
Q-Tube®-Assisted Alkylation and Arylation of Xanthines and Other N-H-Containing Heterocycles in Water


 ,
,  and
and 
Abstract
1. Introduction
2. Materials and Methods
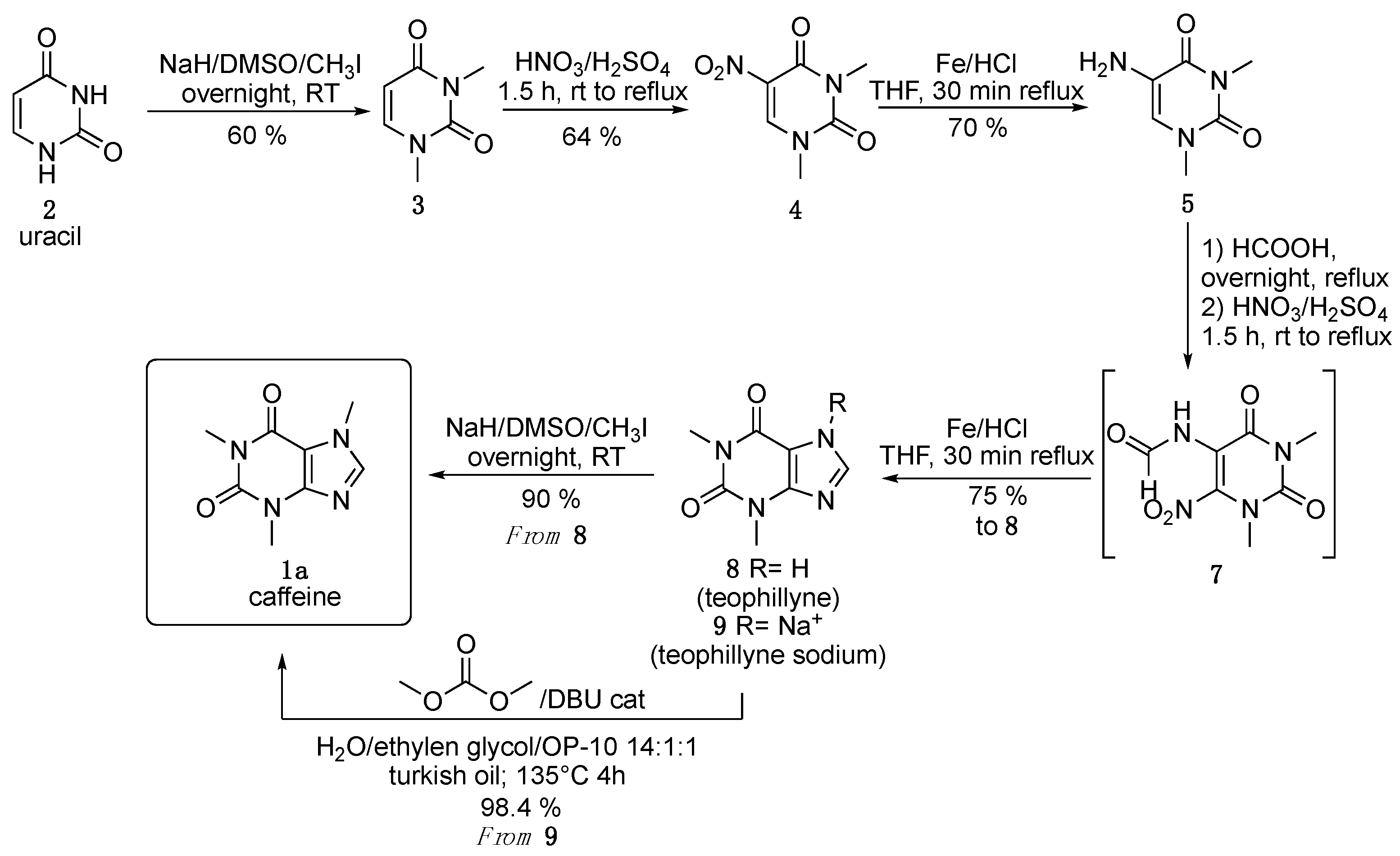
3. Results and Discussion
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Fischer, E.; Ach, L. Neue Synthese der Harnsäure und ihrer Methylderivate. Berichte der Dtsch. Chem. Gesellschaft 1895, 28, 2473–2480. [Google Scholar] [CrossRef]
- Verster, J.C.; Koenig, J. Caffeine intake and its sources: A review of national representative studies. Crit. Rev. Food Sci. Nutr. 2018, 58, 1250–1259. [Google Scholar] [CrossRef]
- Lisko, J.G.; Lee, G.E.; Kimbrell, J.B.; Rybak, M.E.; Valentin-Blasini, L.; Watson, C.H. Caffeine Concentrations in Coffee, Tea, Chocolate, and Energy Drink Flavored E-liquids. Nicotine Tob. Res. 2016, 19, 484–492. [Google Scholar] [CrossRef] [PubMed]
- Singh, N.; Shreshtha, A.K.; Thakur, M.S.; Patra, S. Xanthine scaffold: Scope and potential in drug development. Heliyon 2018, 4, e00829. [Google Scholar] [CrossRef] [PubMed]
- Hosseinzadeh, H.; Bazzaz, B.S.F.; Sadati, M.M. In vitro Evaluation of Methylxanthines and Some Antibiotics: Interaction against Staphylococcus aureus and Pseudomonas aeruginosa. Iran. Biomed. J. 2006, 10, 163–167. [Google Scholar]
- van Dam, R.M.; Hu, F.B.; Willett, W.C. Coffee, Caffeine, and Health. N. Engl. J. Med. 2020, 383, 369–378. [Google Scholar] [CrossRef] [PubMed]
- Ma, Y.J.; Jiang, D.Q.; Meng, J.X.; Li, M.X.; Zhao, H.H.; Wang, Y.; Wang, L.Q. Theophylline: A review of population pharmacokinetic analyses. J. Clin. Pharm. Ther. 2016, 41, 594–601. [Google Scholar] [CrossRef]
- Jiang, X.; Lim, L.Y.; Daly, J.W.; Li, A.H.; Jacobson, K.A.; Roberge, M. Structure-activity relationships for G2 checkpoint inhibition by caffeine analogs. Int. J. Oncol. 2000, 16, 971–979. [Google Scholar] [CrossRef][Green Version]
