What’s in a Name?—A Short History of Coordination Chemistry from Then to Now


Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
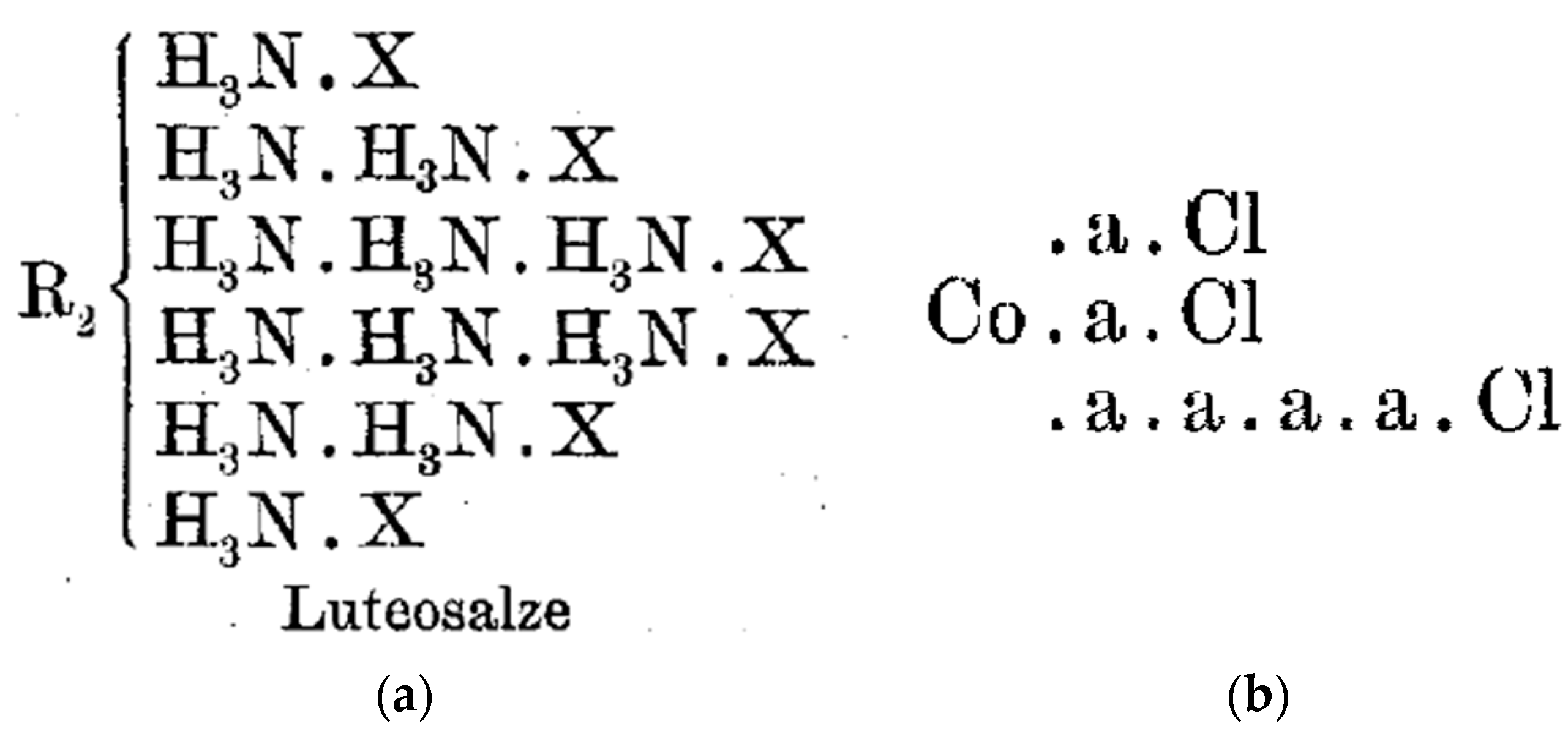{kind=link}
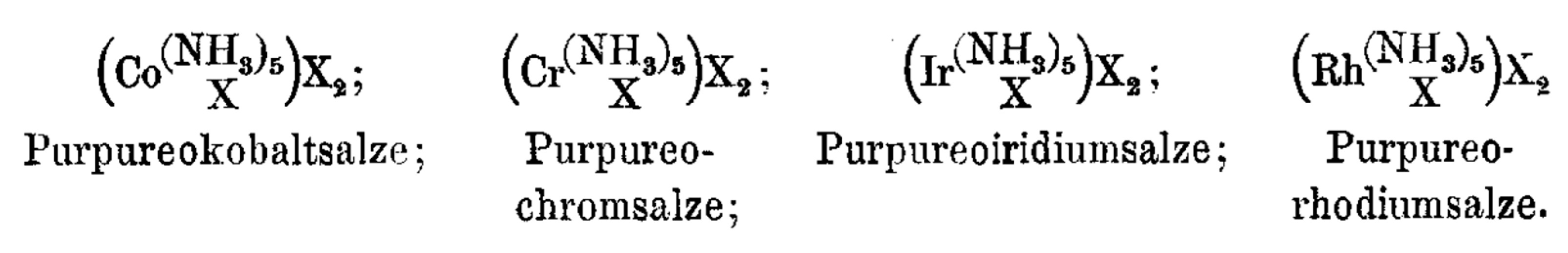{kind=link}
{kind=link}
1. Introduction
2. Early History of Coordination Chemistry
2.1. From Prehistory to History
2.2. Location, Location, Location
2.3. A Neglected Study of Platinum Chemistry
3. The Modern Era
3.1. A New Millennium and New Visions—The Start of the 19th Century of the Common Era
3.2. Some First Approaches to Systematics
Principle 1. That every substance should be denominated by a name, and not by a phrase;Principle 2. That the names should be given according to the nature of the things intended to be signified by them;Principle 3. That when the character of the substance is not sufficiently well known to determine the denomination, a name which has no meaning should be preferred to one which might give an erroneous idea;Principle 4. In the choice of new denominations, those which have their root in the most generally known dead languages should be preferred, in order that the word may be suggested by the sense, and the sense by the word;Principle 5. The denominations should be arranged with care, to suit the genius of the language for which they are proposed.
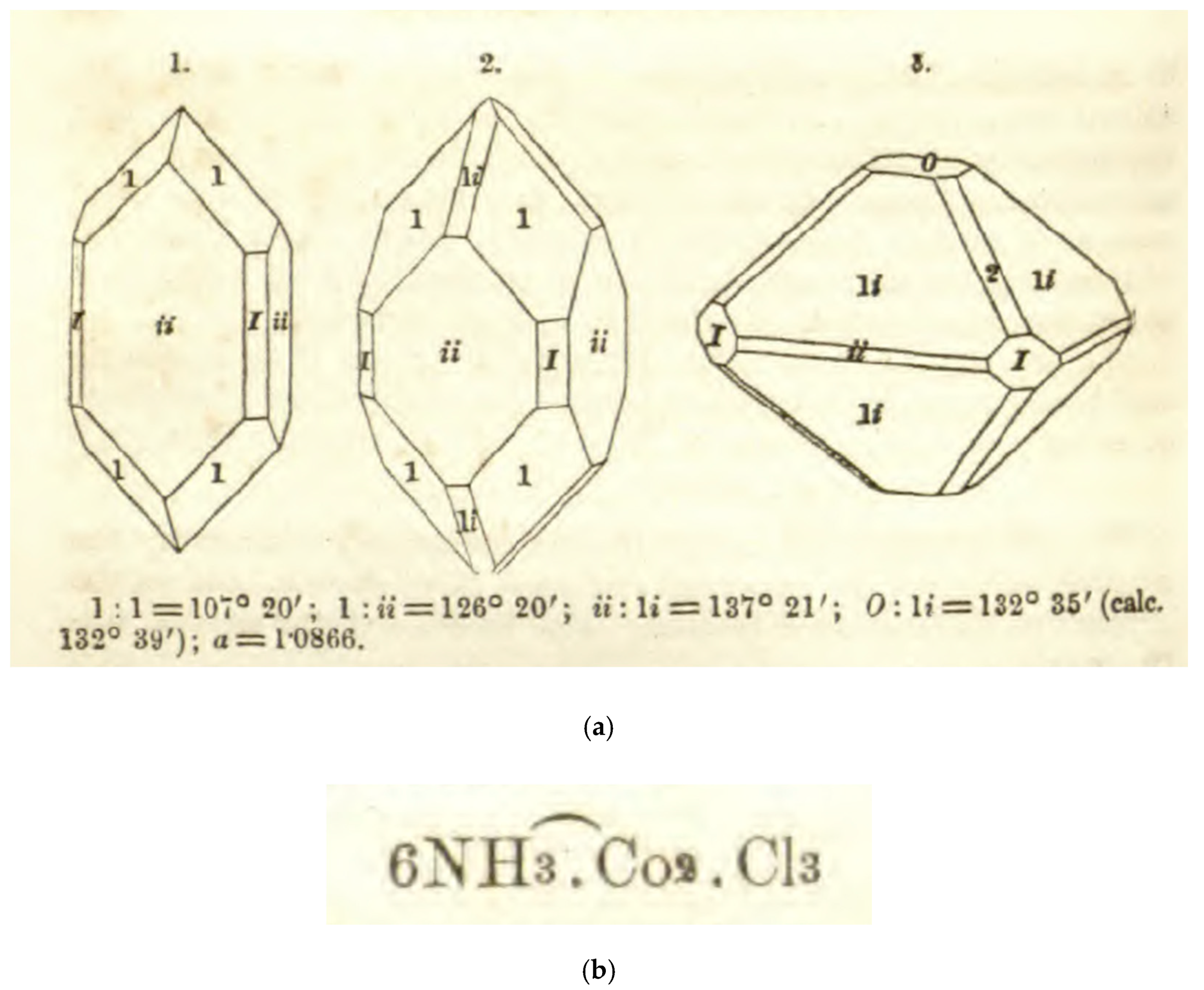
3.3. On the Cusp—Citizen Tassaert and the Beginnings of Cobalt Complex Chemistry
3.4. The Cult of Personality
3.5. When Colour Was King
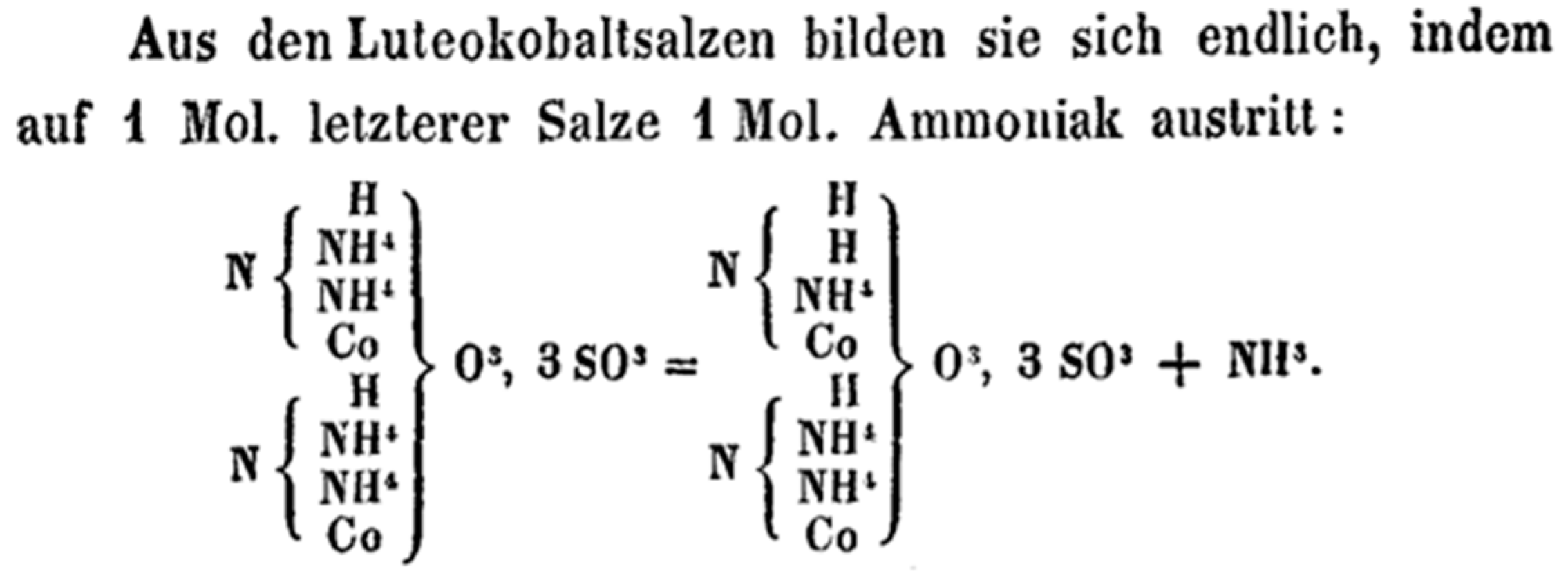
3.6. The Problem of Proportional/Equivalent Weights
4. The Troubled Transition into the Modern Era
4.1. Valence—A Surprisingly Complex Concept
4.2. Variable Valence and the Werner—Jørgensen Controversy
4.3. Werner—Not Just One, But Now Primary and Secondary Valence
4.4. Werner on Nomenclature
- 1.
- Names obtained from the colour of the compounds are to be avoided.
- 2.
- The name of the resulting complex is to be made up by placing side by side the names of the components.
- 3.
- The names of the atoms (or radicles) which are linked to the central metallic atom are to be placed before the name of this central atom. When carrying this out the following order is to be preserved: The names of the acid residues come first, then follow the names of the groups which resemble ammonia, and directly before the name of the metallic atom are to be placed the number of ammonia molecules.
- 4.
- The molecule of ammonia is to be expressed by the word ammine (spelt with a double m), in order to distinguish it from the organic amine. Water, after Palmaer’s suggestion is expressed by aquo.
- 5.
- The names of the acid residues which are not in the first binding zone are placed after the name of the central metallic atom.
| a- | (+1) | o- | (+2) | i- | (+3) | e- | (+4) |
| an- | (+5) | on- | (+6) | in- | (+7) | en- | (+8) |
5. Regulation and Regulations
5.1. Karlsruhe and the First Communion of Chemists
5.2. Paris and Geneva—Rules for Organic Chemistry
5.3. Neologisms and New Terminology
5.4. Preliminary Activities on Inorganic Nomenclature
5.5. Rules for Naming Inorganic Compounds 1940
5.6. The 1951 ACS Symposium
5.7. The 1953 Proposals
5.8. Nomenclature of Inorganic Chemistry. 1957 Rules
5.9. Nomenclature of Inorganic Chemistry. 1970 Rules
5.10. Nomenclature of Inorganic Chemistry. Recommendations 1990
5.11. Nomenclature of Inorganic Chemistry II. Recommendations 2000
5.12. Nomenclature of Inorganic Chemistry. IUPAC Recommendations 2005
5.13. The “Brief Guide”, 2015–2017
5.14. Quo Vadis?
6. Final Thoughts on “the Naming of Complexes”—With Apologies to T.S. Eliot
6.1. Why Bother Naming Things?
6.2. The IUPAC International Chemical Identifier (InChI)
Acknowledgments
Conflicts of Interest
References
- Graham, L. The power of names: in culture and in mathematics. Proc. Am. Philos. Soc. 2013, 157, 229–234. Available online: https://www.jstor.org/stable/24640241 (accessed on 4 August 2019).
- Graham, L. The power of names. Theol. Sci. 2011, 9, 157–164. [Google Scholar] [CrossRef]
- West, A. The rules for longs. TUGboat 2011, 32, 47–55. Available online: https://www.tug.org/TUGboat/tb32-1/tb100west.pdf (accessed on 4 August 2019).
- CAS Source Index (CASSI) Search Tool. Available online: https://cassi.cas.org/search.jsp (accessed on 14 July 2019).
- Kauffman, G.B. General historical survey to 1930. In Comprehensive Coordination Chemistry; Wilkinson, G., Gillard, R.D., McCleverty, J.A., Eds.; Pergamon Elsevier: Oxford, UK, 1987; Volume 1, pp. 1–20. [Google Scholar]
- Kauffman, G.B. Coordination chemistry: history. In Encyclopedia of Inorganic Chemistry; Scott, R.A., Ed.; Wiley: New York, NY, USA, 2006. [Google Scholar]
- Orna, M.V. The Chemical History of Color; Springer: Heidelberg, Germany, 2013. [Google Scholar]
- Singh, H.B.; Bharati, K.A. (Eds.) Mordants and their applications. In Handbook of Natural Dyes and Pigments; Woodhead Publishing India: New Delhi, India, 2014; pp. 18–28. [Google Scholar]
- Abel, A. The history of dyes and pigments: From natural dyes to high performance pigments. In Colour Design; Best, J., Ed.; Woodhead Publishing Elsevier: Duxford, UK, 2012; pp. 557–587. [Google Scholar]
- Ferreira, E.S.B.; Hulme, A.N.; McNab, H.; Quye, A. The natural constituents of historical textile dyes. Chem. Soc. Rev. 2004, 33, 329–399. [Google Scholar] [CrossRef] [PubMed]
- Bucklow, S. Red: The Art and Science of a Colour; Reaktion Books: London, UK, 2016. [Google Scholar]
- Libavius, A.D.O.M.A. Commentationum Metallicarum Libri Quatuor de Natura Metallorum, Mercurio Philosophorum, Azotho, et Lapide seu Tinctura Physicorum Conficienda è RERUM Natura, Experientia, et Autorum Præstantium fide Studio & labore Andreæ Libavii M.D.P. et Physici Rotemburgici Depromti & Expositi, more Veteris Philosophiæ cum Perspicuitate Euidente. In Treatise vi, Tractatus de iudicio aquarum mineralium in tres libros distributus, Part II; Frankfurt, Germany, 1597. [Google Scholar]
- Buser, H.J.; Schwarzenbach, D.; Fetter, W.; Ludi, A. The Crystal Structure of Prussian Blue: Fe4[Fe(CN)6]3.xH2O. Inorg. Chem. 1977, 16, 2704–2710. [Google Scholar] [CrossRef]
- Coleby, L.J.M. A history of Prussian blue. Ann. Sci. 1939, 4, 206–211. [Google Scholar] [CrossRef]
- Kraft, A. On the discovery and history of Prussian blue. Bull. Hist. Chem. 2008, 33, 61–67. Available online: http://acshist.scs.illinois.edu/bulletin_open_access/v33-2/v33-2%20p61-67.pdf (accessed on 4 August 2019).
- Kraft, A. “Notitia Cœrulei Berolinensis Nuper Inventi” on the 300th anniversary of the first publication on Prussian blue. Bull. Hist. Chem. 2011, 36, 3–9. Available online: http://acshist.scs.illinois.edu/bulletin_open_access/v33-2/v33-2%20p61-67.pdf (accessed on 4 August 2019).
- Woodward, J. Praeparatio caerulei Prussiaci ex Germaniâ. Philos. Trans. 1724, 33, 15–17. [Google Scholar] [CrossRef]
- Brown, J. Observations and experiments upon the foregoing preparation. Philos. Trans. 1724, 33, 17–24. [Google Scholar] [CrossRef]
- Sur le Bleue de Prusse. Hist. Acad. R. Sci. Mém. Math. Phys. 1725, 44–51. Available online: https://books.google.ch/books?id=YKgSAAAAYAAJ&lpg=PA44&ots=TbWGUkGQJO&dq=Nouvelles%20observations%20sur%20la%20pr%C3%A9paration%20du%20Bleu%20de%20Prusse&pg=PA44#v=onepage&q=Nouvelles%20observations%20sur%20la%20pr%C3%A9paration%20du%20Bleu%20de%20Prusse&f=false (accessed on 4 August 2019).
- l’Aine, G. Observations sur la préparation du Bleu de Prusse, ou de Berlin. Hist. Acad. R. Sci. Mém. Math. Phys. 1725, 153–172. Available online: https://books.google.ch/books?id=TP_RqzAwoUwC&lpg=PA312&ots=DjK9ing-lL&dq=Observations%20sur%20la%20pr%C3%A9paration%20du%20Bleu%20de%20Prusse&pg=PA312#v=onepage&q=Observations%20sur%20la%20pr%C3%A9paration%20du%20Bleu%20de%20Prusse&f=false (accessed on 4 August 2019).
- l’Aine, G. Nouvelles observations sur la préparation du Bleu de Prusse. Hist. Acad. R. Sci. Mém. Math. Phys. 1725, 220–237. [Google Scholar]
- Chadwick, B.M.; Sharpe, A.G. Transition Metal Cyanides and Their Complexes. Adv. Inorg. Chem. Radiochem. 1966, 8, 83–176. [Google Scholar] [CrossRef]
- Sharpe, A.G. The Chemistry of Cyano Complexes of the Transition Metals; Academic Press: New York, NY, USA, 1976; pp. 99–126. [Google Scholar]
- L.E.L. Ethel Churchill, or, The Two Brides; H. Colburn: London, UK, 1837; Volume 3. [Google Scholar]
- Scheele, C.W. Försök, beträffande det färgande ämnet uti Berlinerblå. Kongl. Vetenskaps Academiens 1782, 3, 264–275. [Google Scholar]
- Miller, L. LEL: The Lost Life and Scandalous Death of Letitia Elizabeth Landon, the Celebrated “Female Byron”; Knopf Doubleday: New York, NY, USA, 2019. [Google Scholar]
- Lewis, W. Commercium Philisophico-Technicum, or, the Philosophical Commerce of Arts: Designed as an Attempt to Improve Arts, Trades, and Manufactures; H. Baldwin: London, UK, 1763. [Google Scholar]
- Buchan, W. Domestic Medicine: or, a Treatise on the Prevention and Cure of Diseases by Regimen and SIMPLE Medicines. with an Appendix, Containing a Dispensatory for the Use of Private Practitioners, 11th ed.; Strahan and Cadell: London, UK, 1790. [Google Scholar]
- Royal College of Surgeons. A Draught for the Reformation of the London Pharmacopoeia, Prepared for the Perusal of the Members of the College of Physicians, by their Committee Appointed to that Purpose; Royal College of Surgeons: London, UK, 1742. [Google Scholar]
- Lewis, W. Pharmacopoeia reformata, or, an Essay for a Reformation of the London Pharmacopoeia, by a Set of Remarks on the Draught for a New One, and on The Brief Account of the Proceedings of the Committee appointed by the College of Physicians to thoroughly reform Their Book. Interspersed with Some Occasional Observations On some of the Most Celebrated Modern Dispensatories and the Present State of Pharmacy; R. Willock: London, UK, 1744. [Google Scholar]
- Duncan, A. The Edinburgh New Dispensatory: Containing I. the Elements of Pharmaceutical Chemistry. II. The Materia Medica; or the Natural, Pharmaceutical and Medical History of the Different Substances Employed IN Medicine. III. The Pharmaceutical Preparations and Compositions. Including Complete and Accurate Translations of the Octavo Edition of the London Pharmacopoeia published in 1791; Dublin Pharmacopoeia, published in 1794; and of the New Edition of the Edinburgh Pharmacopoeia, published in 1803; Bell and Bradfute: Edinburgh, UK, 1804. [Google Scholar]
- Powell, R. The Pharmacopoeia of The Royal College of Physicians of London M.DCCC.IX. Translated into English with Notes, 2nd ed.; Longman, Hurst, Rees, and Orme: London, UK, 1809. [Google Scholar]
- Crosland, M.P. Historical Studies in the Language of Chemistry, 2nd ed.; Dover: New York, NY, USA, 1978. [Google Scholar]
- Thorpe, E. (Ed.) The Scientific Papers of the Honourable Henry Cavendish, FRS.; Cambridge University Press: Cambridge, UK, 1921; Volume 2, p. 326. [Google Scholar]
- Macquer, P.J. Elémens de Chymie Théorique; Jean-Thomas Herissant: Paris, France, 1749. [Google Scholar]
- Macquer, P.J. Elémens de Chymie-Pratique; Jean-Thomas Herissant: Paris, France, 1751. [Google Scholar]
- Macquer, P.J. Dictionnaire de Chymie Contenant la Théorie et la Pratique de Cette Science, Son Application à la Physique, à l’histoire Naturelle, à la Médecine & aux Arts dépendans de la Chymie; P.Fr. Didot jeune: Paris, France, 1766. [Google Scholar]
- Baume, A. Chymie Expérimentale et Raisonée; P.Fr. Didot le jeune: Paris, France, 1773. [Google Scholar]
- Bergman, T. Physical and Chemical Essays Translated from the Original Latin of sir Torbern Bergman Knight of the Order of Wasa, Professor of Chemistry at Upsal, &C. &C. &C. by Edmund Cullen; J. Murray, London, UK: Balfour and Co, W. Dickson and J. Dickson, Edinburgh, UK: L. White, Dublin, Ireland, 1784; Volumes 1 and 2. [Google Scholar]
- Bergman, T. Physical and Chemical Essays translated from the original Latin of Sir Torbern Bergman Knight of the Order of Wasa, Professor of chemistry at Upsal, &c. &c. &c.; 1784, G. Mudie and J. and J. Fairbairn: Edinburgh, UK, 1791; Volume 3. [Google Scholar]
- De Morveau, G. Memoire sur les Dénominations Chymiques, la nécessité d’en perfectionner le Systéme, & les régles pour y parvenir. Obs. Phys. Chim. Hist. Nat. Arts 1782, 19, 370–383. [Google Scholar]
- Ohrström, L.; Reedijk, J. Names and symbols of the elements with atomic numbers 113, 115, 117 and 118 (IUPAC Recommendations 2016). Pure Appl. Chem. 2016, 88, 1225–1229. [Google Scholar] [CrossRef]
- De Morveau, L.; de Fourcroy, B. MÉTHODE DE NOMENCLATURE CHIMIQUE. Proposé par MM. DE MORVEAU, LAVOISIER, BERTHOLET, & DE FOURCROY. ON Y A JOINT Un nouveau Système de Caractères Chimiques, adaptés à cette Nomenclature, par MM. HASSENFRATZ & ADET; Cuchet: Paris, France, 1787. [Google Scholar]
- De Morveau, L.B.B.G.; ADET, P.A.; Berthollet, C.L.; de FOURCROY, A.F.; HASSENFRATZ, J.H.; Lavoisier, A.L.; SAINT JOHN, J. METHOD OF CHYMICAL NOMENCLATURE PROPOSED BY MESSRS. DE MORVEAU, LAVOISIER, BERTHOLET, AND DE FOURCROY. TO WHICH IS ADDED A NEW SYSTEM OF CHYMICAL CHARACTERS, ADAPTED TO THE NOMENCLATURE, by Mess. HASSENFRATZ and ADET. TRANSLATED FROM THE FRENCH, AND THE NEW CHEMICAL NAMES ADAPTED TO THE GENIUS OF THE ENGLISH LANGUAGE; G. Kearsley: London, UK, 1788. [Google Scholar]
- Conant, J.B. The Overthrow of Phlogiston Theory: The Chemical Revolution of 1775–1789; Harvard University Press: Cambridge, USA, 1950. [Google Scholar]
- White, J.H. The History of the Phlogiston Theory; AMS Press Inc.: New York, NY, USA, 1973. [Google Scholar]
- De la, M. Essai sur la nomenclature chimique. Obs. Phys. Chim. Hist. Nat. Arts 1787, 31, 270–285. [Google Scholar]
- Keir, J. The First Part of a Dictionary of Chemistry, &c. By J.K: and S.A.Sc.; Pearson and Rollason: Birmingham, UK, 1789. [Google Scholar]
- Dickson, S. An Essay on Chemical Nomenclature, by Stephen Dickson, M.D. State Physician in Ireland; Professor of the Practice of Medicine in Trinity College, Dublin; Vice President of the College of Physicians; Secretary to the Committee of Science of the R.I.A; F.R.S.S.A. and of Different Medical Societies in London, Edinburgh, Dublin, &c. in which are Comprised Observations on the Same Subject, by Richard Kirwan, LL. D.F.R.S.M.R.I.A and of the Academies of Stockholm, Upsal, Berlin, Manchester, Philadelphia, &c; J. Johnson: Dublin, Ireland, 1796. [Google Scholar]
- Berzelius, J.J. Essai sur la nomenclature chimique. J. Phys. Chim. Hist. Nat. Arts 1811, 73, 253–286. [Google Scholar]
- Tassaert. Analyse du cobalt de Tunaberg suivre de plusleurs moyens d’obtenir ce metal d l’état de pureté, et de quelques-unes de ses propriétés les plus remarquables. Ann. Chim. Phys. 1798, 28, 92–107. [Google Scholar]
- Partington, J.R. A History of Chemistry; MacMillan: London, UK, 1964; Volume 4, p. 920. [Google Scholar]
- Klaproth, M.H. Mémoires de Chimie Contenant des Analyses de Minéraux, 2 Volumes; Buisson: Paris, France, 1807. [Google Scholar]
- Gmelin, L. Ueber Kobaltsäure. J. Chem. Phys. 1822, 6, 235–237. Available online: https://ia800709.us.archive.org/BookReader/BookReaderImages.php?zip=/26/items/journalfrchemie34unkngoog/journalfrchemie34unkngoog_tif.zip&file=journalfrchemie34unkngoog_tif/journalfrchemie34unkngoog_0249.tif&scale=4&rotate=0 (accessed on 4 August 2019).
- Dingler, E.M. Ein Beitrag zur Kenntniss einiger elektronegativen Metalle. Archiv. Ges. Naturlehre 1829, 18, 249–252. [Google Scholar]
- Winkelblech, C. Ueber die Cobaltoxyde. Ann. Pharm. 1835, 13, 148–168. [Google Scholar] [CrossRef]
- Beetz, W. Ueber die Oxyde des Kobalts und einige Verbindungen der selben. Ann. Phys. Chem. 1844, 137, 472–506. [Google Scholar] [CrossRef]
- Rammelsberg, C. Ueber die Verbindungen der Jodmetalle mit Ammoniak. Ann. Phys. Chem. 1839, 124, 151–185. [Google Scholar] [CrossRef]
- Rammelsberg, C. Beiträge zur Kenntniss der Jodsauren und Überjodsauren Salze. Ann. Phys. Chem. 1838, 120, 545–591. [Google Scholar] [CrossRef]
- West, J.B. Joseph Black, carbon dioxide, latent heat, and the beginnings of the discovery of the respiratory gases. Am. J. Physiol. Lung Cell. Mol. Physiol. 2014, 306, L1057–L1063. [Google Scholar] [CrossRef] [PubMed]
- Fischer, N.W. Ueber die salpetrichtsauren Salze. Ann. Phys. Chem. 1848, 150, 115–125. [Google Scholar] [CrossRef] [Green Version]
- Erdmann, O.L. Ueber einige salpetrigsaure Nickel- und Kobalt-verbindungen. J. Prakt. Chem. 1866, 97, 385–413. Available online: https://reader.digitale-sammlungen.de/de/fs1/object/display/bsb10072520_00403.html?contextType=scan&contextSort=score%2Cdescending&contextRows=10&context=einige+salpetrigsaure (accessed on 4 August 2019). [CrossRef]
- Yoshimichi, K. Structures of the Erdmann’s salt, NH4[Co(NH3)2(NO2)4] and some other related nitro-ammine-cobalt (III) complexes. Bull. Chem. Soc. Jpn. 1957, 30, 13–21. [Google Scholar] [CrossRef]
- Vauqelin, N.L. Mémoire sur le Palladium et le Rhodium. Ann. Chim. 1813, 88, 167–186. [Google Scholar]
- Cleve, P.T. Om någar märkliga isomerier uti den oorganiska kemien, Oefversigt. Akad. Förh. 1871, 28, 175–179. [Google Scholar]
- Peyrone, M. Ueber die Einwirkung des Ammoniaks auf Platinchlorür. Ann. Chem. Pharm. 1844, 51, 1–29. [Google Scholar] [CrossRef]
- Zeise, W.C. Von der Wirkung zwischen Platinchlorid und Alkohol, und von den dabei entstehenden neuen Substanzen. Mag. Pharm. 1831, 35, 105–111. [Google Scholar] [CrossRef]
- Zeise, W.C. Von der Wirkung zwischen Platinchlorid und Alkohol, und von den dabei entstehenden neuen Substanzen. Ann. Phys. Chem. 1831, 97, 497–541. [Google Scholar] [CrossRef] [Green Version]
- Zeise, W.C. De chlorido platinae et acohole vini sese invicem permutantibus nec non de novis substantiis inde oriundis. In Anniversary Volume of the University of Copenhagen; 1830. [Google Scholar]
- Gmelin, H.L. Ueber ein besonderes Cyaneisenkalium, und über eine neue Reihe von blausauren Eisensalzen. J. Chem. Phys. 1822, 34, 325–346. Available online: https://books.google.ch/books?id=_-Q4AAAAMAAJ&lpg=PP12&ots=HozIznAfo7&dq=Ueber%20ein%20besonderes%20Cyaneisenkalium%2C%20und%20%C3%BCber%20eine%20neue%20Reihe%20von%20blausauren%20Eisensalzen.&pg=PA325#v=onepage&q=Ueber%20ein%20besonderes%20Cyaneisenkalium,%20und%20%C3%BCber%20eine%20neue%20Reihe%20von%20blausauren%20Eisensalzen.&f=false (accessed on 4 August 2019).
- Reinecke, A. Über Rhodanchromammonium-Verbindungen. Ann. Chem. Pharm. 1863, 126, 113–118. [Google Scholar] [CrossRef]
- Nassar, M.W.I.; Attia, K.A.S.M.; Mohamad, A.A.A.; Said, R.A.M.; Gaber, R.F.A. Atomic absorption spectrometric determination of metoclopramide hydrochloride using ammonium reineckate. World J. Pharm. Res. 2018, 7, 1–12. [Google Scholar] [CrossRef]
- Gros, J. Ueber eine neue Klasse von Platinsalzen. Ann. Pharm. 1838, 36, 241–256. [Google Scholar] [CrossRef]
- Gerhardt, C. Recherches sur les combinaisons ammoniacales du platine. C. R. 1850, 31, 241–244. Available online: https://www.biodiversitylibrary.org/item/16573#page/251/mode/1up (accessed on 4 August 2019).
- Hadow, E.A. The Platinum-bases: The best mode of obtaining and identifying them; some new Compounds. J. Chem. Soc. 1866, 19, 345–352. [Google Scholar] [CrossRef]
- Vortmann, G. Beiträge zur Kenntniss der Kobaltammoniumverbindungen. Monatsh. Chem. 1885, 6, 404–445. [Google Scholar] [CrossRef]
- Tschugajeff, L.; Skanawy-Grigorjewa, M.; Posnjak, A. Über Die Hydrazin-Carbylamin-Komplexe des Platins. Z. Anorg. Allg. Chem. 1925, 148, 37–42. [Google Scholar] [CrossRef]
- Magnus, G. Ueber einige Verbindungen des Platinchlorürs. Ann. Phys. Chem. 1828, 14, 239–242. [Google Scholar] [CrossRef]
- Jörgensen, S.M.; Sörensen, S.P.L. Ueber eine neue, mit Magnus’ grünem Sake isomere, rote Verbindung. Z. Anorg. Allg. Chem. 1905, 48, 441–445. [Google Scholar] [CrossRef]
- Buckton, G.B. On the Platino-tersulphocyanides and the Platino-bisulpho- cyanides, two new series of salts, and their decompositions. Q. J. Chem. Soc. 1855, 7, 22–43. [Google Scholar] [CrossRef]
- Thomson, T. On some of the compounds of chromium. Phil. Trans. 1827, 117, 159–230. Available online: https://www.jstor.org/stable/107874 (accessed on 4 August 2019). [CrossRef]
- Cleve, P.T. Om platinabasernas constitution. Nova Acta Reg. Soc. Sci. Upsal. 1866, 6, 112–118. [Google Scholar]
- Reiset, J. Observations sur une combinaison nouvelle de chlorure de platine et d’ammoniaque, considérée comme le radical des sels de Gros. C. R. 1840, 10, 870–872. Available online: https://www.biodiversitylibrary.org/item/112146#page/873/mode/1up (accessed on 5 August 2019).
- Reiset, J. Mémoire sur les combinaisons de deux nouvelles bases alcalines contenant du platine. Ann. Chim. 1844, 11, 417–433. [Google Scholar]
- Reiset, J. Mémoire sur les combinaisons de deux nouvelles bases alcalines contenant du platine. C. R. 1844, 18, 1100–1105. Available online: https://www.biodiversitylibrary.org/item/21168#page/1115/mode/1up (accessed on 5 August 2019).
- Cossa, A. Sopra alcuni derivati di una nuova base ammoniacale del platino. Atti R. Accad. dei Lincei, Rendiconti 1891, 7, 3–4. Available online: https://www.biodiversitylibrary.org/item/155653#page/9/mode/1up (accessed on 5 August 2019).
- Cossa, A. Sopra un nuovo isomero de sale verde de Magnus. Gazz. Chim. Ital. 1890, 20, 725–753. [Google Scholar]
- Barker, G.F. Frederick Augustus Genth. Proc. Am. Philos. Soc. 1901, 40, x–xxii. Available online: https://www.biodiversitylibrary.org/item/31072#page/204/mode/1up (accessed on 5 August 2019).
- Genth, F.A. Vorlaufige Notiz über Gepaarte Kobalt-Verbindungen. Keller-Tiedem’s Nordamerikanischer Monatsbericht für Natur- und Heilkunde 1851, 2, 8–12. [Google Scholar]
- Claudet, F. On a class of ammoniacal compounds of cobalt. Philos. Mag. 1851, 2, 253–260. [Google Scholar] [CrossRef]
- Claudet, F. Sur une classe de composés ammoniacaux du cobalt. Ann. Chim. Phys. 1851, 33, 483–493. Available online: https://gallica.bnf.fr/ark:/12148/cb343780820/date.r=Annales+de+chimie+et+de+physique.langEN (accessed on 5 August 2019).
- Fremy, E. Recherches sur le cobalt. C. R. 1851, 32, 509–510. Available online: https://www.biodiversitylibrary.org/item/16567#page/511/mode/1up (accessed on 5 August 2019).
- Fremy, E. Recherches sur le cobalt. C. R. 1851, 32, 808–810. Available online: https://www.biodiversitylibrary.org/item/16567#page/818/mode/1up (accessed on 5 August 2019).
- Fremy, E. Note sur les sels ammoniaco-cobaltiques suroxygénés. Ann. Chim. Phys. 1852, 34, 90–91. Available online: https://gallica.bnf.fr/ark:/12148/cb343780820/date.r=Annales+de+chimie+et+de+physique.langEN (accessed on 5 August 2019).
- Fremy, E. Recherches sur le cobalt. Ann. Chim. Phys. 1852, 35, 257–311. Available online: https://gallica.bnf.fr/ark:/12148/cb343780820/date.r=Annales+de+chimie+et+de+physique.langEN (accessed on 5 August 2019).
- Gibbs, W.; Genth, F.A. Researches on the ammonia-cobalt bases. Am. J. Sci. 1857, 23, 234–265. Available online: https://archive.org/details/mobot31753002152509/page/234 (accessed on 5 August 2019).
- Gibbs, W.; Genth, F.A. Researches on the ammonia-cobalt bases. Am. J. Sci. 1857, 23, 319–341. Available online: https://archive.org/details/mobot31753002152509/page/318 (accessed on 5 August 2019).
- Gibbs, W.; Genth, F.A. Researches on the ammonia-cobalt bases. Am. J. Sci. 1857, 24, 86–107. Available online: https://archive.org/details/mobot31753002152517/page/78 (accessed on 5 August 2019).
- Gibbs, W. Researches on the hexatomic compounds of cobalt. Proc. Am. Acad. Arts Sci. 1875, 10, 1–38. Available online: https://www.biodiversitylibrary.org/item/22173#page/9/mode/1up (accessed on 5 August 2019). [CrossRef]
- Gibbs, W. Researches on the hexatomic compounds of cobalt. Am. J. Sci. 1873, 6, 116–126. Available online: https://archive.org/details/mobot31753002152848/page/116 (accessed on 5 August 2019). [CrossRef]
- Gibbs, W. Researches on the hexatomic compounds of cobalt. Proc. Am. Acad. Arts Sci. 1875, 11, 1–51. Available online: https://www.archive.org/download/proceedingsofam11amer/page/n8_w347 (accessed on 5 August 2019). [CrossRef]
- Jörgensen, S.M. Beitrage zur Chemie der Kobaltammoniak-verbindungen. J. Prakt. Chem. 1879, 19, 49–69. [Google Scholar] [CrossRef]
- Jörgensen, S.M. Beitrage zur Chemie der Kobaltammoniak-verbindungen. J. Prakt. Chem. 1879, 19, 209–247. [Google Scholar] [CrossRef]
- Jörgensen, S.M. Zur Constitution der Kobalt-, Chrom- und Rhodiumbasen II. J. Prakt. Chem. 1890, 42, 206–221. [Google Scholar] [CrossRef]
- Jörgensen, S.M. Ueber saure Luteo- und Roseonitrate, Ueber saure Luteo- und Roseonitrate. J. Prakt. Chem. 1891, 44, 63–66. [Google Scholar] [CrossRef]
- Jörgensen, S.M. Beiträge zur Chemie der Rhodiumammoniakverbindungen. Part VI Ueber Roseorhodiumsalze. J. Prakt. Chem. 1886, 34, 394–406. [Google Scholar] [CrossRef]
- Jörgensen, S.M. Zur Konstitution der Kobalt-, Chrom- und Rhodiumbasen XI. Z. Anorg. Chem. 1899, 19, 109–157. [Google Scholar] [CrossRef]
- Werner, A. Ueber stereoisomere Kobaltverbindungen. Ber. Dtsch. Chem. Ges. 1901, 34, 1705–1719. [Google Scholar] [CrossRef]
- Blau, F. Über neue organische Metallverbindungen. Ein Beitrag zur Kenntniss der Metalliake. Monatsh. Chem. 1893, 3, 647–689. [Google Scholar] [CrossRef]
- Weltzien, C. Ueber die Ammoniummoleküle der Metalle. Justus Liebigs Ann. Chem. 1856, 97, 19–33. Available online: https://onlinelibrary.wiley.com/doi/pdf/10.1002/jlac.18560970105 (accessed on 4 August 2019). [CrossRef]
- Weltzien, C. Noch ein Wort über die Ammoniummoleküle der Metalle. Justus Liebigs Ann. Chem. 1856, 97, 108–110. [Google Scholar] [CrossRef]
- Weltzien, C. Ueber die ammoniakalischen Kobaltbasen. Justus Liebigs Ann. Chem. 1862, 121, 247–250. [Google Scholar] [CrossRef]
- Werner, A. Die Ammoniumsalze als einfachste Metallammoniake. Ber. Dtsch. Chem. Ges. 1903, 36, 147–159. [Google Scholar] [CrossRef] [Green Version]
- Hidakas, J.; Yamada, H.; Tsuchi, R. Rotatory Dispersion of Metallic Co-ordination Compounds. I. Rotatory Dispersion in the First and the Second Absorption Bands of Cobaltic Compounds 1. Bull. Chem. Soc. Jpn. 1958, 31, 921–925. [Google Scholar] [CrossRef]
- Hirotaro, K. Physicochemical studies on cobalt salts of higher fatty acids. IX. Cobalt complex soap. Bull. Chem. Soc. Jpn. 1962, 35, 388–930. [Google Scholar] [CrossRef]
- Lepadatu, C.I. On electronic spectra of praseosalts. Rev. Roum. Chim. 1966, 11, 489–495. [Google Scholar]
- Hara, R. Application of high-frequency chemical analysis to the studies of metallic complex salts. V. Reaction of some ammine complex salts of cobalt and chromium with protein. Yakugaku Zasshi 1951, 71, 1144–1152. [Google Scholar] [CrossRef]
- Mohamud, S.; Pagola, S. Revisiting the crystal structure of [Co(NH3)5Cl]Cl2 using synchrotron powder diffraction: to what extent single-crystal diffraction from 1960s got it right? Powder Diffr. 2017, 32, 179–186. [Google Scholar] [CrossRef]
- Odling, W., Translator; A Notation, classification and nomenclature by Auguste Laurent; Harrison and Sons: London, UK, 1855.
- Ramberg, P.J. Chemical Structure and Spatial Arrangement: The Early History of Stereochemistry 1874–1914; Ashgate: Aldershot, UK, 2003. [Google Scholar]
- Palmer, W.G. A History of the Concept of Valency to 1930; Cambridge University Press: Cambridge, UK, 1965. [Google Scholar]
- IUPAC Gold Book. Available online: https://goldbook.iupac.org/terms/view/V06588 (accessed on 24 July 2019).
- Towards a Comprehensive Definition of Valence. Available online: https://iupac.org/projects/project-details/?project_nr=2018-030-2-200 (accessed on 5 August 2019).
- Kekulé, A. Über die sogenannten gepaarten Verbindungen und die Theorie der mehratomigen Radicale. Ann. Chem. Pharm. 1857, 104, 129–150. [Google Scholar] [CrossRef]
- Russell, C.A. The History of Valence; Humanities Press: New York, USA, 1971. [Google Scholar]
- Meyer, V. Zur Valenz und Verbindungsfähigkeit des Kohlenstoffs. Justus Liebigs Ann. Chem. 1876, 180, 192–206. [Google Scholar] [CrossRef]
- Frankland, E. On a New Series of Organic Bodies containing Metals. Philos. Trans. 1852, 142, 417–444. [Google Scholar] [CrossRef]
- Wichelhaus, H. Ueber die Verbindungen des Phosphors. Ann. Chem. Pharm. 1868, (Suppl. 6), 257–280. [Google Scholar]
- Von Hofmann, A.W. Introduction to Modern Chemistry, Experimental and Theoretic, Embodying Twelve Lectures Delivered in the Royal College of Chemistry, London; Walton and Maberley: London, UK, 1865. [Google Scholar]
- Gay-Lussac, T. Extrait de plusieurs notes sur les métaux de la potasse et de la soude, lues à l’Institut depuis le 12 janvier jusqu’au 16 mai. Gazette Nationale, ou le Moniteur Universel 1808, 40, 581–582. Available online: https://gallica.bnf.fr/ark:/12148/cb34452336z/date.item (accessed on 5 August 2019).
- Davy, H. The Bakerian Lecture. An account of some new analytical researches on the nature of certain bodies, particularly the alkalies, phosphorus, sulphur, carbonaceous matter, and the acids hitherto undecomposed; with some general observations on chemical theory. Philos. Trans. 1809, 99, 39–104. [Google Scholar]
- Kekulé, A. Ueber die Constitution und die Metamorphosen der chemischen Verbindungen und über die chemische Natur des Kohlenstoffs. Ann. Chem. Pharm. 1858, 106, 129–159. [Google Scholar] [CrossRef]
- Kekulé, A. Sur l’atomicité des éléments. C. R. 1864, 58, 510–514. Available online: https://www.biodiversitylibrary.org/item/23663#page/518/mode/1up (accessed on 5 August 2019).
- Madan, H.G. Remarks on some Points in the Nomenclature of Salts. J. Chem. Soc. 1870, 23, 22–28. [Google Scholar] [CrossRef]
- Williamson. Remarks on chemical nomenclature and notation. J. Chem. Soc. 1864, 17, 421–432. [Google Scholar] [CrossRef]
- Connelly, N.G.; Damhus, T.; Hartshorn, R.M.; Hutton, A.T. Nomenclature of Inorganic Chemistry. IUPAC Recommendations 2005. Issued by the Division of Chemical Nomenclature and Structure Representation in collaboration with the Division of Inorganic Chemistry; Rule IR-3.3.2; RSC Publishing: Cambridge, UK, 2005. [Google Scholar]
- Favre, H.A.; Powell, W.H. Nomenclature of Organic Chemistry—IUPAC Recommendations and Preferred Names 2013; Royal Society of Chemistry: Cambridge, UK, 2013. [Google Scholar]
- Kragh, K.S.M. Jørgensen and his controversy with A. Werner: A reconsideration. Br. J. Hist. Sci. 1997, 30, 203–219. Available online: https://www.jstor.org/stable/4027717 (accessed on 4 August 2019). [CrossRef]
- Kauffman, G.B. Sophus Mads Jørgensen and the Werner-Jørgensen Controversy. Chymia 1960, 6, 180–204. Available online: https://www.jstor.org/stable/27757198 (accessed on 4 August 2019). [CrossRef]
- Kauffman, G.B. Sophus Mads Jorgensen (1837-1914): A chapter in coordination chemistry history. J. Chem. Educ. 1959, 36, 521–527. [Google Scholar] [CrossRef]
- Whisnant, D.M. Bonding theory/The Werner-Jorgensen controversy. J. Chem. Educ. 1993, 70, 902. [Google Scholar] [CrossRef]
- Gerhardt, C. Sur la constitution des Sels organiques à Acides complexes, et leurs Rapports avec les Sels ammoniacaux. Annales de Chimie et de Physique 1839, 72, 184–214. Available online: https://gallica.bnf.fr/ark:/12148/cb343780820/date1839 (accessed on 5 August 2019).
- Berzelius, J. Ansichten in Betreff der organischen Zusammensetzung. Ann. Phys. Chem. 1846, 78, 161–188. [Google Scholar] [CrossRef]
- Hare, R. Objections to the nomenclature of the celebrated Berzelius: With suggestions respecting a substitute, in a letter to Professor Silliman: first published in 1834, and republished in Silliman’s Journal for 1835, Vol. XXVII. Silliman’s J. 1835, 27, 1–23. Available online: https://archive.org/details/101125880.nlm.nih.gov (accessed on 4 August 2019).
- Berzelius, J.J. Jahres-Bericht über die Fortschritte der Chemie und Mineralogie; Laupp’sche Buchhandlung: Tübingen, Germany, 1842; Volume 21. [Google Scholar]
- Blomstrand, C.W. Die Chemie der Jetztzeit vom Standpunkte der electrochemischen Auffassung. Aus Berzellius lehre entwickelt; Carl Winter’s Universitätsbuchhandlung: Heidelberg, Germany, 1869. [Google Scholar]
- Jörgensen, S.M. Ueber das Verhältniss zwischen Luteo- und Roseosalzen. J. Prakt. Chem. 1884, 29, 409–422. [Google Scholar] [CrossRef]
- Jörgensen, S.M. Zur Constitution der Kobaltbasen I. J. Prakt. Chem. 1890, 41, 429–439. [Google Scholar] [CrossRef]
- Werner, A. Beitrag zur Konstution anorganische Verbindungen. Z. Anorg. Chem. 1893, 3, 267–330. [Google Scholar] [CrossRef]
- Jörgensen, S.M. Zur Konstitution der Kobalt-, Chrom- und Rhodiumbasen V. Z. Anorg. Chem. 1894, 5, 147–196. [Google Scholar] [CrossRef]
- Jörgensen, S.M. Zur Konstitution der Kobalt-, Chrom- und Rhodiumbasen VI. Z. Anorg. Chem. 1894, 7, 289–330. [Google Scholar] [CrossRef]
- Hantzsch, A.; Werner, A. Ueber räumliche Anordnung der Atome in stickstoffhaltigen Molekülen. Ber. Dtsch. Chem. Ges. 1890, 23, 11–30. [Google Scholar] [CrossRef]
- Hantzsch, A.; Werner, A. Bemerkungen über stereochemisch isomere Stickstoffverbindungen. Ber. Dtsch. Chem. Ges. 1890, 23, 1243–1253. [Google Scholar] [CrossRef]
- Hantzsch, A.; Werner, A. Bemerkungen über stereochemisch isomere Stickstoffverbindungen. Ber. Dtsch. Chem. Ges. 1890, 23, 2764–2769. [Google Scholar] [CrossRef]
- Werner, A. Beiträge zur Theorie der Affinität und Valenz. Vierteljahresschr. Naturforsch. Ges. Zürich 1891, 36, 129–161. Available online: https://www.ngzh.ch/archiv/1891_36/36_2/36_10.pdf (accessed on 4 August 2019).
- Werner, A.; Miolati, A. Beiträge zur Konstitution anorganischer Verbindungen. Z. Phys. Chem. 1893, 12, 35–55. [Google Scholar] [CrossRef]
- Werner, A. Beitrag zur Konstitution anorganischer Verbindungen. V. Mitteilung. Die Kobaltammoniakverbindungen und ihre Nomenklatur. Z. Anorg. Chem. 1897, 14, 23–27. [Google Scholar] [CrossRef]
- Werner, A. Neuere Anschauungen auf dem Gebiete der Anorganischen Chemie, 4th ed.; Friedr Vieweg & Sohn: Braunschweig, Germany, 1920. [Google Scholar]
- Hedley, E.P., Translator; New Ideas in Inorganic Chemistry by Dr A. Werner. Translated with the Author’s Sanction, from the Second German Edition; Longmans, Green and Co.: London, UK, 1911.
- Fernelius, W.C. The Nomenclature of Coordination Compounds from Pre-Werner Times to the 1966 IUPAC Report. ACS Ser. 1967, 62, 147–160. [Google Scholar]
- Bailar, J.C., Jr. The chemistry of the coordination compounds; ACS Monograph Series; Reinhold Publishing Corporation: New York, NY, USA; Chapman and Hall: London, UK, 1956. [Google Scholar]
- Nomenclature of Inorganic Chemistry. Definitive Rules 1970, 2nd ed.; Butterworths: London, UK, 1971; p. 39.
- Leigh, G.J. (Ed.) Nomenclature of Inorganic Chemistry. Recommendations Rules 1990; Rule I-10.2.6; Blackwell: Oxford, UK, 1990. [Google Scholar]
- Connelly, N.G.; Damhus, T.; Hartshorn, R.M.; Hutton, A.T. Nomenclature of Inorganic Chemistry. IUPAC Recommendations 2005. Issued by the Division of Chemical Nomenclature and Structure Representation in Collaboration with the Division of Inorganic Chemistry; Rule IR-9.1.2.7; RSC Publishing: Cambridge, UK, 2005. [Google Scholar]
- Ihde, A.J. The Karlsruhe Congress: A Centennial Retrospect. J. Chem. Educ. 1961, 38, 83–86. [Google Scholar] [CrossRef]
- Hudson, J. The History of Chemistry; Palgrave: London, UK, 1992; pp. 122–137. [Google Scholar]
- De Milt, C. The Congress at Karlsruhe. J. Chem. Educ. 1951, 28, 421–425. [Google Scholar] [CrossRef]
- Ball, P. H2O: A Biography of Water; Weidenfeld and Nicolson: London, UK, 1999. [Google Scholar]
- Higgins, W. A Comparative View of the Phlogistic and Antiphlogistic Theories. With Inductions. To Which is Annexed, an Analysis of the Human Calculus, By William Higgins, of Pembroke College, Oxford; J. Murray: London, UK, 1789. [Google Scholar]
- Higgins, W. A Comparative View of the Phlogistic and Antiphlogistic Theories. With Inductions. To Which is Annexed, an Analysis of the Human Calculus, By William Higgins, of Pembroke College, Oxford, 2nd ed.; J. Murray: London, UK, 1791. [Google Scholar]
- Dalton, J. A New System of Chemical Philosophy. Part I; R. Bickerstaff: London, UK, 1808. [Google Scholar]
- Dalton, J. A New System of Chemical Philosophy. Part II; R. Bickerstaff: London, UK, 1810. [Google Scholar]
- Grotthuss, C.J.T. Sur la décomposition de l’eau et des corps qu’elle tient endissolution à l’aide de l’électricité galvanique. Ann. Chim. 1806, 58, 54–74. [Google Scholar]
- Grotthuss, C.J.T. De l’influence de l’électricité galvanique sur les végétations métalliques. Ann. Chim. 1807, 63, 5–34. [Google Scholar]
- Kekulé, A. Lehrbuch der organischen Chemie oder der Chemie der Kohlenstoffverbindungen; Ferdinand Enke: Erlangen, Germany, 1861; Volume 1. [Google Scholar]
- Kekulé, A. Lehrbuch der organischen Chemie oder der Chemie der Kohlenstoffverbindungen; Ferdinand Enke: Erlangen, Germany, 1866; Volume 2. [Google Scholar]
- Kekulé, A. Lehrbuch der organischen Chemie oder der Chemie der Kohlenstoffverbindungen; Ferdinand Enke: Erlangen, Germany, 1867; Volume 3. [Google Scholar]
- Meyer, L. Die Modernen Theorien der Chemie und ihre Bedeutung für die chemische Statik; Maruschke & Barendt: Breslau, Germany, 1864. [Google Scholar]
- Beilstein, F. Handbuch der Organischen Chemie. 1. Abtheilung. Einleitung. Specieller Theil: Fettreihe; Von Leopold Voss: Hamburg and Leipzig, Germany, 1881. [Google Scholar]
- Beilstein, F. Handbuch der Organischen Chemie. 2. Specieller Theil: Aromatische Reihe; Von Leopold Voss: Hamburg/Leipzig, Germany, 1883. [Google Scholar]
- Baskerville, C. International Congresses. Science 1910, 32, 652–659. [Google Scholar] [CrossRef]
- Congrès de nomenclature chimique (Genève, 1892). Bull. Soc. Chim. Fr. 1892, 3, XIII–XXIV.
- Brock, W.H.; Jensen, K.A.; Jørgensen, C.K.; Kauffman, G.B. The origin and dissemination of the term “ligand” in chemistry. Polyhedron 1983, 2, 1–7. [Google Scholar] [CrossRef]
- Stock, A. Siliciumchemie und Kohlenstoffchemie. Ber. Dtsch. Chem. Ges. 1917, 50, 170–182. [Google Scholar] [CrossRef] [Green Version]
- Klement, R. Phosphorsäure als Ligand in komplexen Kobaltverbindungen. Z. Anorg. Allgem. Chem. 1926, 156, 237–244. [Google Scholar] [CrossRef]
- Jensen, K.A. Dipolmessungen an isomeren platokomplexen. Z. Anorg. Allg. Chem. 1935, 225, 97–141. [Google Scholar] [CrossRef]
- Tsuchida, R. Absorption spectra of coordination compounds. I. Nippon Kagaku Kaishi (1921–1947) 1938, 59, 586–596. [Google Scholar] [CrossRef]
- Tsuchida, R. Absorption spectra of coordination compounds. I. Bull. Chem. Soc. Jpn. 1938, 13, 388–400. [Google Scholar] [CrossRef]
- Tsuchida, R. Absorption spectra of coordination compounds. II. Nippon Kagaku Kaishi (1921–1947) 1938, 59, 731–743. [Google Scholar] [CrossRef]
- Tsuchida, R. Absorption spectra of coordination compounds. II. Bull. Chem. Soc. Jpn. 1938, 13, 436–450. [Google Scholar] [CrossRef]
- Tumaki, T. Coordinate valency rings. V. Spectrochemical researches on the inner complex metallic salts of salicylaldehyde-ethylenediimine and related compounds. Bull. Chem. Soc. Jpn. 1938, 13, 583–591. [Google Scholar]
- Tutida, R.; Kobayasi, M.; Kuroya, H. An extended coordination theory of valency. IV. Configuration of compounds of transition elements. Rev. Phys. Chem. Jpn. 1939, 13, 151–165. Available online: http://hdl.handle.net/2433/46556 (accessed on 4 August 2019).
- Ewens, R.V.G.; Bassett, H. Inorganic chemical nomenclature. Chem. Ind. 1949, 131–139. [Google Scholar]
- IUPAC; Commission de Nomenclature de Chimie Inorganique. Tentative Rules for Inorganic Nomenclature; Comptes Rendus de la Dix-Septième Conférence: Stockholm, 1953; pp. 98–119. [Google Scholar]
- Buchner, E.H. Chem. Weekblad. 1955, 51, 295–310.
- IUPAC. Nomenclature of Inorganic Chemistry, 1957 Report of CNIC; Butterworths Scientific Publications: London, UK, 1959. [Google Scholar]
- Martell, A.E.; Calvin, M. Chemistry of the Metal Chelate Compounds; Prentice-Hall Inc.: New York, NY, USA, 1952. [Google Scholar]
- Morgan, G.T.; Drew, H.D.K. Researches on residual affinity and co-ordination. Part II. Acetylacetones of selenium and tellurium. J. Chem. Soc. Trans. 1920, 117, 1456–1465. [Google Scholar] [CrossRef]
- Lowry, T.M. Stability of co-ordination compounds. J. Soc. Chem. Ind. Lond. Rev. Sect. 1923, 42, 711–715. Available online: https://archive.org/details/ost-chemistry-chemistryindustr01soci/page/n735 (accessed on 12 August 2019). [CrossRef]
- Smith, J.D.M. