Effect of Hydrazine Pretreatment on the Activity, Stability and Active Sites of Cobalt Species for Preferential Oxidation (PROX) of CO in H2-Rich Stream

Abstract
:1. Introduction
2. Experimental Section
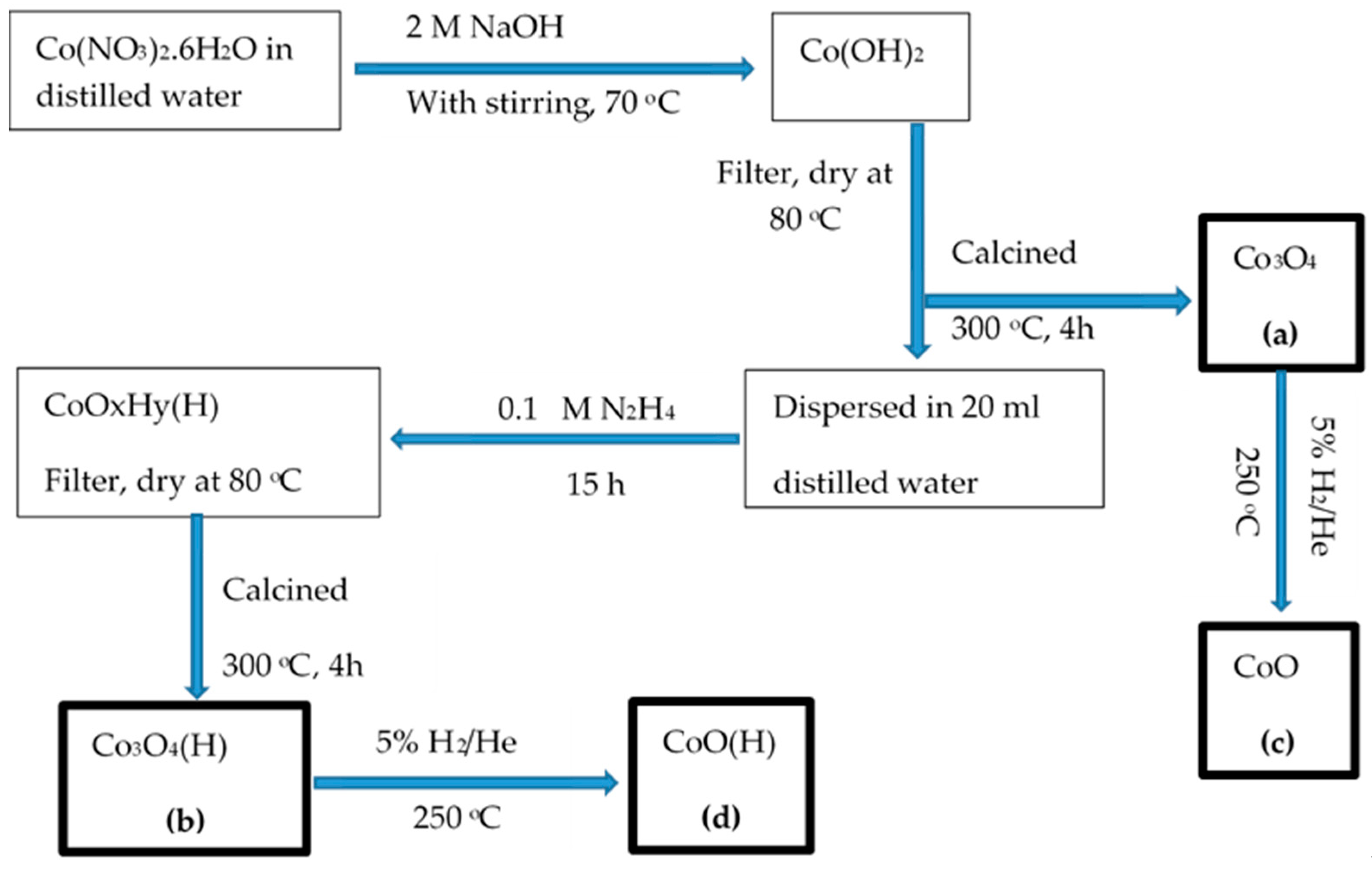
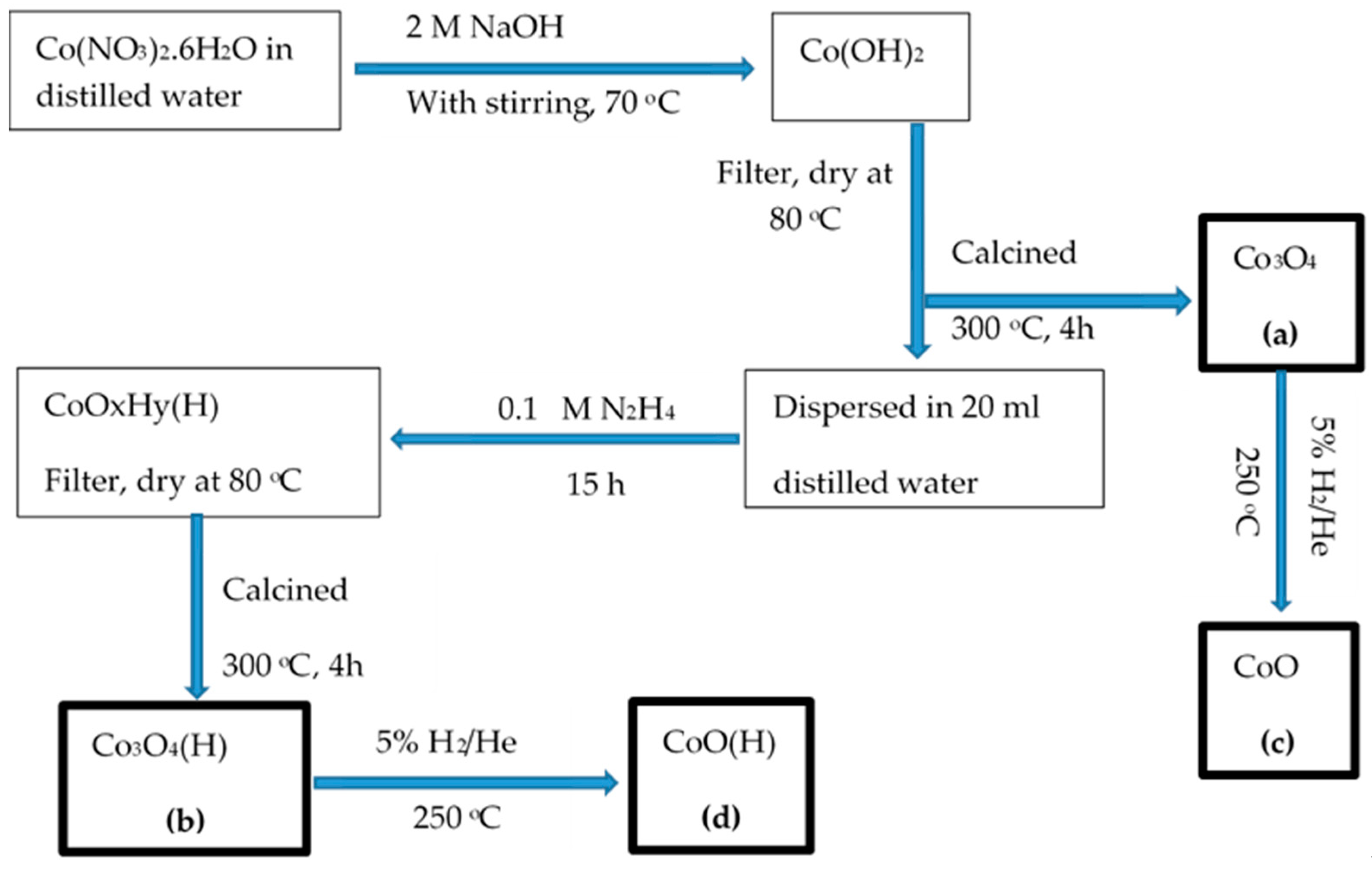
2.1. Preparation of Catalysts
2.2. Characterisation of Catalysts
2.3. Catalytic Activity Tests
3. Results and Discussion
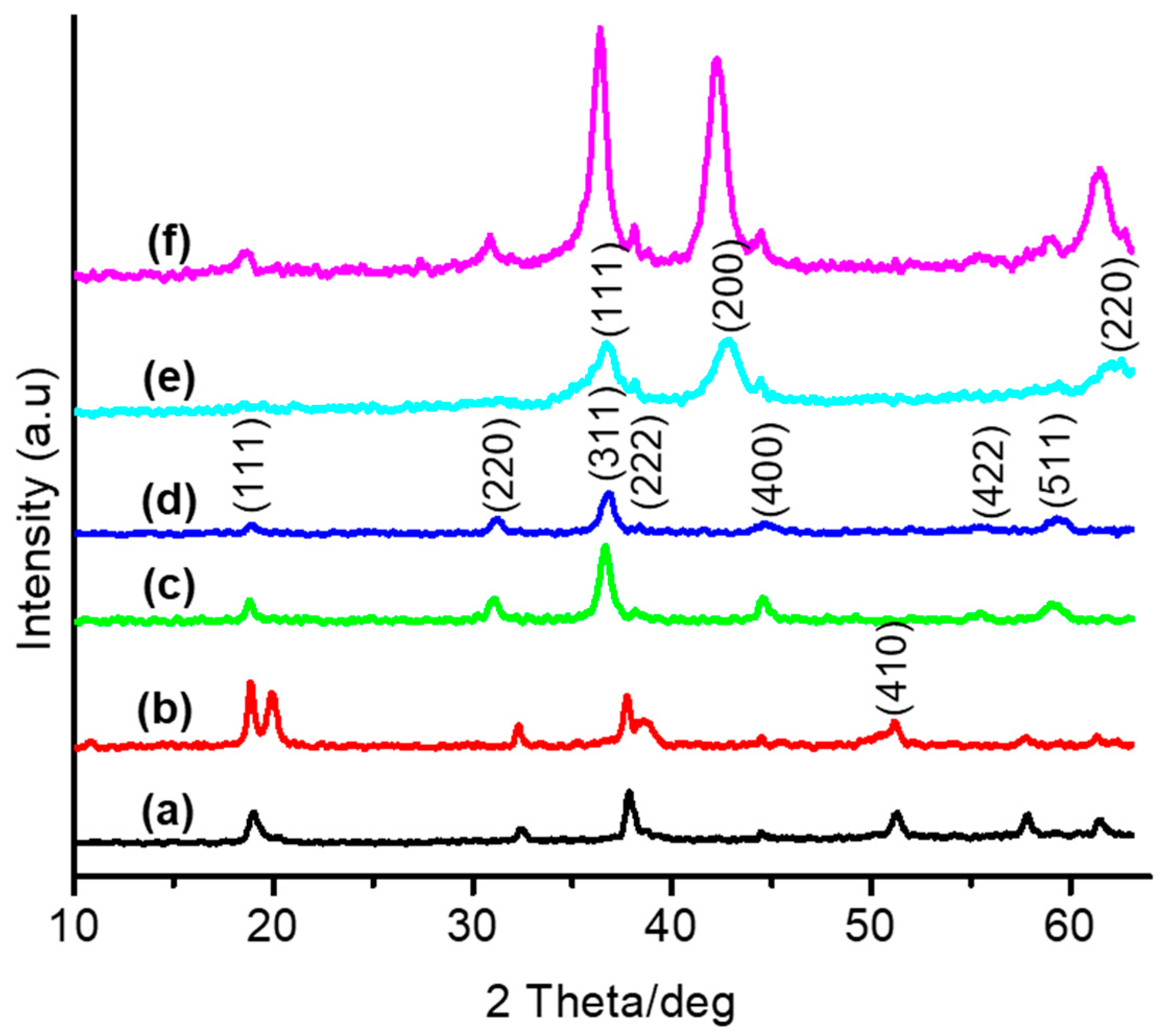
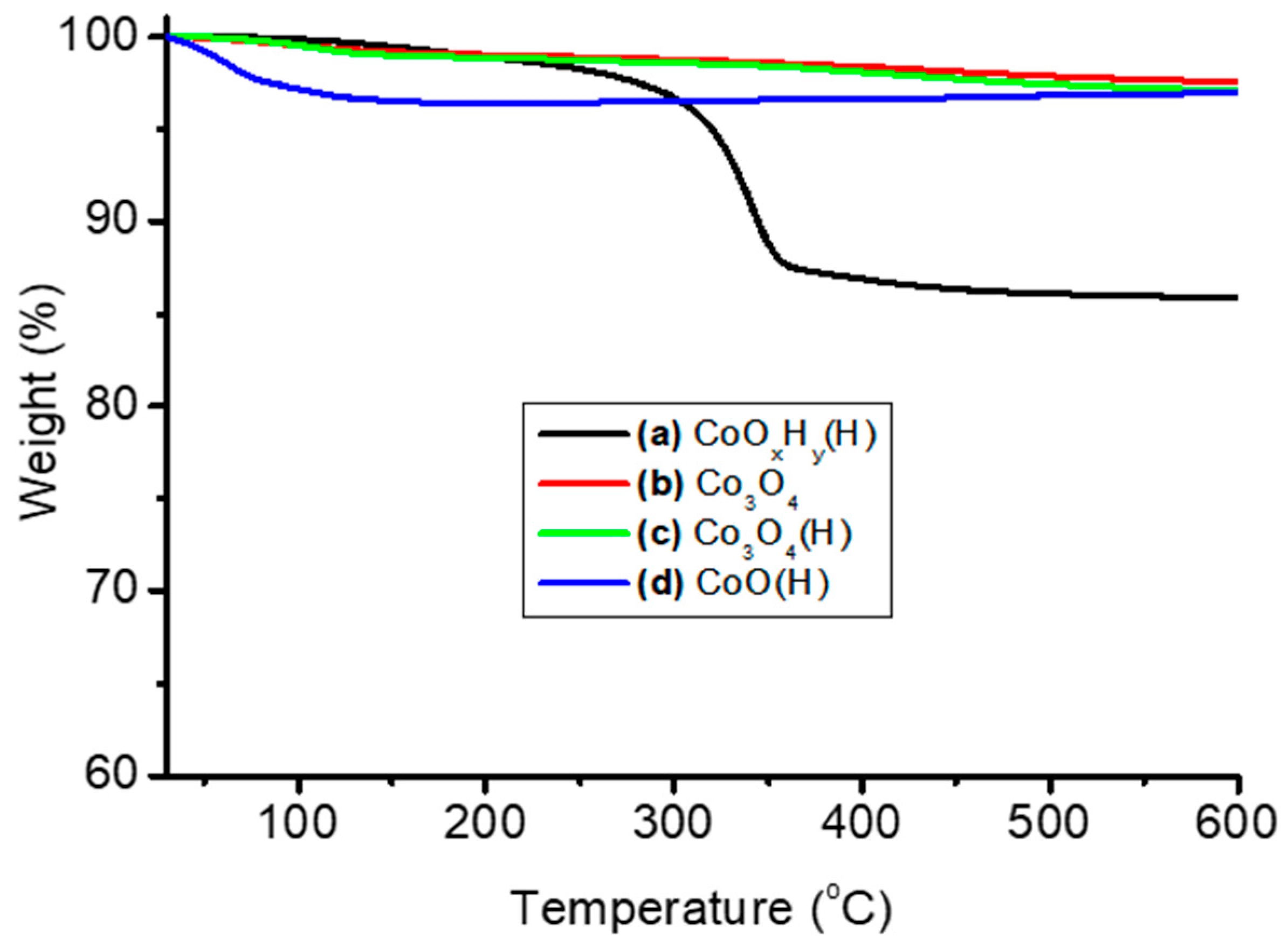
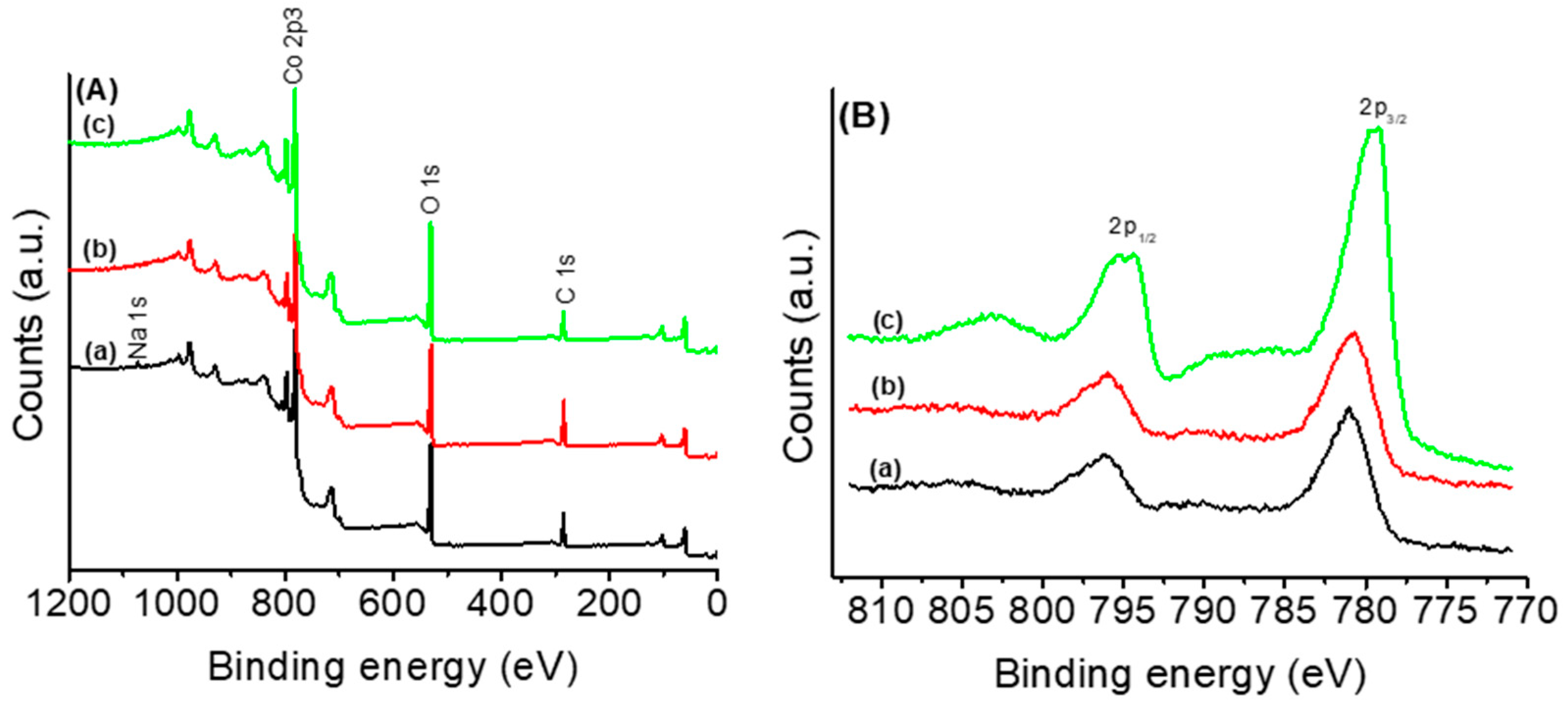
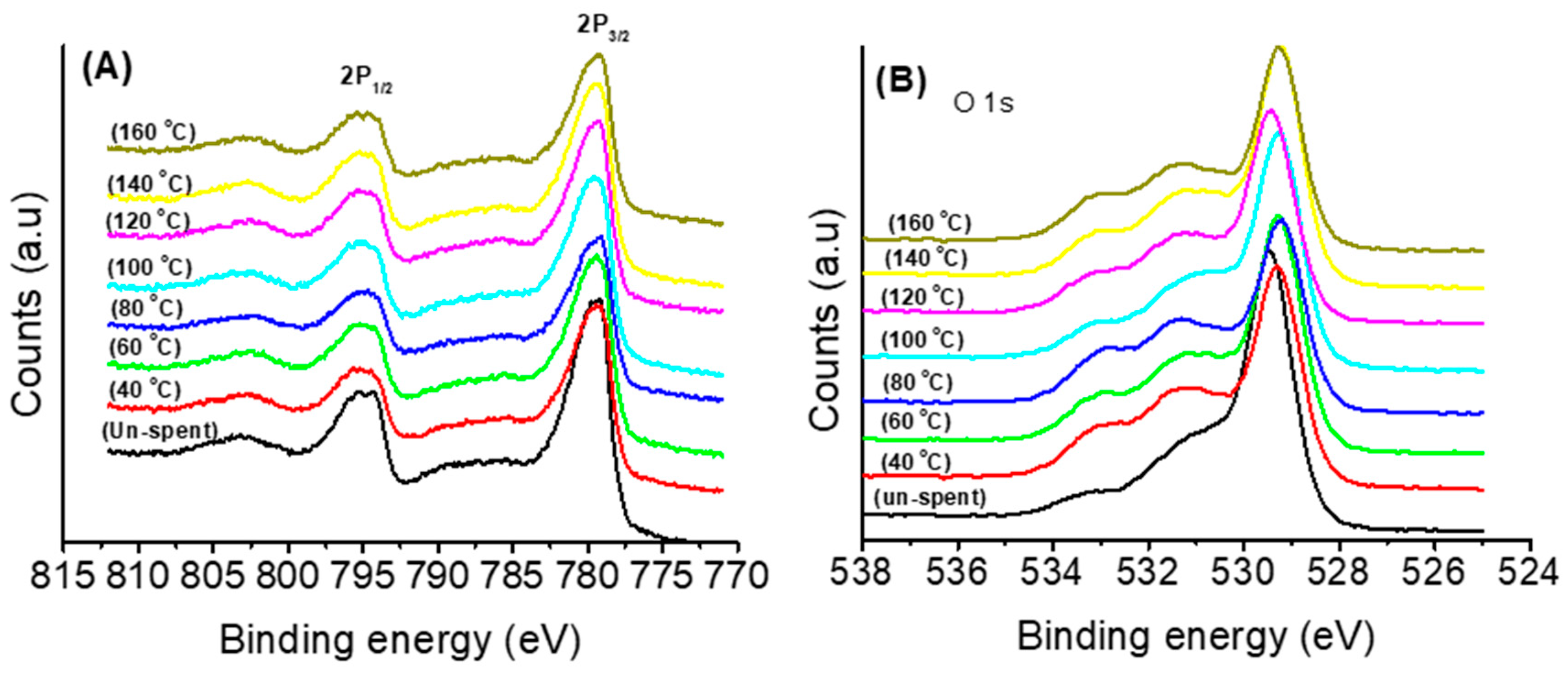
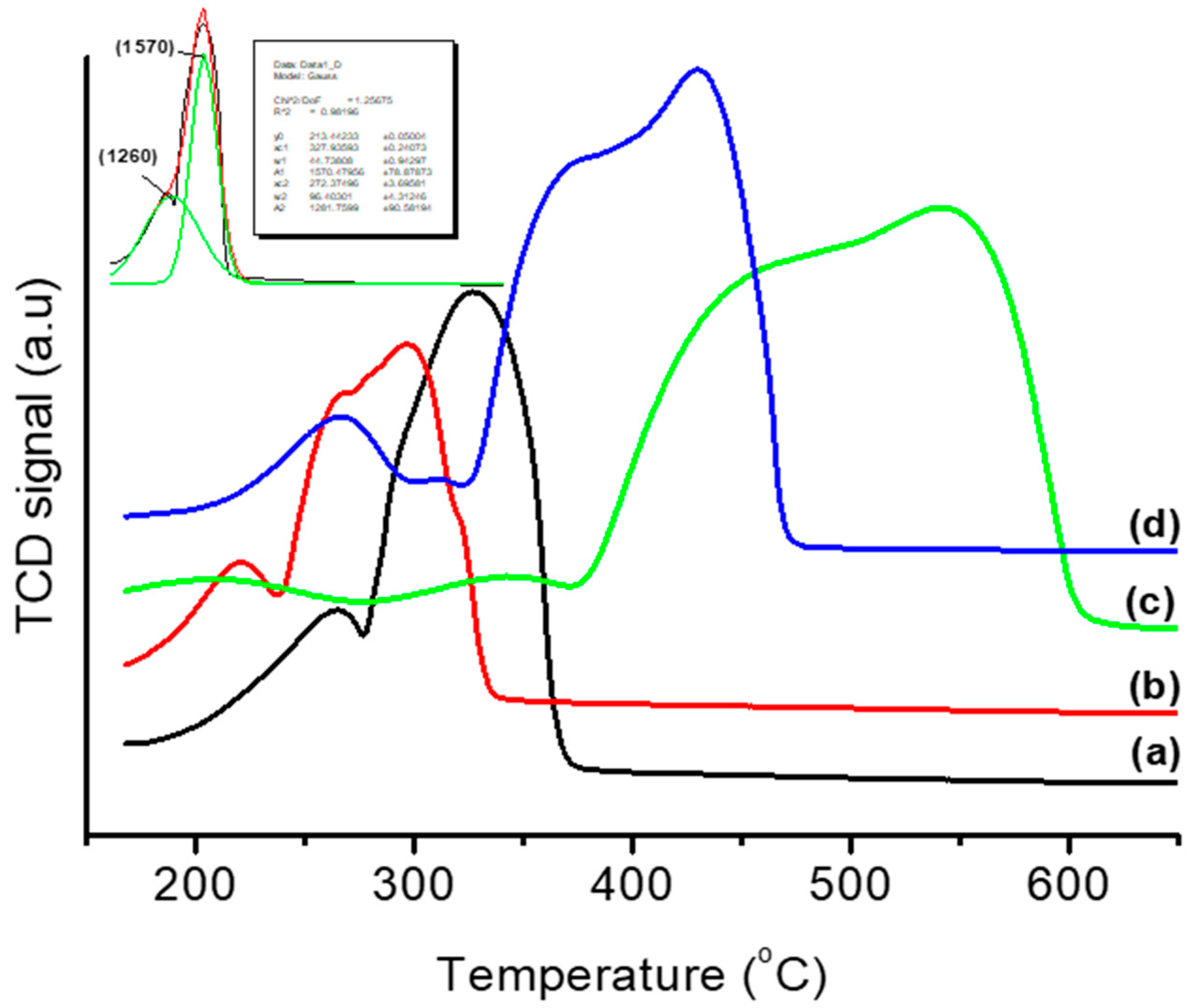
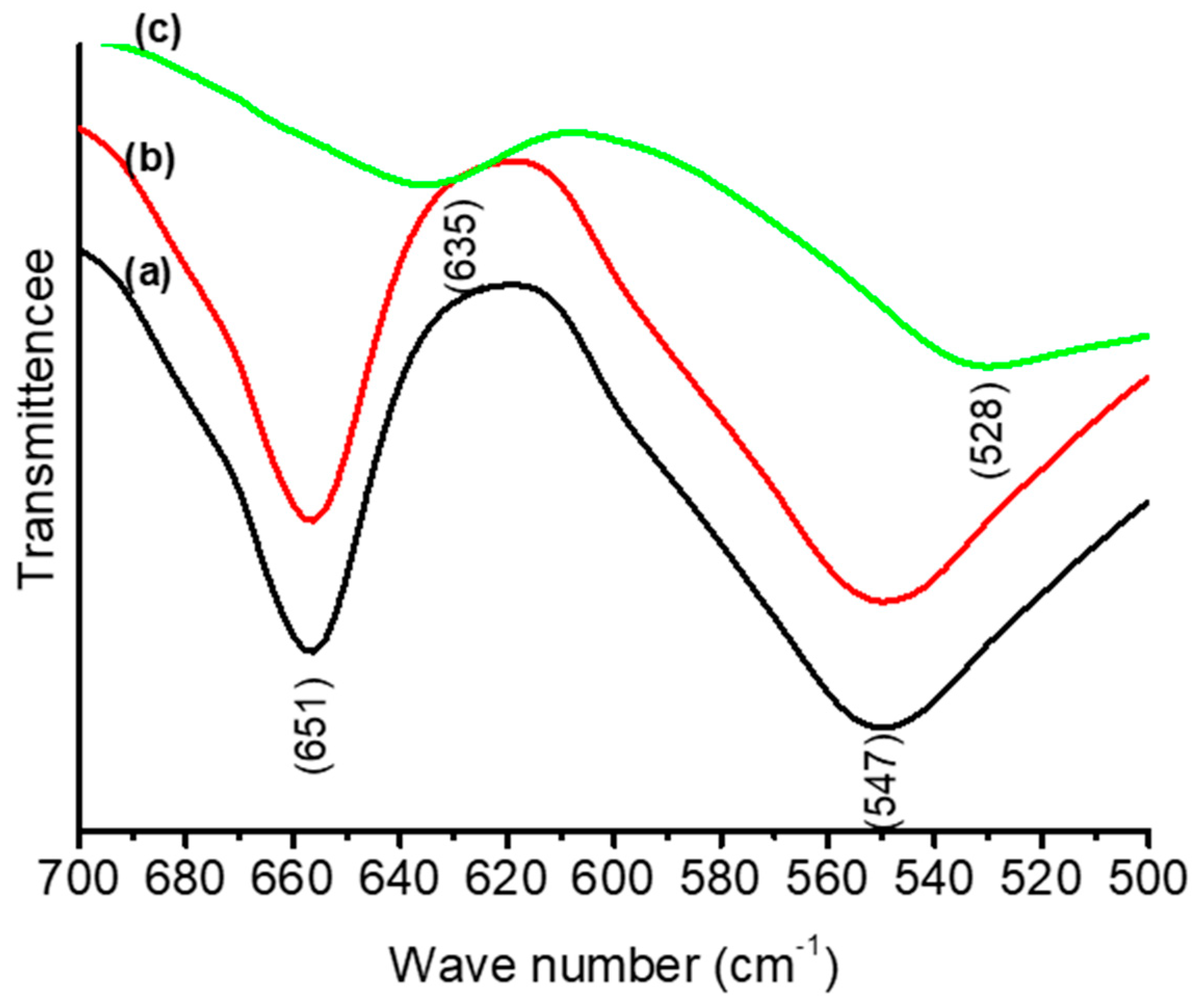
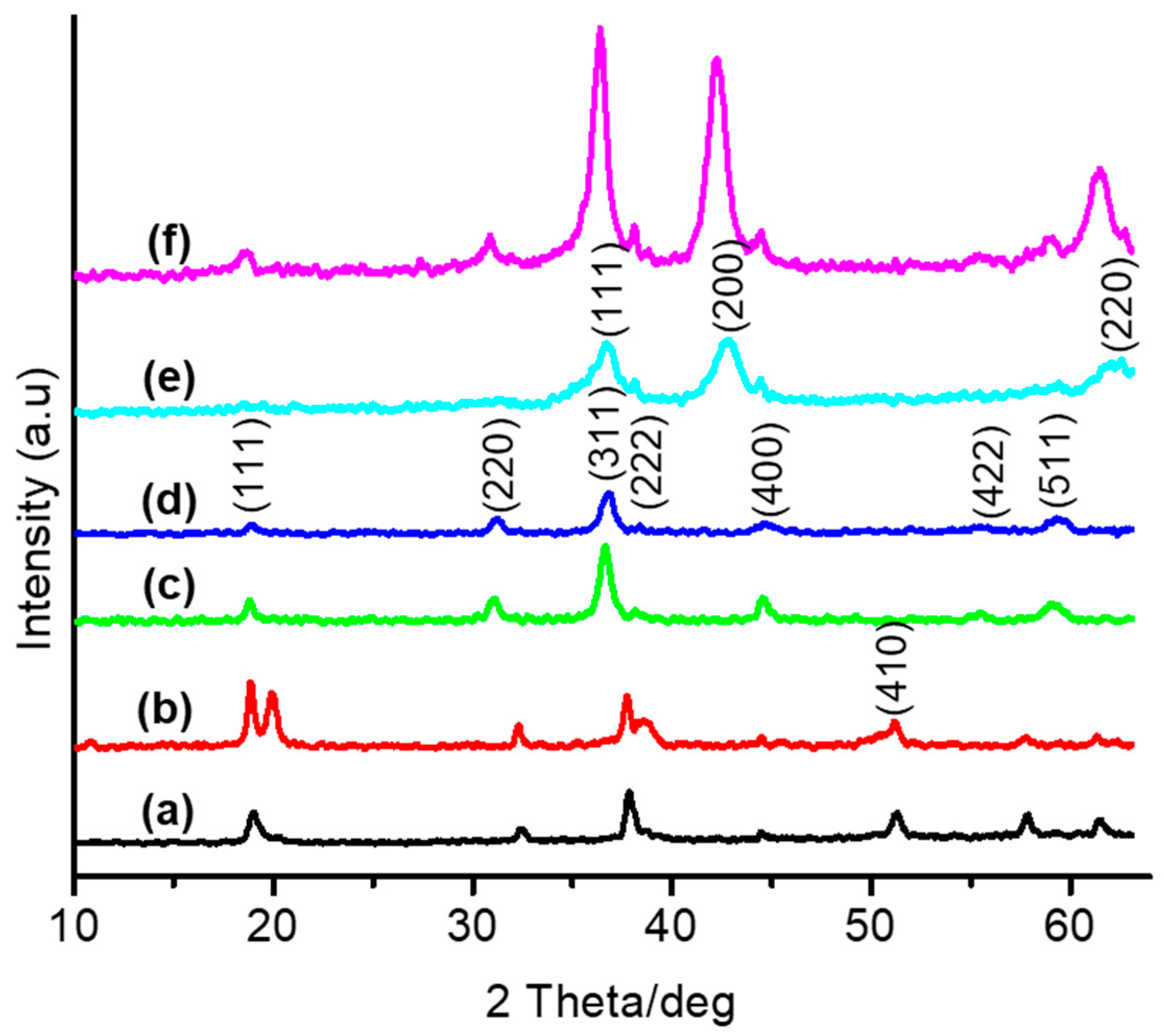
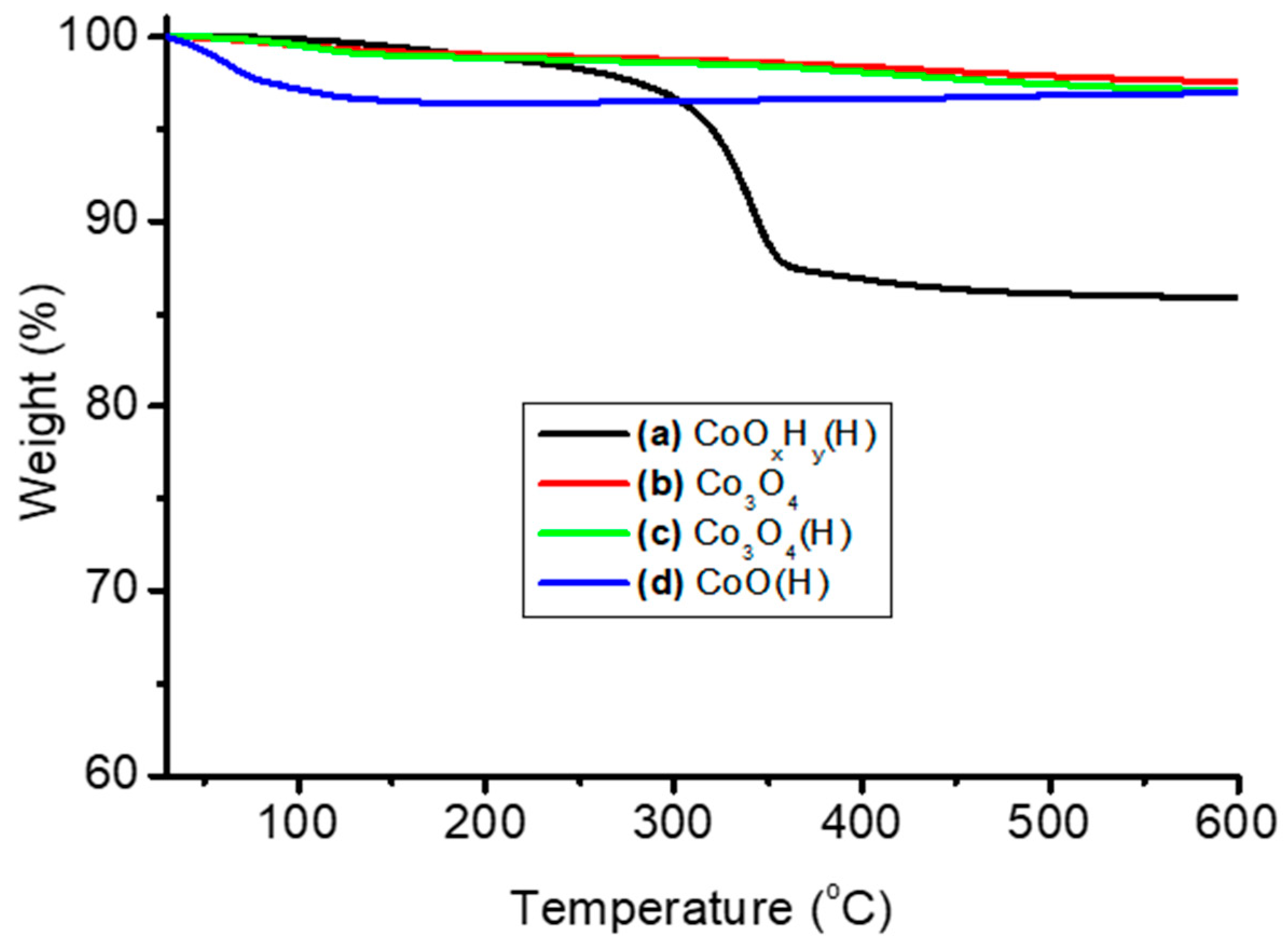
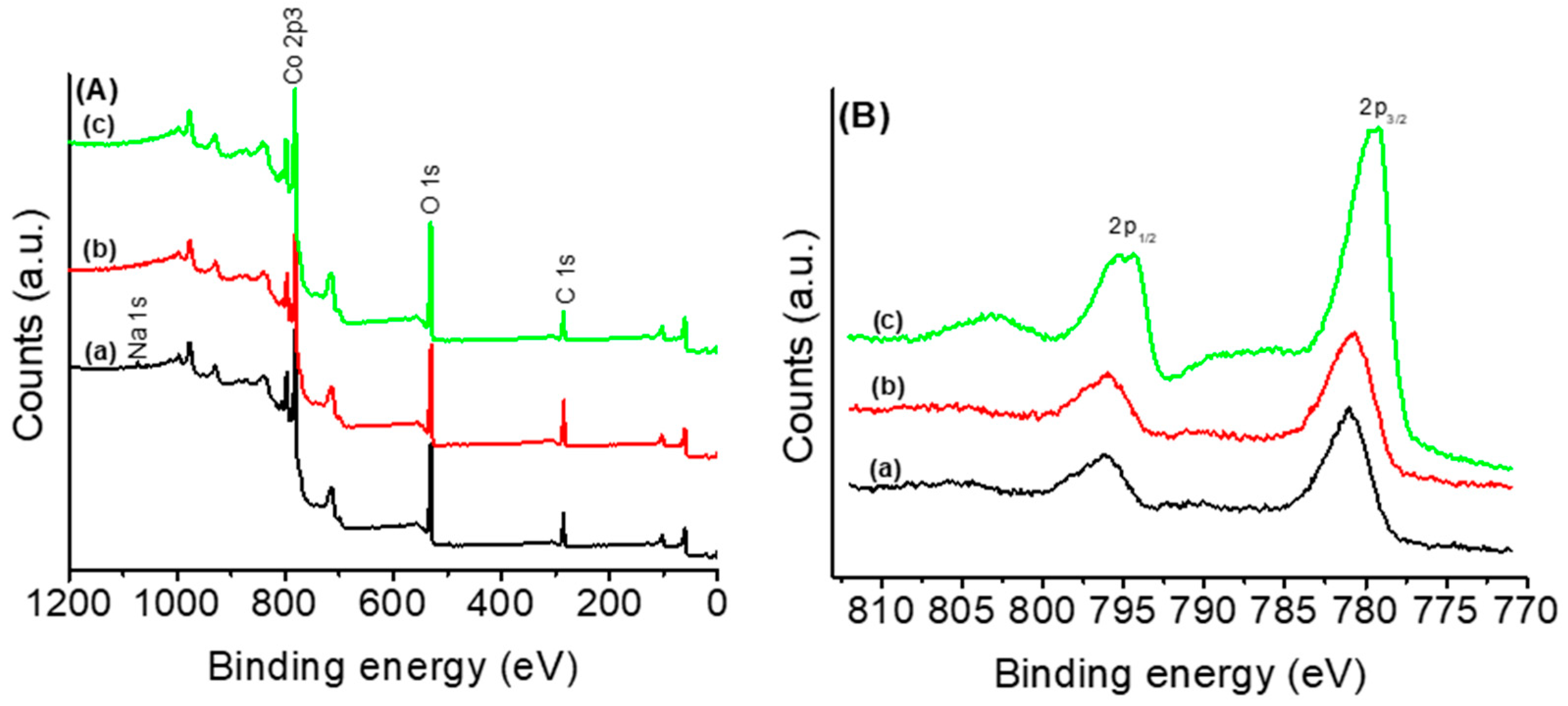
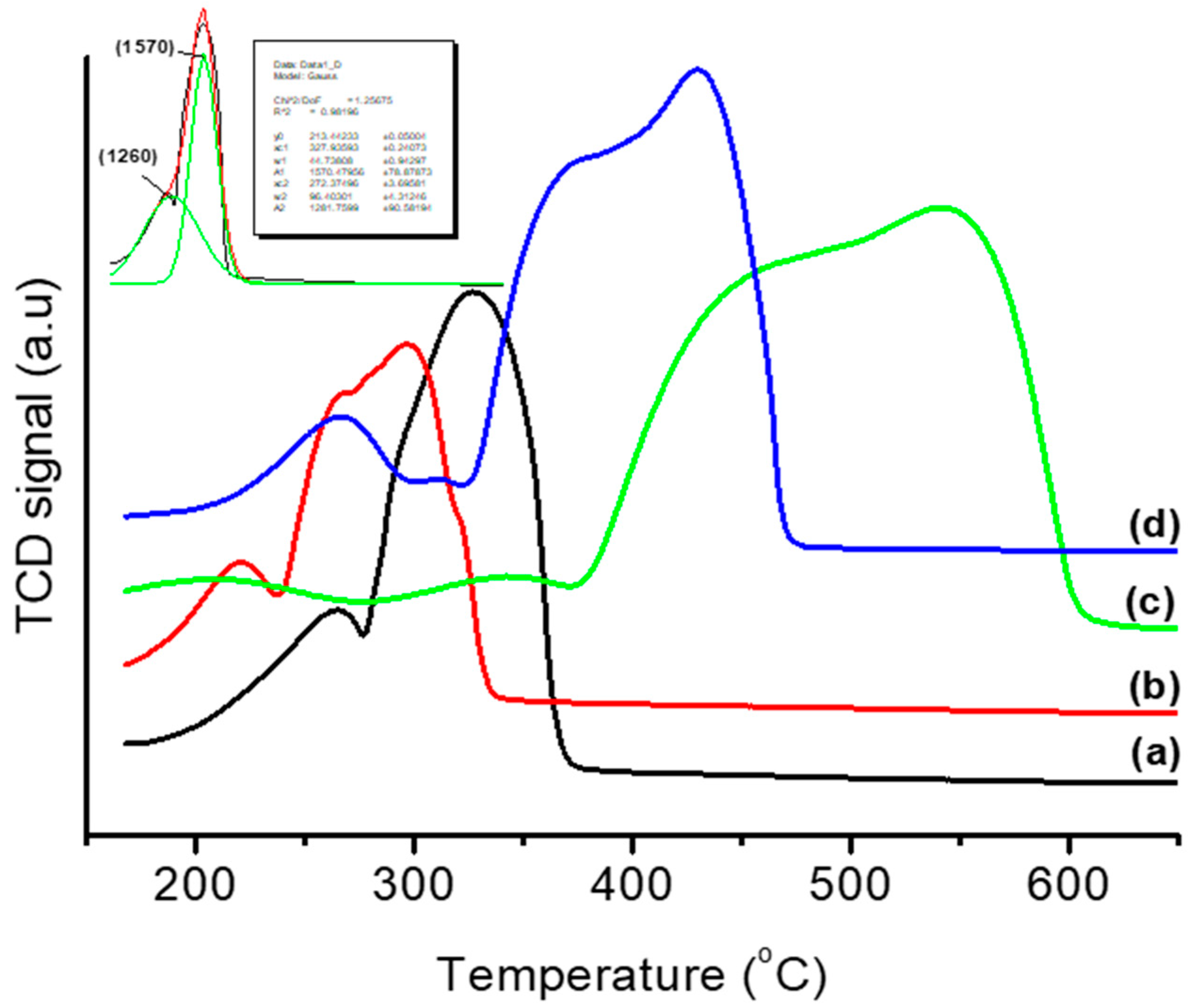
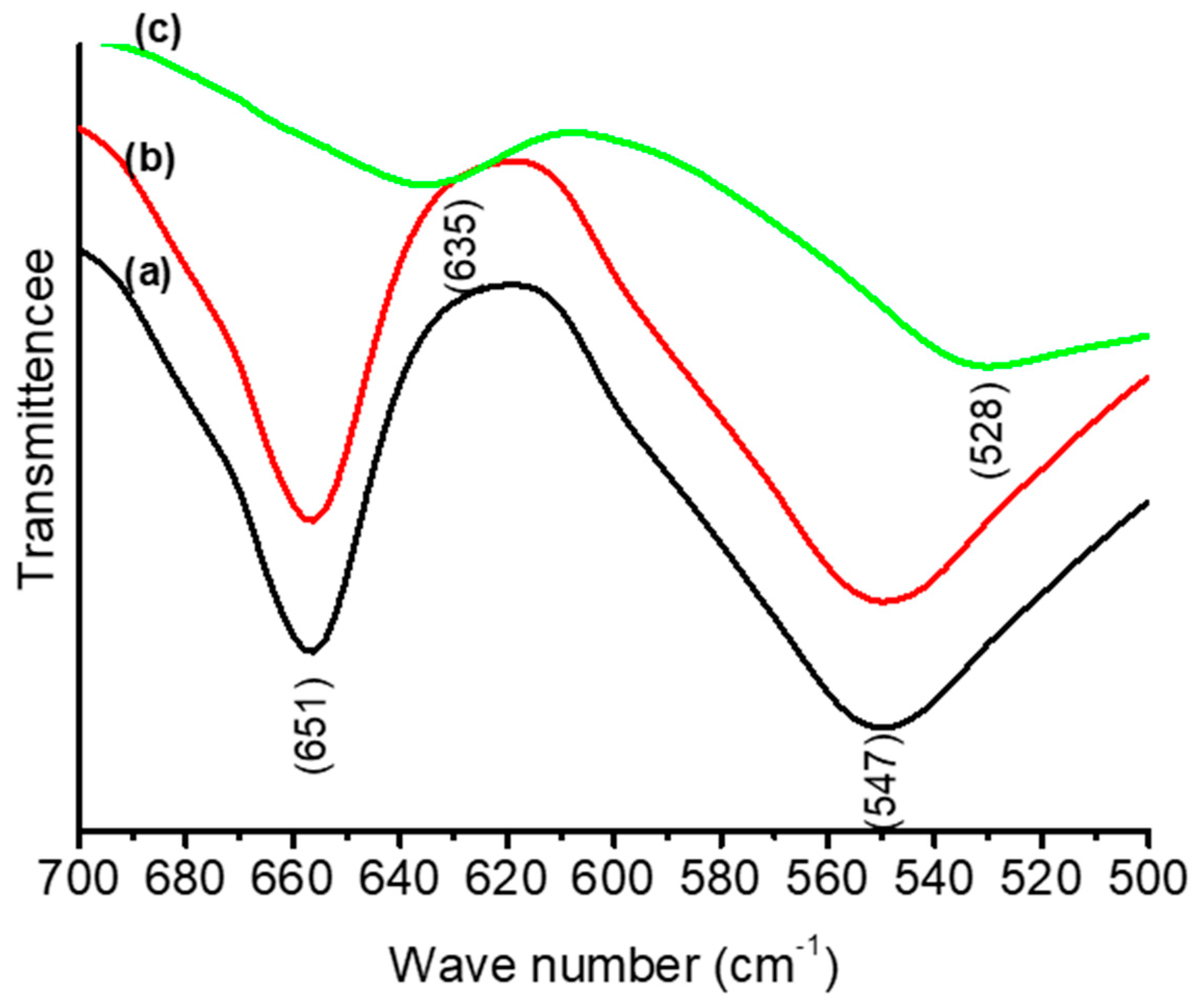
3.1. Thermal and Structural Properties of Catalysts
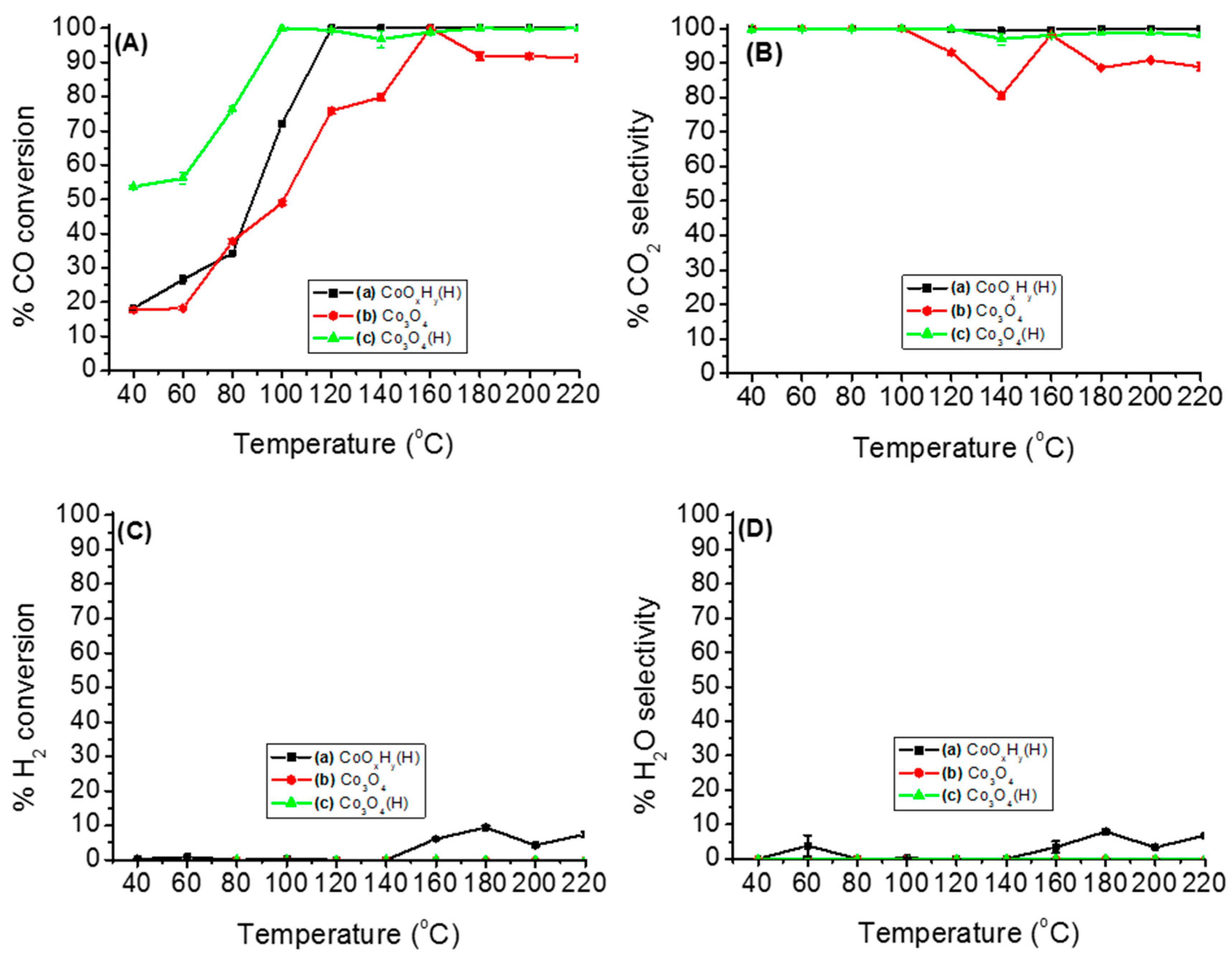
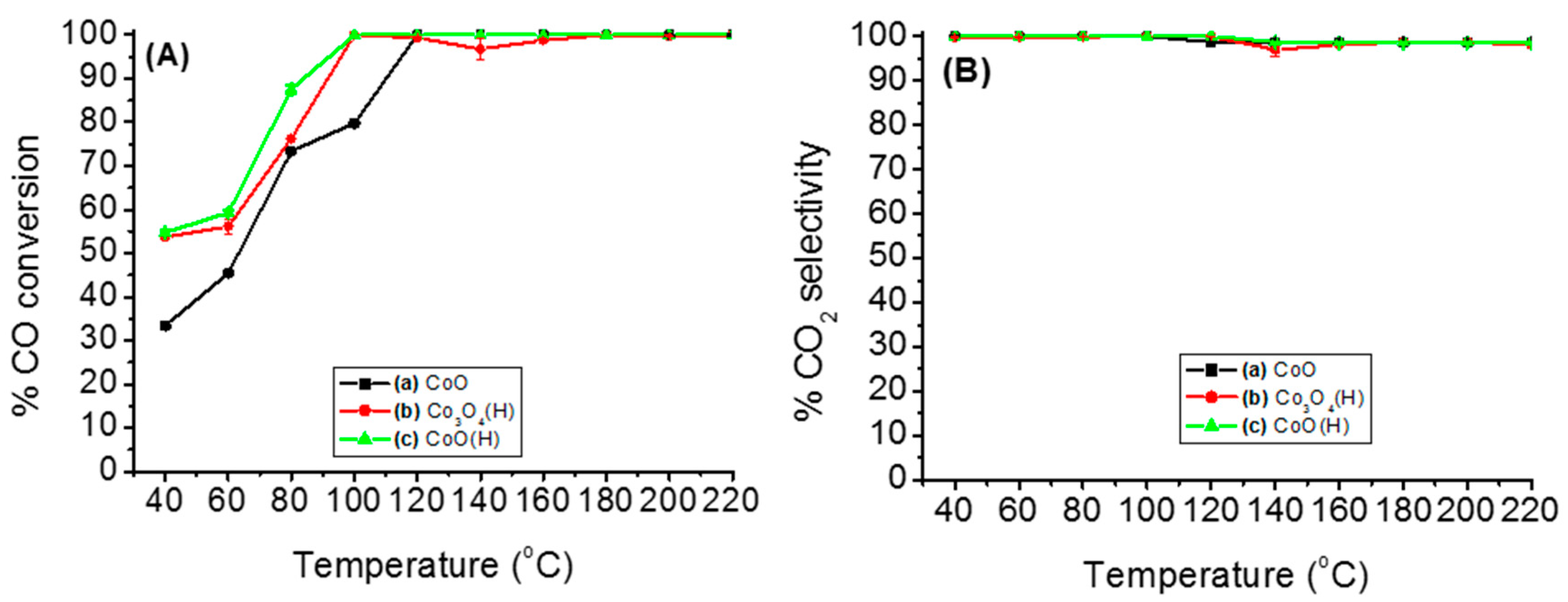
3.2. Catalytic Activity for PROX of CO Reaction
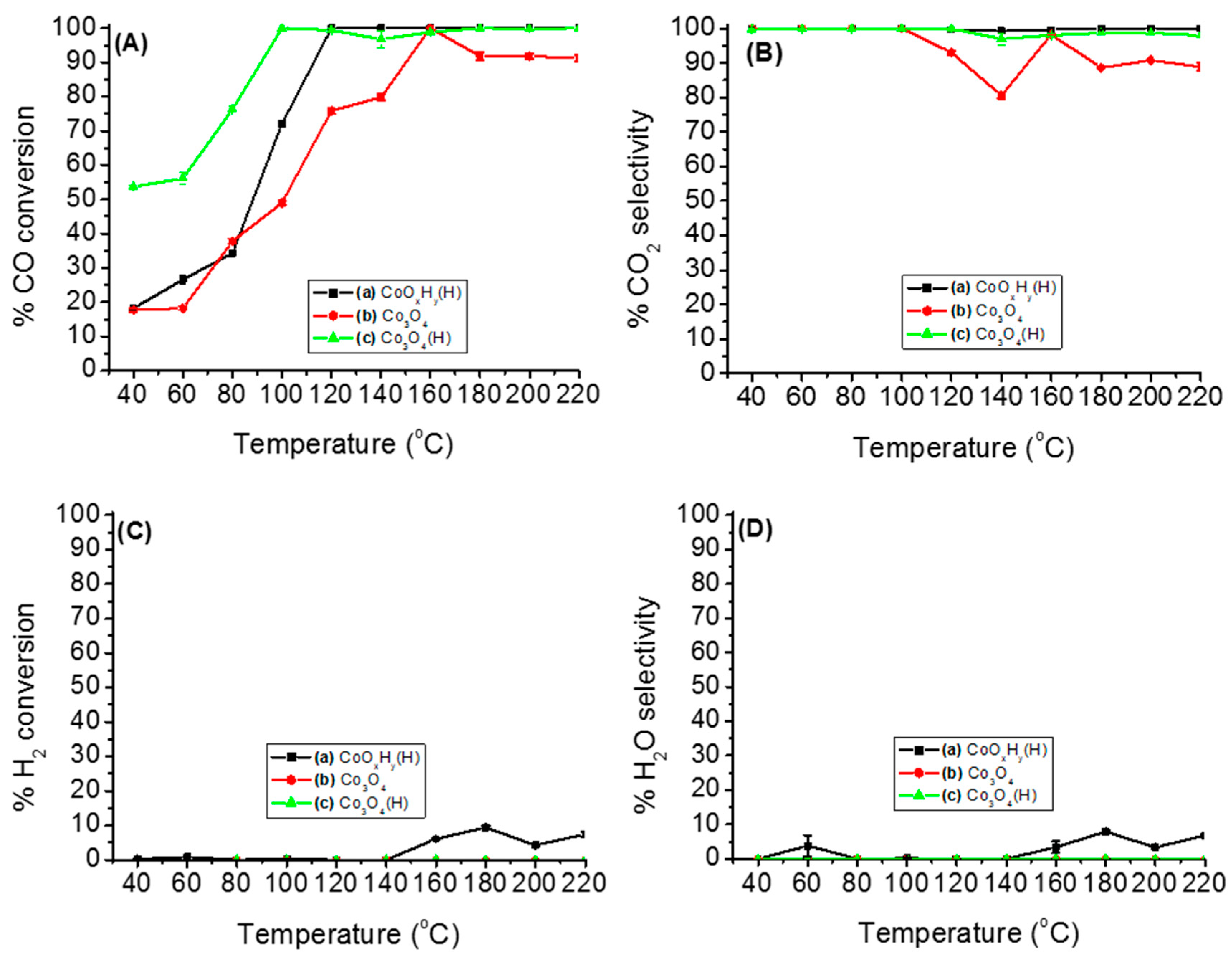
3.2.1. Catalytic Performance of Various Cobalt Species on PROX of CO
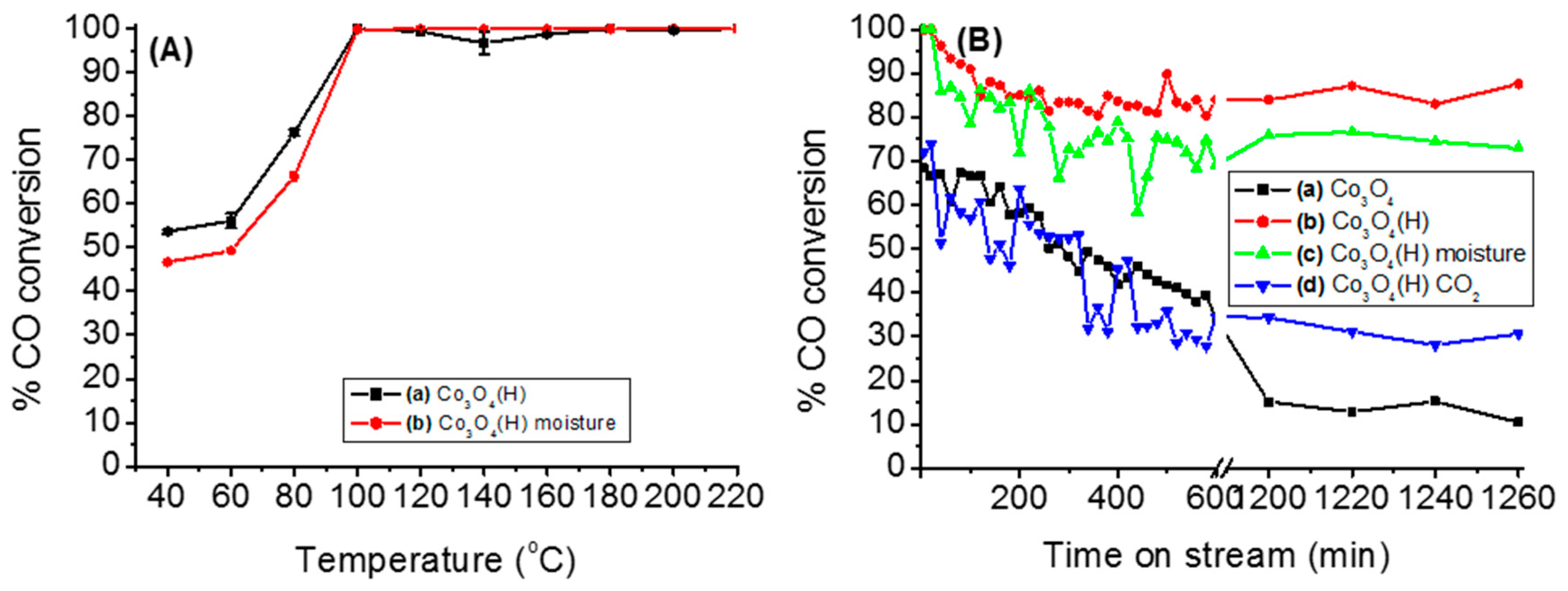
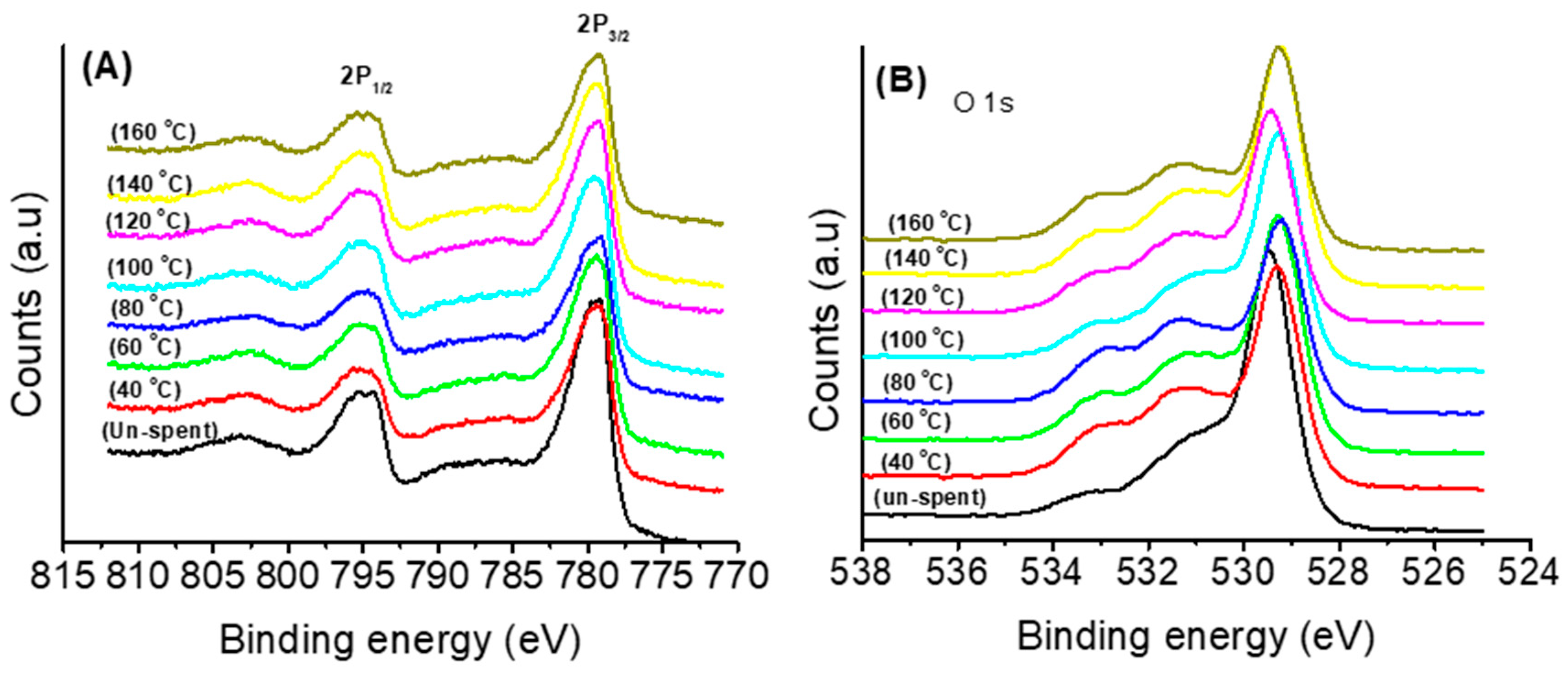
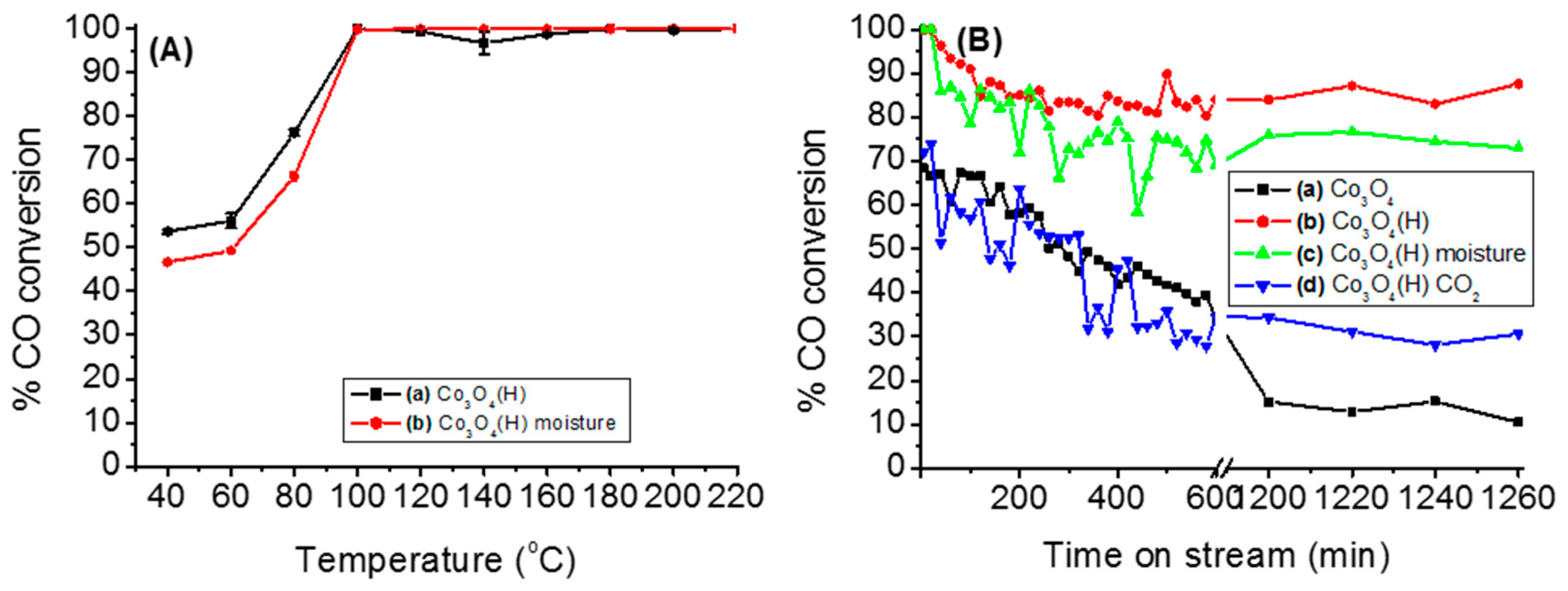
3.2.2. Effects of Temperature, Moisture and CO2 on the Stability of Either Co3O4 or Co3O4(H)
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Abdollah, M.; Yu, J.P.; Liu, K.T.; Ciora, R.; Sahimi, M.; Tsotsis, T.T. Ultra-pure hydrogen production from reformate mixtures using a palladium membrane reactor system. J. Membr. Sci. 2012, 390–391, 32–42. [Google Scholar] [CrossRef]
- Sedmak, G.; Hocevar, S.; Levec, J. Kinetics of selective CO oxidation in excess of H2 over the nanostructured Cu0.1Ce0.9O2−y catalyst. J. Catal. 2003, 213, 135–150. [Google Scholar] [CrossRef]
- Park, J.W.J.; Jeong, H.; Yoon, W.L.C.; Kim, S.D.; Lee, K.; Park, Y.; Rhee, Y. Selective oxidation of CO in hydrogen-rich stream over Cu–Ce catalyst promoted with transition metals. Int. J. Hydrogen Energy 2005, 30, 209–220. [Google Scholar]
