Abstract
In the present study, a quantitative structure–activity relationship (QSAR) and docking studies were accomplished on a series of 1,2,4-oxadiazoles. The results of QSARs are reliable and have high predictive ability for both the internal (q2 = 0.610) and external (pred_r2 = 0.553) datasets with least standard error (SE; i.e., 0.130) and four principal components, which signifies the reliability of the generated model. Molecular docking was also reported by the GOLD docking program, which showed that the hydrogen bonding may be responsible for the activity, and may be further increased upon adding high electronegative substitutions.
1. Introduction
Cancer is the second leading cause of mortality globally and becomes the principal cause of mortality in developing countries [1,2,3]. Recently, several anticancer drugs have become available on the market, acting through different mechanisms; however, the majority of them are associated with serious side effects [4,5]. The lack of targeting ability of these drugs is responsible for their side effects [6]. Large numbers of heterocyclic compounds have been reported for potential anticancer activities [7]. These heterocyclic-based anticancer agents are either under investigation or marketed as potent anticancer agents [8,9,10,11]. Oxadiazole is an important five-membered heterocyclic compound, having one oxygen atom and two nitrogen atoms. Recently, several 1,2,4-oxadiazole derivatives have been shown to possess anticancer activity [12,13].
Nowadays, researchers are focusing on various groups of molecules that are involved in the apoptosis inducing cytotoxicity. Caspases, a group of cysteine proteases, are the executioners of apoptosis. These caspases cleave their substrates after aspartic acid residues. Initiator caspases (caspase 2/8/9/10) and effector caspases (caspase 3/6/7) are the two classes of caspase. Recently, the activation of caspase-3 mediated apoptosis becomes an interesting therapeutic strategy for cancer therapy. Zhang et al. reported a series of 3-Aryl-5-aryl-1,2,4-oxadiazoles as a novel apoptosis inducer through caspase-3 activation. The compounds’ activities have been reported against breast and colorectal cancer cell lines [14].
The quantitative structure–activity relationship (QSARs) is an attempt to correlate the structural features of the compounds quantitatively with their biological activities. Researchers reported thousands of QSAR studies in the search for novel anticancer agents [15,16,17,18,19].
In the search for new anticancer agents, our research group previously reported QSAR studies of 1,2,4-oxadiazole derivatives describing the key structure features responsible for anticancer activities [20,21,22]. In the continuation of our previous work, herein we report the two-dimensional QSAR (2D-QSAR) and molecular docking studies’ outcomes.
The 2D-QSAR studies were done using Step Wise k Nearest Neighbor Molecular Field Analysis [(SW) kNN MFA] using V-Life Molecular Design Software Version 3.0 (V-Life Molecular Design). The docking studies were also performed using GOLD software.
2. Material and Methods
2.1. Dataset
A dataset of twenty eight 3-aryl-5-aryl-1,2,4-oxadiazoles derivatives has been taken for present QSAR study (Table 1). Compounds have high structural diversity with ample range of biological activity [14,23].

Table 1.
1,2,4-Oxadiazole analogues and their experimental caspase-3 activator activity.
2.2. 2D QSAR
2D QSAR studies were performed via Step Wise k Nearest Neighbor Molecular Field Analysis [(SW) kNN MFA] method using V-Life Molecular Design Software Version 3.0 (V-Life Molecular Design) [24,25,26].
The 2D QSAR studies were performed by dividing compounds in the training and test dataset which resulted several QSAR equations. Unicolumn statistics was done to divide training and test data compounds. Twenty-two compounds were positioned in the training set and 6 compounds (4b, 4d, 4e, 10a, 10d and 11h) in the test set.
2.3. Molecular Docking Analysis
Molecular docking was employed to locate the appropriate binding orientations and conformations of these 1,2,4-oxadiazoles interacting with caspase-3 using the docking program GOLD version 3.2. Ten docked conformers were produced for each 1,2,4-oxadiazole derivative. The conformation with the lowest docking energy in the most populated cluster is selected as the possible “active” conformation against the 1RE1 active site. In the present study, 28 compounds were successfully docked into the 1RE1 site.