- Voynikov, Y.; Valcheva, V.; Momekov, G.; Peikov, P.; Stavrakov, G. Theophylline-7-acetic acid derivatives with amino acids as anti-tuberculosis agents. Bioorg. Med. Chem. Lett. 2014, 24, 3043–3045. [Google Scholar] [CrossRef]
- Traube, W. Der synthetische Aufbau der Harnsäure, des Xanthins, Theobromins, Theophyllins und Caffeïns aus der Cyanessigsäure. Berichte der Dtsch. Chem. Gesellschaft 1900, 33, 3035–3056. [Google Scholar] [CrossRef]
- Zajac, M.A.; Zakrzewski, A.G.; Kowal, M.; Narayan, S. A Novel Method of Caffeine Synthesis from Uracil. Synth. Commun. 2003, 33, 3291–3297. [Google Scholar] [CrossRef]
- Yang, S.; Dong, Z.; Yin, C.; Yue, H.; Gao, W.; Yang, F. Green synthesis of caffeine based on methylating reagent dimethyl carbonate and environmental friendly separating method. J. Chin. Chem. Soc. 2020, 67, 1715–1720. [Google Scholar] [CrossRef]
- Sancineto, L.; Tidei, C.; Bagnoli, L.; Marini, F.; Lenardão, E.; Santi, C. Selenium Catalyzed Oxidation of Aldehydes: Green Synthesis of Carboxylic Acids and Esters. Molecules 2015, 20, 10496–10510. [Google Scholar] [CrossRef]
- Vieira, B.M.; Thurow, S.; Brito, J.S.; Perin, G.; Alves, D.; Jacob, R.G.; Santi, C.; Lenardão, E.J. Sonochemistry: An efficient alternative to the synthesis of 3-selanylindoles using CuI as catalyst. Ultrason. Sonochem. 2015, 27, 192–199. [Google Scholar] [CrossRef]
- Krasowska, D.; Begini, F.; Santi, C.; Mangiavacchi, F.; Drabowicz, J.; Sancineto, L. Ultrasound-assisted synthesis of alkali metals diselenides (M2Se2) and their application for the gram-scale preparation of 2,2’-diselenobis(benzoic acid). Arkivoc 2019, 2019, 24–37. [Google Scholar] [CrossRef]
- Begini, F.; Krasowska, D.; Jasiak, A.; Drabowicz, J.; Santi, C.; Sancineto, L. Continuous flow synthesis of 2,2’-diselenobis(benzoic acid) and derivatives. React. Chem. Eng. 2020, 5, 641–644. [Google Scholar] [CrossRef]