- Korotkikh, O.; Farrauto, R. Selective catalytic oxidation of CO in H2: Fuel cell applications. Catal. Today 2000, 62, 249–254. [Google Scholar] [CrossRef]
- Nguyen, T.-S.; Morfin, V.; Aouine, V.; Bosset, V.; Rousset, J.-L.; Piccolo, L. Trends in the CO oxidation and PROX performances of the platinum-group metals supported on ceria. Catal. Today 2015, 253, 106–114. [Google Scholar] [CrossRef]
- Miguel-García, I.; Berenguer-Murcia, Á.; Cazorla-Amorós, D. Preferential oxidation of CO catalyzed by supported polymer-protected palladium-based nanoparticles. Appl. Catal. B Environ. 2010, 98, 161–170. [Google Scholar] [CrossRef]
- Jardim, E.O.; Goncalves, M.; Rico-Francés, S.; Sepúlveda-Escribano, A.; Silvestre-Albero, J. Superior performance of multi-wall carbon nanotubes as support of Pt-based catalysts for the preferential CO oxidation: Effect of ceria addition. Appl. Catal. B Environ. 2012, 113–114, 72–78. [Google Scholar] [CrossRef]
- Rossignol, C.; Arrii, S.; Morfin, F.; Piccolo, L.; Caps, V.; Rousset, J.-L. Selective oxidation of CO over model gold-based catalysts in the presence of H2. J. Catal. 2005, 230, 476–483. [Google Scholar] [CrossRef]
- Reina, T.R.; Megías-Sayago, C.; Florez, A.P.; Ivanova, S.S.; Centeno, S.J.; Odriozola, A. H2 oxidation as criterion for PrOx catalyst selection: Examples based on Au–CoOx-supported systems. J. Catal. 2015, 326, 161–171. [Google Scholar] [CrossRef]
- Nguyen, L.; Zhang, S.; Yoon, S.J.; Tao, F. Preferential Oxidation of CO in H2 on Pure Co3O4-x and Pt/Co3O4-x. ChemCatChem 2015, 7, 2346–2353. [Google Scholar] [CrossRef]
- Pozdnyakoya, O.; Tescher, D.; Wootsch, A.; Krohnert, J.; Steinhauer, B.; Sauer, H.; Toth, L.; Jentoft, F.C.; Knop-Gericke, A.; Paal, Z.; et al. Preferential CO oxidation in hydrogen (PROX) on ceria-supported catalysts, part II: Oxidation states and surface species on Pd/CeO2 under reaction conditions, suggested reaction mechanism. J. Catal. 2006, 237, 17–28. [Google Scholar] [CrossRef]
