Topological Dynamics of a Radical Ion Pair: Experimental and Computational Assessment at the Relevant Nanosecond Timescale †


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. EPR Experiments
2.2. Computer Simulations
2.2.1. Software
2.2.2. Molecular Dynamics Simulations
2.2.3. Thermodynamic Analysis
2.2.4. Kinetic Analysis
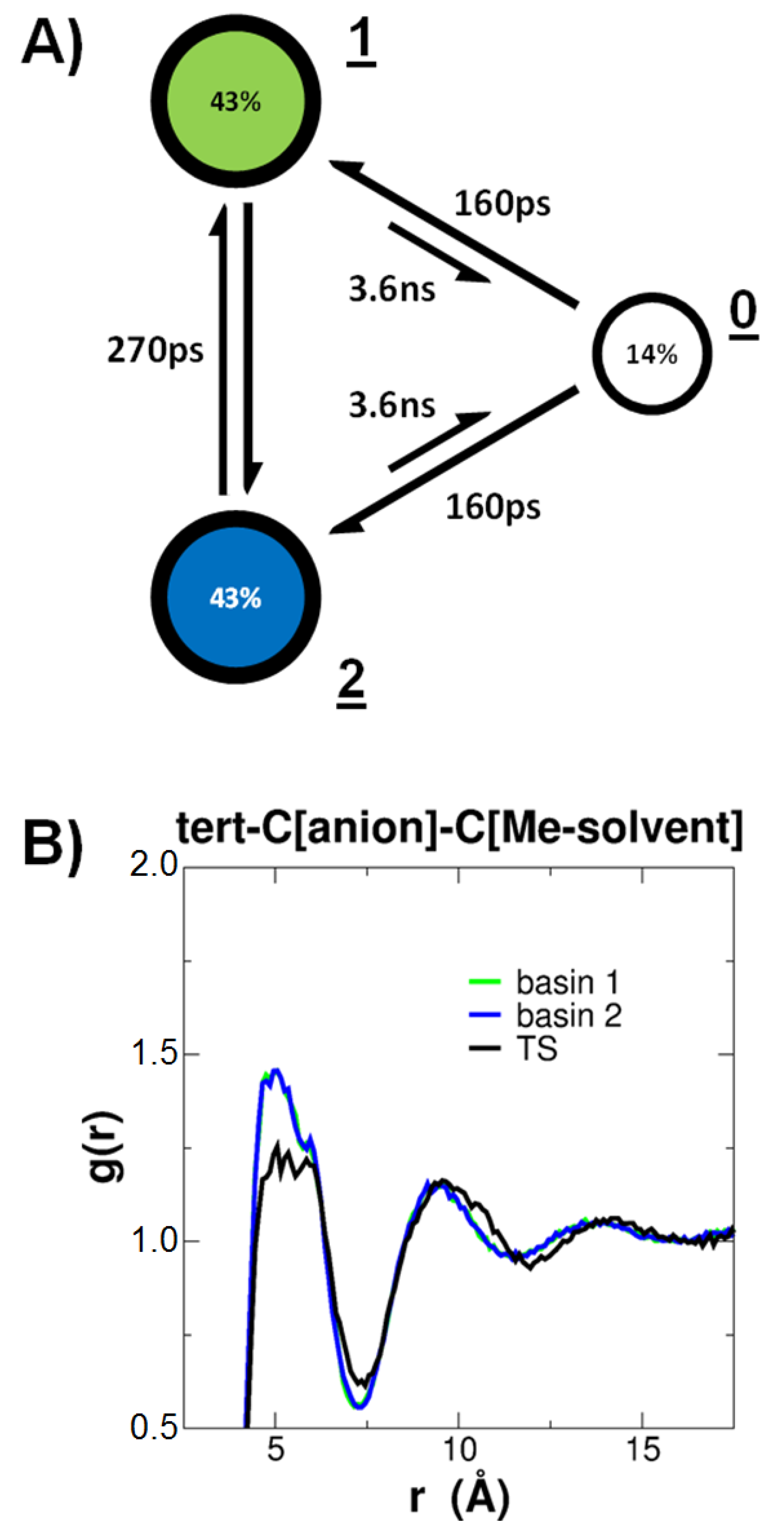
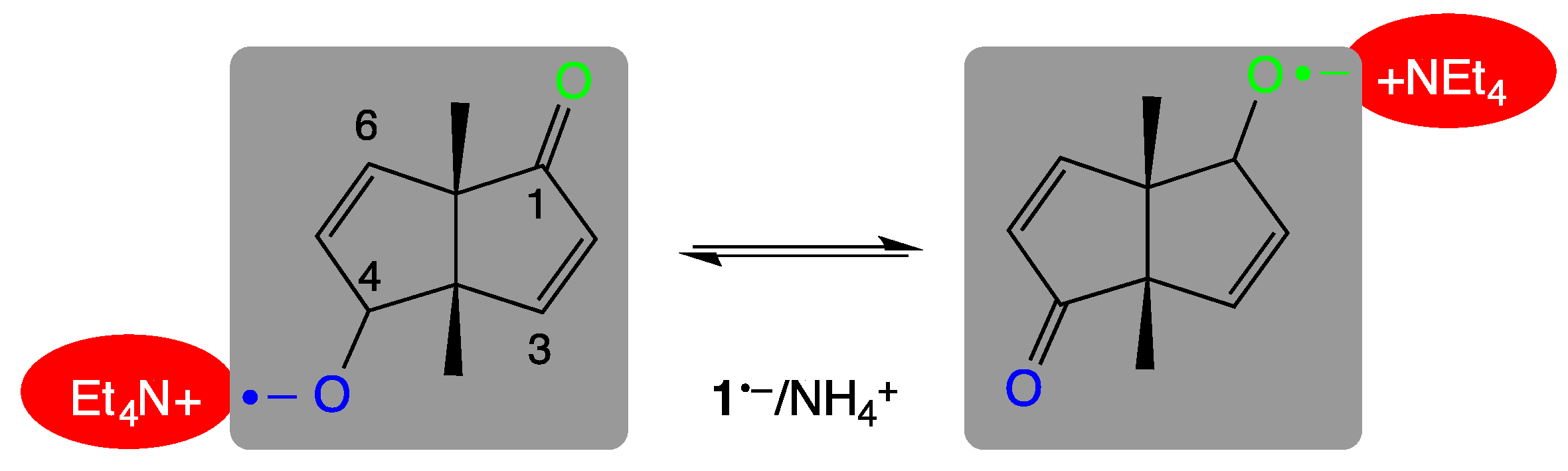
3. Results
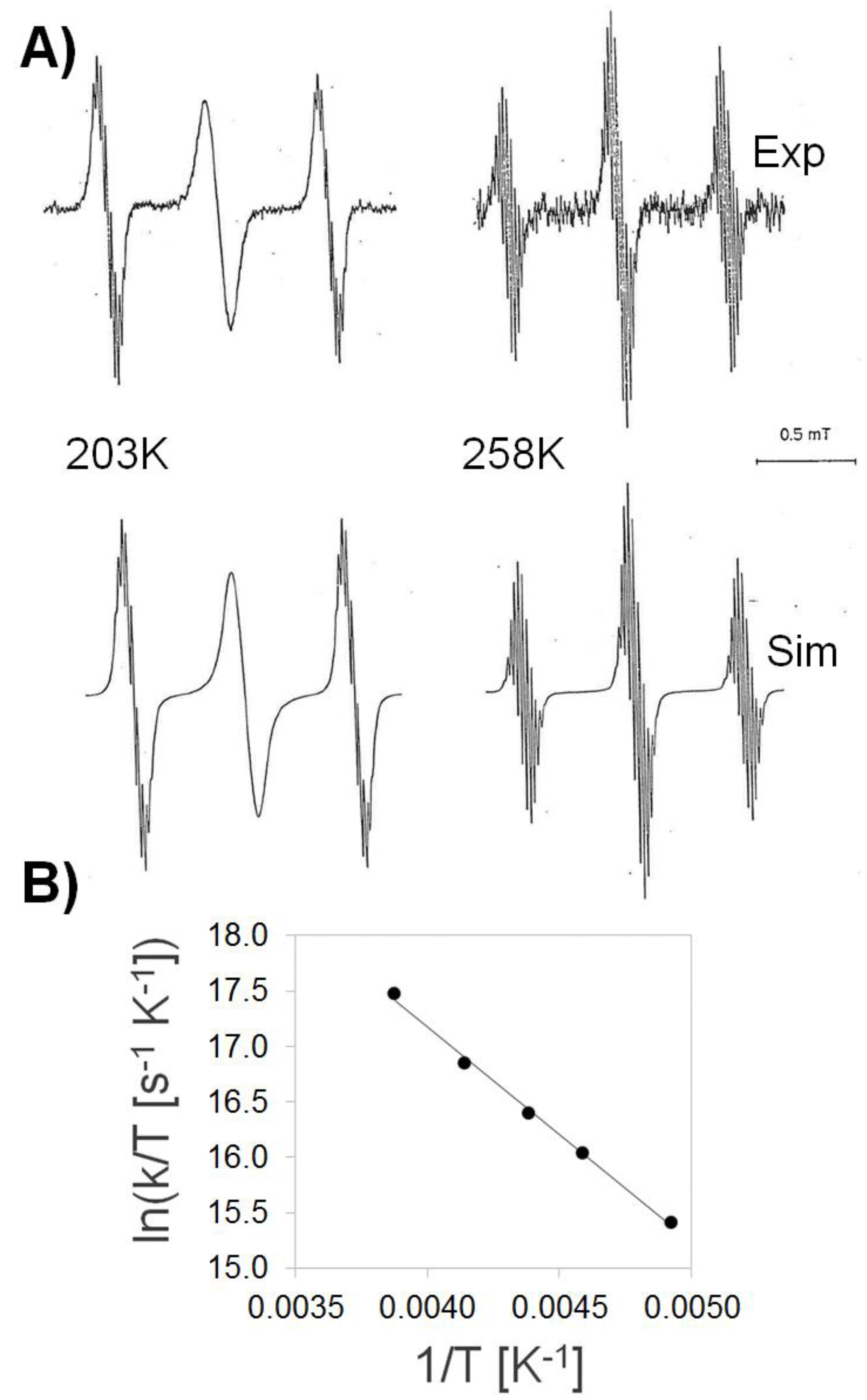
3.1. Temperature-Dependent EPR Experiments
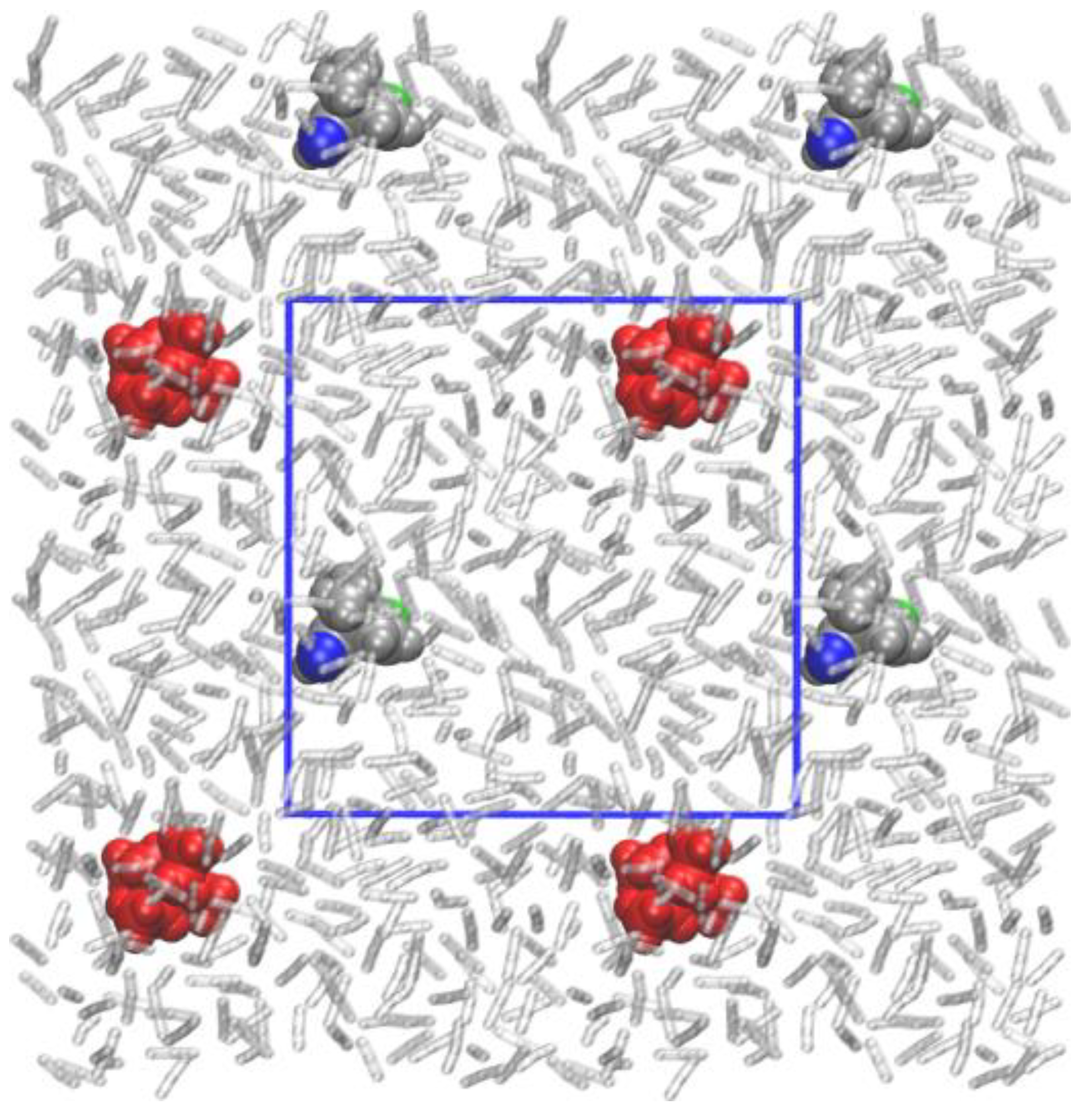
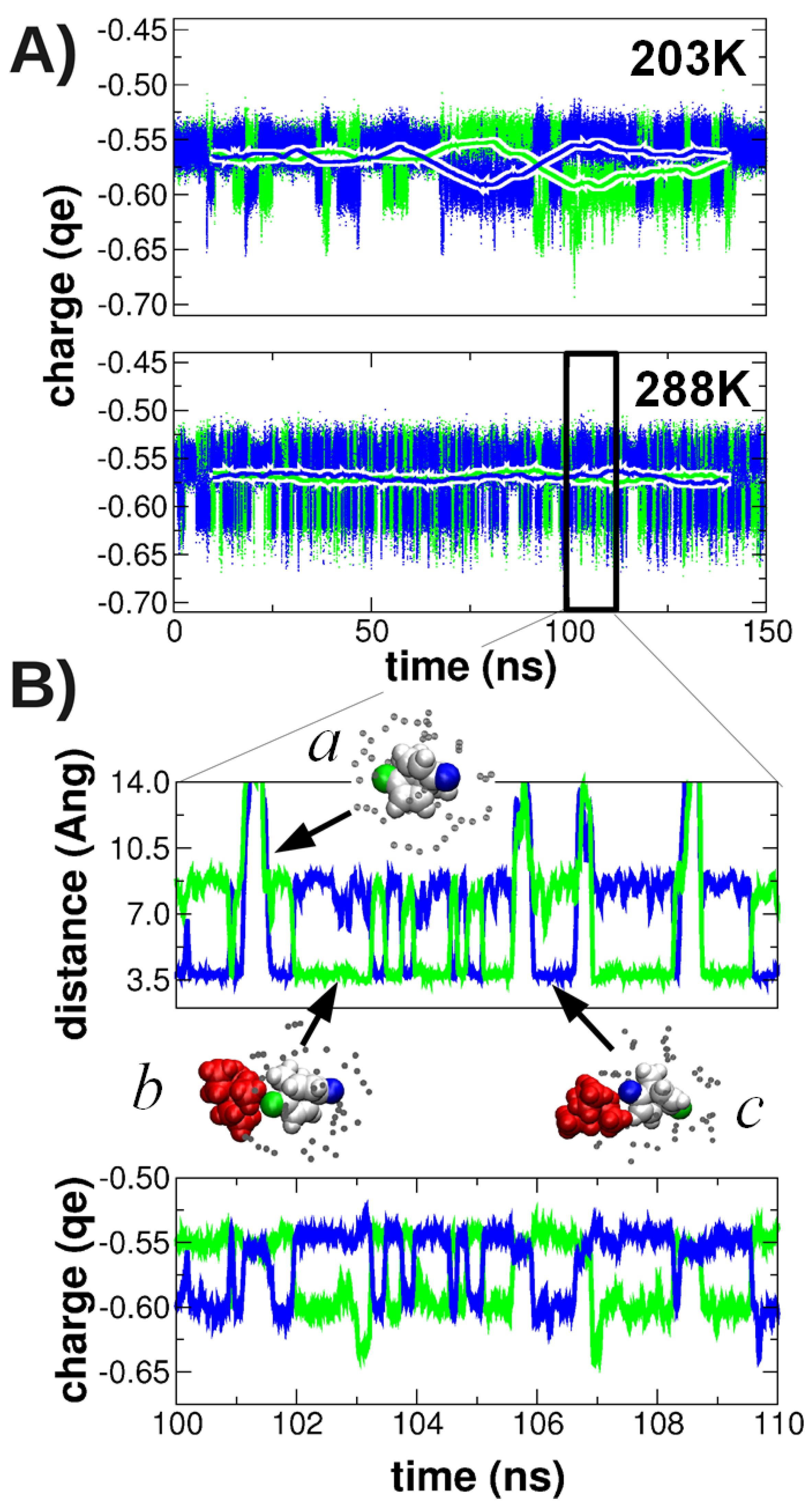
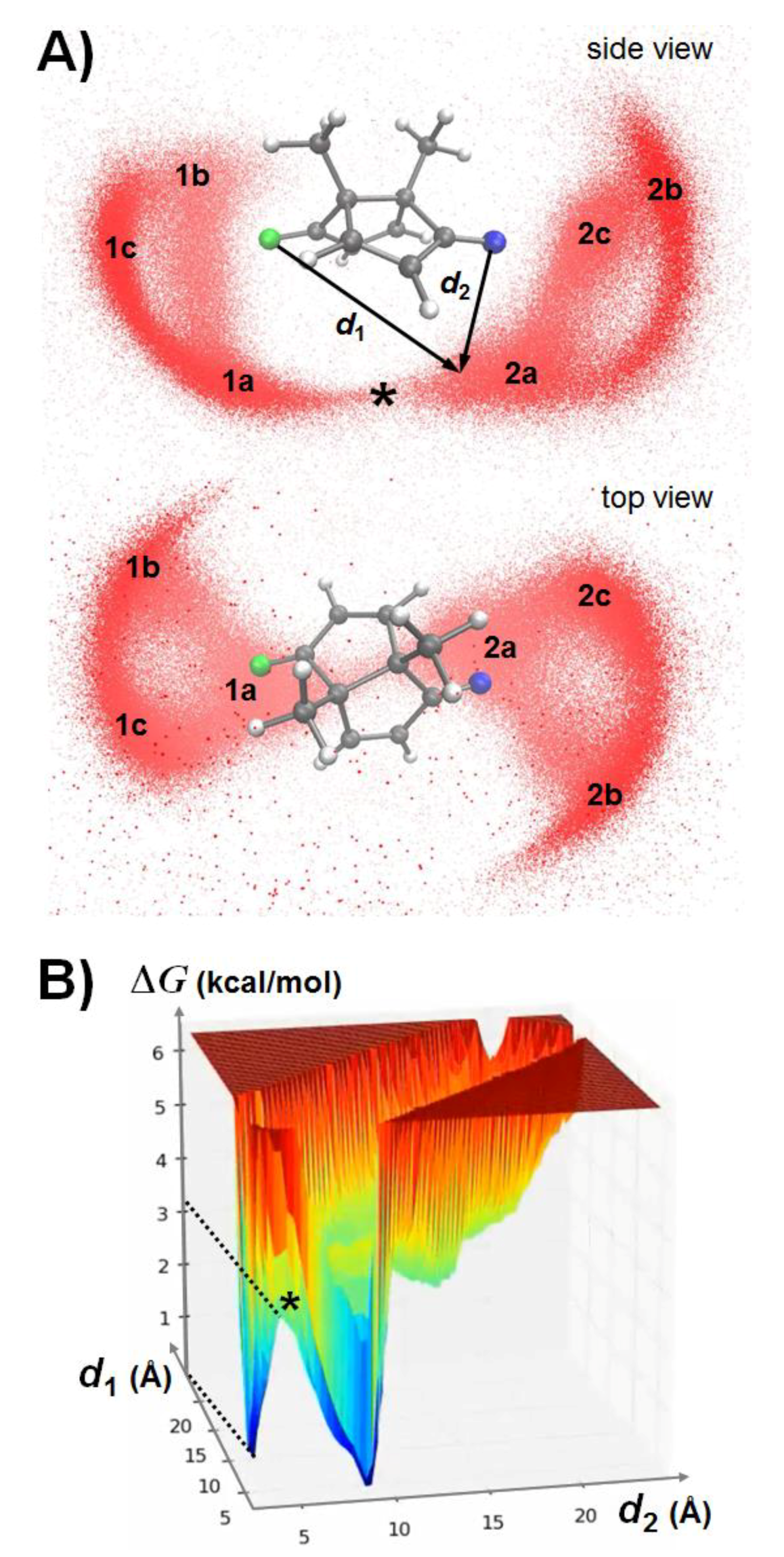
3.2. QM/MM-Based Molecular Dynamics Simulations
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Rockl, J.L.; Pollok, D.; Franke, R.; Waldvogel, S.R. A Decade of Electrochemical Dehydrogenative C,C-Coupling of Aryls. Acc. Chem. Res. 2020, 53, 45–61. [Google Scholar] [CrossRef] [PubMed]
