1. Introduction
Self-assembly is a spontaneous and reversible phenomenon that utilizes the ability of molecules to noncovalently interact with each other to produce supramolecular aggregates [
1]. The assembly of viral capsids or DNA demonstrates the importance of this process in natural systems. On the other hand, supramolecular chemists use the self-assembly process to produce molecular cages or capsules as it allows one to reach such complex systems in relatively fewer steps than conventional synthesis [
2]. In aqueous media, a typical example of self-assembly is the aggregation of amphiphilic compounds driven by hydrophobic forces. This includes common head/tail amphiphiles, which assemble into micelles or bilayers, as well as compounds with complementary hydrophobic areas, which assemble into discrete complexes [
3].
Glycoluril is a versatile building block widely used in supramolecular chemistry for the construction of host molecules, including cucurbit[
n]urils [
4] and bambus[
n]urils [
5]. Many glycoluril-containing host molecules with the ability to self-assemble in organic nonpolar solvents have been described [
6,
7]. However, the self-assembly of glycoluril-based molecules in competitive media such as water is reported to a much lesser extent. In this sense, acyclic containers composed of one to five glycoluril moieties connected by two rows of methylene bridges showed some promising results [
8,
9,
10,
11,
12]. These containers are typically terminated with aromatic sidewalls, and their solubility in water is achieved by installing solubilizing substituents containing, for example, carboxyl or sulfonic groups [
9,
10,
13]. In addition to their ability to act as supramolecular hosts for various guest molecules [
14,
15,
16,
17,
18], glycoluril-based acyclic containers are known to self-associate into dimeric aggregates in water [
19,
20]. This process competes with guest encapsulation; therefore, it is undesirable for applications where strong host–guest binding is required, such as the use of acyclic oligomers to improve the solubility of hydrophobic drugs or to deactivate neuromuscular blocking agents and illicit recreational drugs [
21,
22]. For such purposes, dimerization was suppressed by placing charged (water solubilizing) groups on the aromatic sidewalls, causing the repulsion of these molecules. The aggregation of glycoluril-based acyclic containers in water resulting in large self-assembled structures has been reported sporadically [
23,
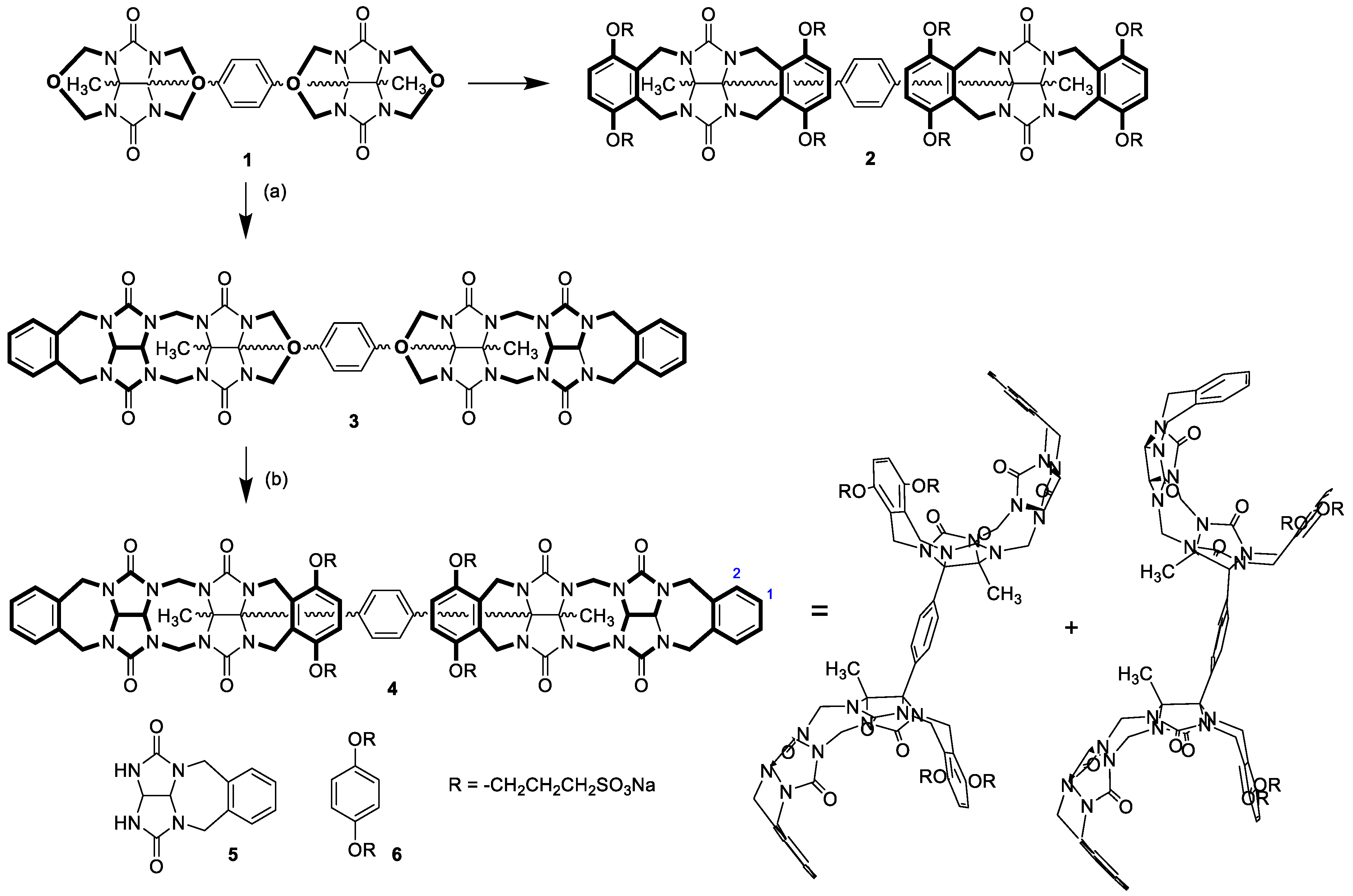
24]. Nolte et al. reported several examples of glycoluril-based clips, that self-assembled into nano-objects of different shapes as a result of a particularly hydrophobic effect. However, glycoluril derivatives with the ability to act as monomers for the formation of water-soluble supramolecular oligomers and polymers have not been described. In this work, we designed and synthesized compound
4 (
Scheme 1) consisting of two connected glycoluril-based containers with the ability to self-associate in water. The formation of supramolecular oligomers induced by the self-association of
4 was investigated.
2. Results and Discussion
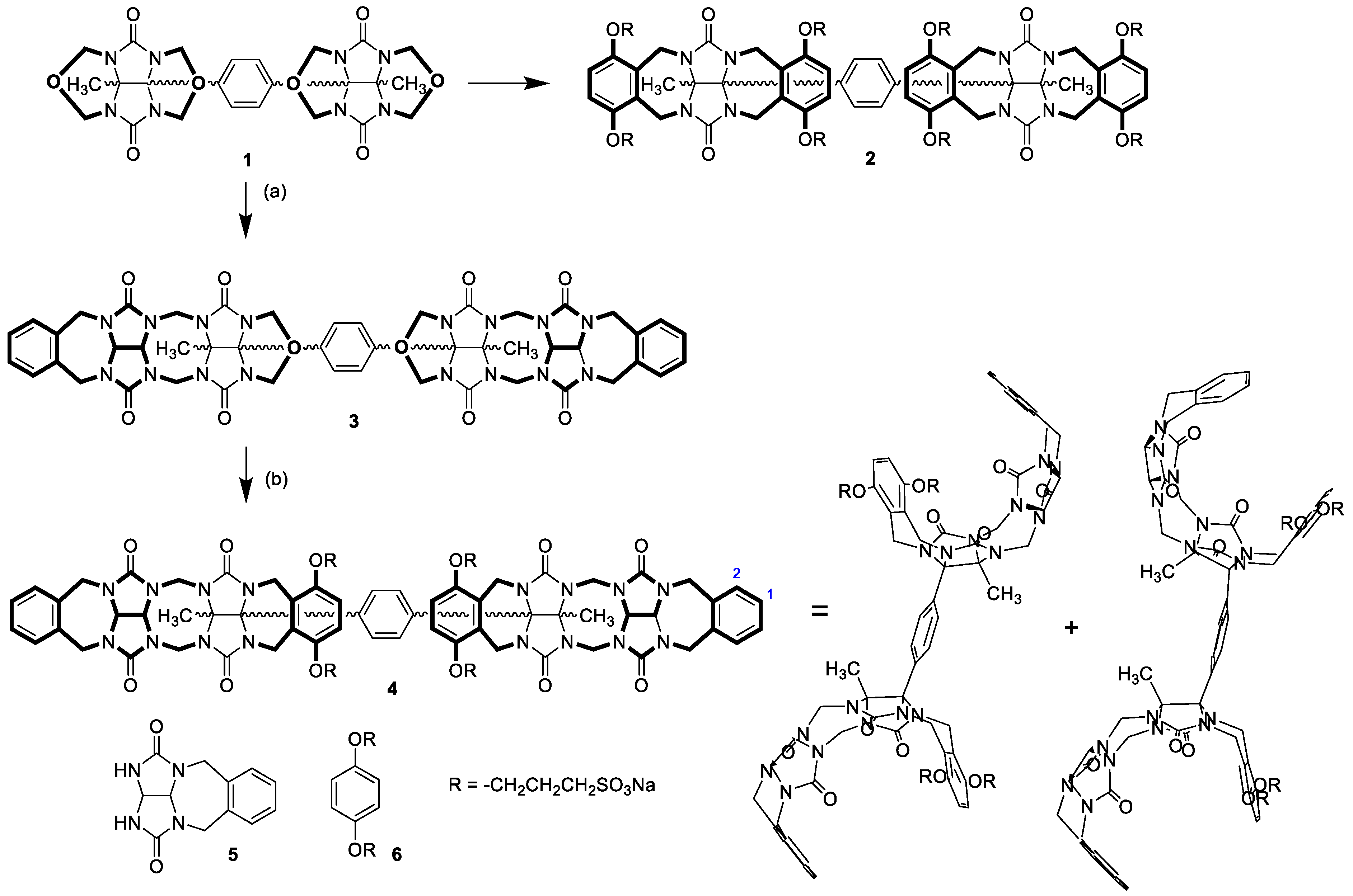
The design of compound
4 was inspired by our previous work [
25], in which we prepared host molecule
2 featuring two coupled glycoluril-based clips (
Scheme 1). The ability of
2 to bind two cationic guests simultaneously in water was reported. However, host
2 had four sulfonate units on each of two connected clips, which prevented the self-association of its molecules. We envisioned, that substitution of the clips in
2 for glycoluril dimer motives in structure
4 might increase the self-association ability required for the formation of a supramolecular polymer. Glycoluril dimer motive was selected in favor of higher glycoluril oligomers as methylene-bridged glycoluril dimers terminated with aromatic side walls showed a higher tendency to self-associate [
19,
20,
26] than monomeric clips [
15,
27,
28], while the self-association of trimers or tetramers was only scarcely reported [
10]. We suggested that each of the two glycoluril dimers of
4 should be terminated by xylylene units, but only one of these xylylenes should be further modified by sulfopropyl substituents. Sulfonate groups were necessary to solubilize
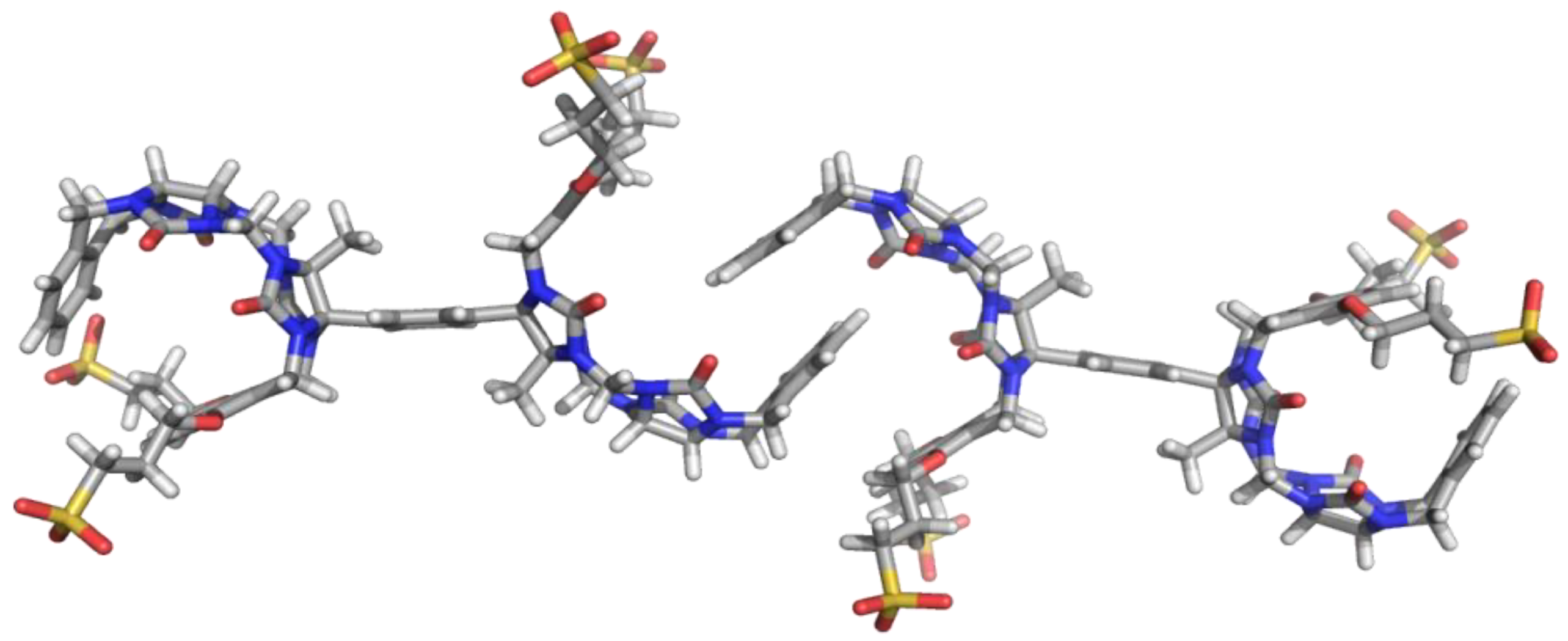
4 in water. On the other hand, the lack of solubilizing substituents on second xylylene units promises a higher tendency toward self-association. Indeed, molecular modeling showed that two molecules of
4 could undergo self-association (
Figure 1). As one molecule of clip
4 possesses two sites capable to self-associate, clip
4 is a promising building block for supramolecular polymers in aqueous solution.
The synthesis of
4 began with the reaction of glycoluril cyclic ether
1 with
o-xylylene-protected glycoluril
5, which was previously designed in our lab as a tool for the synthesis of glycoluril oligomers (
Scheme 1) [
29]. Compound
5 selectively reacted only with those two nitrogen atoms of
1 which were in the proximity of the methyl substituent, leaving the remaining two nitrogen atoms hindered by the phenylene moiety unreacted. The reaction produced compound
3 with a yield of 72%. Compound
3 also reacted with
6 to introduce the other pair of side wall-bearing sulfonate groups, which provide the final compound
4, soluble in water. Compound
4 was prepared with a yield of 45%. The
1H NMR spectra revealed the presence of two conformers of
4 (shown in
Scheme 1) enabled by the rotation of clips around the phenylene unit. We previously described similar conformers for compounds
1 and
2 and studied the energy characteristics of their conformational changes [
25].
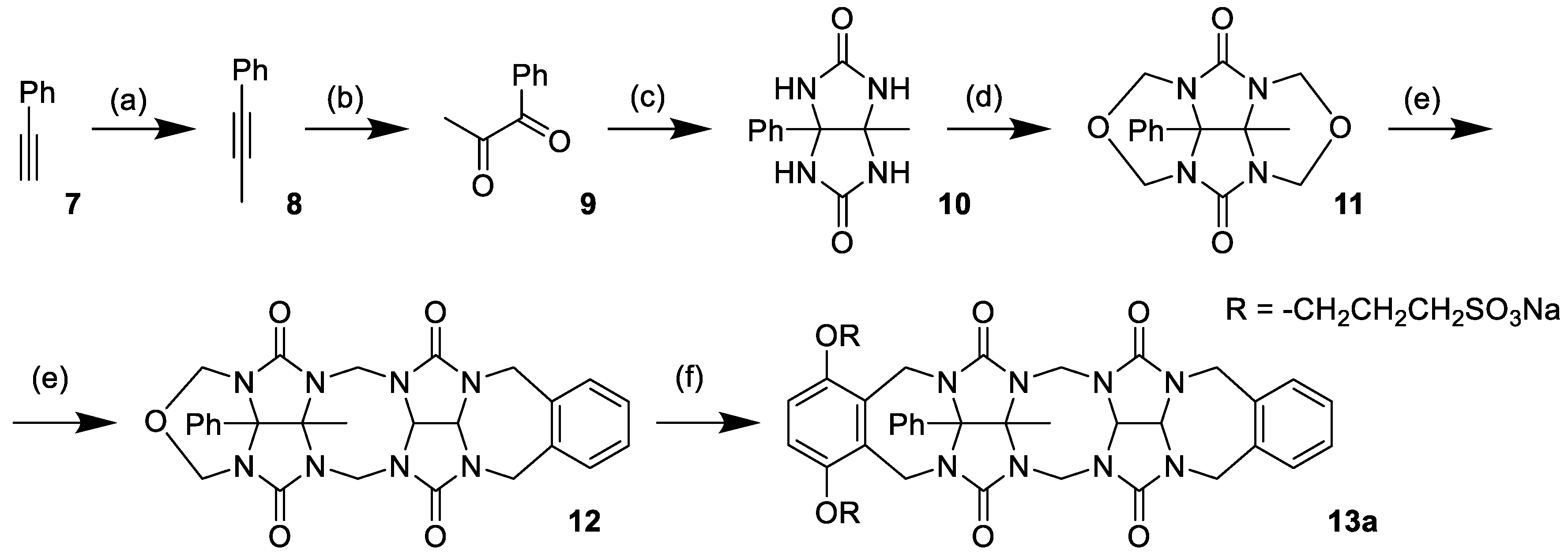
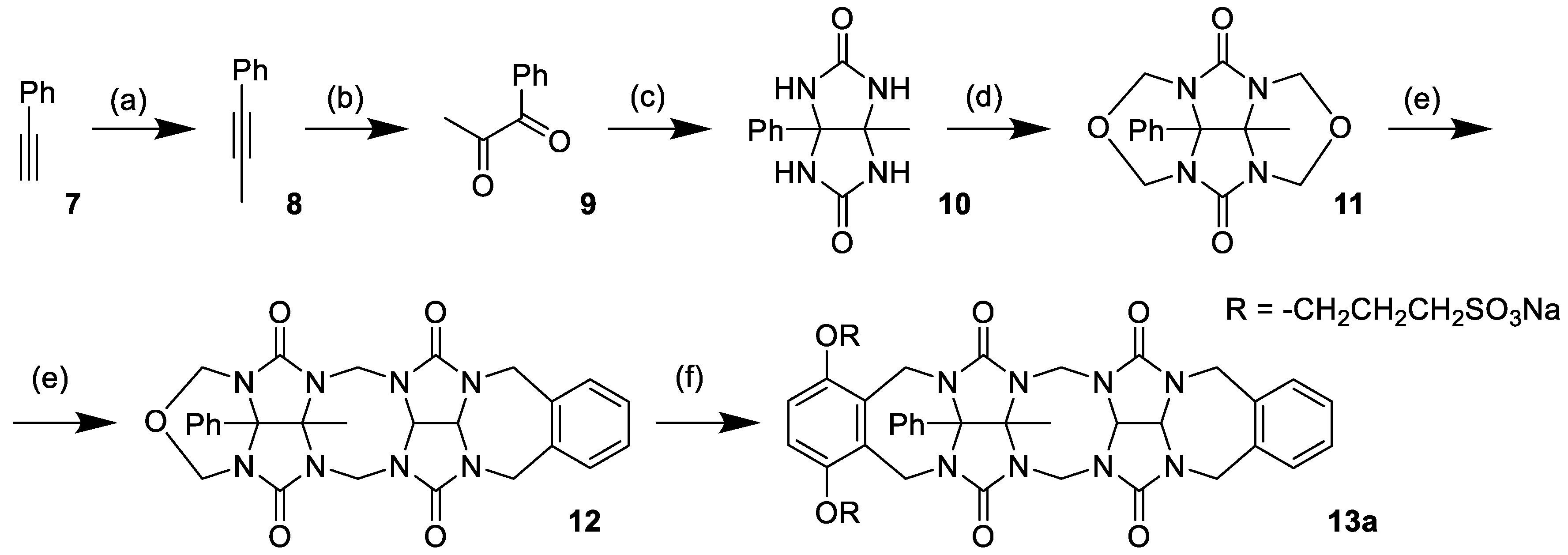
Prior to investigating compound
4, we decided to prepare and test compound
13a, which possesses a single binding site (
Scheme 2). Compound
13a was expected to form the dimeric complex
13a2. We would be able to determine the association constant (
Ka) for
13a2 and use it to estimate the strength of self-association between binding sites of
4. The synthesis of the
13a model compound started from phenylacetylene
7 (
Scheme 2). In the first step, phenylacetylene
7 was deprotonated using
n-butyllithium and subsequently methylated with iodomethane to give prop-1-ynylbenzene
8. The triple bond of
8 was then oxidized using the RuCl
3/NaIO
4 mixture, yielding diketone
9. Compound
9 provided glycoluril
10 after condensation with urea in 0.3 M HCl, and the glycoluril was transformed into cyclic ether
11 by reaction with formaldehyde in 9 M HCl. Condensation of
11 with
5 (for structure, see
Scheme 1) in MeSO
3H provided glycoluril dimer
12. Compound
12 was transformed into the water-soluble glycoluril dimer
13a by reaction with
6.
The self-association of
13a to dimer
13a2 was investigated using dilution experiments using NMR and ITC in pure D
2O/water and in 20 mM and 100 mM phosphate buffer (
Table 1). The association constant of dimerization (
Ka) in the absence of buffer determined by NMR ((2.6 ± 0.1) × 10
3 M
−1) and ITC ((2.1 ± 0.2) × 10
3 M
−1) were in excellent agreement. The
Ka values for
13a2 measured in the presence of 20 mM and 100 mM phosphate buffer were approximately two and four times higher than those measured in pure water (
Table 1). The presence of salt in the solution improved the dimerization of
13a2 either by strengthening the hydrophobic effect or by compensating the charge of the sulfonate groups and weakening their electrostatic repulsion.
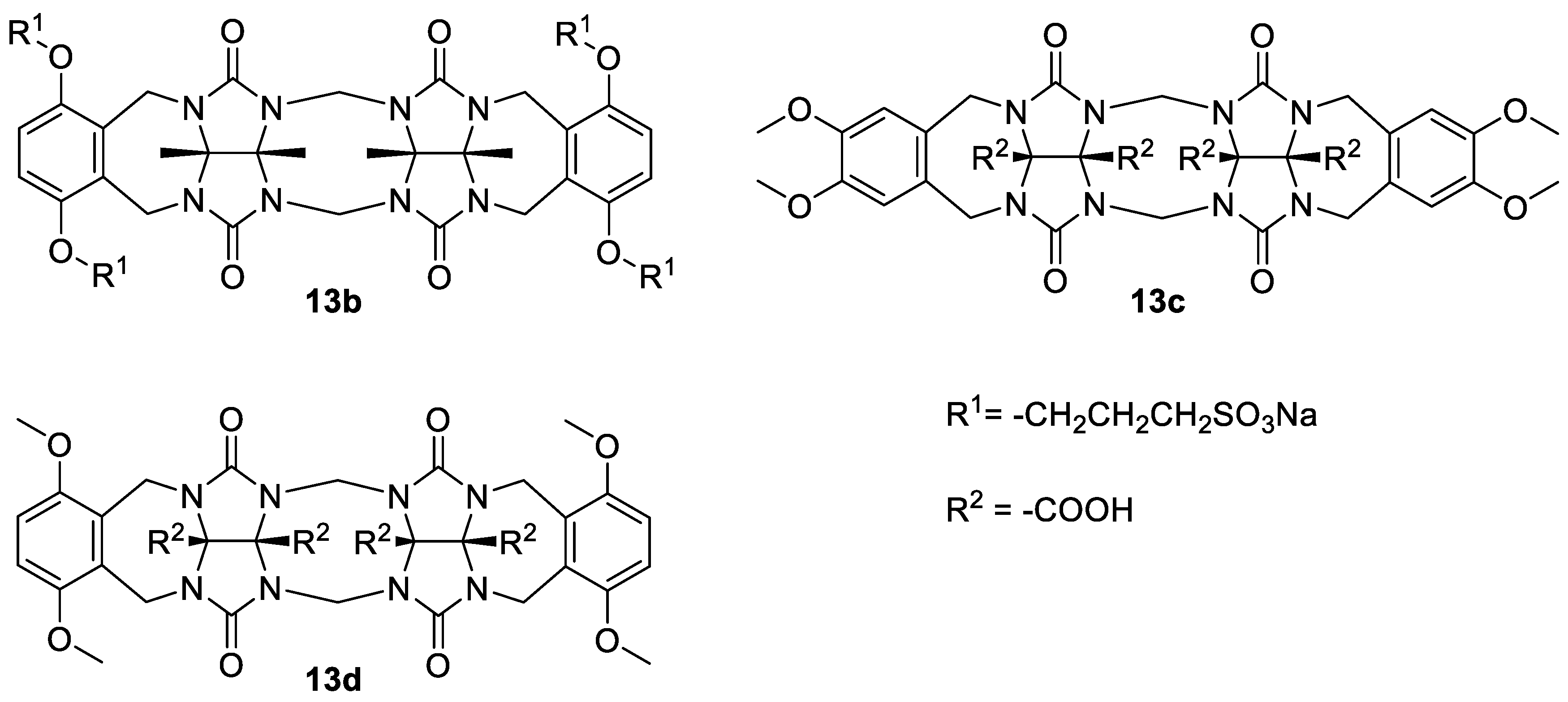
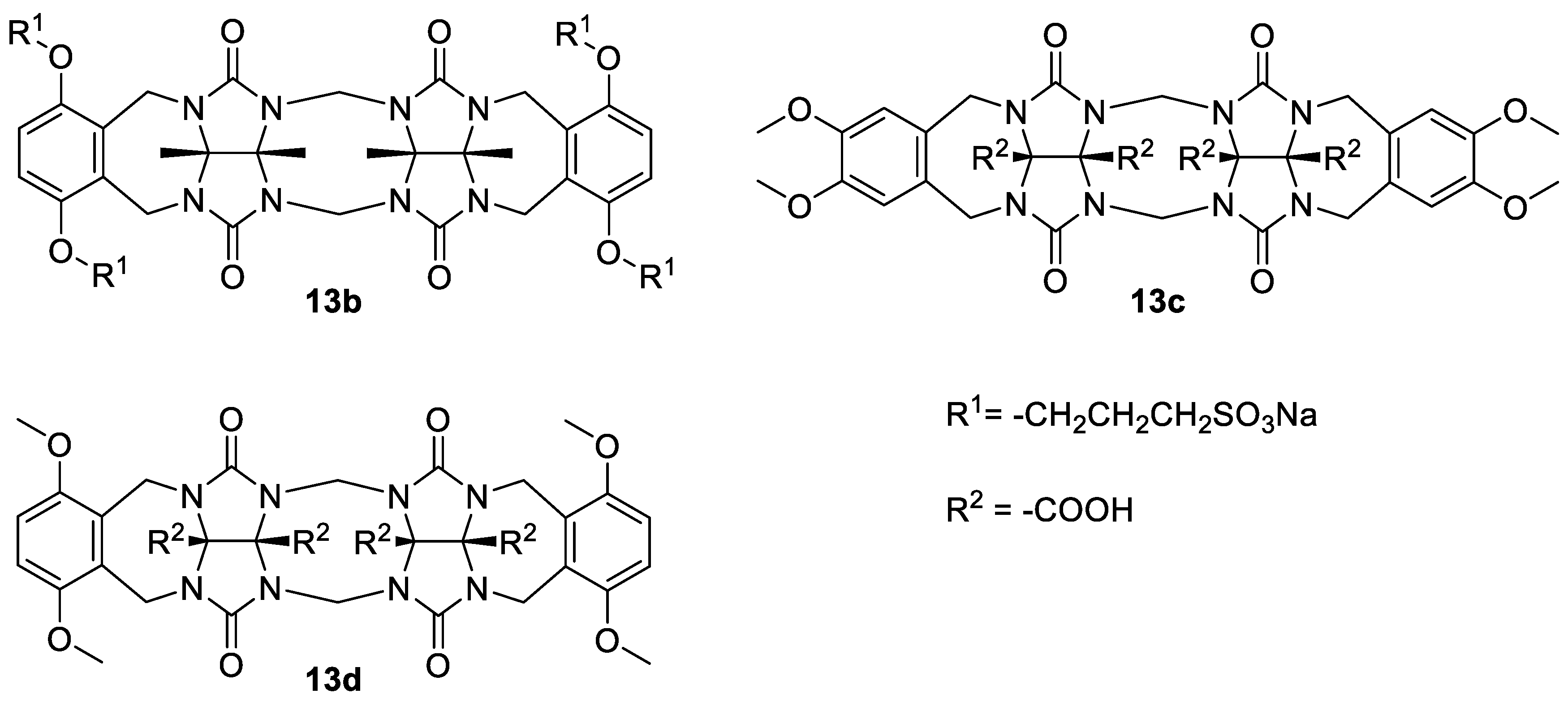
The binding affinities for the dimerization of
13a were compared to the related water-soluble glycoluril dimers
13b–
d (
Figure 2) [
10,
19]. The values showed that
13a dimerizes significantly more strongly than
13b; however, the
Ka of
13a2 is smaller than the
Ka of both
13c2 and
13d2 (
Table 1). Observed differences were probably due to the extended repulsion between the solubilizing groups upon the self-association, which is different among these molecules. Compound
13b has four sulfonate groups close to the binding site, which causes higher repulsion compared to
13a with two sulfonate groups. On the other hand, the carboxylate groups of
13c and
13d are located on the convex side of these molecules; therefore, they are an unlikely source of repulsion that might hinder self-association.
ITC measurements showed that
13a2 dimer formation is an exclusively enthalpically driven process, while entropy is unfavorable (
Table 1). This contrasts with the published thermodynamic data for
13c and
13d, where both favorable enthalpy and entropy of dimerization were reported. Compound
13a has two different types of aromatic side walls, the ‘unsubstituted’ wall originating from
o-xylylene glycoluril and the ‘substituted’ wall that contains electron-donating alkoxy groups. Therefore, we assume that π-π stacking to the dimerization enthalpy of
13a is greater than in the case of the reported compounds
13c and
13d, in which both aromatic walls are electron-rich due to the presence of methoxy groups. This might be the reason for the relatively high enthalpy of the
13a2 dimer formation. Entropy compensation in
13a dimerization, contrasting to the positive entropy term in the case of the
13c2 and
13d2 dimer formation, is probably due to the necessity to adopt an arrangement with the lowest electrostatic repulsion of the sulfonates. It should be noted that the above-mentioned differences among
13b–
d could also be a result of their different hydration [
30,
31].
