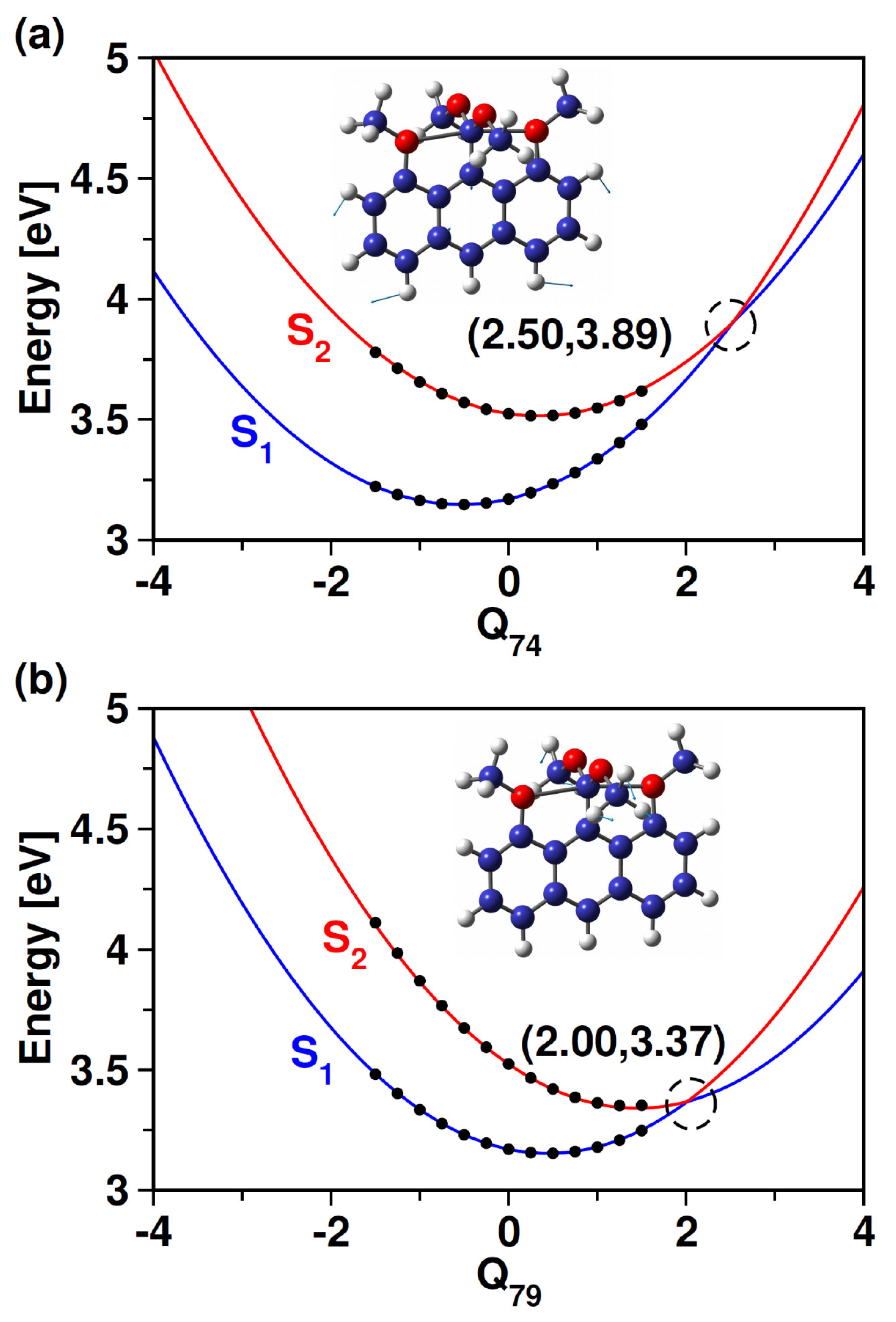
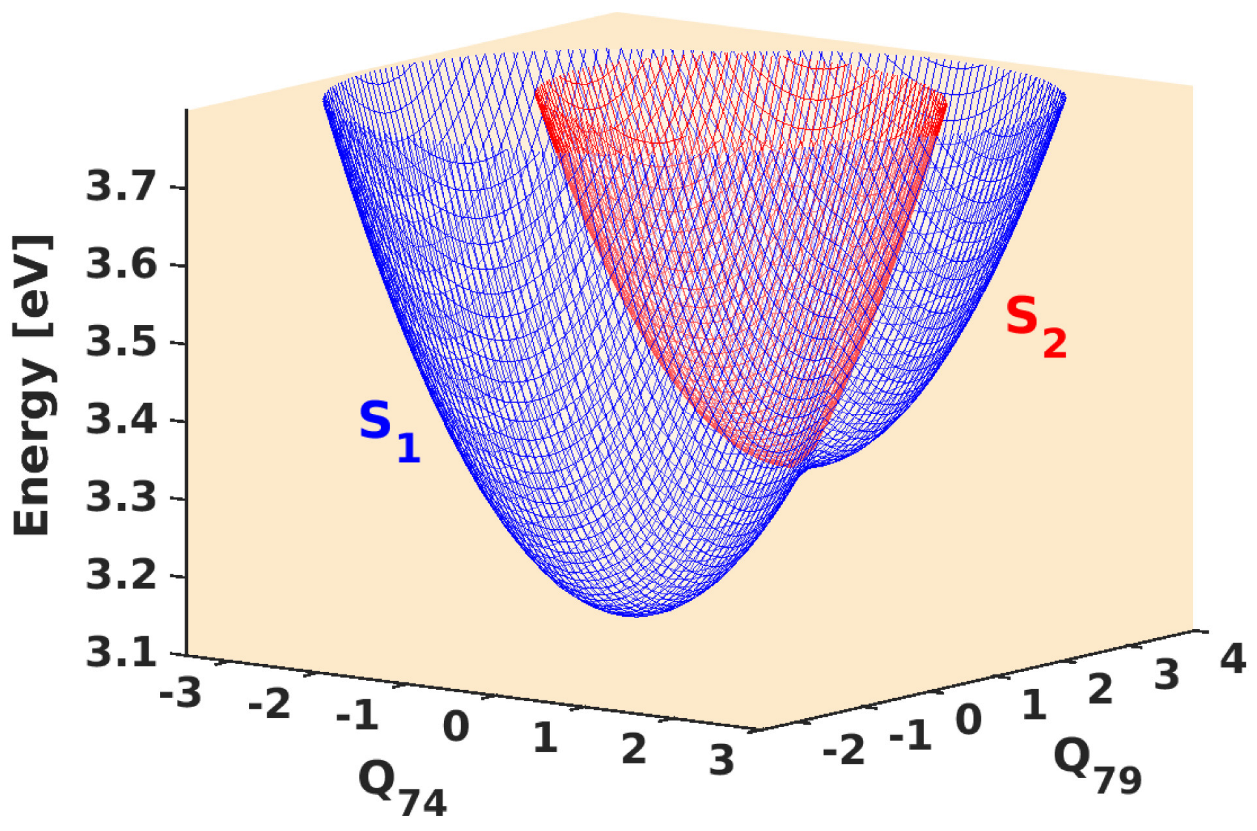
3.1. Stationary Point Analysis
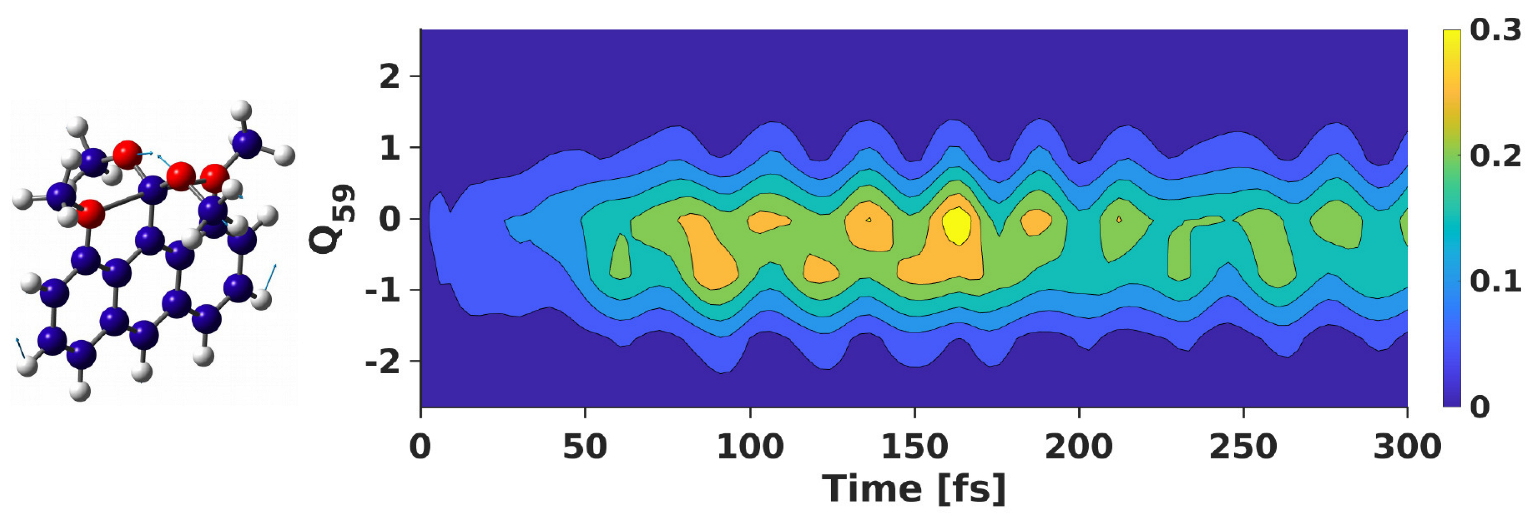
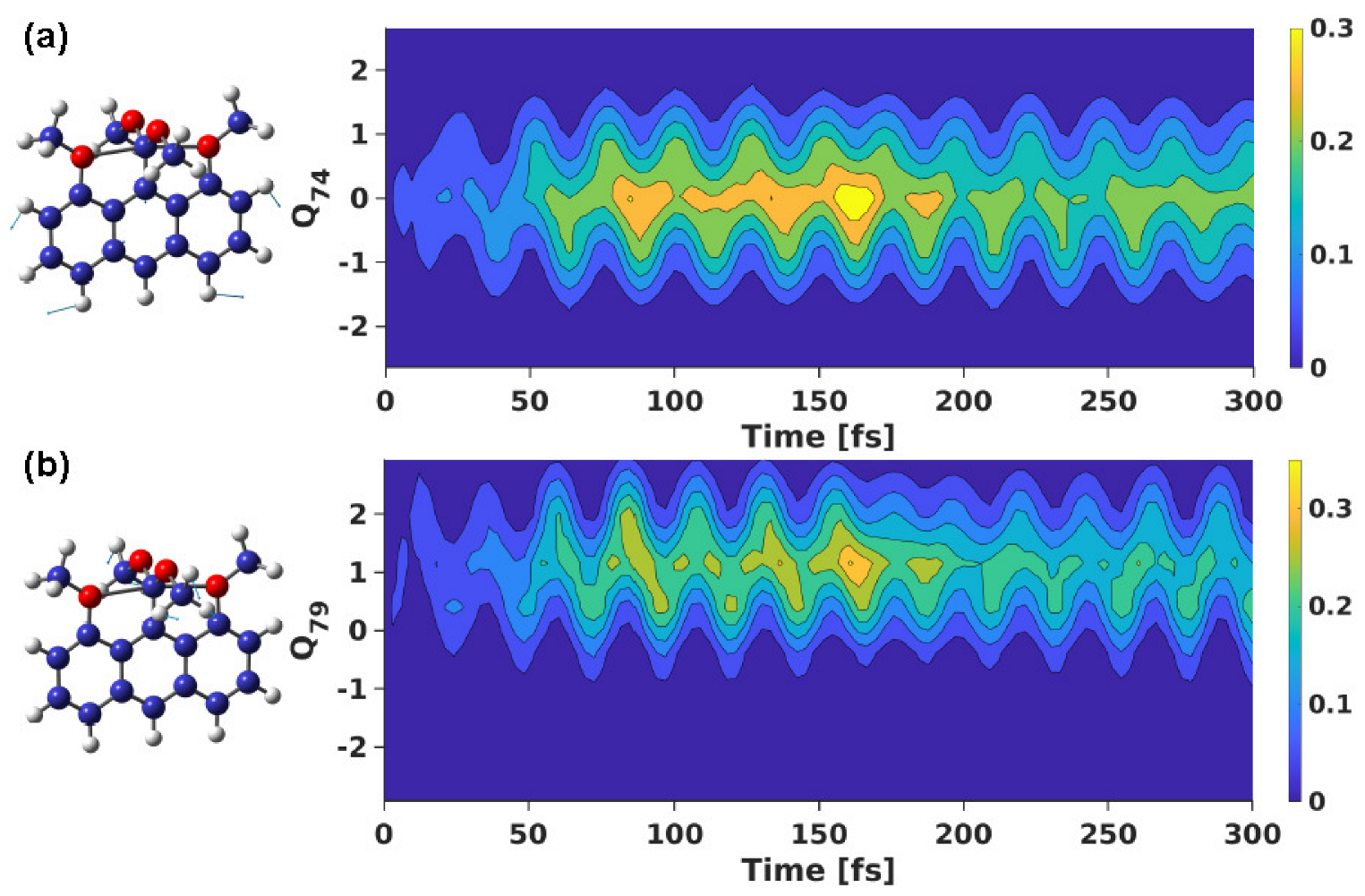
3.2. Wavepacket Simulation Analysis
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| LVC | Linear vibronic coupling |
| PES | Potential energy surface |
| IC | Internal conversion |
| MCTDH | Multiconfiguration time-dependent Hartree |
| TDDFT | Time-dependent density functional theory |
| RI-CC2 | Resolution-of-the-identity second-order approximate coupled-cluster singles and doubles |
| ADC(2) | Algebraic diagrammatic construction method to second-order |
| FC | Franck-Condon |
| MECI | Minimum energy conical intersection |
| GRRM | Global reaction route mapping |
| GP | Gradient projection |
References
- Guélin, M.; Cernicharo, J. Organic Molecules in Interstellar Space: Latest Advances. Front. Astron. Space Sci. 2022, 9, 787567. [Google Scholar] [CrossRef]
- Asvany, O.; Schlemmer, S.; Gerlich, D. Deuteration of CHn+ (n = 3-5) in Collisions with HD Measured in a Low-Temperature Ion Trap. Astrophys. J. 2004, 617, 685. [Google Scholar] [CrossRef]
- Das, P.; Chattaraj, P.K. Structure and Bonding in Planar Hypercoordinate Carbon Compounds. Chemistry 2022, 4, 1723–1756. [Google Scholar] [CrossRef]
- Tal’rose, V.L.; Lyubimova, A.K. Secondary Processes in the Ion Source of the Mass Spectrometer. Dokl. Akad. Nauk SSSR 1952, 86, 909–912. [Google Scholar]
- Stevenson, D.P.; Schissler, D.O. Rate of the Gaseous Reactions, X++YH=XH++Y. J. Chem. Phys. 1955, 23, 1353. [Google Scholar] [CrossRef]
- Jin, Z.; Braams, B.J.; Bowman, J.M. An ab Initio Based Global Potential Energy Surface Describing CH5+ → CH3+ + H2. J. Phys. Chem. A 2006, 110, 1569–1574. [Google Scholar] [CrossRef]
- Tachikawa, H.; Orr-Ewing, A.J. Ab Initio Molecular Dynamics Study on the Electron Capture Processes of Protonated Methane (CH5+). J. Phys. Chem. A 2008, 112, 11575–11581. [Google Scholar] [CrossRef]
- Chabinyc, M.L.; Craig, S.L.; Regan, C.K.; Brauman, J.I. Gas-Phase Ionic Reactions: Dynamics and Mechanism of Nucleophilic Displacements. Science 1998, 279, 1882–1886. [Google Scholar] [CrossRef]
- Akiba, K.y.; Yamashita, M.; Yamamoto, Y.; Nagase, S. Synthesis and Isolation of Stable Hypervalent Carbon Compound (10-C-5) Bearing a 1,8-Dimethoxyanthracene Ligand. J. Am. Chem. Soc. 1999, 121, 10644–10645. [Google Scholar] [CrossRef]
- Yamamoto, Y.; Akiba, K.-y. Synthesis of Hypervalent Pentavalent Carbon and Boron Compounds. J. Synth. Org. Chem. Jpn. 2004, 62, 1128–1137. [Google Scholar] [CrossRef]
- Kikuchi, Y.; Ishii, M.; Akiba, K.-y.; Nakai, H. Discovery of hexacoordinate hypervalent carbon compounds: Density functional study. Chem. Phys. Lett. 2008, 460, 37–41. [Google Scholar] [CrossRef]
- Schleyer, P.v.R.; Wuerthwein, E.U.; Kaufmann, E.; Clark, T.; Pople, J.A. Effectively hypervalent molecules. 2. Lithium carbide (CLi5), lithium carbide (CLi6), and the related effectively hypervalent first row molecules, CLi5-nHn and CLi6-nHn. J. Am. Chem. Soc. 1983, 105, 5930–5932. [Google Scholar] [CrossRef]
- Scherbaum, F.; Grohmann, A.; Müller, G.; Schmidbaur, H. Synthesis, Structure, and Bonding of the Cation [{(C6H5)3PAu}5C]+. Angew. Chem. Int. Ed. Engl. 1989, 28, 463–465. [Google Scholar] [CrossRef]
- Dufour, N.; Schier, A.; Schmidbaur, H. Hypercoordinate carbon in bis(trimethylsilyl)tris[(triphenylphosphine)aurio(I)]methanium tetrafluoroborate. Organometallics 1993, 12, 2408–2410. [Google Scholar] [CrossRef]
