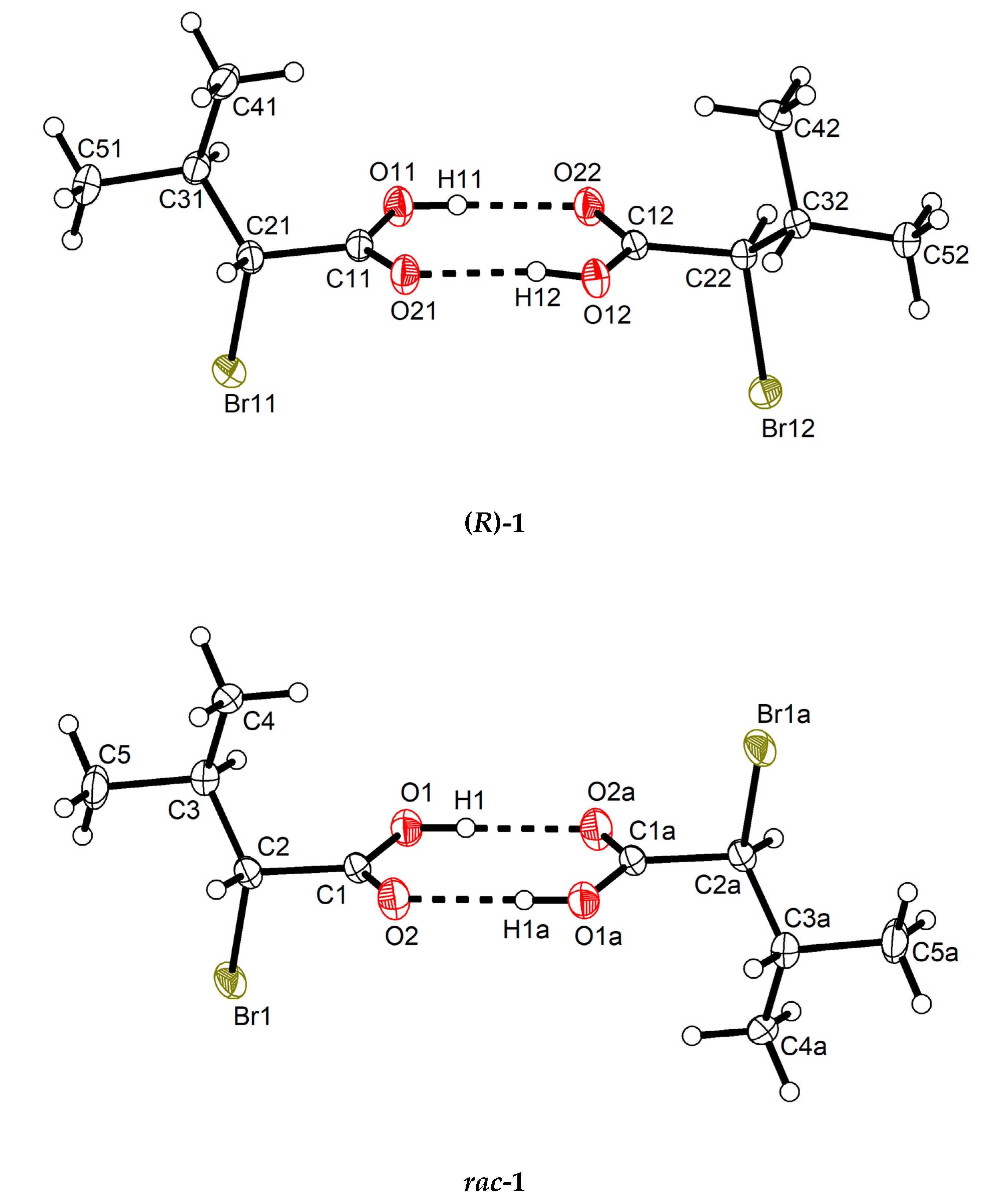
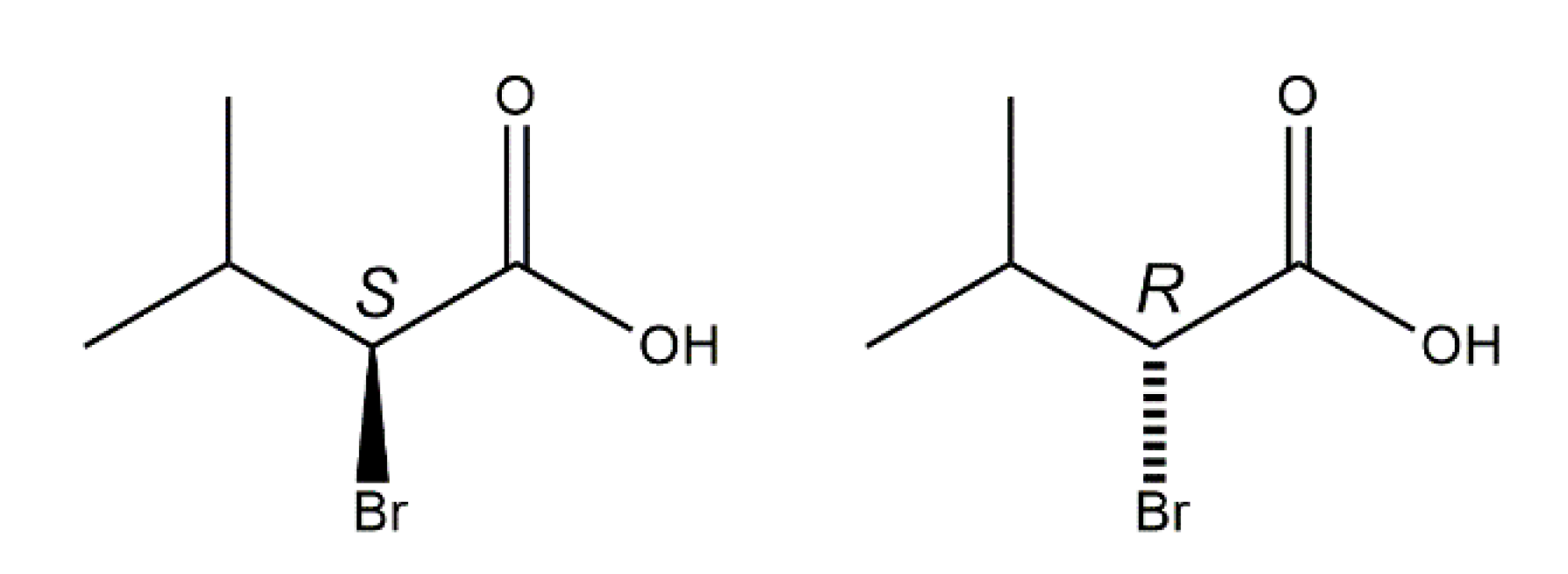
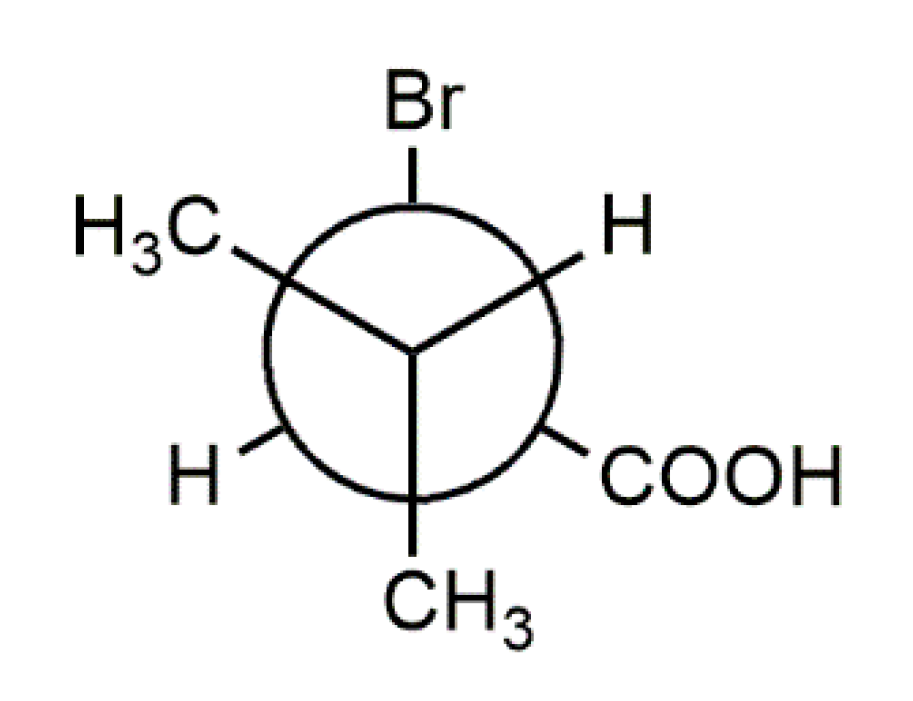
Structural Elucidation of Enantiopure