- Marino, F.; Descorme, F.; Dupre, D. Noble metal catalysts for the preferential oxidation of carbon monoxide in the presence of hydrogen (PROX). Appl. Catal. Environ. 2004, 54, 59–66. [Google Scholar] [CrossRef]
- Zlotea, C.; Morfin, F.; Nguyen, T.S.; Nguyen, N.T.; Nelayah, J.; Ricolleau, C.; Latroche, M.; Piccolo, L. Nanoalloying bulk-immiscible iridium and palladium inhibits hydride formation and promotes catalytic performances. Nanoscale 2014, 6, 9955–9959. [Google Scholar] [CrossRef] [PubMed]
- Marba’n, G.; Fuertes, A.B. Highly active and selective CuOx/CeO2 catalyst prepared by a single-step citrate method for preferential oxidation of carbon monoxide. Appl. Catal. B Environ. 2005, 57, 43–53. [Google Scholar] [CrossRef]
- Gong, X.; Liu, B.; Kang, B.; Xu, G.; Wang, Q.; Jia, C.; Zhang, J. Boosting Cu-Ce interaction in CuxO/CeO2 nanocube catalysts for enhanced catalytic performance of preferential oxidation of CO inH2-rich gases. Mol. Catal. 2017, 436, 90–99. [Google Scholar] [CrossRef]
- Zhu, C.; Ding, T.; Gao, W.; Ma, K.; Tian, Y.; Li, X. CuO/CeO2 catalysts synthesized from Ce-UiO-66 metal-organic framework for preferential CO oxidation. Int. J. Hydrogen Energy 2017, 42, 17457–17465. [Google Scholar] [CrossRef]
- Di Benedetto, A.; Landi, G.; Lisi, L. Improved CO-PROX performance of CuO/CeO2 catalysts by using nanometric ceria as support. Catalysts 2018, 8, 209. [Google Scholar] [CrossRef]
- Gu, D.; Jia, C.-J.; Weidenthaler, C.; Bongard, H.-J.; Spliethoff, B.; Schmidt, W.; Schüth, F. Highly Ordered Mesoporous Cobalt-Containing Oxides: Structure, Catalytic Properties, and Active Sites in Oxidation of Carbon Monoxide. J. Am. Chem. Soc. 2015, 137, 11407–11418. [Google Scholar] [CrossRef]
- Teng, Y.; Skurai, H.; Ueda, A.; Kobayashi, T. Oxidative removal of CO contained in hydrogen by using metal oxide catalysts. Int. J. Hydrogen Energy 1999, 24, 355–358. [Google Scholar] [CrossRef]