- Jimenez-Halla, J.O.C.; Wu, Y.B.; Wang, Z.X.; Islas, R.; Heine, T.; Merino, G. CAl4Be and CAl3Be2−: Global minima with a planar pentacoordinate carbon atom. Chem. Commun. 2010, 46, 8776–8778. [Google Scholar] [CrossRef]
- Leyva-Parra, L.; Diego, L.; Inostroza, D.; Yañez, O.; Pumachagua-Huertas, R.; Barroso, J.; Vásquez-Espinal, A.; Merino, G.; Tiznado, W. Planar Hypercoordinate Carbons in Alkali Metal Decorated CE32− and CE22− Dianions. Chem.—A Eur. J. 2021, 27, 16701–16706. [Google Scholar] [CrossRef]
- Becke, A.D. Density-functional thermochemistry. III. The role of exact exchange. J. Chem. Phys. 1993, 98, 5648–5652. [Google Scholar] [CrossRef]
- Dunning, T.H. Gaussian basis sets for use in correlated molecular calculations. I. The atoms boron through neon and hydrogen. J. Chem. Phys. 1989, 90, 1007–1023. [Google Scholar] [CrossRef]
- Yanai, T.; Tew, D.P.; Handy, N.C. A new hybrid exchange–correlation functional using the Coulomb-attenuating method (CAM-B3LYP). Chem. Phys. Lett. 2004, 393, 51–57. [Google Scholar] [CrossRef]
- Kendall, R.A.; Dunning, T.H.; Harrison, R.J. Electron affinities of the first-row atoms revisited. Systematic basis sets and wave functions. J. Chem. Phys. 1992, 96, 6796–6806. [Google Scholar] [CrossRef]