and Racemic 2-Bromo-3-Methylbutyric Acid †


Abstract
1. Introduction
2. Materials and Methods
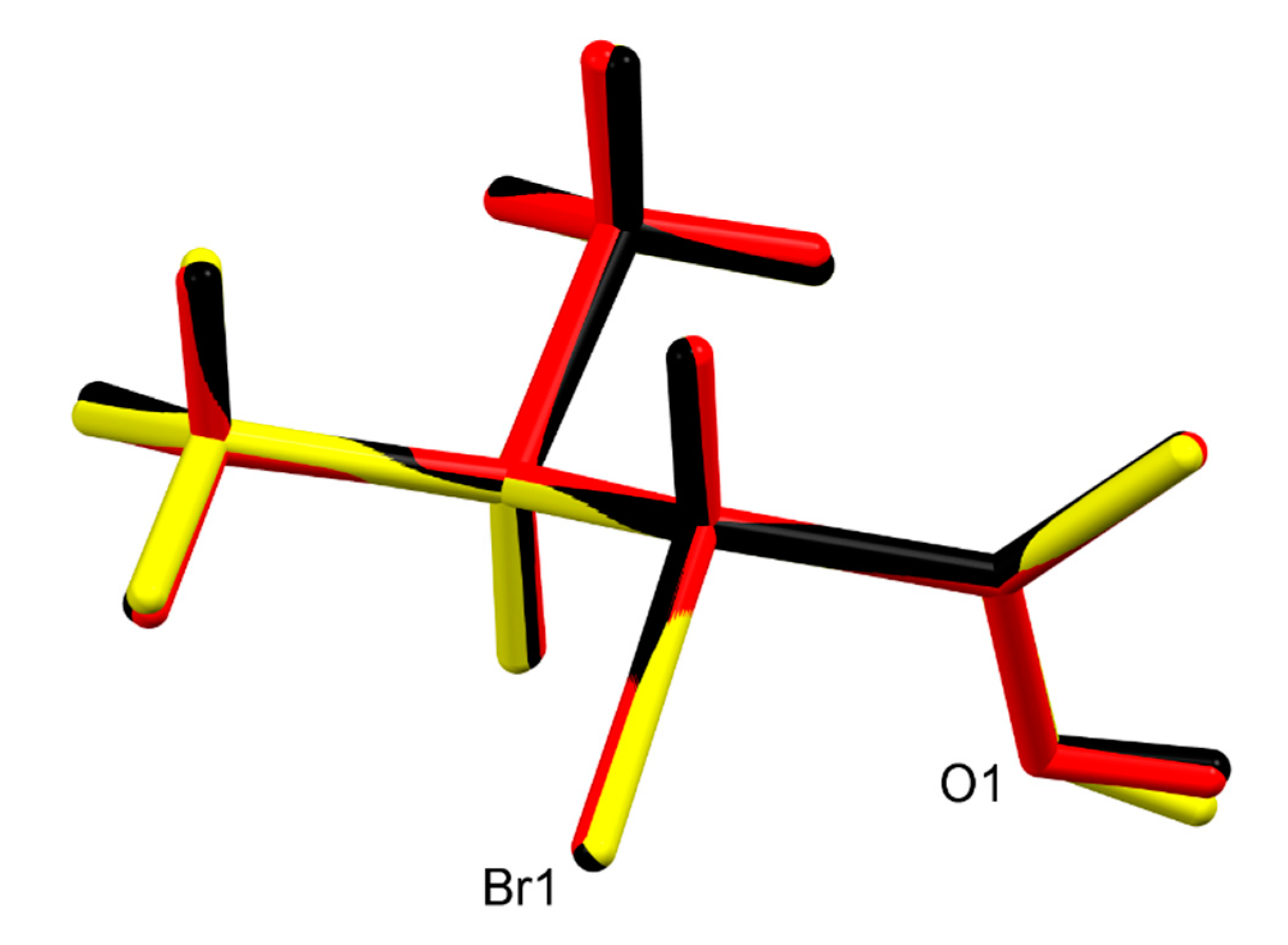
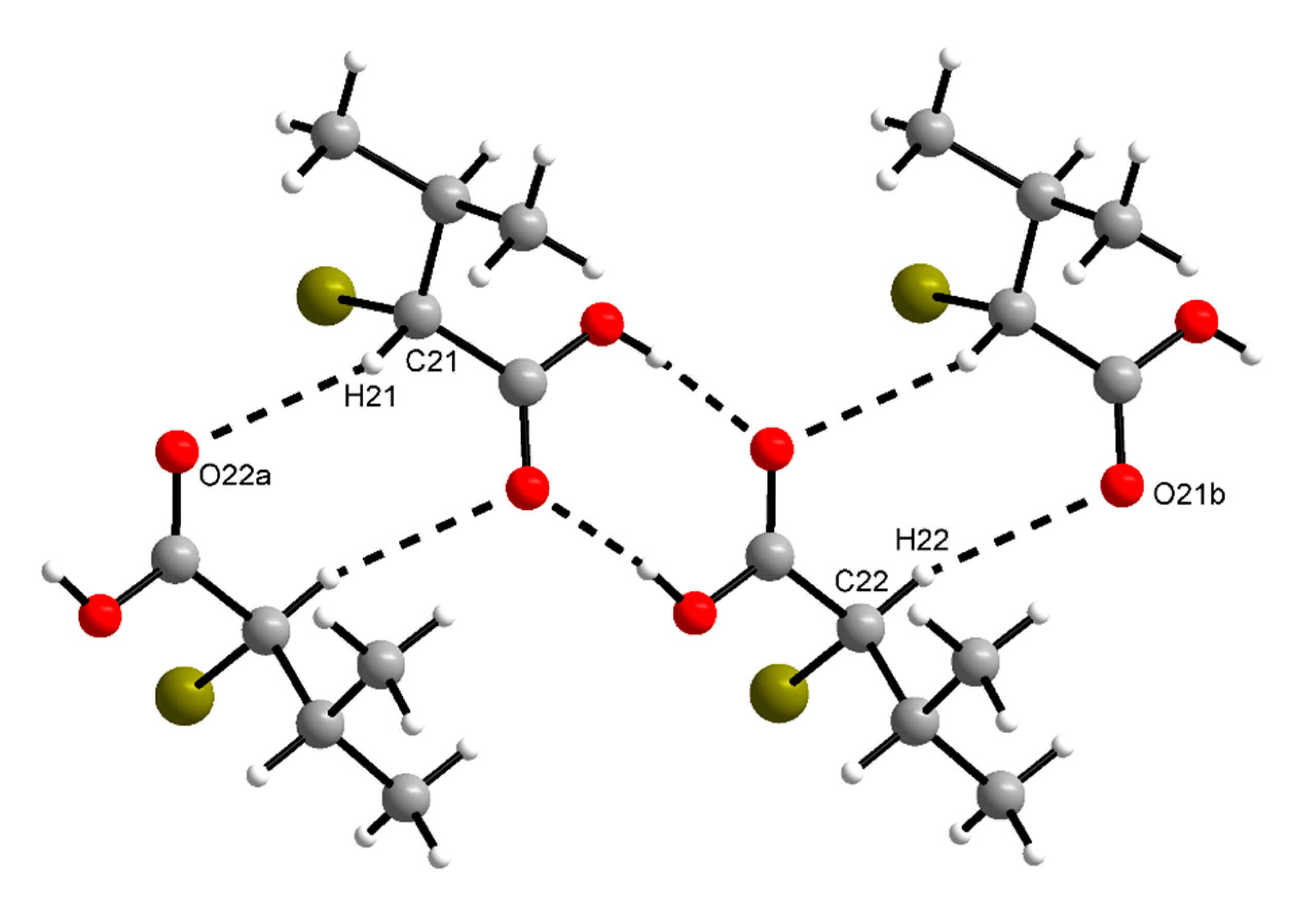