- Lukashuk, L.; Föttinger, K.; Kolar, E.; Rameshan, C.; Teschner, D.; Hävecker, M.; Knop-Gericke, A.; Yigit, N.; Li, H.; McDermott, E.; et al. Operando XAS and NAP-XPS studies of preferential CO oxidation on Co3O4 and CeO2-Co3O4 catalysts. J. Catal. 2016, 344, 1–15. [Google Scholar] [CrossRef]
- Broqvist, P.; Panas, I.; Persson, H.A. A DFT Study on CO Oxidation over Co3O4. J. Catal. 2002, 210, 198–206. [Google Scholar] [CrossRef]
- Umegaki, T.; Inoue, T.; Kojima, Y. Fabrication of hollow spheres of Co3O4 for catalytic oxidation of carbon monoxide. J. Alloys Compd. 2016, 663, 68–76. [Google Scholar] [CrossRef]
- Jansson, J.; Palmqvist, A.E.C.; Fridell, E.; Skoglundh, M.; Österlund, L.; Thormählen, P.; Langer, V. On the Catalytic Activity of Co3O4 in Low-Temperature CO Oxidation. J. Catal. 2002, 211, 387–397. [Google Scholar] [CrossRef]
- Arango-Diaz, A.; Cecilia, J.A.; Marrero-Jerez, J.; Nuñez, P.; Jiménez-Jiménez, J.; Rodríguez-Castellón, E. Freeze-dried Co3O4–CeO2 catalysts for the preferential oxidation of CO with the presence of CO2 and H2O in the feed. Ceram. Int. 2016, 42, 7462–7474. [Google Scholar] [CrossRef]
- Boyd, D.; Golunski, S.; Hearne, G.R.; Magadzu, T.; Mallick, K.; Raphulu, M.C.; Venugopal, A.; Scurrell, M.S. Reductive routes to stabilized nano gold and relation to catalysis by supported gold. Appl. Catal. A Gen. 2005, 292, 76–81. [Google Scholar] [CrossRef]
- Lakshmanan, P.; Park, J.E.; Kim, B.; Park, E.D. Preferential oxidation of CO in a hydrogen-rich stream over Au/MOx/Al2O3(M = La, Ce, and Mg) catalysts. Catal. Today 2016, 265, 19–26. [Google Scholar] [CrossRef]
- Li, G.; Li, L.; Yuan, Y.; Shi, J.; Yuan, Y.; Li, Y.; Zhao, W.; Shi, J. Highly efficient mesoporous Pd/CeO2catalyst for low temperature CO oxidation especially under moisture condition. Appl. Catal. B Environ. 2014, 158–159, 341–347. [Google Scholar] [CrossRef]
