1. Introduction
In line with the formulation of the ligand field theory [
1,
2], or as it was originally called by Bethe, crystal-field theory [
3], it was realized that an open-shell metal with at least two valence electrons in a specific chemical environment could exist with either high-spin or low-spin configuration [
4]. Following van Vleck’s approach to magnetism [
5], Pauling perceptively recognized that it would be feasible to obtain systems in which two spin states could be present simultaneously, while their ratio should depend on the energy difference between them [
6,
7]. The discovery of thermal spin-state equilibria operating in Fe
III dithiocarbamate by Cambi et al. [
8,
9,
10] at the same period, indeed confirmed these predictions. Since then, a myriad of metal coordination complexes and polymeric materials have been shown to display spin transitions, often referred to as spin-crossover (SCO) materials. These have been studied in detail and extensively reviewed over the last two decades [
11,
12,
13,
14,
15,
16,
17,
18,
19,
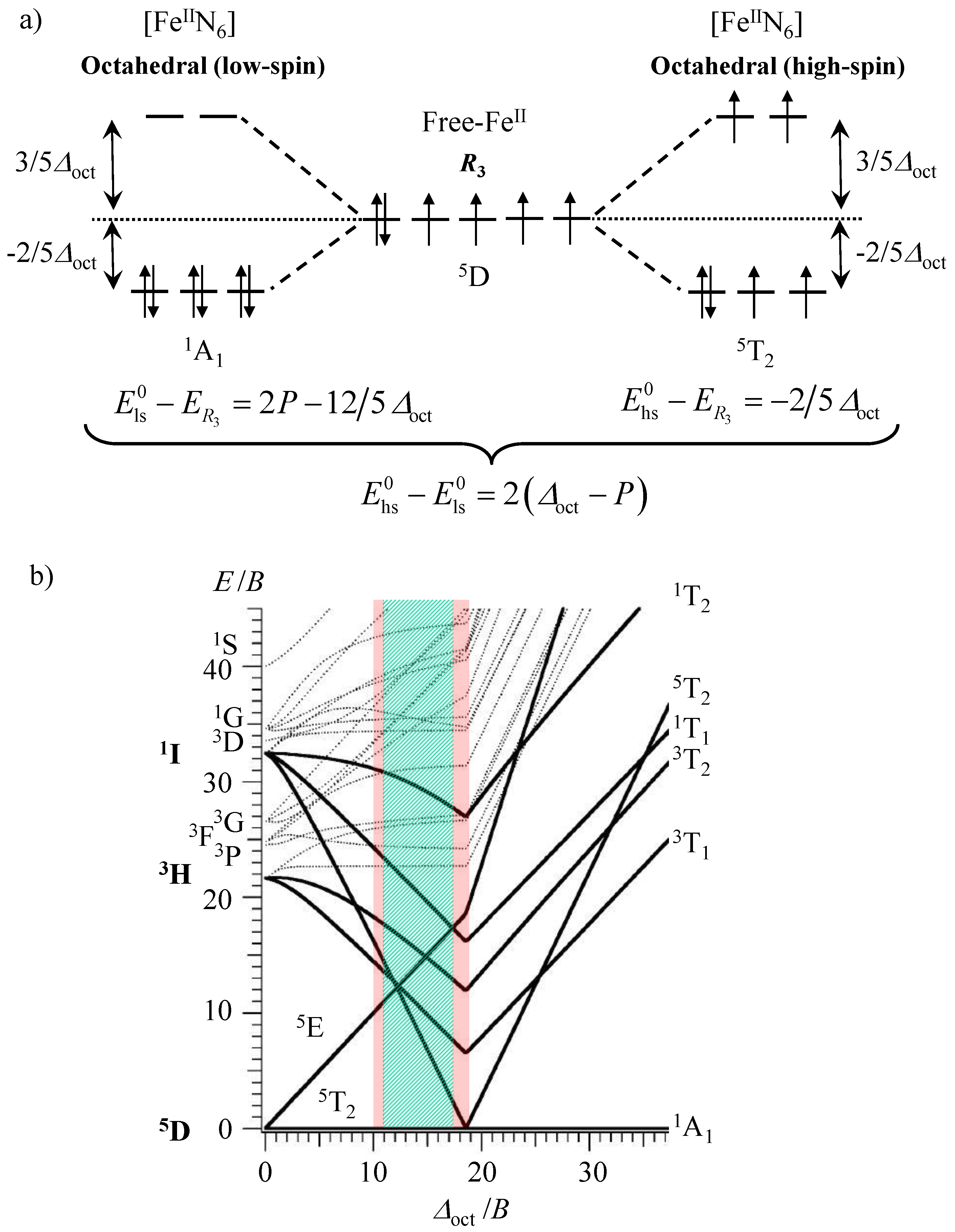
20]. Due to the ‘on–off’ switching of the magnetic properties accompanying the spin transition from the low-spin diamagnetic configuration (
1A
1 label in octahedral symmetry) to the high-spin paramagnetic form (
5T
2 label in octahedral symmetry) for d
6 transition metals in pseudo-octahedral geometry (
Scheme 1a), the ‘magic’ [Fe
IIN
6] chromophores, where N is a heterocyclic nitrogen donor atom, have been intensively investigated [
11,
12,
13,
14,
15,
16,
17,
18,
19,
20]. Various external stimulations such as changes in temperature [
21,
22], pressure [
23,
24], magnetic field [
25] or light-irradiation [
26,
27] can be used for inducing the SCO processes, which makes these microscopic magneto-optical switches very attractive for their introduction into responsive macroscopic materials [
12,
13,
16,
28,
29,
30,
31]. The most common and accepted approach for rationalizing the design of spin-crossover pseudo-octahedral Fe
II complexes relies on the energetic balance
between the ligand-field stabilization energy as measured by ∆
oct = 10 Dq and the spin pairing energy modeled with the Racah parameters
B and
C with
P = 2
B + 4
C ≈ 19
B (
Scheme 1a) [
31]. When ∆
oct >>
P,
and the pseudo-octahedral Fe
II complex adopts a low spin configuration with a diamagnetic
1A
1 electronic ground state, as shown in the right part of the Tanabe–Sugano diagram built for the electronic d
6 configuration (∆
oct/
B > 20 in
Scheme 1b). The reverse situation occurs when
P >> ∆
oct, which leads to
and the existence of the paramagnetic high-spin
5T
2 ground state (left part of Tanabe–Sugano diagram with ∆
oct/
B < 10 in
Scheme 1b). Finally, for intermediate values
(
m ≤ 10), the two spin states coexist and are thermally populated at accessible temperatures. However, the latter statement is misleading and physically unsound since both ∆
oct and
P change during the spin transition as a result of the population of the antibonding orbitals in the high-spin form. For pseudo-octahedral spin-crossover [Fe
IIN
6] complexes, the Fe–N bond lengths extend by approximately 10% upon the low-spin to high-spin transition and ∆
oct consequently decreases according to a 1/
rn dependence with
n = 5–6 (Equation (1)) [
31].
Taking typical Fe–N bond distances of
rLS = 2.0 Å and
rHS = 2.2 Å [
11,
12,
13,
14,
15,
16,
17,
18,
19,
20] leads to
≈ 1.75 accompanying the spin transition, whereas
P changes very little (
), except for a faint reduction of the nephelauxetic effect with larger bond lengths [
32].
Taking into account the 10% bond length expansion accompanying the spin transition, the simplistic zero-point energy differences between the two states summarized in
Scheme 1a (i.e.,
) should be replaced with Equation (2), which is transformed into Equations (3) and (4) upon introducing
≈ 1.75.
Solving Equations (3) and (4) for ∆
EHL = 0 provides
and
as the lower limit of the existence of thermally accessible spin state equilibria, while the higher limits can be estimated for
∆EHL = 2000 cm
−1, which gives
and
(red areas in
Scheme 1b). More sophisticated calculations using Racah parameters
B and
C, reduced by 70–80% of their free ion values, predict narrow ranges of ligand field strengths 11,000 ≤
≤ 12,500 cm
−1 and 19,000 ≤
≤ 22,000 cm
−1, for which the phenomenon of a thermal spin transition can be expected in Fe
II coordination complexes [
31]. Following this theoretical approach, the toolkit of coordination chemists for programming and tuning the thermodynamic spin transition parameters in molecular [Fe
IIN
6] complexes logically relied on the manipulation of ∆
oct and
B via (i) some controlled distortions of the coordination geometry from a perfect octahedron by using chelating ligands with fixed bite angles [
33] and (ii) specific programming of metal–ligand bonding interactions via ligand design [
15,
19,
34]. Benefiting from the huge amount of experimental data collected during the last decades for [Fe(N
∩N)
3]
2+ complexes, where N
∩N is an α,α’-diimine chelate ligand possessing two N-heterocyclic donor atoms, it was shown that the connection of a six-membered heterocycle to a five-membered heterocycle in N
∩N provides favorable ligand-field strengths around Fe
II for promoting spin-state equilibria (Equation (5)) with transition temperatures
T1/2 =
∆HSCO/
∆SSCO (i.e., the temperature at which
∆GSCO = 0 and
xhs =
xls = 0.5) within the 30–500 K range [
14,
15,
33].
The didentate ligands
L1 and
L2 match the latter criteria and the associated pseudo-octahedral complexes [Fe(
Lk)
3]
2+ indeed exhibit spin-crossover behaviors in acetonitrile solutions, the transition temperatures of which reveal the stronger Fe–N bonds induced by the strong-accepting pyrazine units in [Fe(
L2)
3]
2+ (
T1/2 ~350 K) compared with pyridine units in [Fe(
L1)
3]
2+ (
T1/2 ~310 K,
Scheme 2) in the absence of sterical constraints [
35].