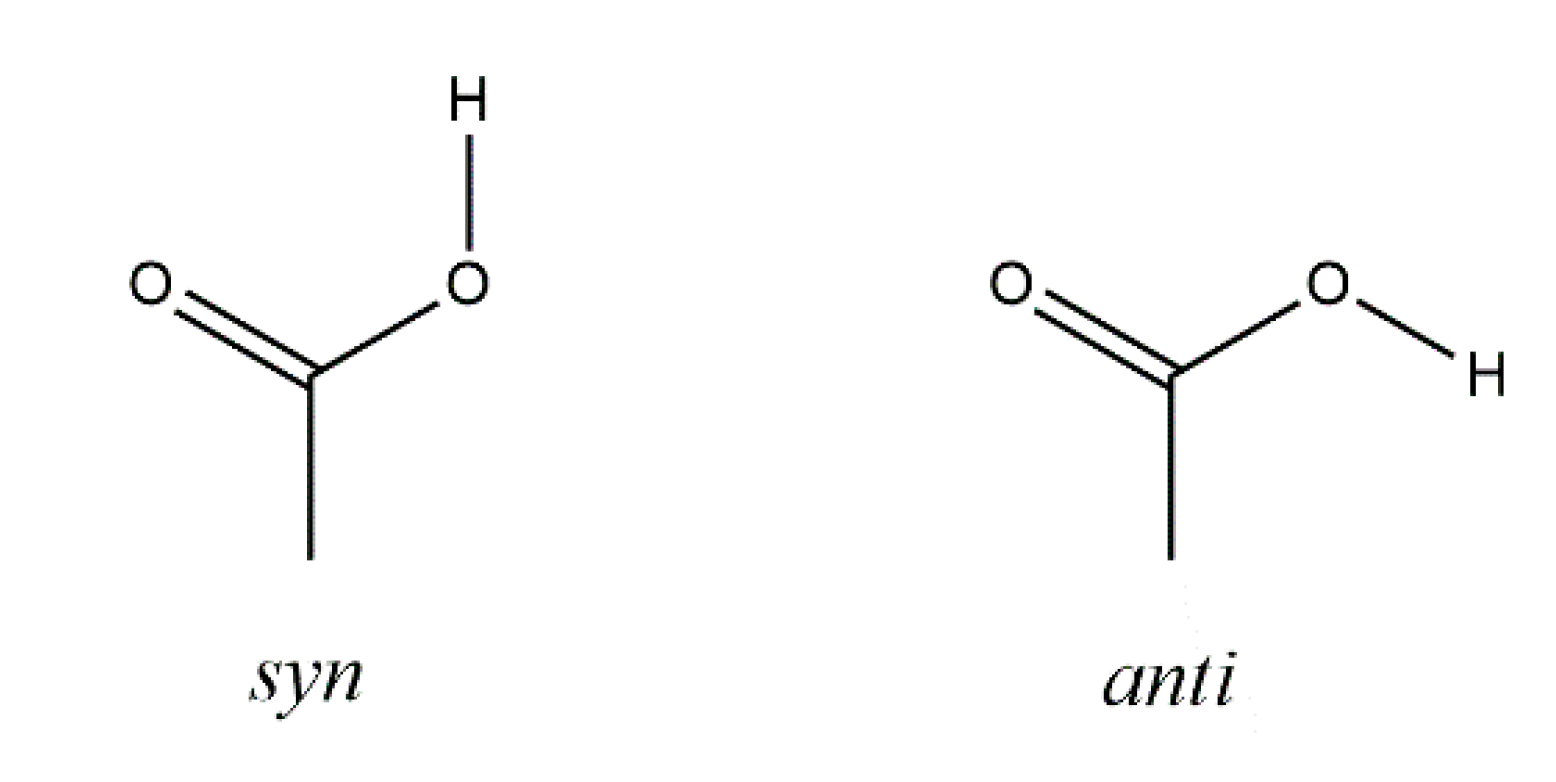
3. Results
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References and Note
- Czekelius, C.; Tzschucke, C.C. Synthesis of Halogenated Carboxylic Acids and Amino Acids. Synth. Stuttg. 2010, 543–566. [Google Scholar] [CrossRef]
- Kubitschke, J.; Lange, H.; Strutz, H. Carboxylic Acids, Aliphatic. Ullmann’s Encycl. Ind. Chem. 2014, 1–18. [Google Scholar] [CrossRef]
- Ullmann’s Fine Chemicals; Wiley: New York, NY, USA, 2014.