The X-ray crystal structure (pdb: 1RE1) of caspase-3 was obtained from the Protein Data Bank. Initially, for protein preparation, water molecules were removed, hydrogen atoms added and AMBER7FF99 charges to the protein were applied. The ligands were docked inside a cubic GRID box (within 5 A° surrounding to the cocrystallized ligand) centered at the midpoint between the Cys205 and Gly238. Ten docking runs were performed for each compound in the dataset. In most cases the chosen pose was the top ranked solution.
3. Results and Discussion
3.1. 2D QSAR Results
The results of the unicolumn statistics are summarized in Table 2, which showed that the test is interpolative i.e., both test and training dataset contain compounds of high structure diversity with variation in biological activity. The test and the training set contained a diverse set of compounds with low, moderate and high biological activity.
Table 2.
Unicolumn statistical data of training and test set in 2D quantitative structure–activity relationship (QSAR) models.
Finally, the following model was selected.
pEC50 = 0.243 * IP − 0.139 * BC + 0.155 * DM + 0.008 * PSA + 0.0005
The obtained model showed a high correlation coefficient (r = 0.862) between descriptors including ionization potential (IP), bromine count (BC), dipole moment (DM), polar surface area (PSA) and anticancer activities. The squared correlation coefficient (r2) of 0.743, explains 74.29% of the variance in biological activity. The obtained model is statistical significant with F values F(4,21) = 11.561. The obtained model showed both good internal and external predictive ability with cross-validated squared correlation coefficient for internal dataset (q2) value 0.610 and for external dataset (pred_r2) value 0.553 with a standard error (SE) of 0.130 (Table 3).
Table 3.
Parameters value for the best 2D QSAR model generated.
In the model, the contribution of the descriptors is presented in the contribution chart (Figure 1), signifying the positive contribution of the ionization potential (IP), dipole moment (DM) and polar surface area (PSA) towards the biological activity. The addition of substitution that increases the polarity of the compounds results in increased anticancer activity. The negative contribution of the bromine count signifies the lower number of bromine encouraging biological activities.
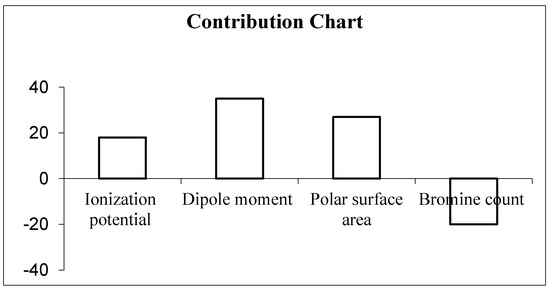
Figure 1.
Contribution chart of descriptors in 2D QSAR model.
The correlation between the experimental and predicted activity of the compounds is shown in Table 4 and represented in Figure 2.
Table 4.
Experimental, predicted and residual activities of the compounds obtained in 2D QSAR and GOLD score.
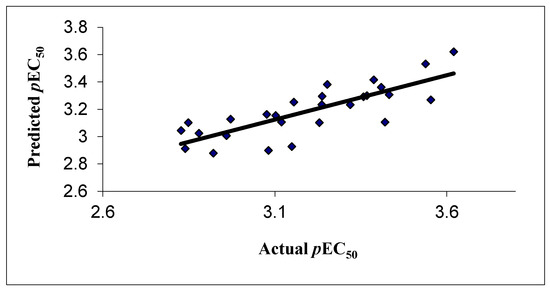
Figure 2.
Correlation of experimental and predicted activity in 2D QSAR model.
3.2. GOLD Docking Studies
All 28, 1,2,4-oxadiazoles derivatives were docked into the binding site of caspase-3 and the energy scores of the activators are also shown in Table 4. A precise correlation was observed in between docking scores and pIC50 values.
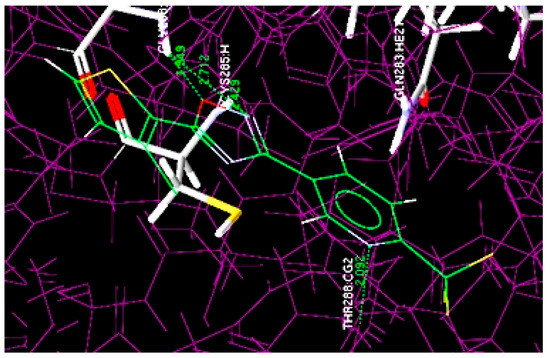
Figure 3.
Overlay of docked highest potent oxadiazole compound (4m) at the active site of 1RE1.