- Chen, Y.; Liu, D.; Yang, L.; Meng, M.; Zhang, J.; Zheng, L.; Chu, S.; Hu, T. Ternary composite oxide catalysts CuO/Co3O4–CeO2 with wide temperature-window for the preferential oxidation of CO in H2-rich stream. Chem. Eng. J. 2013, 234, 88–98. [Google Scholar] [CrossRef]
- Liu, Y.-C.; Koza, J.A.; Switzer, J.A. Conversion of electrodeposited Co(OH)2 to CoOOH and Co3O4, and comparison of their catalytic activity for the oxygen evolution reaction. Electrochim. Acta 2014, 140, 359–365. [Google Scholar] [CrossRef]
- Farhadi, S.; Pourzare, K.; Sadeghinejad, S. Synthesis and characterization of Co3O4 nanoplates by simple thermolysis of the [Co(NH3)6]2(C2O4)3-4H2O complex. Polyhedron 2014, 67, 104–110. [Google Scholar] [CrossRef]
- Kaviyarasu, K.; Raja, A.; Devarajan, P.A. Structural elucidation and spectral characterizations of Co3O4 nanoflakes. Spectrochim. Acta Part A Mol. Biomol. Spectrosc. 2013, 114, 586–591. [Google Scholar] [CrossRef] [PubMed]
- Sharifi, S.L.; Shakur, H.R.; Mirzaei, A.; Salmani, A.; Hosseini, M.H. Characterization of Cobalt Oxide Co3O4 Nanoparticles Prepared by Various Methods: Effect of Calcination Temperatures on Size, Dimension and Catalytic Decomposition of Hydrogen Peroxide. Int. J. Nanosci. Nanotechnol. 2013, 9, 51–58. [Google Scholar]
- Li, G.; Li, L.; Shi, J.; Yuan, Y.; Li, Y.; Zhao, W.; Shi, J. One-pot pyrolytic synthesis of mesoporous MCo2O4(4.5) (M = Mn, Ni, Fe, Cu) spinels and its high efficient catalytic properties for CO oxidation at low temperature. J. Mol. Catal. A Chem. 2014, 390, 97–104. [Google Scholar] [CrossRef]
- Tang, C.-W.; Hsu, L.-C.; Yu, S.-W.; Wang, C.-B.; Chien, S.-H. In situ FT-IR and TPD-MS study of carbon monoxide oxidation over a CeO2/Co3O4 catalyst. Vib. Spectrosc. 2013, 65, 110–115. [Google Scholar] [CrossRef]
- Ozkaya, T.; Baykal, A.; Toprak, M.S.; Koseoglu, Y.; Durmus, Z. Reflux synthesis of Co3O4 nanoparticles and magnetic characterisation. J. Magn. Magn. Mater. 2009, 321, 2145–2149. [Google Scholar] [CrossRef]