- Mangiavacchi, F.; Crociani, L.; Sancineto, L.; Marini, F.; Santi, C. Continuous Bioinspired Oxidation of Sulfides. Molecules 2020, 25, 2711. [Google Scholar] [CrossRef]
- Cerra, B.; Mangiavacchi, F.; Santi, C.; Lozza, A.M.; Gioiello, A. Selective continuous flow synthesis of hydroxy lactones from alkenoic acids. React. Chem. Eng. 2017, 2, 467–471. [Google Scholar] [CrossRef]
- Nacca, F.G.; Merlino, O.; Mangiavacchi, F.; Krasowska, D.; Santi, C.; Sancineto, L. The Q-tube System, A Nonconventional Technology for Green Chemistry Practitioners. Curr. Green Chem. 2017, 4, 58–66. [Google Scholar] [CrossRef]
- Malakar, C.C.; Schmidt, D.; Conrad, J.; Beifuss, U. Double C−H Activation: The Palladium-Catalyzed Direct C-Arylation of Xanthines with Arenes. Org. Lett. 2011, 13, 1378–1381. [Google Scholar] [CrossRef] [PubMed]
- Ruddarraju, R.R.; Murugulla, A.C.; Kotla, R.; Tirumalasetty, M.C.B.; Wudayagiri, R.; Donthabakthuni, S.; Maroju, R.; Baburao, K.; Parasa, L.S. Design, synthesis, anticancer, antimicrobial activities and molecular docking studies of theophylline containing acetylenes and theophylline containing 1,2,3-triazoles with variant nucleoside derivatives. Eur. J. Med. Chem. 2016, 123, 379–396. [Google Scholar] [CrossRef] [PubMed]
- Bhalla, H.L.; Vavia, P.R.; Samuel, G.; Sivaprasad, N. Development of radioimmunoassay. J. Radioanal. Nucl. Chem. 1997, 220, 73–76. [Google Scholar] [CrossRef]
- Kondo, K.; Kuwata, K.; Takemoto, K. Functional monomers and polymers. IX. Synthesis of 2′,3′-epoxypropyl derivatives of adenine and theophylline. Makromol. Chem. 1972, 160, 341–346. [Google Scholar] [CrossRef]
- Hartman, T.; Cibulka, R. Photocatalytic Systems with Flavinium Salts: From Photolyase Models to Synthetic Tool for Cyclobutane Ring Opening. Org. Lett. 2016, 18, 3710–3713. [Google Scholar] [CrossRef]