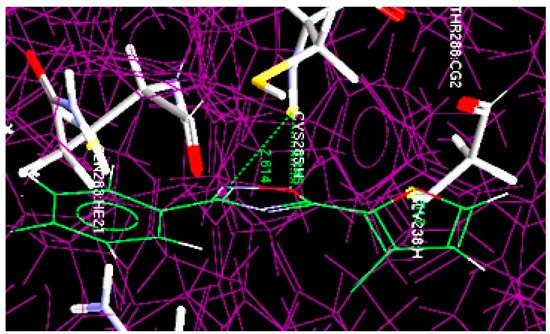
Figure 4.
Overlay of docked least potent oxadiazole compound (10b) at the active site of 1RE1.
The docking results revealed that most active compound 4m is properly located at the binding site of the Cys205 and Gly238 amino acid residues and numerous interactions occur between it and the binding region of the enzyme. The four key hydrogen bond interactions occur: (1) between the NH of Gly238 and the O of the oxadiazole ring; (2) between the NH of Gly238 and the N of the oxadiazole ring; (3) between the NH of Cys285 and the N of the oxadiazole ring; (4) between the NH of THR288 and the N of the pyridine ring residue (Figure 3). The hydrogen bonding distances observed were 1.549 Å (O···H-NH-Gly238), 2.712 Å (N···H-NH-Gly238), 2.429 Å (N···H-NH-Cys285) and 2.092 Å between the N of the pyridine ring and NH of THR288 (N···H-NH-THR288).
Akin to compound 4m, compound 10b was also docked at the same binding pockets having Cys205 and Gly238 amino acid residues (Figure 4). The result showed the formation of two hydrogen bonds: (1) between the NH of Cys205 and the O of the oxadiazole ring (O···H-NH-Cys205), having 2.145 Å bond distance; (2) between the NH of Cys205 and the N of the oxadiazole ring (N···H-NH-Cys205) with 2.614 Å bond length.
The docking results revealed that the hydrogen bonding may be responsible for biological activity, which may be further increase upon adding more electronegative substitutions. The correlation between the dock score and the experimental activity is shown graphically in Figure 5, which shows a linear correlation between the dock score and biological activity.
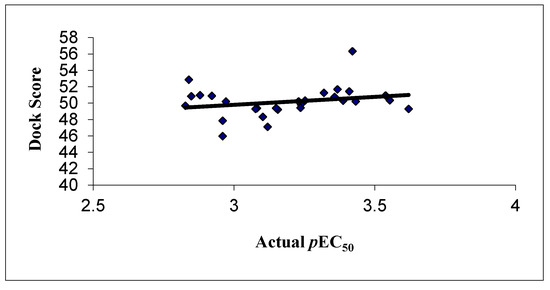
Figure 5.
Correlation between the experimental activities and dock score in GOLD docking.
The results of the QSAR analysis clearly show that upon increasing the polarity in terms of theionization potential (IP), dipole moment (DM) and polar surface area (PSA), biological activity will also be enhanced. The docking results also support the QSAR outcomes.
4. Conclusions
In conclusion, the current QSAR studies established a reliable QSAR model with high predictive ability with q2 = 0.610, r2 = 0.743 and low standard error (SE) = 0.130 and four principal components. The predicted value of the external test set (pred_r2) was also high (i.e., 0.553). The developed model was reliable, which indicated the importance of substitution in 1,2,4-oxadiazoles at their respective positions to improve anticancer activity. The positive contribution of ionization potential (IP), dipole moment (DM) and polar surface area (PSA) is conducive for biological activity, and further addition of these substitutions increases anticancer activity, while the negative contribution of the bromine count signifies the lower number of bromine encouraging the biological activities. The docking results explore the binding mode between the ligands and the receptor.
Funding
This research received no external funding.
Data Availability Statement
Data available in article and raw data are available from the corresponding authors upon request.
Acknowledgments
We would like to thank G. Narahari Sastry, former head of the department, Molecular Modeling Group, IICT Hyderabad, India, for providing access to computational resources and for their valuable help during the modeling studies.
Conflicts of Interest
The authors declare that they have no conflict of interest.