- Jayashree, R.S.; Kamath, V.P. Electrochemical synthesis of a-cobalt hydroxide. J. Chem. 1999, 9, 961–963. [Google Scholar] [CrossRef]
- Barde´, F.; Palacin, M.-R.; Beaudoin, B.; Delahaye-Vidal, A.; Tarascon, J.-M. New Approaches for Synthesizing çIII-CoOOH by Soft Chemistry. Chem. Mater. 2004, 16, 299–306. [Google Scholar] [CrossRef]
- Tang, C.W.; Wang, C.B.; Chien, S.H. Characterization of cobalt oxides studied by FT-IR, Raman, TPR and TG-MS. Thermochim. Acta 2008, 473, 68–73. [Google Scholar] [CrossRef]
- Natile, M.M.; Glisenti, A. CoOx/CeO2 Nanocomposite Powders: Synthesis, Characterization, and Reactivity. Chem. Mater. 2005, 17, 3403–3414. [Google Scholar] [CrossRef]
- Mate, V.R.; Jha, A.; Joshi, U.D.; Patil, K.R.; Shirai, M.; Rode, C.V. Effect of preparation parameters on characterization and activity of Co3O4 catalyst in liquid phase oxidation of lignin model substrates. Appl. Catal. A Gen. 2014, 487, 130–138. [Google Scholar] [CrossRef]
- Zhang, S.; Shan, J.-J.; Zhu, Y.; Frenkel, A.I.; Patlolla, A.; Huang, W.; Yoon, S.J.; Wang, L.; Yoshida, H.; Takeda, S.; et al. WGS Catalysis and in Situ Studies of CoO1−x, PtCon/Co3O4, and PtmCom′/CoO1−x Nanorod Catalysts. J. Am. Chem. Soc. 2013, 135, 8283–8293. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Zhang, S.; Zhu, Y.; Patlolla, A.; Shan, J.; Yoshida, H.; Takeda, S.; Frenkel, A.I.; Tao, F. Catalysis and In Situ Studies of Rh1/Co3O4 Nanorods in Reduction of NO with H2. Acta Catal. 2013, 3, 1011–1019. [Google Scholar] [CrossRef]
- Tang, Y.; Ma, L.; Dou, J.; Andolina, C.M.; Li, Y.; Ma, H.; House, S.D.; Zhan, X.; Yang, J.; Tao, F. Transition of surface phase of cobalt oxide during CO oxidation. Phys. Chem. Chem. Phys. 2018, 20, 6440–6449. [Google Scholar] [CrossRef] [PubMed]
- Chang, T.; Shen, Z.; Huang, Y.; Lu, J.; Ren, D.; Cao, J.; Liu, H. Post-plasma-catalytic removal of toluene using MnO2–Co3O4 catalysts and their synergistic mechanism. Chem. Eng. J. 2018, 348, 15–25. [Google Scholar] [CrossRef]
- Nidhin, M.; Sreeram, K.J.; Nair, B.U. Green synthesis of rock salt CoO nanoparticles for coating applications by complexation and passivation with starch surface. Chem. Eng. J. 2012, 185–186, 352–357. [Google Scholar] [CrossRef]
- de la Pena O’Shea, V.A.; Homs, N.; Pereira, E.B.; Nafria, R.; de la Piscina, P.R. X-ray diffraction study of Co3O4 activation under ethanol steam-reforming. Catal. Today 2007, 126, 148–152. [Google Scholar] [CrossRef]
- Xu, Z.P.; Zeng, H.C. Interconversion of Brucite-like and Hydrotalcite-like Phases in Cobalt Hydroxide Compounds. Chem. Mater. 1999, 11, 67–74. [Google Scholar] [CrossRef]
- Chen, H.; Yang, M.; Tao, S.; Chen, G. Oxygen vacancy enhanced catalytic activity of reduced Co3O4 toward sp-nitrophenol reduction. Appl. Catal. B Environ. 2017, 209, 648–656. [Google Scholar] [CrossRef]
- Yang, J.; Liu, H.; Martens, V.; Frost, R.L. Synthesis and characterization of Cobalt Hydroxide, Cobalt Oxyhydroxide, and Cobalt Oxide Nanodiscs. J. Phys. Chem. C 2010, 114, 111–119. [Google Scholar] [CrossRef]
- Yang, J.; Quaresma, S.; Mei, S.; Ferreira, J.M.F. Hydrothermal synthesis of free-standing Co3O4 nanocubes. Key Eng. Mater. 2005, 280–283, 713–716. [Google Scholar]
- Mahammadunnisa, S.K.; Akanksha, T.; Krushnamurty, K.; Subrahmanyam, C.H. Catalytic decomposition of N2O over CeO2 supported Co3O4 catalysts. J. Chem. Sci. 2016, 128, 1795–1804. [Google Scholar] [CrossRef]
- Jia, C.-J.; Schwickardi, M.; Weidenthaler, C.; Schmidt, W.; Korhonen, S.; Weckhuysen, B.M.; Schüth, F. Co3O4-SiO2 Nanocomposite: A Very Active Catalyst for CO Oxidation with Unusual Catalytic Behavior. J. Am. Chem. Soc. 2011, 133, 11279–11288. [Google Scholar] [CrossRef] [PubMed]
- Liotta, L.F.; Di Carlo, G.; Pantaleo, G.; Venezia, A.M.; Deganello, G. Co3O4/CeO2 composite oxides for methane emissions abatement: Relationship between Co3O4–CeO2 interaction and catalytic activity. Appl. Catal. B Environ. 2006, 66, 217–227. [Google Scholar] [CrossRef]