- Farney, E.P.; Chapman, S.J.; Swords, W.B.; Torelli, M.D.; Hamers, R.J.; Yoon, T.P. Discovery and Elucidation of Counteranion Dependence in Photoredox Catalysis. J. Am. Chem. Soc. 2019, 141, 6385–6391. [Google Scholar] [CrossRef] [PubMed]
- Chiarotto, I.; Mattiello, L.; Feroci, M. The Electrogenerated Cyanomethyl Anion: An Old Base Still Smart. Acc. Chem. Res. 2019, 52, 3297–3308. [Google Scholar] [CrossRef] [PubMed]
- Li, G.C.; Brady, M.D.; Meyer, G.J. Visible Light Driven Bromide Oxidation and Ligand Substitution Photochemistry of a Ru Diimine Complex. J. Am. Chem. Soc. 2018, 140, 5447–5456. [Google Scholar] [CrossRef] [PubMed]
- Jiang, C.; Chen, W.; Zheng, W.-H.; Lu, H. Advances in asymmetric visible-light photocatalysis, 2015–2019. Org. Biomol. Chem. 2019, 17, 8673–8689. [Google Scholar] [CrossRef] [PubMed]
- Morack, T.; Muck-Lichtenfeld, C.; Gilmour, R. Bioinspired Radical Stetter Reaction: Radical Umpolung Enabled by Ion-Pair Photocatalysis. Angew. Chem.-Int. Ed. 2019, 58, 1208–1212. [Google Scholar] [CrossRef]
- Ravelli, D.; Protti, S.; Fagnoni, M. Carbon-Carbon Bond Forming Reactions via Photogenerated Intermediates. Chem. Rev. 2016, 116, 9850–9913. [Google Scholar] [CrossRef]
- Lim, C.H.; Ryan, M.D.; McCarthy, B.G.; Theriot, J.C.; Sartor, S.M.; Damrauer, N.H.; Musgrave, C.B.; Miyake, G.M. Intramolecular Charge Transfer and Ion Pairing in N,N-Diaryl Dihydrophenazine Photoredox Catalysts for Efficient Organocatalyzed Atom Transfer Radical Polymerization. J. Am. Chem. Soc. 2017, 139, 348–355. [Google Scholar] [CrossRef]