- Toma, G.; Yamaguchi, R. Cobalt-Catalyzed C-N Bond-Forming Reaction between Chloronitrobenzenes and Secondary Amines. European J. Org. Chem. 2010, 2010, 6404–6408. [Google Scholar] [CrossRef]
- Ibata, T.; Isogami, Y.; Toyoda, J. Aromatic Nucleophilic Substitution of Halobenzenes with Amines under High Pressure. Bull. Chem. Soc. Jpn. 1991, 64, 42–49. [Google Scholar] [CrossRef]
- Watson, A.J.A.; Maxwell, A.C.; Williams, J.M.J. Borrowing Hydrogen Methodology for Amine Synthesis under Solvent-Free Microwave Conditions. J. Org. Chem. 2011, 76, 2328–2331. [Google Scholar] [CrossRef]
- Esmaeili, A.A.; Darbanian, M. Reaction between alkyl isocyanides and dialkyl acetylenedicarboxylates in the presence of N-alkyl isatins: Convenient synthesis of γ-spiro-iminolactones. Tetrahedron 2003, 59, 5545–5548. [Google Scholar] [CrossRef]
- Lacour, J.; Vial, L.; Herse, C. Efficient NMR Enantiodifferentiation of Chiral Quats with BINPHAT Anion. Org. Lett. 2002, 4, 1351–1354. [Google Scholar] [CrossRef]
- Sancineto, L.; Monti, B.; Merlino, O.; Rosati, O.; Santi, C. Q-Tube © assisted MCRs for the synthesis of 2,3-dihydroquinazolin-4(1H)-ones. Arkivoc 2018, 2018, 270–278. [Google Scholar] [CrossRef]
- Mordini, A.; Reginato, G.; Calamante, M.; Zani, L. Stereoselective Synthesis of Polysubstituted Piperazines and Oxopiperazines. Useful Building Blocks in Medicinal Chemistry. Curr. Top. Med. Chem. 2014, 14, 1308–1316. [Google Scholar] [CrossRef] [PubMed]
- Patel, R.; Park, S. An Evolving Role of Piperazine Moieties in Drug Design and Discovery. Mini-Reviews Med. Chem. 2013, 13, 1579–1601. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
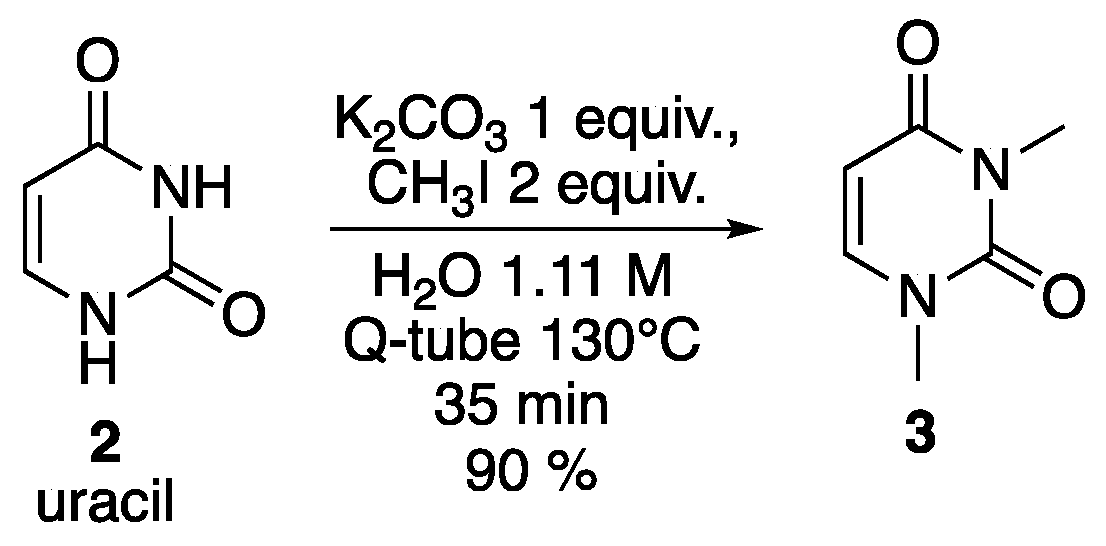{kind=link}
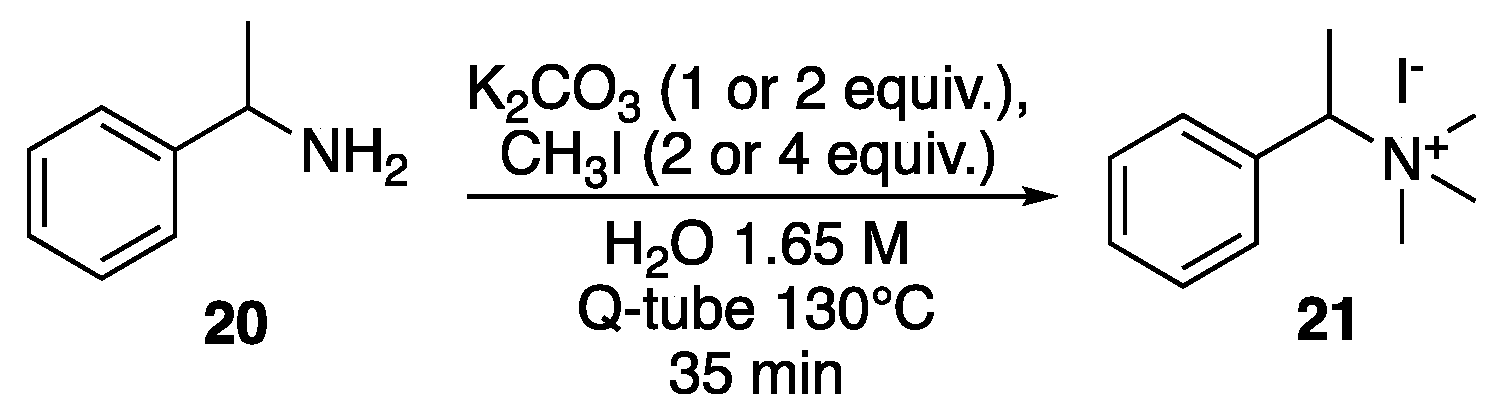{kind=link}
 | |||
|---|---|---|---|
| Entry | Base | Method a | Conversion b (Isolated Yield) |
| 1 | Cs2CO3 | A | 85% |
| 2 | K2CO3 | A | 91% |
| 3 | KOH | A | 77% |
| 4 | NaOH | A | 65% |
| 5 | LiOH | A | 55% |