- Kirk, K.L. Biochemistry of halogenated organic compounds. In PATAI’S Chemistry of Functional Groups; Rappoport, Z., Ed.; Wiley: New York, NY, USA, 2009. [Google Scholar] [CrossRef]
- Yue, Y.; Chen, J.; Bao, L.; Wang, J.; Li, Y.; Zhang, Q. Fluoroacetate dehalogenase catalyzed dehalogenation of halogenated carboxylic acids: A QM/MM approach. Chemosphere 2020, 254, 126803. [Google Scholar] [CrossRef] [PubMed]
- Whitehouse, S.; Cooper, R.H.; Randle, P.J. Mechanism of activation of pyruvate dehydrogenase by dichloroacetate and other halogenated carboxylic acids. Biochem. J. 1974, 141, 761–774. [Google Scholar] [CrossRef] [PubMed]
- Kirk, K.L. Biochemistry of Halogenated Carboxylic Acids. In Biochemistry of Halogenated Organic Compounds; Kirk, K.L., Ed.; Springer: Boston, MA, USA, 1991. [Google Scholar] [CrossRef]
- Groom, C.R.; Bruno, I.J.; Lightfoot, M.P.; Ward, S.C. The Cambridge Structural Database. Acta Crystallogr. B Struct. Sci. Cryst. Eng. Mater. 2016, 72, 171–179. [Google Scholar] [CrossRef] [PubMed]
- Vor der Bruck, O.; Leiserowitz, L. Molecular packing modes. Two crystalline modifications of bromoacetic acid, C2H3BrO2. Cryst. Struct. Commun. 1975, 647–651. [Google Scholar]
- Murakami, Y.; Iitaka, Y. Determination of the Absolute Configuration of (-)-2-Bromosuccinamic Acid by X-Ray Diffraction Method. Chem. Pharm. Bull. 1969, 17, 2397–2404. [Google Scholar] [CrossRef]
- Thong, P.Y.; Lo, K.M.; Ng, S.W. 2,3-Dibromo-3-phenylpropionic acid. Acta Crystallogr. Sect. E 2008, 64, o1946. [Google Scholar] [CrossRef]
- Howard, T.R.; Mendez-deMello, K.A.; Cardenas, A.J.P. 2,3-Dibromo-3-phenylpropanoic acid: A monoclinic polymorph. IUCrData 2016, 1, x161885. [Google Scholar] [CrossRef]
- D’Ascenzo, L.; Auffinger, P. A comprehensive classification and nomenclature of carboxyl-carboxyl(ate) supramolecular motifs and related catemers: Implications for biomolecular systems. Acta Crystallogr. Sect. B 2015, 71, 164–175. [Google Scholar] [CrossRef]
- Levene, P.A.; Mori, T.; Mikeska, L.A. On Walden inversion: X. On the oxidation of 2-thiolcarboxylic acids to the corresponding sulfonic acids and on the Walden inversion in the series of 2-hydroxycarboxylic acids. J. Biol. Chem. 1927, 75, 337–365. [Google Scholar]
- Auterhoff, H.; Lang, W. Darstellung und Eigenschaften der optisch aktiven Bromisovale. Arch. Pharm. 1970, 303, 49–52. [Google Scholar] [CrossRef] [PubMed]
- Thomas, I.R.; Bruno, I.J.; Cole, J.C.; Macrae, C.F.; Pidcock, E.; Wood, P.A. WebCSD: The online portal to the Cambridge Structural Database. J. Appl. Crystallogr. 2010, 43, 362–366. [Google Scholar] [CrossRef] [PubMed]
- Steed, K.M.; Steed, J.W. Packing problems: High Z’ crystal structures and their relationship to cocrystals, inclusion compounds, and polymorphism. Chem. Rev. 2015, 115, 2895–2933. [Google Scholar] [CrossRef] [PubMed]
- Thompson, A.L.; Watkin, D.J. X-ray crystallography and chirality: Understanding the limitations. Tetrahedron Asymmetry 2009, 20, 712–717. [Google Scholar] [CrossRef]
- Rekis, T. Crystallization of chiral molecular compounds: What can be learned from the Cambridge Structural Database? Acta Crystallogr. Sect. B 2020, 76, 307–315. [Google Scholar] [CrossRef]
- A preliminary X-ray analysis of the crystals obtained from the melt revealed a severely disordered structure with unit cell parameters similar to those of rac-1 crystallized from ethyl acetate.
- SADABS; Bruker AXS Inc.: Madison, WI, USA, 2012.