- Zollitsch, T.M.; Jarocha, L.E.; Bialas, C.; Henbest, K.B.; Kodali, G.; Dutton, P.L.; Moser, C.C.; Timmel, C.R.; Hore, P.J.; Mackenzie, S.R. Magnetically Sensitive Radical Photochemistry of Non-natural Flavoproteins. J. Am. Chem. Soc. 2018, 140, 8705–8713. [Google Scholar] [CrossRef]
- Zelenka, J.; Cibulka, R.; Roithova, J. Flavinium Catalysed Photooxidation: Detection and Characterization of Elusive Peroxyflavinium Intermediates. Angew. Chem.-Int. Ed. 2019, 58, 15412–15420. [Google Scholar] [CrossRef]
- Glatthar, R.; Spichty, M.; Gugger, A.; Batra, R.; Damm, W.; Mohr, M.; Zipse, H.; Giese, B. Mechanistic studies in the radical induced DNA strand cleavage-Formation and reactivity of the radical cation intermediate. Tetrahedron 2000, 56, 4117–4128. [Google Scholar] [CrossRef]
- Horibe, T.; Ohmura, S.; Ishihara, K. Structure and Reactivity of Aromatic Radical Cations Generated by FeCl3. J. Am. Chem. Soc. 2019, 141, 1877–1881. [Google Scholar] [CrossRef] [PubMed]