Moving the methyl group bound to the pyridine ring from the 5-position in
L1 to the 3-position in
L3 and to the 6-position in
L4 (
Scheme 2) is well-known to stepwise decrease the ligand-field strengths in the resulting [Fe(
Lk)
3]
2+ complexes because the operation of additional sterical constraints, produced by intra-strand interactions in [Fe(
L3)
3]
2+ [
37] and by inter-strand interactions in [Fe(
L4)
3]
2+, extends the Fe–N bond lengths (see Equation (1)) [
38,
39,
40]. The associated trend ∆
oct(
L1) ≈ ∆
oct(
L2) > ∆
oct(
L3) > ∆
oct(
L4) observed for the isostructural [Ni(
Lk)
3]
2+ complexes (
Scheme 2), for which the determination of ligand field ∆
oct and Racah
B parameters are not complicated by any SCO behavior, are in line with the observation of pure high-spin configurations for the [Fe(
L3)
3]
2+ and [Fe(
L4)
3]
2+ complexes in solution (
Scheme 2) [
35]. Whereas the connection of methyl groups adjacent to the donor nitrogen atom in the bound 6-methyl-pyridine groups in [Fe(
L4)
3]
2+ produces such large inter-strand interactions that the contraction accompanying the high-spin to low-spin transition cannot be envisioned [
41], the situation with the remote 3-methyl substituted pyridine units in [Fe(
L3)
3]
2+ is less clear and a sophisticated triple-stranded heterometallic LaFe helicate containing the facial [Fe(
L3)
3]
2+ chromophore has been shown to display partial SCO behavior at low temperature (
T1/2 ~50 K) [
36]. Taking into account that (i) the replacement of a pyridine with a pyrazine ring in going from [Fe(
L1)
3]
2+ and [Fe(
L2)
3]
2+ stabilizes the low-spin state by
T1/2 = 40 K (
Scheme 2) and (ii) moving the methyl group from the 5-position in [Fe(
L1)
3]
2+ to the 3-position in
fac-[Fe(
L3)
3]
2+ (as found in the related LaFe helicate) produces an opposite trend with the stabilization of the high-spin form by
T1/2 ≈ 50–310 = −260 K [
36], we thus ingenuously explored the possibility of combining both aspects in the didentate ligand
L5 where the methyl group is now connected to the 3-position of a pyrazine ring (
Scheme 3a) with the hope of pushing the transition temperature toward cryoscopic temperatures for [Fe(
L5)
3]
2+ around
T1/2 ~
T1/2 ([Fe(
L2)3]
2+) − 260 = 350 − 260 = 90 K (3-methyl-pyrazine). This effort is justified by our long-term quest for designing a pseudo-octahedral spin-crossover [Fe(
Lk)
3]
2+ unit that can modulate the luminescence of adjacent emissive lanthanides in (supra)molecular assemblies via energy transfers within a temperature domain (77–150 K) accessible to optical reading and addressing [
36]. Finally, since minor structural variations may induce large changes in ligand-field strength, the systematic exploration of unpredictable intermolecular packing interactions [
42,
43] operating in crystalline samples of [Fe(
L5)
3]X
2 complexes (X
- = monoanionic counter-ions) may contribute to the lucky search for some ‘ideal’ Fe
II complexes, which additionally exhibit hysteretic behavior and bistability [
17,
44,
45].
2. Experimental
Chemicals were purchased from Sigma-Aldrich (Gmbh, Buchs) and Acros and used without further purification unless otherwise stated. Dichloromethane, 1,2-dichloroethane, tert-butylmethyl ether, and N,N-dimethylformamide were dried through an alumina cartridge. Silica-gel plates (Merck, 60 F254) were used for thin-layer chromatography, SiliaFlash® silica gel P60 (0.04–0.063 mm,) and Acros silica gel 60 (0.035–0.07 mm) were used for preparative column chromatography.
Preparation ofN-methyl-2-nitroaniline(4). 1-chloro-2-nitrobenzene (3, 31.75 g, 201.5 mmol, 1.0 eq) and methylamine (198 mL, 40% weight in H2O, 2295.6 mmol, 11.4 eq) were introduced into a Carius tube equipped with a magnetic stirrer and heated at 120 °C for 48 h. Excess of methylamine was rotatory evaporated and the residual brown oil was partitioned between CH2Cl2 (300 mL) and half sat. aq. NH4Cl (300 mL). The organic layer was separated and the aq. phase was further extracted with CH2Cl2 (3 × 150 mL). The combined organic extracts were dried over anhydrous Na2SO4, filtered, and the solvent evaporated to dryness. The resulting red oil was purified by column chromatography (Silica, CH2Cl2) to give 28.99 g of N-methyl-2-nitroaniline (4, 190.5 mmol, yield 94%) as a deep red orange oil, which slowly crystallized within hours. 1H NMR (CDCl3, 400 MHz, 298 K) δ/ppm: 8.13 (1H, dd, 3J = 8.8 Hz, 4J = 1.6 Hz), 8.00 (1H, bs), 7.43 (1H, ddd, 3J = 8.8 Hz, 3J = 7.2 Hz, 4J = 1.6 Hz), 6.81 (1H, dd, 3J = 8.6 Hz, 4J = 1.0 Hz), 6.62 (1H, ddd, 3J = 8.4 Hz, 3J = 7.2 Hz, 4J = 1.2 Hz), 2.99 (3H, s).
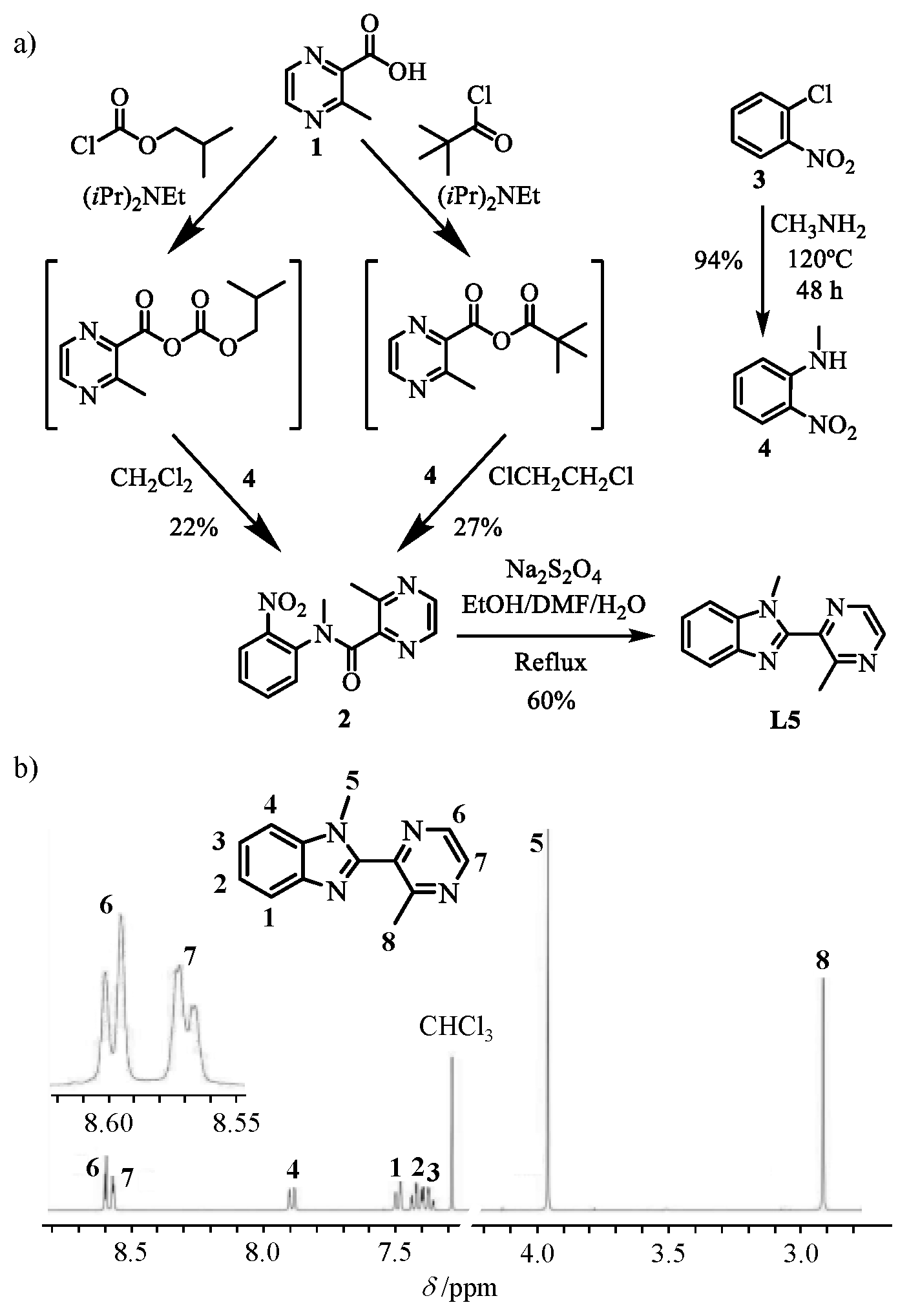
Preparation of N,3-dimethyl-N-(2-nitrophenyl) pyrazine-2-carboxamide (
2, left pathway in
Scheme 3a). A suspension of 3-methyl-pyrazine-2-carboxylic acid (
1, 5 g, 35.5 mmol, 1 eq) and di-isopropyl-ethylamine (7.2 mL, 5.5 g, 45 mmol, 1.26 eq) in CH
2Cl
2 (4 mL) was added dropwise into a two-necked flask containing isobutyl chloroformate (5.4 mL, 5.408 g, 45 mmol, 1.26 eq) dissolved in CH
2Cl
2 (1 mL). The mixture was stirred at −15 °C for 120 min, after which a solution of
N-methyl-2-nitroaniline (
4, 5.5 g, 0.0352 mol, 1 eq) in CH
2Cl
2 was added. After stirring for 15 h at room temperature, the solution was partitioned between CH
2Cl
2 (100 mL) and half-sat. aq. NH
4Cl (250 mL). The organic layer was separated and the aqueous phase was further extracted with CH
2Cl
2 (3 × 100 mL). The organic fractions were dried over anhydrous Na
2SO
4, concentrated under vacuum, and purified by column chromatography (Silica, CH
2Cl
2/MeOH 99.2:0.8) to yield
N,3-dimethyl-
N-(2-nitrophenyl) pyrazine-2-carboxamide (
2, 8.22 mmol, yield 22%).
1H NMR (CDCl
3, 400 MHz, 298 K)
δ/ppm: mixture of two rotamers A (72.5%) and B (27.5 %): 2.67 (3H, s, A), 2.72 (3H, s, B), 3.58 (3H, s, A), 3.33 (3H, s, B), 7.37–7.80 (3H, m, A and B), 8.29 (1H, d,
3J = 2.5 Hz, A), 8.60 (1H, d,
3J = 2.5 Hz, B), 8.0 (1H, dd,
3J = 2.5 Hz,
5J = 0.6 Hz, A), 8.50 (1H, dd,
3J = 2.5 Hz,
5J = 0.6 Hz, B), 7.86 (1H, dd,
3J = 8.5 Hz,
4J = 1.5 Hz, A), 8.13 (1H, dd,
3J = 8.5 Hz,
4J = 1.5 Hz, B). ESI-MS (soft-positive mode; MeOH+CHCl
3+HCOOH):
m/
z =273.0 ([2 + H]
+), 295.1 ([2 + Na]
+).
Preparation of N,3-dimethyl-N-(2-nitrophenyl) pyrazine-2-carboxamide (2, right pathway in
Scheme 3a). A suspension of 3-methyl-pyrazine-2-carboxylic acid (
1, 0.2 g, 1.44 mmol, 1 eq) and di-isopropyl-ethylamine (0.37 mL, 0.28 g, 2.16 mmol, 1.5 eq) in 1,2-dichloroethane (4 mL) was added dropwise into a two-necked flask containing trimethylacetyl chloride (0.194 mL, 0.19 g, 1.58 mmol, 1.1 eq) in 1,2-dichloroethane (1 mL). The mixture was stirred at −20 °C for 60 min, after which a solution of
N-methyl-2-nitroaniline (4, 0.329 g, 2.16 mmol, 1.5 eq) in 1,2-dichloroethane (5 mL) was added. After refluxing for 16 h, the solution was concentrated under vacuum and partitioned between CH
2Cl
2 (80 mL) and half-saturated aqueous solution of NH
4Cl (200 mL). The organic layer was separated and the aqueous phase was further extracted using CH
2Cl
2 (3 × 80 mL). The combined organic fractions were concentrated under vacuum after drying with anhydrous Na
2SO
4. Ultimate purification using column chromatography (Silica, CH
2Cl
2/MeOH 99.2:0.8) yielded 0.106 g of
N, 3-dimethyl-
N-(2-nitrophenyl) pyrazine-2-carboxamide (
2, 0.387 mmol, yield 27%).
Preparation of 1-methyl-2-(3-methylpyrazin-2-yl)-1H-benzo[d]imidazole (L5). N,3-dimethyl-N-(2-nitrophenyl)pyrazine-2-carboxamide (2, 2.56 g, 7.58 mmol, 1 eq) was dissolved in EtOH:DMF (20 mL:25 mL). Sodium dithionite (8.0 g, 40 mmol, 5.2 eq) was added to the mixture and the temperature of the system was raised to 80 °C when 20 mL of water was added. After refluxing for 36 h, the mixture was neutralized using aqueous ammonia and the solvents were removed in vacuo. The concentrate was dissolved in CH2Cl2 (50 mL) and washed with water (3 × 200 mL). The aqueous layers were collectively further extracted with CH2Cl2 (3 × 50 mL). The combined organic fraction was then concentrated under vacuum and purified by column chromatography (silica, CH2Cl2/MeOH 98:2) to yield L5 (7.67 mmol, yield 60%). The compound was crystallized as needles by slow diffusion of n-hexane into a concentrated CH2Cl2 solution of L5. 1H NMR (CDCl3, 400 MHz, 298 K) δ/ppm: 2.91 (3H, s), 3.96 (3H, s), 7.35–7.44 (2H, m), 7.49 (1H, d, 3J = 7 Hz), 7.88 (1H, d, 3J = 7 Hz), 8.57 (1H-pz, dd, 3J = 2.4 Hz, 5J = 0.5 Hz), 8.60 (1H-pz, dd, 3J = 2.4 Hz). 13C NMR (CDCl3, 101 MHz, 298K) δ/ppm: 155.68 (Cq), 149.12 (Cq), 144.54 (Cq), 143.84 (CHpz), 142.45 (Cq), 140.86 (CHpz), 136.15 (Cq), 123.85 (CH), 122.90 (CH), 120.45 (CH), 110.01 (CH), 31.83 (CH3), 23.26 (CH3). ESI-MS (soft-positive mode; MeOH+CHCl3+HCOOH): m/z 225.1([L5 + H]+). Elemental analysis calculated for C13H12N4 (%): C 69.62, H 5.39, N 24.98; Found (%): C 69.46, H 5.09, N 25.20.
Preparation of mononuclear FeII, ZnII, and NiII complexes with 1-methyl-2-(3-methylpyrazin-2-yl)-1H-benzo[d]imidazole (L5). In a typical synthesis, 0.3 mmol (3 eq) of the ligand
L5 dissolved in acetonitrile (2 mL) was added to 0.1 mmol (1 eq) of Fe(ClO
4)
2∙6H
2O or Fe(CF
3SO
3)
2 or Ni(BF
4)
2∙6H
2O or Zn(CF
3SO
3)
2 in acetonitrile (2 mL). The resulting mixture was stirred under an inert atmosphere for 3 h, then evaporated to dryness under vacuum to yield microcrystalline powders of the respective complexes. These powders were dissolved in acetonitrile and allowed to crystallize by evaporation or by slow diffusion of tert-butyl methyl ether to give 64–78% of primary [Fe(
L5)
3](CF
3SO
3)
2∙1.5H
2O, [Ni(
L5)
3](BF
4)
2∙1.5H
2O∙1.5CH
3CN and [Zn(
L5)
3](BF
4)
2∙4H
2O complexes (
Table S1). Single crystals suitable for characterization by X-ray diffraction could be obtained by slow evaporation of acetonitrile solution containing 10 eq of (
nBu)
4NClO
4 or (
nBu)
4NBF
4 to give [Fe(
L5)
3](ClO
4)
2 (I), [Ni(
L5)
3](ClO
4)
2 (II), [Ni(
L5)
3](BF
4)
2∙H
2O (III), [Zn(
L5)
3](ClO
4)
2 (IV), [Ni(
L5)
3](BF
4)
2∙1.75CH
3CN (V) and [Zn(
L5)
3](BF
4)
2∙1.5CH
3CN (VI).
Caution! Dry perchlorates may explode and should be handled in small quantities and with the necessary precautions [
46,
47].
2.1. Spectroscopic and Analytical Measurements
1H and
13C NMR spectra were recorded at 298 K on a Bruker Avance 400 MHz spectrometer. Chemical shifts are given in ppm with respect to tetramethylsilane. Spectrophotometric titrations were performed with a J&M diode array spectrometer (Tidas series) connected to an external computer. In a typical experiment, 25 cm
3 of ligand in acetonitrile (2 × 10
−4 M) was titrated at 298 K with a solution of Fe(CF
3SO
3)
2 or Ni(BF
4)2∙6H
2O or Zn(CF
3SO
3)
2 (2 × 10
−3 M) in acetonitrile under an inert atmosphere. After each addition of 33 μL, the absorbance was recorded using Hellma optrodes (optical path length 0.1 cm) immersed in the thermostated titration vessel and connected to the spectrometer. Mathematical treatment of the spectrophotometric titrations was performed with factor analysis [
48,
49,
50] and with ReactLab
TM Equilibria (previously Specfit/32) [
51,
52,
53]. Pneumatically-assisted electrospray (ESI-MS) mass spectra were recorded from 10
−4 M (ligands) and 10
−3 M (complexes) solutions on an Applied Biosystems API 150EX LC/MS System equipped with a Turbo Ionspray source. Elemental analyses were performed by K. L. Buchwalder from the Microchemical Laboratory of the University of Geneva. Elemental analysis was not conducted for perchlorate salts for security reasons, while crystals of the tetrafluoroborate salts lost their solvent upon separation from the mother liquor and were not further characterized. Electronic spectra in the UV–Vis region were recorded at 293 K from solutions in CH
3CN with a Perkin-Elmer Lambda 1050 using quartz cells of a 0.1 or 1.0 mm path length. Solid-state absorption spectra were recorded with a Perkin-Elmer Lambda 900 using capillaries. Solid-state magnetic data were recorded on a MPMS 3 or MPMS 5 QUANTUM DESIGN magnetometers using magnetic fields of 1000–5000 Oe at 1 K/min rates within the 5–300 K range. The magnetic susceptibilities were corrected for the magnetic response of the sample holder and for the diamagnetism of the compounds by using the approximation
cm
3·mol
−1 [
54].
2.2. X-Ray Crystallography
Summary of crystal data, intensity measurements, and structure refinements for compounds
L5, [Fe(
L5)
3](ClO
4)
2 (
I), [Ni(
L5)
3](ClO
4)
2 (
II), [Ni(
L5)
3](BF
4)
2∙H
2O (
III), and [Zn(
L5)
3](ClO
4)
2 (
IV) is presented in
Tables S2–S4. Pertinent bond lengths, bond angles, and interplanar angles are collected in
Tables S5–S14 together with ORTEP views and pertinent numbering schemes gathered in
Figures S1–S5. The crystals were mounted on MiTeGen kapton cryoloops with protection oil. X-ray data collection was performed with an Agilent SuperNova Dual diffractometer equipped with a CCD Atlas detector (Cu[Kα] radiation). The structures were solved by using direct methods [
55,
56] or dual-space methods [
57]. Full-matrix least-square refinements on
F2 were performed with SHELX2014 [
58]. CCDC 1988655-1988659 contained the supplementary crystallographic data. These data can be obtained free of charge from the Cambridge Crystallographic Data Centre via
www.ccdc.cam.ac.uk/. Single crystals of [Ni(
L5)
3](BF
4)
2∙1.75CH
3CN (
V) and [Zn(
L5)
3](BF
4)
2∙1.5CH
3CN (
VI) could also be obtained as inversion twins. The two complexes were isostructural and crystallized in the trigonal system (
P3
c1 space group) with five independent complexes in the asymmetric unit, all located on three-fold rotation axes (the metal content of the asymmetric unit is 5/3) and
Z = 10 (
Table S15). Although there is no doubt that the three ligands adopt facial arrangements around the metal to give exclusively
fac-[M(
L5)
3]
2+ cations (
Figures S6–S7), we were only able to locate unambiguously three BF
4- counter-anions in the asymmetric unit. Despite numerous efforts, we were not able to obtain a satisfying model for the last third of a BF
4− counter-anion and gave up to further discuss these structures and to deposit the cif files.