Supramolecular studies performed with the model compound
13a revealed that its dimerization occurs at a submillimolar concentration in water. Compound
4 contains very similar binding motives as
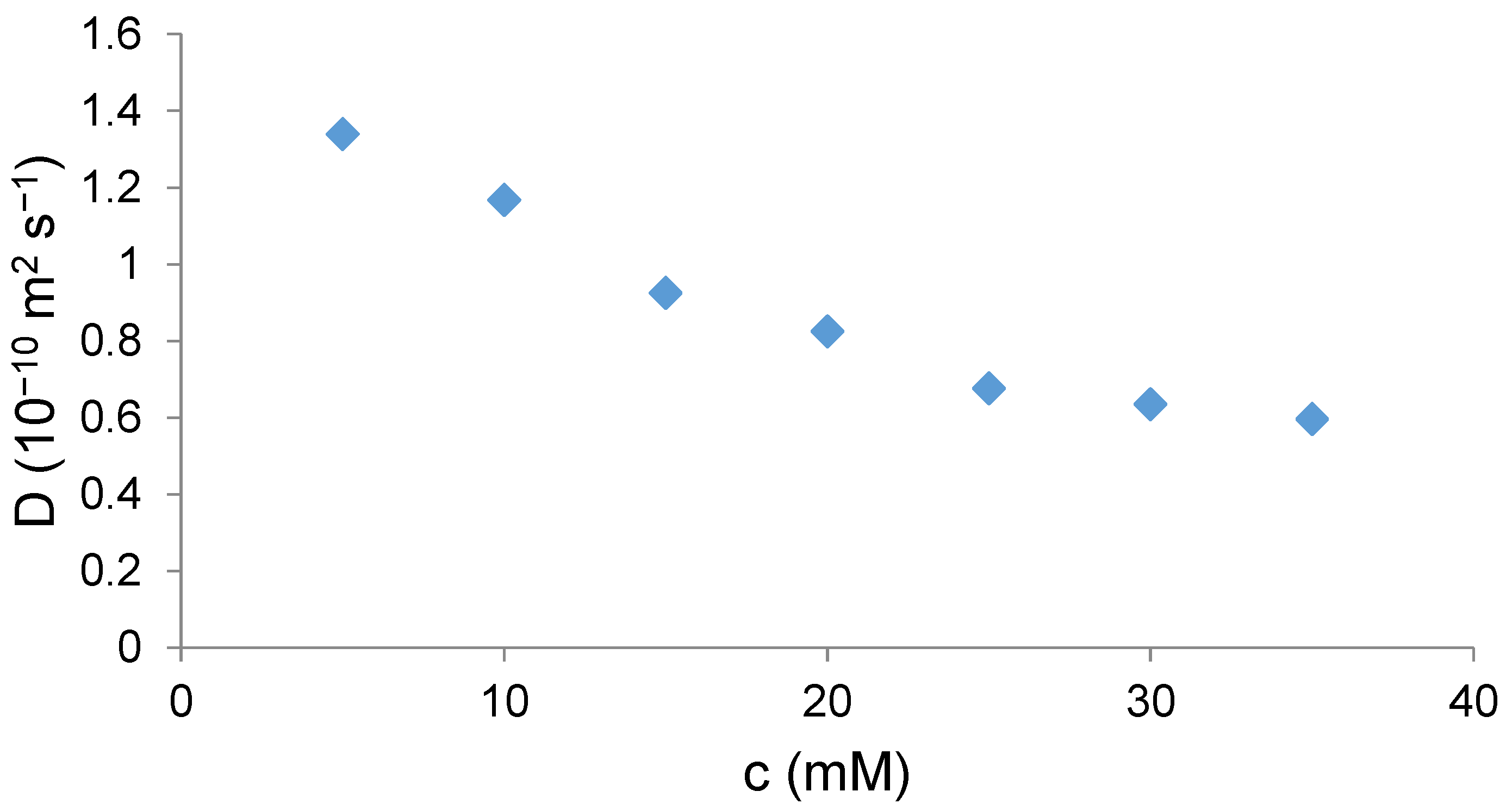
13a. Therefore, the formation of supramolecular polymers under similar conditions was expected. We decided to monitor the formation of supramolecular polymers by measuring diffusion coefficients of
4 at various concentrations using DOSY in D
2O. The measurement gave a monotonous decrease in the diffusion coefficient with an increasing concentration of
4 (
Figure 3). This trend indeed indicates that self-assembly occurred.
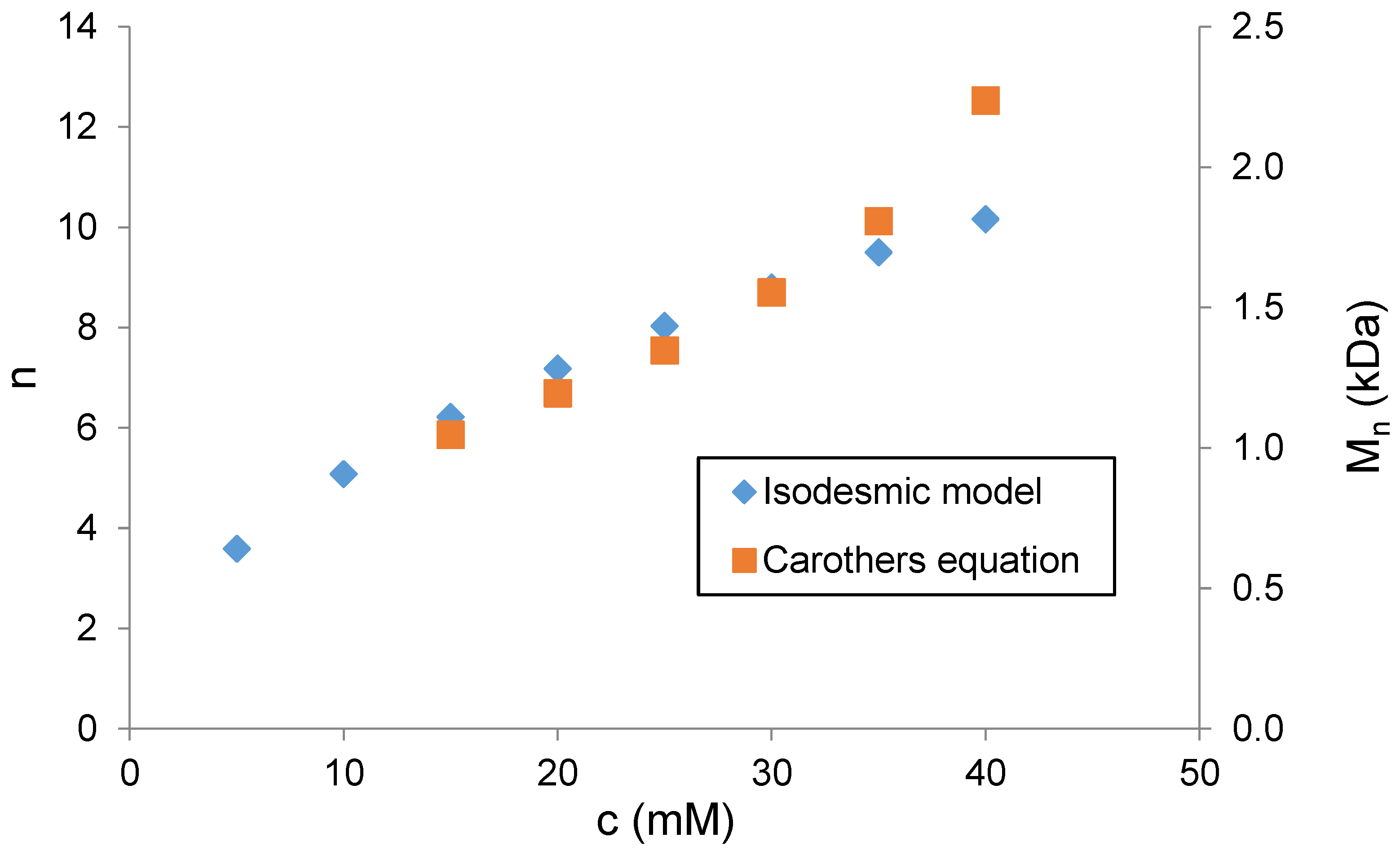
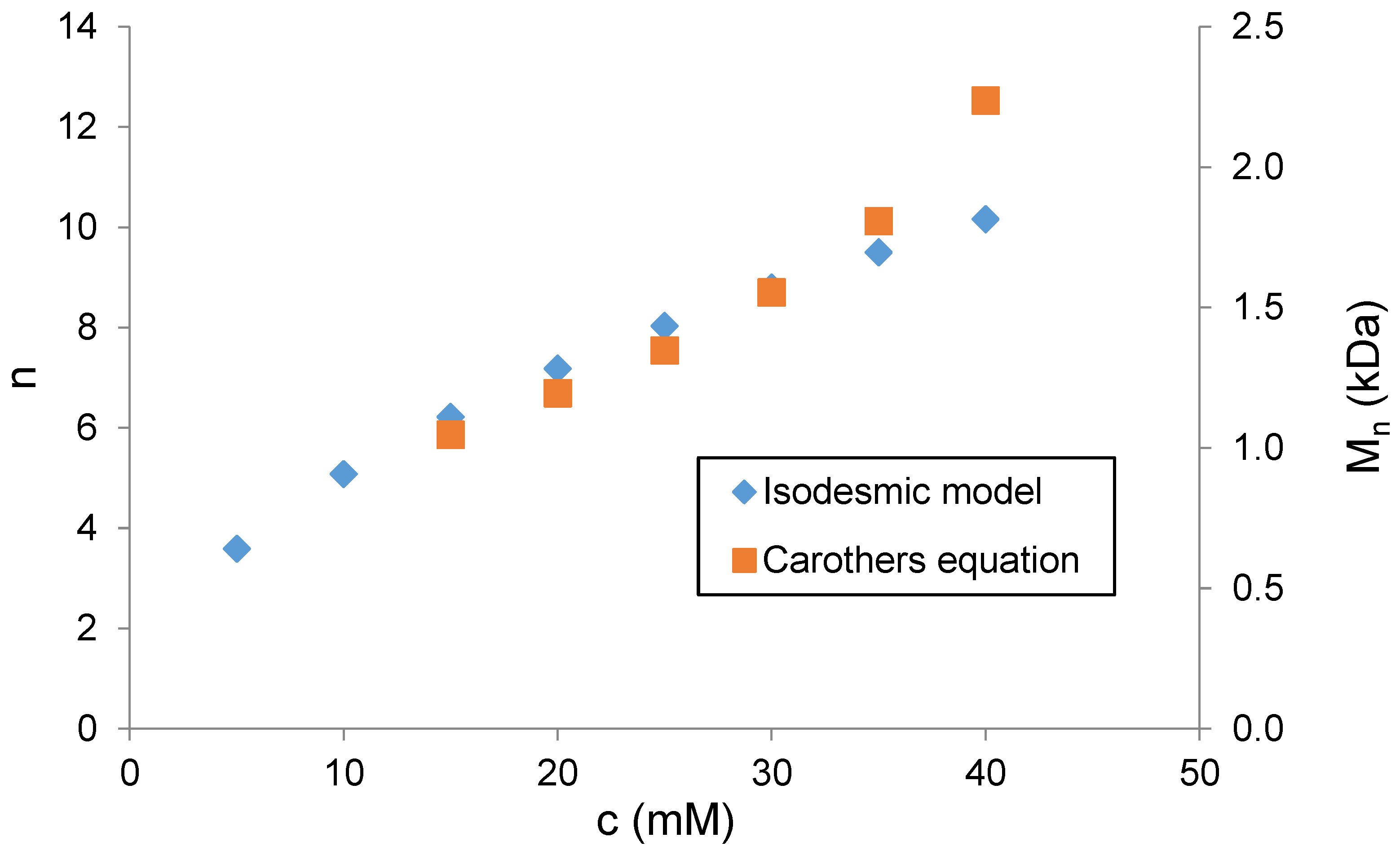
The degree of polymerization (
n) was then determined. With the
Ka of the compound of model
13a, it is possible to predict the degree of polymerization
of
4 using the isodesmic model [
32]. This model assumes that the binding sites of
13a and
4 are essentially the same, and all the binding events occur with the same association constant. The model is described by Equation (1):
where
c is the molar concentration of
4 and
Ka is the dimerization constant of
13a determined by NMR. The results presented in
Figure 4 show that the length of the supramolecular polymer increases from 3.6 repeating units at 5 mM concentrations of
4 to 10 units at 40 mM concentrations of
4 in D
2O.
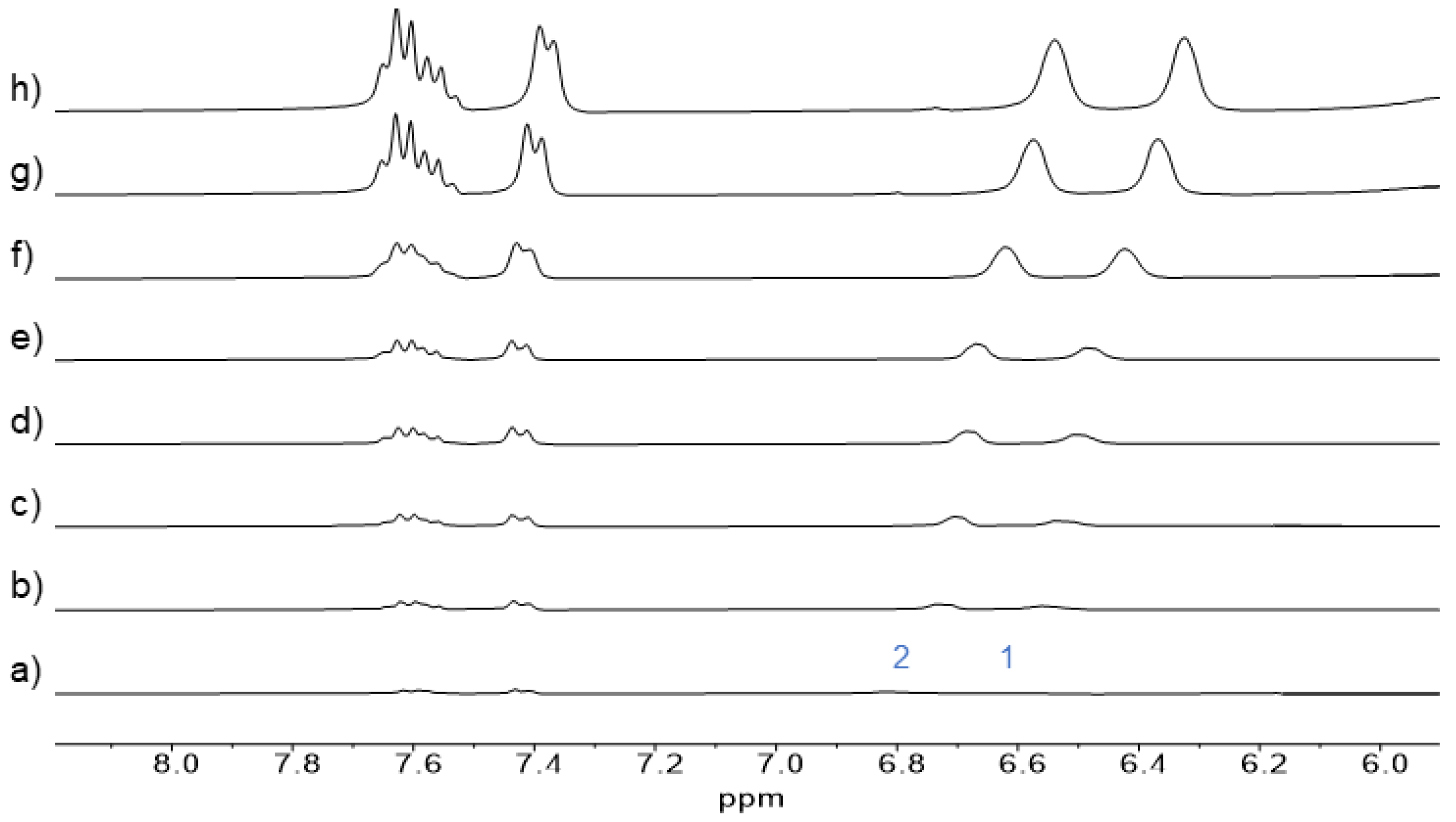
The second method for determining the degree of polymerization of
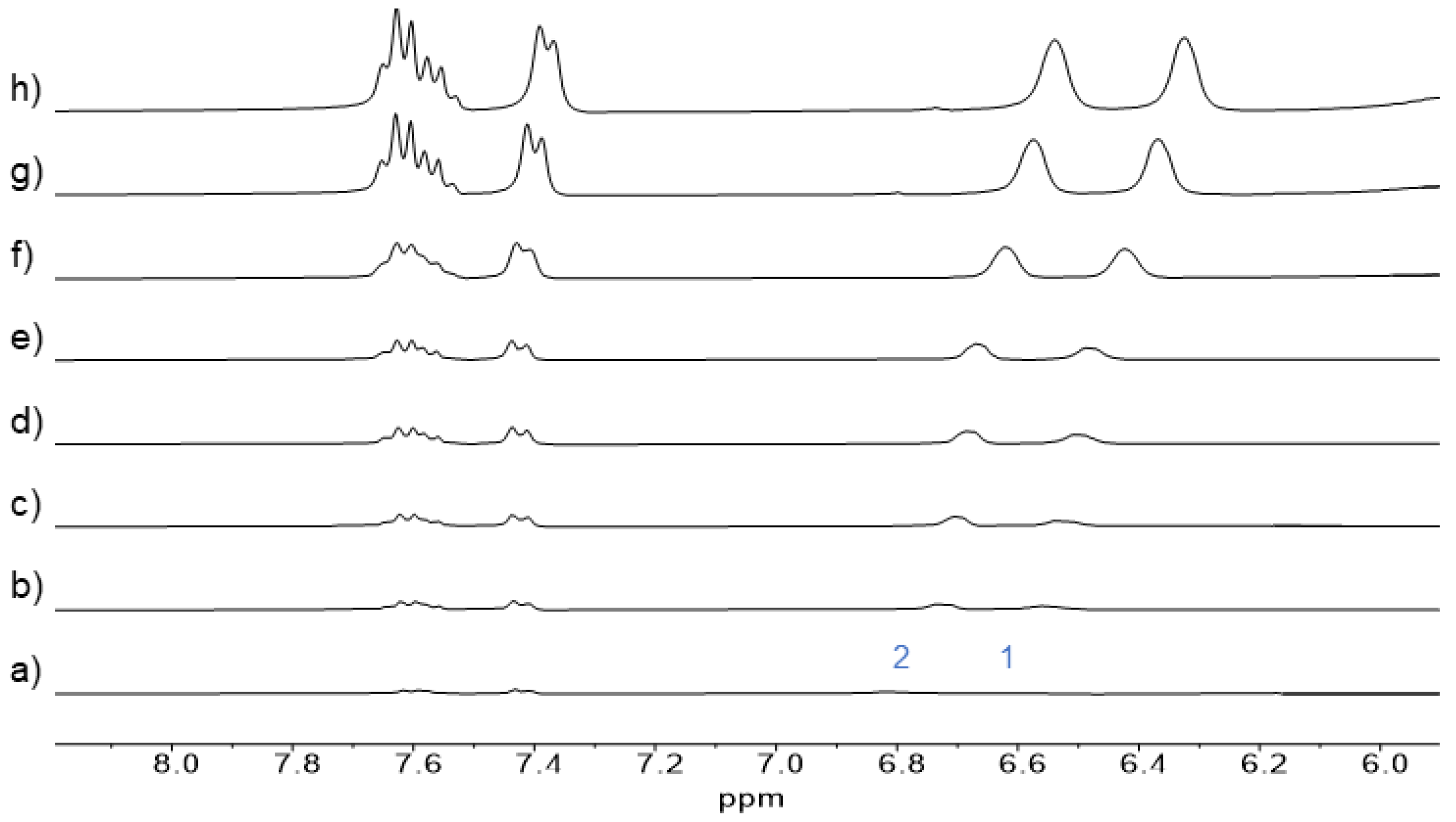
4 utilized NMR spectroscopy, specifically the chemical shift of protons of
4 induced by its self-association. Self-association of the
13a model compound was necessary for it to be monitored by
1H NMR (
Figure 5). We particularly monitored the chemical shift of aromatic protons H1 and H2 of
13a (for proton assignment, see
Scheme 1), which shifted to lower ppm with an increasing concentration of
13a in D
2O. From these measurements, chemical shifts corresponding to monomeric and dimeric forms of
13a were calculated using the non-linear curve-fitting of the data. The calculation yielded the percentage of free and complexed forms of
13a at various concentrations. The to lower ppm shift of aromatic protons similar to those of
13a was also observed for the solution of
4 in D
2O with its increasing concentration (
Figure S10). If we consider that the chemical shifts of aromatic protons in
4 and
13a with their increasing concentrations are essentially identical, we can use data obtained from the dimerization of
13a to calculate the chemical shifts of the free and dimeric compound
4. Subsequently, we determined the percentage of binding sites of
4, which undergo self-association at different concentrations (for calculation, see
Supplementary Materials).
Assuming that only linear aggregates are formed, the percentage of complexed sites can be associated with the monomer conversion
p the Carothers equation [
33], which allows one to calculate the degree of polymerization:
Figure 4 shows good agreement between the degrees of polymerization calculated by two different methods in the concentration range from 15 mM to 35 mM. However, at a higher concentration of
4 (>30 mM), the polymerization degree values obtained from the isodesmic model are significantly lower compared to those obtained from the Carothers equation. We assume that this deviation is due to increasing ionic strength, which influences the self-association process. As compound
4 is a sodium salt of sulfonic acid, the ionic strength increases with the increasing concentration of
4. The Carothers equation uses data of chemical shift from real solutions; thus, calculated values also reflect the increase in ionic strength. On the other hand, the isodesmic model presumes that self-association affinity is concentration independent, which causes deviation of calculated values from those obtained by Carothers equation above 30 mM of
4.
3. Experimental Section
All chemicals were purchased from commercial suppliers and used without further purification. THF was dried over 4 Å molecular sieves. NMR spectra were recorded on a Bruker Avance 300 spectrometer with a working frequency of 300.13 MHz for 1H and 75.48 MHz for 13C or a Bruker Avance 500 spectrometer with a working frequency of 500.13 MHz for 1H and 125.77 MHz for 13C. Both spectrometers were equipped with a BBFO probe. NMR spectra were recorded in D2O, DMSO-d6, or 95% DCOOD. Chemical shifts are provided in parts per million (ppm), and coupling constants (J) are presented in Hertz (Hz). The multiplicities of signals are reported as singlet (s), doublet (d), triplet (t), or multiplet (m). Data from the NMR titration experiments were analyzed by non-linear regression using an appropriate binding model. HypNMR2008 software was used for the analysis. HR-MS spectra were recorded on an Agilent 6224 Accurate-Mass TOF mass spectrometer. Samples were ionized by electrospray ionization (ESI) or by atmospheric pressure chemical ionization (APCI). Melting points were determined using an MPM-HV2 melting point apparatus in an open-end capillary tube.
ITC experiments were performed on a VP-ITC instrument (Microcal, GEHealthcare). Experiments were carried out at 303.15 ± 0.1 K. In a typical experiment, 29 aliquots (10 μL) of 5.0 mM solution of 13a were injected into ITC cells containing either ultrapure water, 20 mM, or 100 mM sodium phosphate-buffered water at pH = 7.0. Integrated heat effects were analyzed by non-linear regression using a dissociation model. Microcal Origin 7 software was used for the analysis.
3.1. Synthesis of Compound 3
Cyclic ether
1 [
25] (1.112 g, 2.01 mmol) was dissolved in methanesulfonic acid (17 mL).
o-Xylyleneglycoluril
5 (1.014 g, 4.15 mmol) was added and the solution was stirred under a nitrogen atmosphere at 60 °C for 4 h. The solution was allowed to cool down to RT, and it was poured into acetone (100 mL). The resulting precipitate was collected by filtration and washed with acetone. The crude product was dissolved in formic acid (15 mL) and precipitated with water (100 mL). The precipitate was collected by filtration and washed with water, acetone, and diethyl ether to afford compound 3 as a white powder (1.622 g, 72%).
NMR spectra of this compound indicate the presence of distinct conformers. 1H NMR (500 MHz, 95% DCOOD): δ (ppm) = 7.69 (s, 4H); 7.38 (m, 4H); 7.31 (m, 4H); 5.93 (d, J = 8.1 Hz, 2H); 5.89 (d, J = 16.0 Hz, 4H); 5.77 (d, J = 8.1 Hz, 2H); 5.49 (d, J = 11.0 Hz, 4H); 4.87 (d, J = 15.9 Hz, 4H); 4.70 (d, J = 15.9 Hz, 4H); 4.59 (m, 4H); 4.53 (m, 4H); 1.43, 1.39 (s, 6H). 13C NMR (125 MHz, 95% DCOOD): δ (ppm) = 157.44; 157.17; 136.65; 135.16; 134.96; 129.48; 129.10; 129.07; 128.44; 79.42; 78.25; 71.81; 70.53; 70.17; 48.56; 45.66; 17.70. HR-MS (MALDI, HCCA): calculated: [C48H46N16O10 + H]+ = 1007.3656 observed 1007.3667. Melting point > 300 °C.