References
- Thun, M.J.; Henley, S.J.; Burns, D.; Jemal, A.; Shanks, T.G. Lung cancer death rates in lifelong nonsmokers. J. Natl. Cancer Inst. 2006, 98, 691–699. [Google Scholar] [CrossRef]
- Jain, S.; Pathak, K.; Vaidya, A. Molecular therapy using siRNA: Recent trends and advances of multi target inhibition of cancer growth. Int. J. Biol. Macromol. 2018, 116, 880–892. [Google Scholar] [CrossRef] [PubMed]
- Vaidya, A.; Jain, S.; Sahu, S.; Jain, P.K.; Pathak, K.; Pathak, D.; Kumar, R.; Jain, S.K. Anticancer agents based on vulnerable components in a signalling pathway. Mini Rev. Med. Chem. 2020, 20, 886–907. [Google Scholar] [CrossRef] [PubMed]
- Ayati, A.; Emami, S.; Asadipour, A.; Shafiee, A.; Foroumadi, A. Recent applications of 1,3-thiazole core structure in the identification of new lead compounds and drug discovery. Eur. J. Med. Chem. 2015, 97, 699–718. [Google Scholar] [CrossRef]
- Vaidya, A.; Jain, S.; Jain, A.K.; Agrawal, A.; Kashaw, S.K.; Jain, S.K.; Agrawal, R.K. Metabotropic Glutamate Receptors: A Review on Prospectives and Therapeutic Aspects. Mini Rev. Med. Chem. 2013, 12, 1967–1981. [Google Scholar] [CrossRef]
- Jain, S.; Vaidya, A.; Jain, A.K.; Agrawal, R.K.; Kashaw, S.K. Computational analysis of benzyl vinylogous derivatives as potent PDE3B inhibitors. Arab. J. Chem. 2017, 10, S109–S113. [Google Scholar] [CrossRef]
- Jain, A.K.; Vaidya, A.; Ravichandran, V.; Kashaw, S.K. Recent Developments and Biological Activities of Thiazolidinone Derivatives: A Review. Bioorg. Med. Chem. 2012, 20, 3378–3395. [Google Scholar] [CrossRef]
- Jain, A.K.; Sharma, S.; Vaidya, A.; Ravichandran, V.; Agrawal, R.K. 1,3,4-Thiadiazole and Its Derivatives: A Review on Recent Progress in Biological Activities. Chem. Biol. Drug. Des. 2013, 81, 557–576. [Google Scholar] [CrossRef] [PubMed]
- Jain, S.; Pattnaik, S.; Pathak, K.; Kumar, S.; Pathak, D.; Jain, S.K.; Vaidya, A. Anticancer Potential of Thiazole Derivatives: A Retrospective Review. Mini Rev. Med. Chem. 2017, 18, 640–655. [Google Scholar] [CrossRef]
- Vaidya, A.; Jain, S.; Jain, A.K.; Prashanthakumar, B.R.; Kashaw, S.K.; Agrawal, R.K. Computational Analysis of Quinoline Derivatives as Potent Topoisomerase-II Inhibitors. Med. Chem. Res. 2015, 24, 383–393. [Google Scholar] [CrossRef]
- Jain, S.; Chandra, V.; Pankaj Kumar, J.; Pathak, K.; Pathak, D.; Vaidya, A. Comprehensive review on current developments of quinoline-based anticancer agents. Arab. J. Chem. 2019, 12, 4920–4946. [Google Scholar] [CrossRef]
- Anjos, J.V.; Ricardo, A.W.; Filho, N.; Nascimento, S.C.; Srivastava, R.M.; Melo, S.; Sinou, D.J. Synthesis and cytotoxic profile of glycosyl-triazole linked to 1,2,4-oxadiazole moiety at C-5 through a straight-chain carbon and oxygen atoms. Eur. J. Med. Chem. 2009, 44, 3571–3576. [Google Scholar] [CrossRef]
- Vaidya, A.; Jain, S.; Jain, P.; Jain, P.; Tiwari, N.; Jain, R.; Jain, R.; Jain, A.K.; Agrawal, R.K. Synthesis and Biological Activities of Oxadiazole Derivatives: A Review. Mini Rev. Med. Chem. 2016, 16, 825–845. [Google Scholar] [CrossRef]