- Karplus, M. Development of Multiscale Models for Complex Chemical Systems: From H+H2 to Biomolecules (Nobel Lecture). Angew. Chem. Int. Ed. 2014, 53, 9992–10005. [Google Scholar] [CrossRef] [PubMed]
- Levitt, M. Birth and Future of Multiscale Modeling for Macromolecular Systems (Nobel Lecture). Angew. Chem. Int. Ed. 2014, 53, 10006–10018. [Google Scholar] [CrossRef] [PubMed]
- Warshel, A. Multiscale Modeling of Biological Functions: From Enzymes to Molecular Machines (Nobel Lecture). Angew. Chem. Int. Ed. 2014, 53, 10020–10031. [Google Scholar] [CrossRef]
- Allendoerfer, R.D.; Martinchek, G.A.; Bruckenstein, S. Simultaneous electrochemical-electron spin resonance measurements with a coaxial microwave cavity. Anal. Chem. 1975, 47, 890–894. [Google Scholar] [CrossRef]
- Stoll, S.; Schweiger, A. EasySpin, a comprehensive software package for spectral simulation and analysis in EPR. J. Magn. Reson. San Diego Calif 1997 2006, 178, 42–55. [Google Scholar] [CrossRef]
- Zalibera, M.; Jalilov, A.S.; Stoll, S.; Guzei, I.A.; Gescheidt, G.; Nelsen, S.F. Monotrimethylene-bridged bis-p-phenylenediamine radical cations and dications: Spin states, conformations, and dynamics. J. Phys. Chem. A 2013, 117, 1439–1448. [Google Scholar] [CrossRef]
- Binsch, G. Unified theory of exchange effects on nuclear magnetic resonance line shapes. J. Am. Chem. Soc. 1969, 91, 1304–1309. [Google Scholar] [CrossRef]
- Heinzer, J. Fast computation of exchange-broadened isotropic E.S.R. spectra. Mol. Phys. 1971, 22, 167–177. [Google Scholar] [CrossRef]
- Gerson, F.; Huber, W. Electron Spin Resonance Spectroscopy of Organic Radicals; John Wiley & Sons: Weinheim, Germany, 2003. [Google Scholar]
- Spichty, M.; Giese, B.; Matsumoto, A.; Fischer, H.; Gescheidt, G. Conformational dynamics in a methacrylate-derived radical: A computational and EPR study. Macromolecules 2001, 34, 723–726. [Google Scholar] [CrossRef]
- Brooks, B.R.; Brooks III, C.L.; Mackerell, A.D.; Nilsson, L.; Petrella, R.J.; Roux, B.; Won, Y.; Archontis, G.; Bartels, C.; Boresch, S.; et al. CHARMM: The biomolecular simulation program. J. Comput. Chem. 2009, 30, 1545–1614. [Google Scholar] [CrossRef] [PubMed]
- Seeber, M.; Cecchini, M.; Rao, F.; Settanni, G.; Caflisch, A. Wordom: A program for efficient analysis of molecular dynamics simulations. Bioinformatics 2007, 23, 2625–2627. [Google Scholar] [CrossRef] [PubMed]
- Rao, F. PYNORAMIX Is a Fast and Efficient Python Library to Analyze Water Structure and Dynamics. Available online: https://github.com/ruvido/Pynoramix (accessed on 14 May 2016).