3. Results and Discussion
Synthesis, characterization, and solid-state structures were obtained for the didentate ligand
L5 and its pseudo-octahedral complexes [M(
L5)
3]X
2 (M = Fe, Ni, Zn and X = BF
4, ClO
4). Compared with pyridine-carboxylic acids, which are easily activated via their transformation into acyl chloride with the help of thionyl chloride or oxalyl chloride [
59], the electron-rich pyrazine analogue
1 produced only negligible yield (<1%) of the target amide product
2 under these standard conditions [
60]. The 3-methylpyrazine-2-carboxylic acid
1 was thus activated as its anhydride through reaction with either isobutyl chloroformate (left path in
Scheme 3a) or pivaloyl chloride (right path in
Scheme 3a). Subsequent nucleophilic attack with
N-methyl-2-nitroaniline
4 yielded the ortho-nitroamide compound
2 in moderate yield. A subsequent reductive cyclisation reaction provided ligand
L5, which was characterized by its
1H-NMR spectrum (
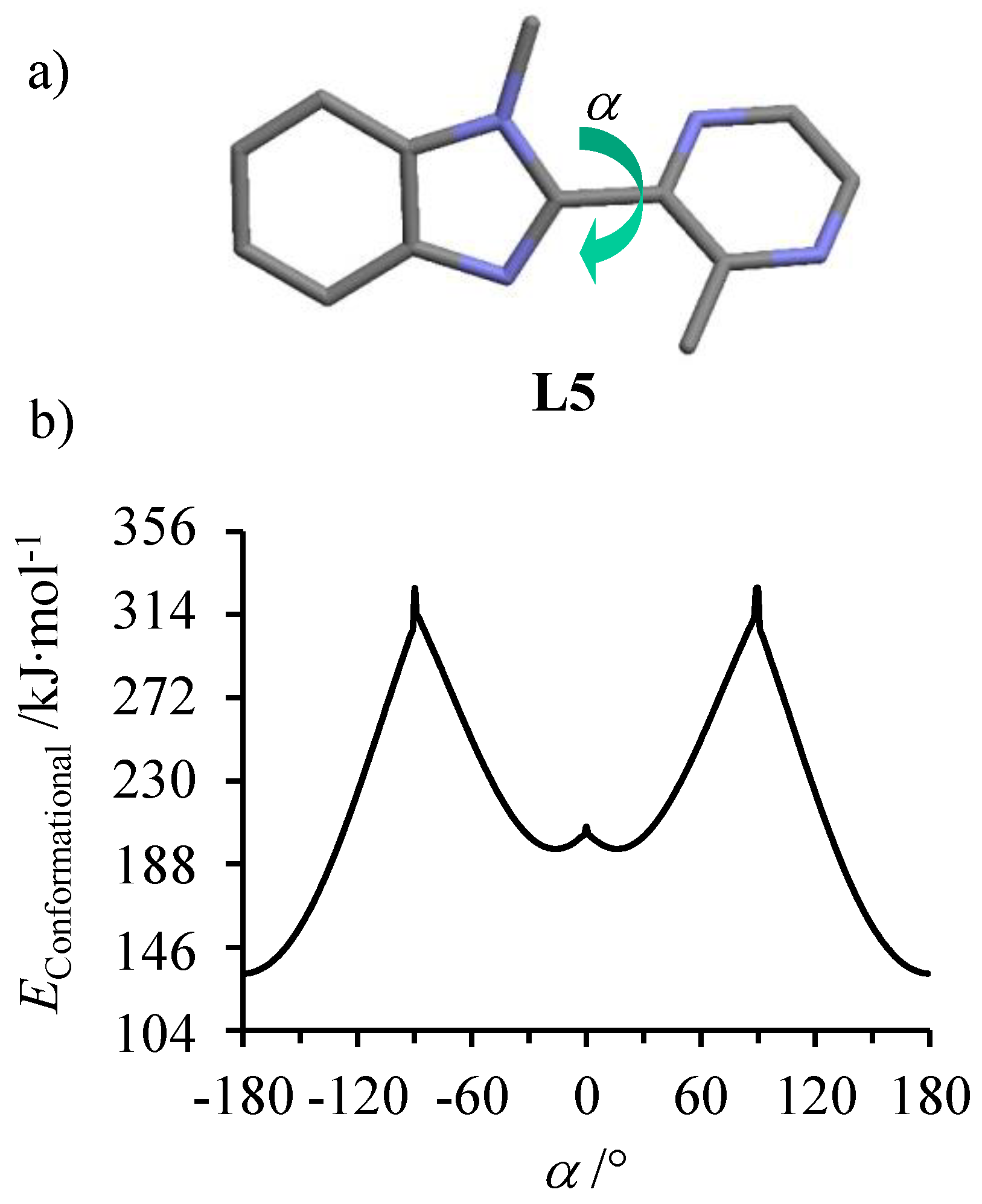
Scheme 3b). The lack of NOE effect observed between the methyl groups in positions 5 and 8 indicates an anti-conformation for the α,α’-diimine chelate unit, which was confirmed by (i) the crystal structure of
L5 (
Figure 1a) and (ii) gas-phase calculations predicting a global energy minimum for the planar anti-conformation (interplanar angle between the two aromatic rings
α = 180°,
Figure 1b) [
61]. Given that the same anti-conformations are (i) found in the solid state (
Figure S8) and (ii) predicted in the gas phase for the ligands
L2 [
60] and
L5, their computed EHMO frontiers orbitals are comparable (
Figure S9a) and lead to akin electronic absorption spectra dominated by intense π*←π covering the near UV range (
Figure S9b).
Interestingly, the gas-phase energy of
L5 displayed two additional local energy minima for
α = ±16° (
Figure 1b), which shifted from
α = 0° previously reported for the second local minimum in
L2 (
Figure S10) [
60]. The larger interplanar angle of 16° in the optimized syn-conformation of
L5 was the result of the sterical crowding between the close methyl groups connected to the adjacent aromatic rings (positions 5 and 8 in the numbering of
Scheme 3b). Taking the latter conformation as a limiting structural model when
L5 is bound to a metal cation provides
dN∙∙∙N = 2.83 Å between the two nitrogen donor atoms of the α,α’-diimine chelate. According to Phan et al. [
33], the latter separation matches the 2.78 ≤
dN∙∙∙N ≤ 2.93 Å range for which a diimine ligand might be used to achieve spin-crossover behavior in tris-homoleptic Fe
II complexes.
Stoichiometric mixing of
L5 (3 eq.) with Fe(CF
3SO
3)
2, Ni(BF
4)
2∙6H
2O or Zn(BF
4)
2∙6H
2O (1 eq,) in acetonitrile gave fair yields of microcrystalline primary precipitates of [Fe(
L5)
3](CF
3SO
3)
2∙1.5H
2O, [Ni(
L5)
3](BF
4)
2∙1.5H
2O∙1.5CH
3CN, and [Zn(
L5)
3](BF
4)
2∙4H
2O complexes (
Table S1). A series of isostructural complexes [Fe(
L5)
3](ClO
4)
2 (
I), [Ni(
L5)
3](ClO
4)
2 (
II), and [Zn(
L5)
3](ClO
4)
2 (
IV) could be obtained as single crystals by recrystallization in acetonitrile containing 10 eq of (
nBu)
4NClO
4.