3.2. Synthesis of Compound 4
Compound 3 (1.401 g, 1.39 mmol) and compound 6 (2.251 g, 5.65 mmol) were dissolved in a mixture of TFA (15 mL) and acetic anhydride (15 mL). The solution was heated to 95 °C for 3 h. Solvents were removed by rotary evaporation, and the resulting solid was triturated with methanol (200 mL). The crude product was dissolved in water (20 mL) and precipitated with acetone (150 mL). The precipitation was repeated twice with the same volume of solvents. The precipitate was then again dissolved in water (20 mL); the pH of the solution was adjusted to 7 using 1 M NaOH, and the water was removed by rotary evaporation. The resulting solid was washed with methanol and diethyl ether and dried in vacuo to afford compound 4 as a beige solid (1.101 g, 45%).
NMR spectra of this compound indicate the presence of distinct conformers. 1H NMR (300 MHz, DMSO-d6): δ (ppm) = 7.46, 7.39 (s, 4H); 7.22 (m, 8H); 5.54 (m, 8H); 5.36 (m, 4H); 4.57 (m, 8H); 4.13 (m, 4H); 3.92 (m, 8H); 3.80 (m, 4H); 2.62 (m, 8H); 2.00 (m, 8H); 1.08 (m, 6H). 13C NMR (125 MHz, DMSO-d6): δ (ppm) = 155.00; 154.89; 149.87; 137.38; 134.78, 134.69; 129.24; 128.73; 127.58; 127.21; 127.03; 113.37; 83.27, 83.09; 77.54; 77.18; 68.81; 68.35; 48.33; 48.29; 45.15; 36.02; 25.55; 17.81. HR-MS (ESI): calculated: [C72H74N16Na4O24S4 + 2 Na]2+ = 906.1660 observed = 906.1593. Melting point > 300 °C.
3.3. Synthesis of Compound 8
Phenylacetylene 7 (5.013 g, 48.95 mmol) was dissolved in anhydrous THF (150 mL) under a nitrogen atmosphere, the solution was cooled to −78 °C, and n-butyllithium (1.6 M in hexanes, 32.0 mL, 51.20 mmol) was added dropwise. The reaction mixture was stirred for 40 min, and it was heated up to 0 °C. Iodomethane (6.0 mL, 96.37 mmol) was added, and the reaction mixture was allowed to warm to room temperature and was stirred overnight. The reaction mixture was then treated with water (100 mL); the product was extracted with CH2Cl2 (3 × 80 mL); combined organic extracts were washed with water (40 mL) and brine (40 mL) dried over anhydrous MgSO4, and the solvent was removed by rotary evaporation to afford 8 as a yellowish oil (5.031 g, 89%).
The spectroscopic data matches those reported in the literature [
34].
3.4. Synthesis Compound 9
Alkyne 8 (4.721 g, 50.0 mmol) was dissolved in a mixture of CCl4 (61 mL) and CH3CN (61 mL). A mixture of NaIO4 (25.94 g, 121.3 mmol), NaHCO3 (0.270 g, 3.2 mmol), and MgSO4 (1.220 g, 10.1 mmol) in water (82 mL) was added to the solution. Aqueous 0.01 M RuCl3 (14 mL, 0.14 mmol) was added, and the reaction mixture was stirred at room temperature for 1.5 h. The mixture was diluted with ethyl acetate (400 mL), and the organic phase was separated from the precipitated salts by decantation. The organic phase was dried over MgSO4, filtered and evaporated. The product was purified by silica gel column chromatography using ethyl acetate as a mobile phase to give compound 9 as a yellow oil (5.47 g, 74%).
The spectroscopic data matches those reported in the literature [
35].
3.5. Synthesis of Glycoluril 10
Diketone 9 (4.60 g, 31.0 mmol) was dissolved in 0.3 M HCl, urea (7.6 g, 12.7 mmol) was added, and the mixture was stirred at 60 °C for 24 h. The suspension was filtered and the solid product was washed with water, acetone, and diethyl ether to afford glycoluril 10 as a white powder (4.102 g, 57%).
The spectroscopic data matches those reported in the literature [
36].
3.6. Synthesis of Compound 11
Paraformaldehyde (2.55 g, 85.0 mmol) was dissolved in 9 M HCl (100 mL) at 60 °C; the solution was cooled down to room temperature. Glycoluril 10 (3.854 g, 16.6 mmol) was added, and the solution was stirred for 24 h. Water (100 mL) was then added to the mixture, and it was stirred for another 12 h. The resulting precipitate was collected by filtration, washed with water and dried in vacuo to provide ether 11 as a white powder (2.751 g, 52%).
The spectroscopic data matches those reported in the literature [
36].
3.7. Synthesis of Compound 12
Cyclic ether 11 (2.451 g, 7.64 mmol) was dissolved in methanesulfonic acid (25 mL). o-Xylyleneglycoluril 5 (1.956 g, 8.02 mmol) was added, and the solution was stirred under a nitrogen atmosphere at 60 °C for 4 h. The solution cooled down to room temperature, and it was poured into water (100 mL). The resulting precipitate was collected by filtration, washed with water, and dried in vacuo to afford compound 12 as a white powder (3.804 g, 90%).
1H NMR (300 MHz, DMSO-d6): δ (ppm) = 7.50 (m, 3H); 7.36 (m, 2H); 7.27 (m, 4H,) 5.66 (d, J = 8.1 Hz, 1H); 5.59 (d, J = 8.1 Hz, 1H); 5.56 (d, J = 15.2 Hz, 2H); 5.25 (d, J = 11.00 Hz, 2H); 4.65 (d, J = 15.6 Hz, 2H); 4.59 (d, J = 15.6 Hz, 2H); 4.37 (d, J = 11.00 Hz, 2H); 4.24 (d, J = 15.2 Hz, 2H); 1.18 (s, 3H). 13C NMR (125 MHz, DMSO-d6): δ (ppm) = 155.01; 154.64; 137.64; 133.75; 129.63; 129.19; 129.07; 127.65; 127.56; 77.98; 76.52; 71.33; 68.73; 68.61; 48.50; 45.07; 17.87. HR-MS (APCI): calculated: [C27H26N8O5 + Cl]− = 577.1720, observed = 577.1719. Melting point > 290 °C (decomposition).
3.8. Synthesis of Compound 13a
Compound 12 (3.022 g, 5.56 mmol) and compound 6 (4.507 g, 11.31 mmol) were dissolved in a mixture of TFA (30 mL) and acetic anhydride (30 mL). The solution was heated to 95 °C for 3 h. Solvents were removed by rotary evaporation, and the resulting solid was triturated with methanol (500 mL). The crude product was dissolved in water (55 mL) and precipitated with acetone (400 mL). The precipitate was then again dissolved in water (30 mL); the pH of the solution was adjusted to 7 using 1 M NaOH, and the water was removed by rotary evaporation. The resulting solid was washed with methanol and diethyl ether and dried in vacuo to afford compound 13a as a yellowish solid (2.621 g, 51%).
1H NMR (500 MHz, DMSO-d6): δ (ppm) = 7.54 (t, J = 7.7 Hz, 2H); 7.46 (t, J = 7.4 Hz, 1H); 7.29 (d, J = 7.5 Hz, 2H); 7.23 (m, 2H); 7.19 (m, 2H); 6.74 (s, 2H); 5.57, (d, J = 8.2 Hz, 1H); 5.50 (d, J = 15.2 Hz, 2H); 5.49 (d, J = 8.2 Hz, 1H); 5.38 (d, J = 15.7 Hz, 2H); 4.59 (d, J = 15.5 Hz, 2H); 4.53 (d, J = 15.5 Hz, 2H); 4.12 (d, J = 15.2 Hz, 2H); 3.91 (m, 4H); 3.72 (d, J = 15.7 Hz, 2H); 2.65 (m, 2H); 2.59 (m, 2H); 1.99 (m, 4H); 1.06 (s, 3H). 13C NMR (125 MHz, DMSO-d6): δ (ppm) = 154.91; 154.84; 149.92; 137.33; 133.47; 129.45; 129.32; 129.24; 127.72; 127.61; 127.17; 113.55; 83.31; 77.32; 68.83; 68.54; 48.40; 48.28; 45.18; 35.86; 25.69; 17.82. HR-MS (ESI): calculated: [C39H40N8Na2O12S2-2 Na + H]− = 877.2291, observed: = 877.2289. Melting point > 300 °C.






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}