- Zhang, H.Z.; Kaisbhatla, S.; Kuemmerle, J.; Kemnitzer, W.; Mason, K.O.; Qui, L.; Grundy, C.C.; Tseng, B.; Drew, J.; Cai, S.X. Discovery and Structure−Activity Relationship of 3-Aryl-5-aryl-1,2,4-oxadiazoles as a New Series of Apoptosis Inducers and Potential Anticancer Agents. J. Med. Chem. 2005, 48, 5215–5223. [Google Scholar] [CrossRef]
- Bhatiya, R.; Vaidya, A.; Kashaw, S.K.; Jain, A.K.; Agrawal, R.K. QSAR analysis of furanone derivatives as potential COX-2 inhibitors: kNN MFA approach. J. Saudi Chem. Soc. 2014, 18, 977–984. [Google Scholar] [CrossRef]
- Vaidya, A.; Jain, S.; Jain, S.; Jain, A.K.; Agrawal, R.K. Quantitative Structure-Activity Relationships: A Novel Approach of Drug Design and Discovery. J. Pharm. Sci. Pharmacol. 2014, 1, 219–232. [Google Scholar] [CrossRef]
- Agrawal, R.K.; Jain, A.K.; Veerasamy, R.; Vaidya, A.; Kashaw, S.; Mourya, V.K.; Agrawal, R.K. QSAR analysis of B-ring-modified diaryl ether derivatives as a InhA inhibitors. Med. Chem. Res. 2012, 21, 145–151. [Google Scholar]
- Vaidya, A.; Jain, A.K.; Kumar, P.; Kashaw, S.K.; Agrawal, R.K. Predicting anti-cancer activity of quinoline derivatives: CoMFA and CoMSIA approach. J. Enzyme Inhib. Med. Chem. 2011, 26, 854–861. [Google Scholar] [CrossRef] [PubMed]
- Jain, A.K.; Veerasamy, R.; Vaidya, A.; Mourya, V.; Agrawal, R.K. QSAR analysis of some novel sulfonamides incorporating 1,3,5-triazine derivatives as carbonic anhydrase inhibitors. Med. Chem. Res. 2010, 19, 1191–1202. [Google Scholar] [CrossRef]
- Vaidya, A.; Jain, A.K.; Kumar, B.R.P.; Sastry, G.N.; Kashaw, S.K.; Agrawal, R.K. CoMFA, CoMSIA, kNN MFA and Docking studies of 1.;2.;4-Oxadiazole derivatives as potent Caspase-3 activators. Arab. J. Chem. 2017, 10, S3936–S3946. [Google Scholar] [CrossRef]
- Vaidya, A.; Jain, S.; Kumar, B.R.P.; Singh, S.K.; Kashaw, S.K.; Agrawal, R.K. Synthesis of 1,2,4-oxadiazole derivatives: Anticancer and 3D QSAR studies. Mon. Chem. 2020, 151, 385–395. [Google Scholar] [CrossRef]
- Vaidya, A.; Pathak, D.; Shah, K. 1,3,4-oxadiazole and its Derivatives: A Review on Recent Progress in Anticancer Activities. Chem. Biol. Drug Des. 2020, 97, 572–591. [Google Scholar] [CrossRef] [PubMed]
- Kemnitzer, W.; Kuemmerle, J.; Zhang, H.Z.; Kaisbhatla, S.; Tseng, B.; Drew, J.; Cai, S.X. Discovery of 3-aryl-5-aryl-1,2,4-oxadiazoles as a new series of apoptosis inducers. 2. Identification of more aqueous soluble analogs as potential anticancer agents. Bioorg. Med. Chem. Lett. 2009, 19, 4410–4415. [Google Scholar] [PubMed]
- Roy, K.; Das, R.N. A review on principles, theory and practices of 2D-QSAR. Curr. Drug. Metab. 2014, 15, 346–379. [Google Scholar] [CrossRef] [PubMed]
- Adhikari, C.; Mishra, B.K. Quantitative Structure-Activity Relationships of Aquatic Narcosis: A Review. Curr. Comput. Aided Drug Des. 2018, 14, 7–28. [Google Scholar] [CrossRef] [PubMed]
- Lewis, R.A.; Wood, D. Modern 2D QSAR for drug discovery. Wiley Interdiscip. Rev. Comput. Mol. Sci. 2014, 4, 505–522. [Google Scholar] [CrossRef]






























































Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).