- Zhao, Y.; Truhlar, D.G. Comparative DFT Study of van der Waals Complexes: Rare Gas Dimers, Alkaline Earth Dimers, Zinc Dimer, and Zinc Rare Gas Dimers. J. Phys. Chem. A 2006, 110, 5121–5129. [Google Scholar] [CrossRef]
- Chai, J.D.; Head-Gordon, M. Long-range corrected hybrid density functionals with damped atom–atom dispersion corrections. Phys. Chem. Chem. Phys. 2008, 10, 6615–6620. [Google Scholar] [CrossRef]
- Hättig, C.; Weigend, F. CC2 excitation energy calculations on large molecules using the resolution of the identity approximation. J. Chem. Phys. 2000, 113, 5154–5161. [Google Scholar] [CrossRef]
- Trofimov, A.; Krivdina, I.; Weller, J.; Schirmer, J. Algebraic-diagrammatic construction propagator approach to molecular response properties. Chem. Phys. 2006, 329, 1–10. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 16 Revision C.01; Gaussian Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
- TURBOMOLE V7.4 2019, a Development of University of Karlsruhe and Forschungszentrum Karlsruhe GmbH, 1989–2007, TURBOMOLE GmbH, Since 2007. Available online: http://www.turbomole.com (accessed on 11 August 2021).