- Li, J.-B.; Jiang, Z.-Q.; Qianb, K.; Huang, W.-X. Effect of calcination temperature on surface oxygen vacancies and catalytic performance towards CO oxidation of Co3O4 nanoparticles supported on SiO2. Chin. J. Chem. Phys. 2012, 25, 103–109. [Google Scholar] [CrossRef]
- Mei, J.; Ke, Y.; Yu, Z.; Hu, X.; Qu, Z.; Yan, N. Morphology-dependent properties of Co3O4/CeO2 catalysts for low temperature dibromomethane (CH2Br2) oxidation. Chem. Eng. J. 2017, 320, 124–134. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Catalyst | SBET (m2/g) | Pore Volume (cm3·g−1) | Pore Size (nm) | Average Crystallite Size (nm) a | Tmax (°C) b | Maximum CO Conversion (%) b |
|---|---|---|---|---|---|---|
| Co3O4(H) | 62.2 | 0.158 | 9.95 | 20.0 | 120 | 98.1 |
| CoO(H) | 56.6 | 0.184 | 13.0 | 11.4 | 140 | 98.5 |
| Co3O4 | 49.7 | 0.137 | 11.3 | 23.2 | 160 | 96.7 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mhlaba, R.; Mosuang, T.; Magadzu, T. Effect of Hydrazine Pretreatment on the Activity, Stability and Active Sites of Cobalt Species for Preferential Oxidation (PROX) of CO in H2-Rich Stream. Chemistry 2019, 1, 164-179. https://doi.org/10.3390/chemistry1010011
Mhlaba R, Mosuang T, Magadzu T. Effect of Hydrazine Pretreatment on the Activity, Stability and Active Sites of Cobalt Species for Preferential Oxidation (PROX) of CO in H2-Rich Stream. Chemistry. 2019; 1(1):164-179. https://doi.org/10.3390/chemistry1010011
Chicago/Turabian StyleMhlaba, Reineck, Thuto Mosuang, and Takalani Magadzu. 2019. "Effect of Hydrazine Pretreatment on the Activity, Stability and Active Sites of Cobalt Species for Preferential Oxidation (PROX) of CO in H2-Rich Stream" Chemistry 1, no. 1: 164-179. https://doi.org/10.3390/chemistry1010011
APA StyleMhlaba, R., Mosuang, T., & Magadzu, T. (2019). Effect of Hydrazine Pretreatment on the Activity, Stability and Active Sites of Cobalt Species for Preferential Oxidation (PROX) of CO in H2-Rich Stream. Chemistry, 1(1), 164-179. https://doi.org/10.3390/chemistry1010011