Monocrystals suitable for x-ray diffractions were also obtained for [Ni(
L5)
3](BF
4)
2∙H
2O (
III), [Ni(
L5)
3](BF
4)
2∙1.75CH
3CN (
V), and [Zn(
L5)
3](BF
4)
2∙1.5CH
3CN (
VI) using the same method except for the replacement of (
nBu)
4NClO
4 with (
nBu)
4NBF
4 (
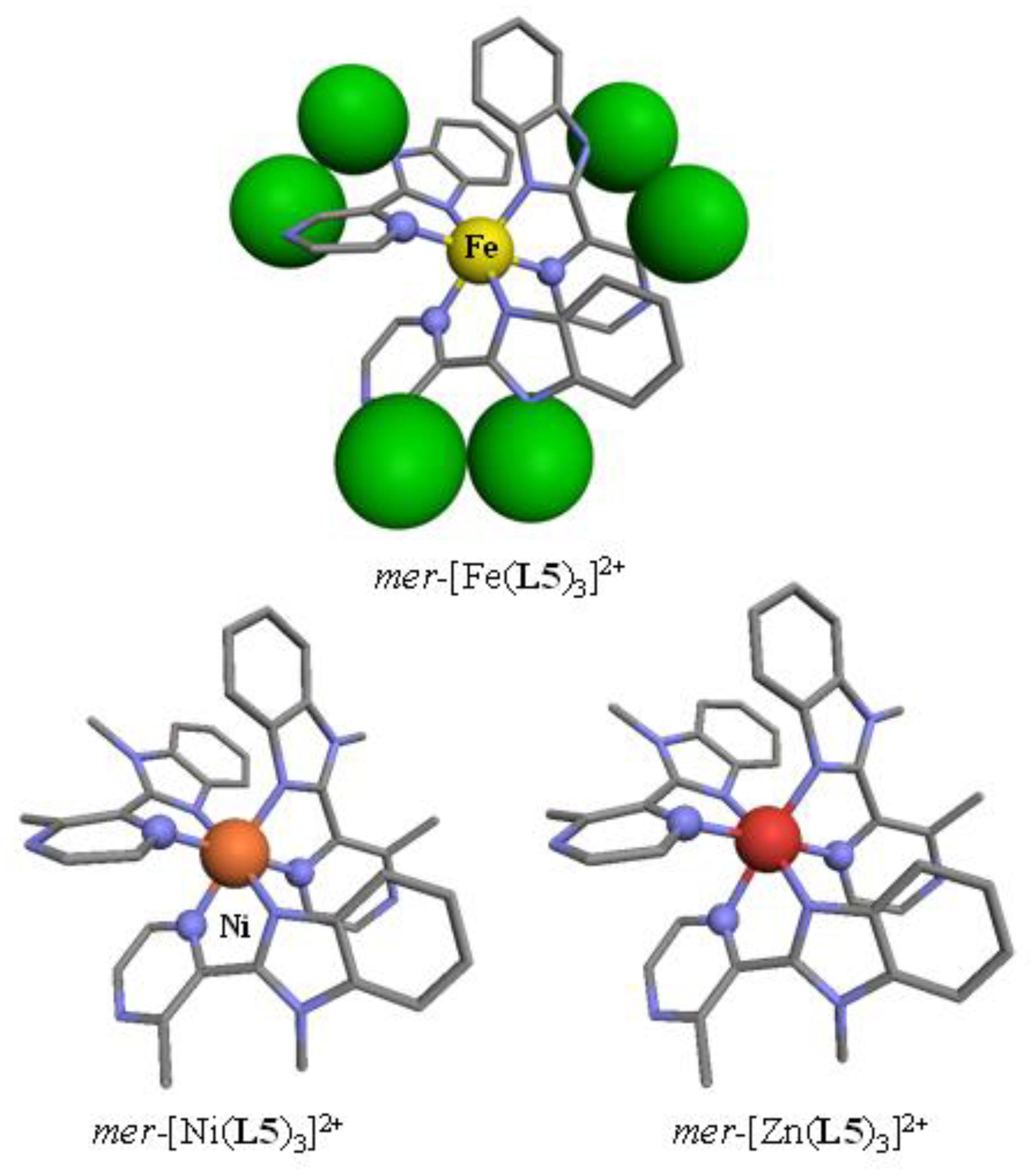
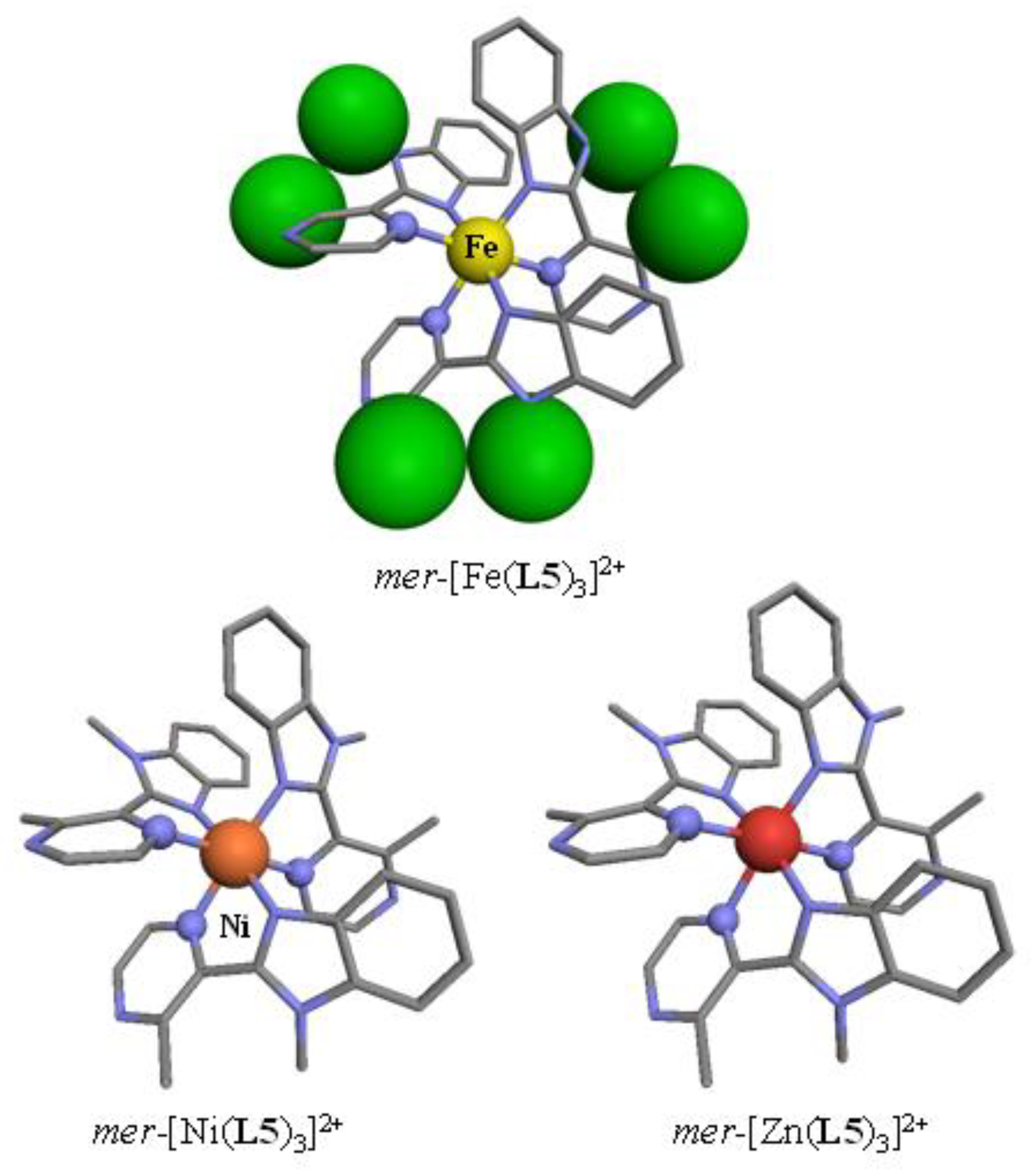
Tables S3 and S4). The crystal structures of the perchlorate salts systematically displayed the formation of
mer-[M(
L5)
3]
2+ cations (
Figure 2), in which the [MN
6] chromophores adopted a geometry close to the perfect octahedron as ascertained by SHAPE’s scores close to zero (
Table 1) [
62,
63,
64,
65,
66].
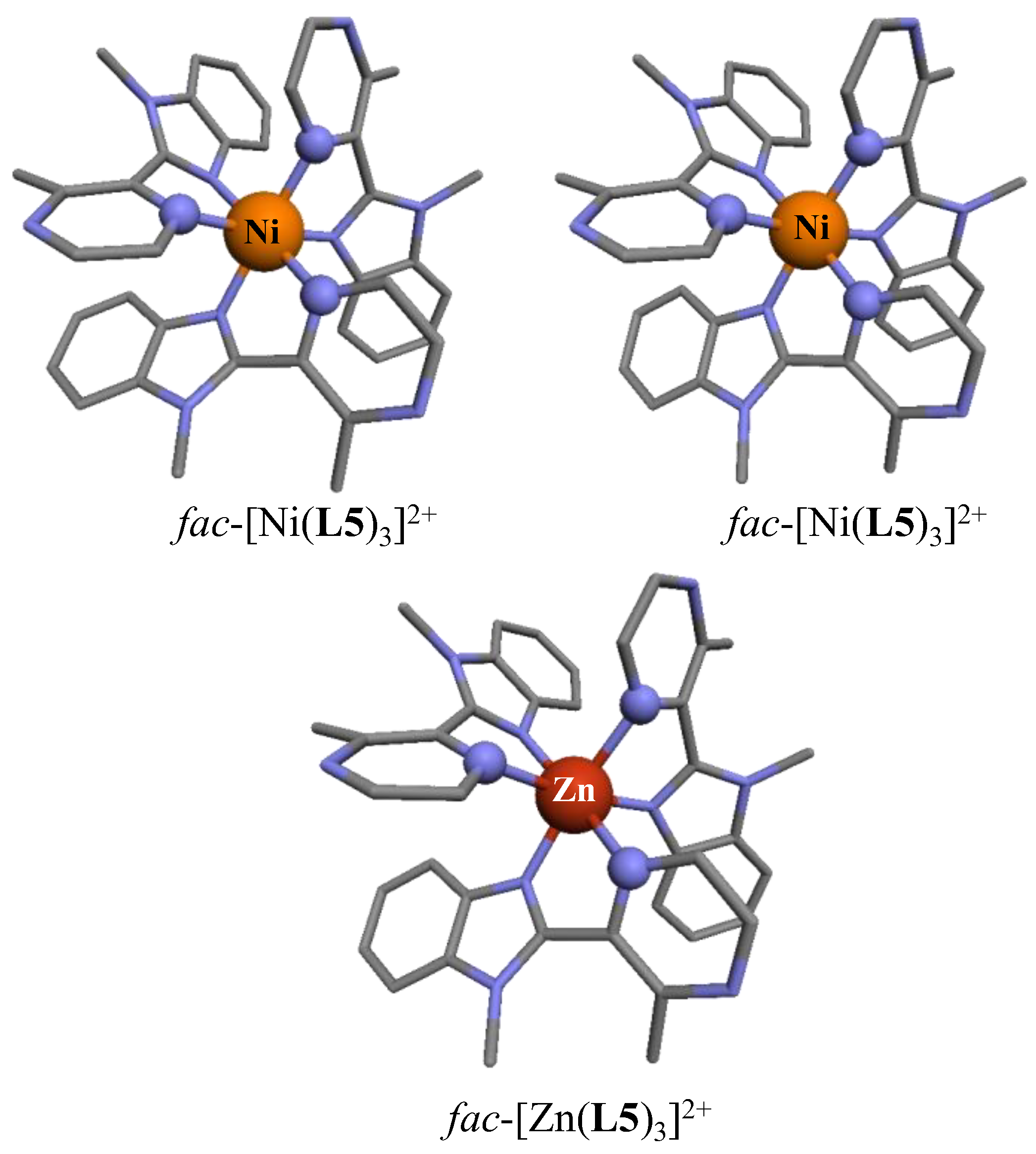
On the contrary, the crystal structures of the tetrafluoroborate salts showed the existence of
fac-[M(
L5)
3]
2+, in which the three didentate ligands adopted the same orientation along the pseudo-threefold axis passing through the metal (
Figure 3). Having previously established that the energy gap between the facial (
C3-symmetry) and meridional (
C1-symmetry) geometries in [Zn(
L1)
3]
2+ and [Zn(
L2)
3]
2+ roughly followed a pure statistical (i.e., entropic) trend and does not overcome thermal energy at room temperature [
60], we concluded that packing forces, specific to the use of perchlorate or tetrafluoroborate counter anions, are more than enough for the quantitative and selective crystallization of pure meridional, respectively facial isomers. For the [Ni(
L5)
3]
2+ and [Zn(
L5)
3]
2+ chromophores, the M–N bonds are systematically shorter for the more basic benzimidazole nitrogen donor (
Table 1, entry 4; p
Ka(bzim) = 5.68) than with its pyrazine counterpart (
Table 1, entry 5; p
Ka(pyrazine) = 0.65) [
67], a trend in complete agreement with that reported for the analogous complex [Zn(
L2)
3]
2+ (
Table 1, column 8) [
60]. Moreover, the shift of the methyl group bound to the pyrazine ring from the 5-position in
L2 to the 3-position in
L5 has globally no geometric influence on the [ZnN
6] coordination sphere, thus leading to Zn–N bond distances surrounding the standard value of Zn–N = 0.74 + 1.46 = 2.20 Å deduced from the effective ionic radii [
68]. In other words, the close methyl groups found in the bound didentate ligand
L5 do not induce major intramolecular steric constraints in [Zn(
L5)
3]
2+ and only a slight increase of the interannular intraligand angles can be detected in going from [Zn(
L2)
3]
2+ (
α = 21(13)°) to [Zn(
L5)
3]
2+ (
α = 38(4)°, entry 5 in
Table 1). The molecular structures of [Ni(
L5)
3]
2+ were very similar to those observed for the Zn
II analogues (
Figure 2 and
Figure 3), except for the slightly shorter Ni–N bond distances, a trend in line with the contraction of Shannon’s effective ionic radii predicted to be 0.74 Å for six-coordinate Zn
2+ and 0.69 Å for six-coordinate Ni
2+ [
68]. The detection of long Fe–N bond distances in [Fe(
L5)
3]
2+ (
d(Fe-N
bz) = 2.14(1) Å and
d(Fe-N
pz) = 2.24(3) Å, entries 3–4 in
Table 1) is more remarkable since it suggests that the Fe
II metal center adopts a pure high spin configuration at 180 K as previously found for the analogous 3-methylpyridine-benzimidazole ligand in [Fe(
L3)
3]
2+ (
d(Fe-N
bz) = 2.14(2) Å and
d(Fe-N
py) = 2.26(5) Å) [
35].