- Köppel, H.; Domcke, W.; Cederbaum, L.S. Multimode Molecular Dynamics Beyond the Born-Oppenheimer Approximation. Adv. Chem. Phys. 1984, 57, 59–246. [Google Scholar]
- Maeda, S.; Ohno, K.; Morokuma, K. Systematic exploration of the mechanism of chemical reactions: The global reaction route mapping (GRRM) strategy using the ADDF and AFIR methods. Phys. Chem. Chem. Phys. 2013, 15, 3683–3701. [Google Scholar] [CrossRef]
- Banerjee, A.; Adams, N.; Simons, J.; Shepard, R. Search for stationary points on surfaces. J. Phys. Chem. 1985, 89, 52–57. [Google Scholar] [CrossRef]
- Bearpark, M.J.; Robb, M.A.; Bernhard Schlegel, H. A direct method for the location of the lowest energy point on a potential surface crossing. Chem. Phys. Lett. 1994, 223, 269–274. [Google Scholar] [CrossRef]
- Maeda, S.; Ohno, K.; Morokuma, K. Updated Branching Plane for Finding Conical Intersections without Coupling Derivative Vectors. J. Chem. Theory Comput. 2010, 6, 1538–1545. [Google Scholar] [CrossRef]
- Meyer, H.D.; Manthe, U.; Cederbaum, L.S. The multi-configurational time-dependent Hartree approach. Chem. Phys. Lett. 1990, 165, 73–78. [Google Scholar] [CrossRef]
- Manthe, U.; Meyer, H.D.; Cederbaum, L.S. Wave-packet dynamics within the multiconfiguration Hartree framework: General aspects and application to NOCl. J. Chem. Phys. 1992, 97, 3199–3213. [Google Scholar] [CrossRef]
- Beck, M.H.; Jäckle, A.; Worth, G.A.; Meyer, H.D. The multiconfiguration time-dependent Hartree (MCTDH) method: A highly efficient algorithm for propagating wavepackets. Phys. Rep. 2000, 324, 1–105. [Google Scholar] [CrossRef]
- Meyer, H.D.; Gatti, F.; Worth, G.A. Multidimensional Quantum Dynamics: MCTDH Theory and Applications; Wiley-VCH: Weinheim, Germany, 2009. [Google Scholar]
- Worth, G.A.; Beck, M.H.; Jäckle, A.; Meyer, H.D. The MCTDH Package, Version 8.5, 2019. Available online: http://mctdh.uni-hd.de (accessed on 21 April 2021).








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Methods | S (A) | S (A) | |
|---|---|---|---|
| TD-CAM-B3LYP | 3.17 (0.10) | 3.52 (0.00) | ∼0.35 |
| TD-B97XD | 3.20 (0.10) | 3.60 (0.00) | ∼0.40 |
| TD-M062X | 3.19 (0.09) | 3.60 (0.00) | ∼0.41 |
| ADC(2) | 3.12 (0.10) | 3.37 (0.00) | ∼0.25 |
| RI-CC2 | 3.16 (0.09) | 3.43 (0.00) | ∼0.27 |
| Stationary Point | Energy |
|---|---|
| S | 2.89 |
| S | 2.90 |
| MECI | 3.02 (3.09) |
| Ground-State Geometry | MECI Geometry 1 | % Change | |
|---|---|---|---|
| C-C | 1.50 | 1.47 | 2.00 |
| C-O | 2.46 | 2.54 | 3.25 |
| C-O | 1.29 | 1.32 | 2.33 |
| C-O | 1.29 | 1.32 | 2.33 |
| C-O | 2.46 | 2.54 | 3.25 |
| C-C-O | 85.76 | 85.16 | 0.70 |
| O-C-O | 92.38 | 92.79 | 0.44 |
| O-C-O | 111.61 | 110.51 | 0.99 |
| O-C-O | 92.38 | 92.74 | 0.39 |
| C-C-O | 85.76 | 85.16 | 0.70 |
| C-C-O-C | 180 | 179.97 | 0.02 |
| C-C-O-C | 360 | 360.06 | 0.02 |
| C-C-O-C | 360 | 360.05 | 0.01 |
| C-C-O-C | 180 | 179.98 | 0.01 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nag, P.; Vennapusa, S.R. Theoretical Study of Excited-State Dynamics of Hypercoordinated Carbon Molecule. Chemistry 2023, 5, 269-280. https://doi.org/10.3390/chemistry5010021
Nag P, Vennapusa SR. Theoretical Study of Excited-State Dynamics of Hypercoordinated Carbon Molecule. Chemistry. 2023; 5(1):269-280. https://doi.org/10.3390/chemistry5010021
Chicago/Turabian StyleNag, Probal, and Sivaranjana Reddy Vennapusa. 2023. "Theoretical Study of Excited-State Dynamics of Hypercoordinated Carbon Molecule" Chemistry 5, no. 1: 269-280. https://doi.org/10.3390/chemistry5010021
APA StyleNag, P., & Vennapusa, S. R. (2023). Theoretical Study of Excited-State Dynamics of Hypercoordinated Carbon Molecule. Chemistry, 5(1), 269-280. https://doi.org/10.3390/chemistry5010021