| 6 | K2CO3 | B | 99% (64%) |
 | |||||
|---|---|---|---|---|---|
| Entry | R1 | R2 | Electrophile (E) | Products (1a–g) | Isolated Yield |
| 1 | Me | H | MeI |  | 64% |
| 2 | Me | H |  |  | 60% |
| 3 | Me | H |  |  | 30% a |
| 4 | Me | H |  |  | 42% |
| 5 | Me | H |  |  | 22% b |
| 6 | Me | H |  |  | 55% c |
| 7 | Me | H |  |  | 55% |
| 8 | H | Me | MeI |  | 76% |
 | ||||
|---|---|---|---|---|
| Entry | Heterocycles (11–13) | Electrophile (E) | Products (14–19) | Isolated Yield |
| 1 |  |  |  | 70% |
| 2 |  |  |  | 37% b |
| 3 |  |  |  | 78% |
| 4 a |  |  |  | 65% |
| 5 |  |  |  | 12% c |
| 6 |  | MeI |  | 27% d |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Scimmi, C.; Cardinali, M.; Abenante, L.; Amatista, M.; Nacca, F.G.; Lenardao, E.J.; Sancineto, L.; Santi, C. Q-Tube®-Assisted Alkylation and Arylation of Xanthines and Other N-H-Containing Heterocycles in Water. Chemistry 2021, 3, 1126-1137. https://doi.org/10.3390/chemistry3040082
Scimmi C, Cardinali M, Abenante L, Amatista M, Nacca FG, Lenardao EJ, Sancineto L, Santi C. Q-Tube®-Assisted Alkylation and Arylation of Xanthines and Other N-H-Containing Heterocycles in Water. Chemistry. 2021; 3(4):1126-1137. https://doi.org/10.3390/chemistry3040082
Chicago/Turabian StyleScimmi, Cecilia, Margherita Cardinali, Laura Abenante, Marina Amatista, Francesca Giulia Nacca, Eder J. Lenardao, Luca Sancineto, and Claudio Santi. 2021. "Q-Tube®-Assisted Alkylation and Arylation of Xanthines and Other N-H-Containing Heterocycles in Water" Chemistry 3, no. 4: 1126-1137. https://doi.org/10.3390/chemistry3040082
APA StyleScimmi, C., Cardinali, M., Abenante, L., Amatista, M., Nacca, F. G., Lenardao, E. J., Sancineto, L., & Santi, C. (2021). Q-Tube®-Assisted Alkylation and Arylation of Xanthines and Other N-H-Containing Heterocycles in Water. Chemistry, 3(4), 1126-1137. https://doi.org/10.3390/chemistry3040082