Spin-state, magnetic, and electronic properties of the pseudo-octahedral complexes [M(
L5)
3]X
2 (M = Fe, Ni and X = BF
4, ClO
4) were obtained in the solid state. Molar magnetic susceptibilities (
χM), corrected for diamagnetism of solid state samples of [Fe(
L5)
3](ClO
4)
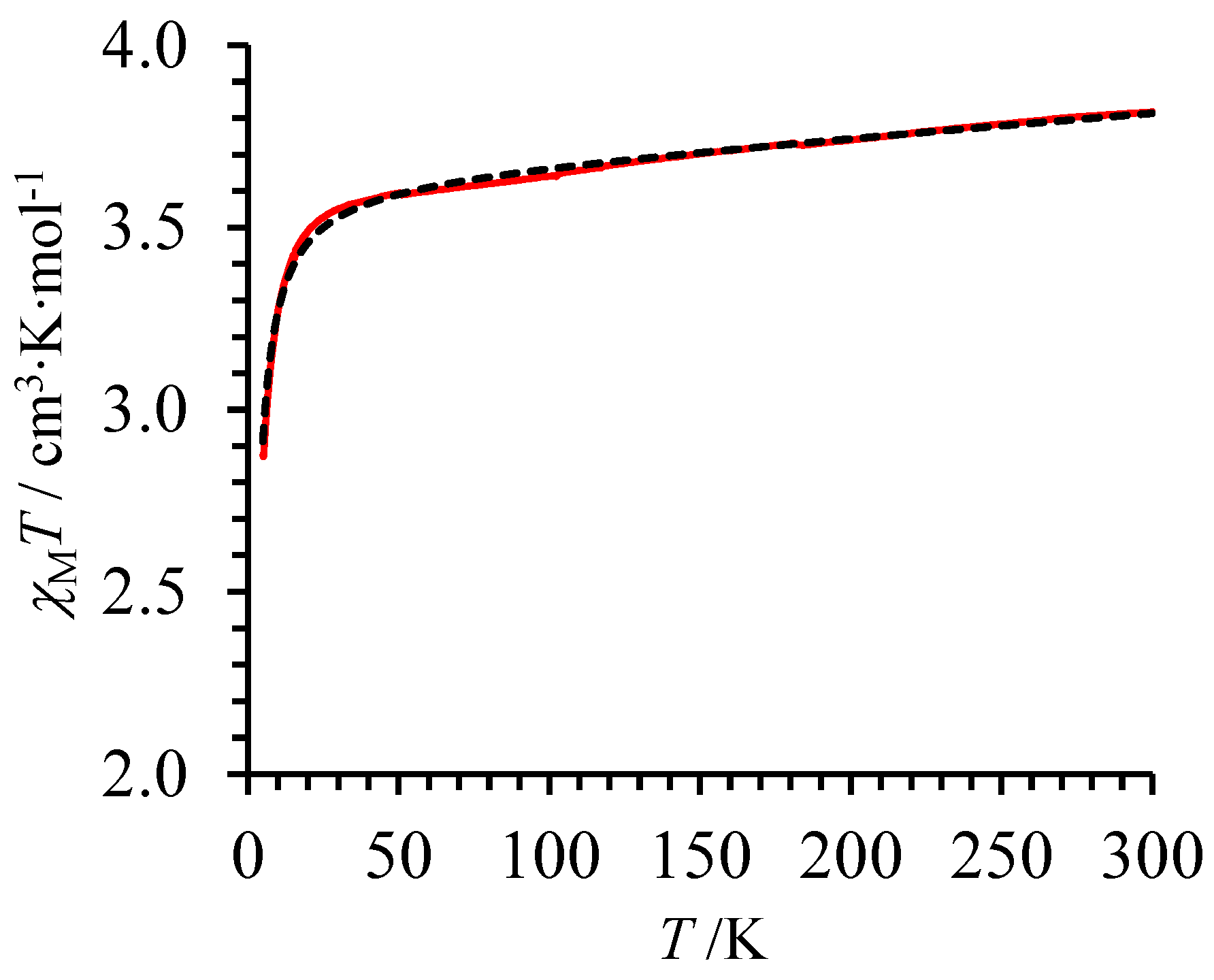
2 (I) were recorded at variable temperatures in a constant magnetic field of 5000 Oe. The
χMT versus
T plot shows a smooth and regular increase of the
χMT product in the 50–300 K range (red trace in
Figure 4), which can be fitted to Equation (6) using a high Curie constant
C = 3.5708(9) cm
3·K·mol
−1 and a non-negligible temperature independent paramagnetism TIP = 848(5) × 10
−6 cm
3·mol
−1, a behavior diagnostic for a high-spin Fe(II) complex [
69].
The additional abrupt decrease in the magnetic susceptibility occurring at low temperature (
T < 40 K) can be assigned to zero-field splitting (ZFS) of high-spin Fe(II), which was modeled with Equation (7), where
D and
E are the axial and rhombic ZFS parameters, respectively [
70,
71,
72,
73].
The pseudo-threefold axis characterizing the [FeN
6] chromophore in [Fe(
L5)
3](ClO
4)
2 implies that
E can be neglected (
E ~ 0). Consequently, the electron–electron interaction splits the
S = 2 manifold at zero magnetic field into three energy levels located at
(
ms = 0),
(
ms = ±1), and
(
ms = ±2). Application of the van Vleck Equation (8), where
kB = 0.695039 cm
−1∙K
−1 is the Boltzmann constant and
NA is Avogadro number,
are first-order spin-only Zeeman effects given in Equation (9), where
μB = −4.6686 × 10
−5 cm
−1·G
−1 is the Bohr magneton, and
stands for the second-order Zeeman effects, leads to a satisfying fit (dotted black trace in
Figure 4 with agreement factor AF = 3.73 × 10
−3) with Landé factor
g = 2.20(1),
D = 0.52(1) cm
−1 and
= 319(4) × 10
−6 cm
3·mol
−1.
The latter magnetic data closely matched those reported for the analogous [Fe(
L3)
3](CF
3SO
3)
2 complex (
g = 2.20(2),
D = 0.85(1) cm
−1 [
36]), and demonstrate that our novel [Fe(
L5)
3](ClO
4)
2 complex, in which the 3-methyl-pyridine group of
L3 is replaced with a 3-methyl-pyrazine group in
L5, is also purely high-spin within the 5–300 K range with no trace of SCO behavior. A careful inspection of the experimental curve around 80 K (
Figure 4) showed a very minor deviation from the theoretical model, which could be tentatively assigned to traces of trapped low-spin form as previously reported for
fac-[Fe(
L3)
3]
2+ when it is incorporated into a LaFe triple-stranded helicate [
36].