- Sheldrick, G.M. SHELXT—Integrated space-group and crystal-structure determination. Acta Crystallogr. A Found. Adv. 2015, 71, 3–8. [Google Scholar] [CrossRef]
- Sheldrick, G.M. Crystal structure refinement with SHELXL. Acta Crystallogr. C Struct. Chem. 2015, 71, 3–8. [Google Scholar] [CrossRef]
- Flack, H. On enantiomorph-polarity estimation. Acta Crystallogr. Sect. A 1983, 39, 876–881. [Google Scholar] [CrossRef]
- Parsons, S.; Flack, H.D.; Wagner, T. Use of intensity quotients and differences in absolute structure refinement. Acta Crystallogr. B Struct. Sci. Cryst. Eng. Mater. 2013, 69, 249–259. [Google Scholar] [CrossRef]
- Hooft, R.W.W.; Straver, L.H.; Spek, A.L. Determination of absolute structure using Bayesian statistics on Bijvoet differences. J. Appl. Crystallogr. 2008, 41, 96–103. [Google Scholar] [CrossRef] [PubMed]
- Hooft, R.W.W.; Straver, L.H.; Spek, A.L. Probability plots based on Student’s t-distribution. Acta Crystallogr. Sect. A 2009, 65, 319–321. [Google Scholar] [CrossRef] [PubMed]
- Hooft, R.W.W.; Straver, L.H.; Spek, A.L. Using the t-distribution to improve the absolute structure assignment with likelihood calculations. J. Appl. Crystallogr. 2010, 43, 665–668. [Google Scholar] [CrossRef]
- Spek, A.L. checkCIF validation ALERTS: What they mean and how to respond. Acta Crystallogr. E Crystallogr. Commun. 2020, 76, 1–11. [Google Scholar] [CrossRef]
- Brandenburg, K. DIAMOND, 3.2k3; Crystal Impact GbR: Bonn, Germany, 2018. [Google Scholar]
- Macrae, C.F.; Sovago, I.; Cottrell, S.J.; Galek, P.T.A.; McCabe, P.; Pidcock, E.; Platings, M.; Shields, G.P.; Stevens, J.S.; Towler, M.; et al. Mercury 4.0: From visualization to analysis, design and prediction. J. Appl. Crystallogr. 2020, 53, 226–235. [Google Scholar] [CrossRef]
- Bernstein, J.; Davis, R.E.; Shimoni, L.; Chang, N.L. Patterns in hydrogen bonding—Functionality and graph set analysis in crystals. Angew. Chem. Int. Ed. 1995, 34, 1555–1573. [Google Scholar] [CrossRef]
- Kitajgorodskij, A.I. Molecular Crystals and Molecules; Academic Press: New York, NY, USA, 1973. [Google Scholar]
- Parsons, S. Determination of absolute configuration using X-ray diffraction. Tetrahedron Asymmetry 2017, 28, 1304–1313. [Google Scholar] [CrossRef]
- Flack, H.D.; Bernardinelli, G. Absolute structure and absolute configuration. Acta Crystallogr. Sect. A 1999, 55, 908–915. [Google Scholar] [CrossRef]
- Flack, H.D.; Bernardinelli, G. Reporting and evaluating absolute-structure and absolute-configuration determinations. J. Appl. Crystallogr. 2000, 33, 1143–1148. [Google Scholar] [CrossRef]
- Spek, A. Absolute structure determination: Pushing the limits. Acta Crystallogr. Sect. B 2016, 72, 659–660. [Google Scholar] [CrossRef]
- Collet, A.; Ziminski, L.; Garcia, C.; Vigné-Maeder, F. Chiral Discrimination in Crystalline Enantiomer Systems: Facts, Interpretations, and Speculations. In Supramolecular Stereochemistry; Siegel, J.S., Ed.; Springer: Dordrecht, The Netherlands, 1995; pp. 91–110. [Google Scholar] [CrossRef]
- Brock, C.P.; Schweizer, W.B.; Dunitz, J.D. On the validity of Wallach’s rule: On the density and stability of racemic crystals compared with their chiral counterparts. J. Am. Chem. Soc. 1991, 113, 9811–9820. [Google Scholar] [CrossRef]
- Leiserowitz, L. Molecular packing modes. Carboxylic acids. Acta Crystallogr. Sect. B 1976, 32, 775–802. [Google Scholar] [CrossRef]
- Corpinot, M.K.; Bučar, D.-K. A Practical Guide to the Design of Molecular Crystals. Cryst. Growth Des. 2018, 19, 1426–1453. [Google Scholar] [CrossRef]
- Etter, M.C. Encoding and decoding hydrogen-bond patterns of organic-compounds. Acc. Chem. Res. 1990, 23, 120–126. [Google Scholar] [CrossRef]
- Thakuria, R.; Sarma, B.; Nangia, A. 7.03—Hydrogen Bonding in Molecular Crystals. In Comprehensive Supramolecular Chemistry II; Atwood, J.L., Ed.; Elsevier: Oxford, UK, 2017; pp. 25–48. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| (R)-1 | Rac-1 | |
|---|---|---|
| empirical formula | C5H9BrO2 | C5H9BrO2 |
| Mr | 181.03 | 181.03 |
| T (K) | 100(2) | 100(2) |
| λ (Å) | 0.71073 | 0.71073 |
| crystal system | triclinic | triclinic |
| space group | P1 | P |
| a (Å) | 6.0261(11) | 6.5849(14) |
| b (Å) | 6.7000(16) | 7.5490(16) |
| c (Å) | 9.900(2) | 7.7328(17) |
| α (°) | 102.144(17) | 112.283(4) |
| β (°) | 102.477(15) | 92.655(4) |
| γ (°) | 107.20(3) | 101.085(3) |
| V (Å3) | 356.34(14) | 346.03(13) |
| Z, Z’ | 2, 2 | 2, 1 |
| ρcalc (mg m−3) | 1.687 | 1.737 |
| μ (mm−1) | 5.685 | 5.854 |
| F(000) | 180 | 180 |
| crystal size (mm) | 0.350 × 0.180 × 0.100 | 0.279 × 0.226 × 0.128 |
| θ range for data collection (°) | 3.411–38.060 | 2.872–37.221 |
| reflections collected/unique | 18,005/7664 | 12,972/3401 |
| Rint | 0.0237 | 0.0371 |
| observed reflections [I > 2σ(I)] | 6888 | 2743 |
| Tmax/Tmin | 0.58973/0.2342 | 0.61929/0.32069 |
| data/restraints/parameters | 7664/5/155 | 3401/0/75 |
| Goodness-of-fit on F2 | 1.089 | 1.041 |
| R1 [I > 2σ(I)] | 0.0287 | 0.0421 |
| wR2 (all data) | 0.0694 | 0.1116 |
| Flack x parameter (refined) | 0.000(8) | - |
| Flack x parameter (from quotients) | −0.006(5) [3068 quotients] | - |
| Hooft parameter | −0.011(4) | - |
| Δρmax/Δρmin (e Å−3) | 1.30/−0.72 | 2.74/−0.56 |
| D–H···A | d(D–H) | d(H···A) | d(D···A) | ˂(DHA) |
|---|---|---|---|---|
| (R)-1 | ||||
| O11–H11···O22 | 0.82(2) | 1.82(2) | 2.636(3) | 170(4) |
| O12–H12···O21 | 0.82(2) | 1.82(2) | 2.635(3) | 171(4) |
| rac-1 | ||||
| O1–H1···O2a | 0.84 | 1.82 | 2.658(2) | 175 |
| (R)-1 | Rac-1 | ||
|---|---|---|---|
| Molecule 1 | Molecule 2 | ||
| C2–Br1 | 1.969(3) | 1.966(2) | 1.972(2) |
| C1–O1 | 1.291(3) | 1.282(3) | 1.302(3) |
| C1–O2 | 1.242(3) | 1.248(3) | 1.217(3) |
| O2–C1–O1 | 124.5(2) | 124.4(2) | 123.84(19) |
| O1–C1–C2 | 116.06(19) | 116.07(18) | 115.35(19) |
| O2–C1–C2 | 119.4(2) | 119.51(19) | 120.8(2) |
| C1–C2–C3–C4 | −59.2(3) | −61.1(2) | −59.3(2) |
| C1–C2–C3–C5 | 179.7(2) | 176.70(19) | 179.18(19) |
| O1–C1–C2–C3 | −44.8(3) | −46.7(3) | −46.3(3) |
| Br1–C2–C3–C4 | −177.52(15) | −179.30(15) | −177.72(13) |
| Br1–C2–C3–C5 | 61.3(2) | 58.5(2) | 60.8(2) |
| Br1–C2–C1–O1 | 78.1(2) | 75.9(2) | 76.86(19) |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Seidel, R.W.; Nöthling, N.; Goddard, R.; Lehmann, C.W. Structural Elucidation of Enantiopure and Racemic 2-Bromo-3-Methylbutyric Acid. Chemistry 2020, 2, 691-699. https://doi.org/10.3390/chemistry2030044
Seidel RW, Nöthling N, Goddard R, Lehmann CW. Structural Elucidation of Enantiopure and Racemic 2-Bromo-3-Methylbutyric Acid. Chemistry. 2020; 2(3):691-699. https://doi.org/10.3390/chemistry2030044
Chicago/Turabian StyleSeidel, Rüdiger W., Nils Nöthling, Richard Goddard, and Christian W. Lehmann. 2020. "Structural Elucidation of Enantiopure and Racemic 2-Bromo-3-Methylbutyric Acid" Chemistry 2, no. 3: 691-699. https://doi.org/10.3390/chemistry2030044
APA StyleSeidel, R. W., Nöthling, N., Goddard, R., & Lehmann, C. W. (2020). Structural Elucidation of Enantiopure and Racemic 2-Bromo-3-Methylbutyric Acid. Chemistry, 2(3), 691-699. https://doi.org/10.3390/chemistry2030044