- Humphrey, W.; Dalke, A.; Schulten, K. VMD–Visual Molecular Dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef]
- Elstner, M.; Porezag, D.; Jungnickel, G.; Elsner, J.; Haugk, M.; Frauenheim, T.; Suhai, S.; Seifert, G. Self-consistent-charge density-functional tight-binding method for simulations of complex materials properties. Phys. Rev. B 1998, 58, 7260–7268. [Google Scholar] [CrossRef]
- Cui, Q.; Elstner, M.; Kaxiras, E.; Frauenheim, T.; Karplus, M. A QM/MM implementation of the self-consistent charge density functional tight binding (SCC-DFTB) method. J. Phys. Chem. B 2001, 105, 569–585. [Google Scholar] [CrossRef]
- Vanommeslaeghe, K.; Hatcher, E.; Acharya, C.; Kundu, S.; Zhong, S.; Shim, J.; Darian, E.; Guvench, O.; Lopes, P.; Vorobyov, I.; et al. CHARMM General Force Field (CGenFF): A force field for drug-like molecules compatible with the CHARMM all-atom additive biological force fields. J. Comput. Chem. 2010, 31, 671–690. [Google Scholar]
- Van Dongen, S. Graph. Clustering by Flow Simulation. Ph.D. Thesis, University of Utrecht, Utrecht, The Netherlands, 2000. [Google Scholar]
- Gfeller, D.; Rios, P.D.L.; Caflisch, A.; Rao, F. Complex network analysis of free-energy landscapes. Proc. Natl. Acad. Sci. USA 2007, 104, 1817–1822. [Google Scholar] [CrossRef]
- Furderer, P.; Gerson, F.; Heinzer, J.; Mazur, S.; Ohyanishiguchi, H.; Schroeder, A.H. Esr and Endor Studies of a Radical-Anion with Formally Nonconjugated Keto Groups - Cis-10,11-Dimethyldiphensuccindan-9,12-Dione. J. Am. Chem. Soc. 1979, 101, 2275–2281. [Google Scholar] [CrossRef]
- Redfield, A.G. On the Theory of Relaxation Processes. IBM J. Res. Dev. 1957, 1, 19–31. [Google Scholar] [CrossRef]
- Eyring, H. The Activated Complex in Chemical Reactions. J. Chem. Phys. 1935, 3, 107–115. [Google Scholar] [CrossRef]
- Giese, B. Electron transfer in DNA. Curr. Opin. Chem. Biol. 2002, 6, 612–618. [Google Scholar] [CrossRef]
- Cordes, M.; Giese, B. Electron transfer in peptides and proteins. Chem. Soc. Rev. 2009, 38, 892–901. [Google Scholar] [CrossRef] [PubMed]
- Marcus, R.A. Electron Transfer Reactions in Chemistry: Theory and Experiment (Nobel Lecture). Angew. Chem. Intl. Ed. 1993, 32, 1111–1121. [Google Scholar] [CrossRef]






© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Quast, H.; Gescheidt, G.; Spichty, M. Topological Dynamics of a Radical Ion Pair: Experimental and Computational Assessment at the Relevant Nanosecond Timescale. Chemistry 2020, 2, 219-230. https://doi.org/10.3390/chemistry2020014
Quast H, Gescheidt G, Spichty M. Topological Dynamics of a Radical Ion Pair: Experimental and Computational Assessment at the Relevant Nanosecond Timescale. Chemistry. 2020; 2(2):219-230. https://doi.org/10.3390/chemistry2020014
Chicago/Turabian StyleQuast, Helmut, Georg Gescheidt, and Martin Spichty. 2020. "Topological Dynamics of a Radical Ion Pair: Experimental and Computational Assessment at the Relevant Nanosecond Timescale" Chemistry 2, no. 2: 219-230. https://doi.org/10.3390/chemistry2020014
APA StyleQuast, H., Gescheidt, G., & Spichty, M. (2020). Topological Dynamics of a Radical Ion Pair: Experimental and Computational Assessment at the Relevant Nanosecond Timescale. Chemistry, 2(2), 219-230. https://doi.org/10.3390/chemistry2020014