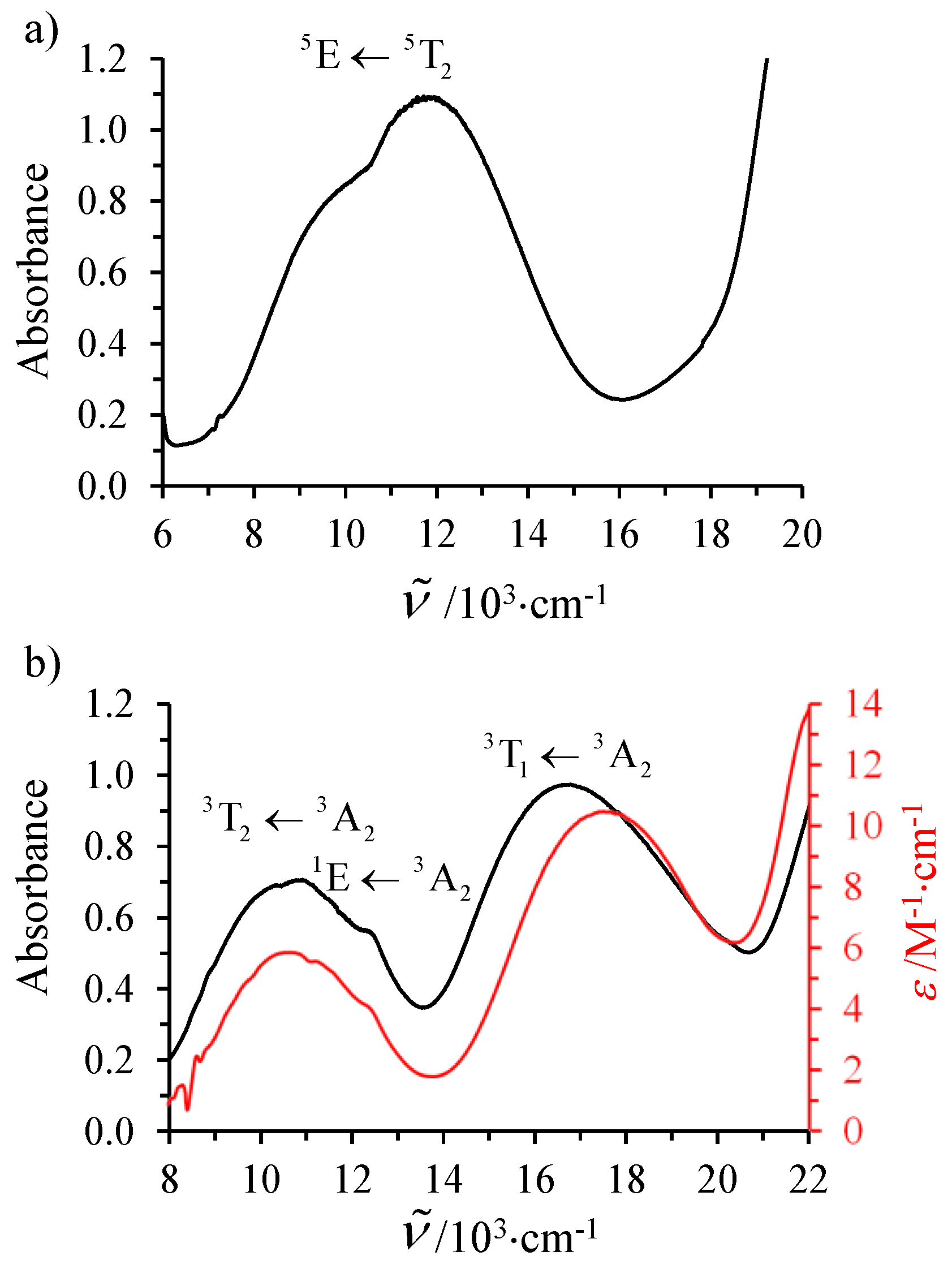
The electronic absorption spectrum recorded for [Fe(
L5)
3](ClO
4)
2 (
I) in the solid state shows the expected Jahn–Teller split Fe
II(
5E←
5T
2) ligand-field transition (
Figure 5a) [
31]. A deconvolution using two Gaussian functions gives
= 8881 cm
−1 and 11,887 cm
−1, thus leading to a barycenter at 10,384 cm
−1, which provides a direct estimation of
= 10 Dq, a value only 700 cm
−1 below the minimum of
≈ 11,000 cm
−1 suggested to be the lower limit for inducing spin state equilibria [
31]. However, the latter criterion strongly depends on the choice of the Racah parameter
B, which is not easily extracted from the single intrashell d–d transition observed in the electronic spectra of high-spin Fe
II complexes. For this reason, we have recorded the electronic absorption spectrum of the analogous Ni
II complex [Ni(
L5)
3](BF
4)
2∙H
2O (
III), for which the combination of two spin-allowed d–d transitions Ni(
3T
2←
3A
2) and Ni(
3T
1←
3A
2) and one spin-forbidden transition Ni(
1E←
3A
2) (
Figure 5b) allows a complete characterization of the electronic parameters
,
B(Ni
II), and
C(Ni
II) with the help of Equations (10)–(13) [
74,
75].
A Gaussian deconvolution of the visible part of the absorption spectrum into three peaks yielded two broad bands, diagnostic for the spin-allowed, but parity-forbidden, transitions at 10,672 cm
−1 (
3T
2←
3A
2) and 16,763 cm
−1 (
3T
1←
3A
2;
Table S16), together with a third weaker band at 12,584 cm
−1, which can be ascribed to the spin-forbidden
1E←
3A
2 component (
Figure 5b). Subsequent non-linear least-squares fits of the energies of these transitions with Equations (10)–(13) provides a first rough set of ligand field strength Δ
oct = 10,672 cm
−1 and Racah parameters
B = 760 cm
−1 and
C = 3413 cm
−1 (
Table S16). However, the mixing of the spin-allowed
3T
2←
3A
2 transition with the spin-forbidden
1E←
3A
2 transition via spin-orbit coupling for apparent ligand field strengths around 11,000–12,000 cm
−1, as found for [Ni(
L5)
3](BF
4)
2∙H
2O, requires further refinements [
76]. A detailed analysis of a series of Ni
II complexes led
Hancock and coworkers to propose three empirical Equations (14)–(16) to obtain more reliable ligand field strengths Δ
oct and Racah parameters
B and
C in cm
−1 units (ε
1 and ε
2 are the extinction coefficients at the observed frequencies of the
1E←
3A
2 transition and
3T
2←
3A
2 transition, respectively) [
76]. The analysis of the experimental absorption spectra using this model gives the corrected parameters gathered in
Table 2 for the [Ni(
L5)
3]
2+ and [Ni(
L2)
3]
2+ chromophores.
The refined ∆
oct,
B and
C parameters computed for [Ni(
L5)
3]
2+ (
Table 2) almost exactly matched those previously reported for the unconstrained [Ni(
L2)
3]
2+ complex, for which the related [Fe(
L2)
3]
2+ complex displayed spin-crossover behavior above room temperature (
T1/2 ~400 K in the solid state,
T1/2 ~ 350 K in acetonitrile solution [
35]). Moreover ∆
oct ([Ni(
L5)
3]
2+) = 11,630 cm−
1 is compatible with the ligand field range 11,200 ≤ ∆
oct (Ni
II) ≤ 12,400 cm
−1 established by Busch and coworkers [
78] as a reliable and useful benchmark for predicting and rationalizing the spin-crossover of the related Fe
II complexes [
40]. The absence of SCO behavior depicted by [Fe(
L5)
3](ClO
4)
2 is thus difficult to assign to some inadequate electronic properties of the [FeN
6] chromophore, but more probably to the impossibility of the coordination sphere to shrink for adopting short-enough Fe–N bonds compatible with low-spin Fe
II. This pure sterical limitation can be tentatively assigned to the intraligand sterical constraints programmed to occur between the methyl groups bound to the pyrazine and benzimidazole rings in each coordinated syn-
L5 ligand in [Fe(
L5)
3]
2+. However, packing forces operating in the solid state may be as important, or even much larger than intramolecular constraints and a definitive assessment requires the extension of our analysis to isolated complexes in solution, where intermolecular interactions are significantly reduced.
Stabilities and electronic properties of the pseudo-octahedral complexes [M(
L5)
3]X
2 (M = Fe, Ni, Zn and X = BF
4, CF
3SO
3) were obtained in acetonitrile solutions. Following the procedure previously detailed for analogous [Zn(
Lk)
3]
2+ [
60] and [Fe(
Lk)
3]
2+ [
35] with the didentate ligands
L1 and
L2, spectrophotometric titrations of submillimolar concentrations of
L5 with M(CF
3SO
3)
2 (M = Ni, Zn) in dry acetonitrile (
Figure 6a,b and
Figure S11a,b) showed the successive formation of two absorbing complexes [M(
L5)
n]
2+ (
n = 2, 1; equilibria (17)–(18)) as ascertained by their independent eigenvectors found in the factor analyses (
Figure 6c and
Figures S11c–S12c) [
48,
49,
50] and their satisfying re-constructed absorption spectra (
Figure 6d and
Figure S11d) [
51,
52,
53].
The 4000 cm−1 red-shift of the ligand-centered π*←π transition observed upon complexation to M
II (
Figure 6a and
Figure S11a) is diagnostic for the anti→syn conformational change of the α,α’diimine unit accompanying the coordination of
L5 to M
II [
79,
80]. Non-linear least-square fits [
51,
52,
53] of the spectrophotometric data to equilibria (17)–(18) provide the macroscopic cumulative formation constants gathered in
Table 3 (entries 3–4) together with speciation curves [
81] showing a maximum formation of ca. 50% of the ligand speciation under the form of [M(
L5)
2]
2+ at submillimolar concentrations (
Figure 6e and
Figure S11e). Attempts to consider the formation of an additional [M(
L5)
3]
2+ complex according to equilibrium (19) only failed in our hands, which suggests that the
constant is too low for providing significant quantities of the latter complex at this concentration.
According to the site-binding model [
82,
83], the first stability constant
reflects the simple intermolecular affinity
between the didentate ligand
Lk and the entering M
II cation (including the change in solvation, Equation (20)) modulated by a pure entropic contribution
ω1,1 = 24 [
60] produced by the change in rotational entropies accompanying the transformation of the reactants into products, a parameter often referred to as the statistical factor [
84,
85].
Applying Equation (20) to the stability constants
collected in
Table 3 (entry 3) leads to intrinsic free energy affinities
(entry 6), which are roughly reduced by a factor two in going from the 5-methyl-pyrazine ligand
L2 to the 3-methyl-pyrazine analogue
L5, as illustrated for [Zn(
L2)]
2+ (
= −31.1(3) kJ∙mol
−1) and [Zn(
L5)]
2+ (
= −17.1(7) kJ∙mol
−1).
The fixation of two ligands to give [M(
Lk)
2]
2+ obeying equilibrium (18) requires twice the intermolecular metal-ligand affinity, a statistical factor of
ω1,2 = 120, which takes into account all the possible geometric isomers [
60] and the operation of allosteric cooperativity factors
measuring the extra energy cost (
< 1), respectively, energy benefit (
> 1) produced by the binding of two ligands to the same metal (Equation (21)) [
82,
83,
86].
Applying Equation (21) to the stability constants
(entry 4 in
Table 3) with the help of the intrinsic affinities
deduced from
(entry 6 in
Table 3) provides interligand free energies interactions
(entry 7 in
Table 3) close to zero (non-cooperativity) for [M(
L2)
2]
2+ complexes, but negative (positive cooperativity) for [M(
L5)
2]
2+. However, the neglect of the expected [M(
L5)
3]
2+ for modeling the spectrophotometric titrations results in a slight overestimation of the second cumulative constant
, fully compatible with the apparent, but probably not pertinent, minor positive cooperativity observed for the successive binding of
L5 ligands to M
2+ centers. Finally, the introduction of the estimated intrinsic affinities and interligand interactions into Equation (22) allows some predictions concerning the inaccessible third cumulative stability constants [
35].
The resulting values of
≈ 12.0 (
Table 3, entry 5) correspond to a reduction by five orders of magnitude with respect to
≈ 17.0 experimentally found with the less constrained ligand
L2. There is no doubt that the hindered planar arrangement of the two aromatic heterocycles in
L5 has a deep impact on the strength of the M–N bonds because of the misalignment of the nitrogen lone pairs with metal d-orbitals in pseudo-octahedral geometry. Nevertheless, at 1 M concentration of
L5 in acetonitrile, the ligand speciation curve computed [
81] by using
(
n = 1–3) gathered in
Table 3 shows that [M(
L5)
3]
2+ (M = Ni, Zn) corresponds to more than 90% of the distribution at the stoichiometric M:
L5 = 1/3 ratio (
Figure S12). At a total ligand concentration of 0.1 M in acetonitrile, [Ni(
L5)
3]
2+ stands for 84% of the ligand speciation and its absorption spectrum closely matches that recorded for related solid state samples (
Figure 5). Repeating the detailed analysis described in the previous section (Equations (10)–(16)) provides ligand-field strengths (∆
oct) and Racah parameters (
B,
C) similar to those found in the solid state (
Table 2), especially for the crucial ratio ∆
oct/
B = 13.3, which is identical for [Ni(
L2)
3]
2+ and [Ni(
L5)
3]
2+, both in solution and in the solid state. In other words, the moving of the methyl group connected to the pyrazine ring from the 5-position in
L2 to the 3-position in
L5 indeed strongly reduces the affinity of the didentate ligand for Ni(II) in solution, but only has a weak effect on the ligand field strengths and on the Ni–N (or Zn–N) bond length. Interestingly, the variable-temperature
1H NMR spectra recorded for diamagnetic [Zn(
L5)
3]
2+ (>80% for a total concentration of 0.1 M in CD
3CN in the 233–333 K range,
Figure S13) showed a single set of signals compatible with the exclusive formation of an averaged
C3-symmetrical species with no contribution from either blocked facial and meridional isomers or from partial decomplexation to give [Zn(
L5)
2]
2+ +
L5 (equilibrium (19)). These observations are in contrast with the detection at low temperature in CD
3CN of two well-resolved spectra characteristics of a slow exchange operating between
fac-[Zn(
L2)
3]
2+ and
mer-[Zn(
L2)
3]
2+ [
60], and suggests that the weaker stability constants are accompanied by faster ligand exchange processes around Zn
2+ in [Zn(
L5)
3]
2+. This decrease in affinity reaches its paroxysm for the coordination of Fe
2+ since the spectrophotometric titration of
L5 with Fe(CF
3SO
3)2 conducted at submillimolar concentration displays only a minor drift of the absorption spectra with no pronounced end point (
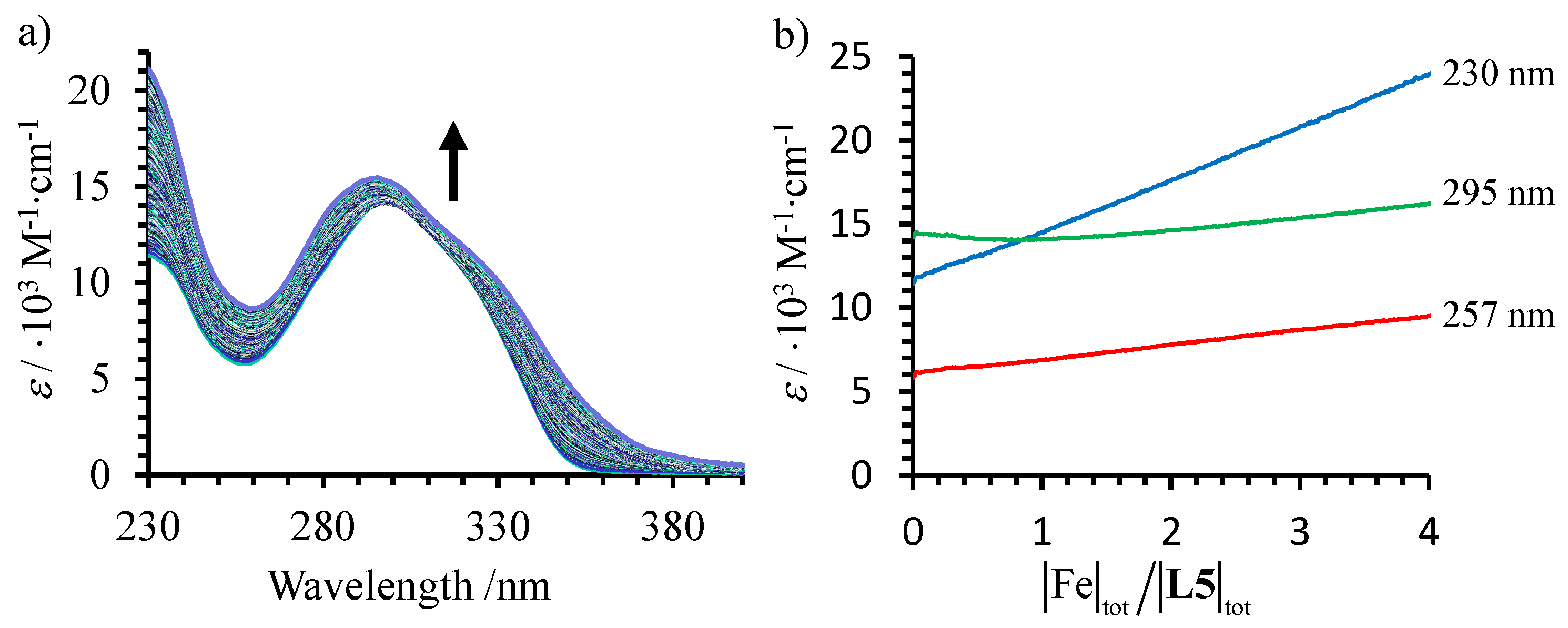
Figure 7).
Attempts to model these limited variations within the frame of equilibria (17)–(18) only failed. The amount of [Fe(
L5)
n]
2+ in solution is strongly limited by (very) low cumulative stability constants, a situation produced by the impossibility for Fe
II to adopt a compact low-spin configuration in the sterically constrained complex [
35]. In the absence of a significant amount of [Fe(
L5)
3]
2+ complexes in solution, no spin state equilibria could be investigated for this system.











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
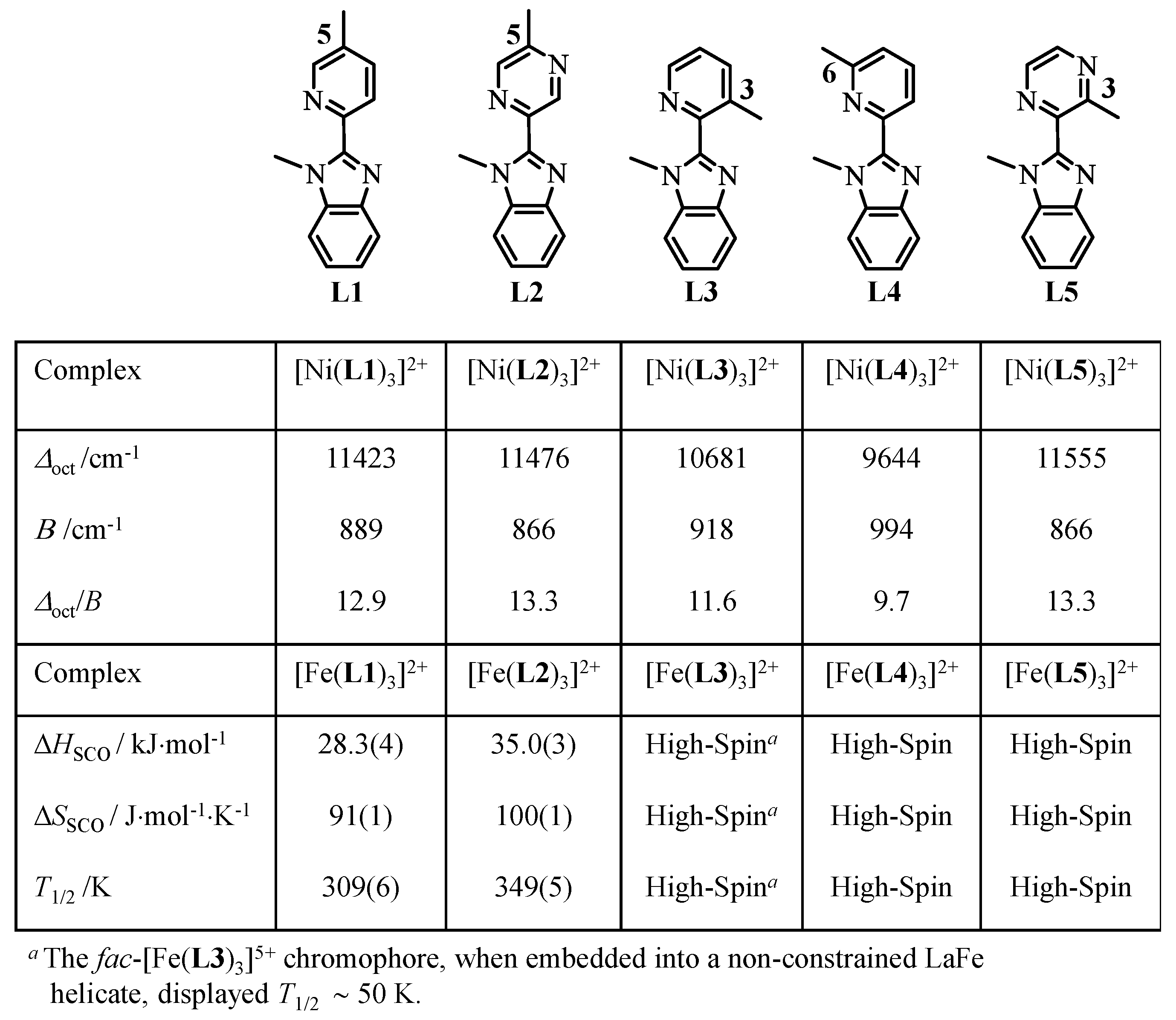{kind=link}
{kind=link}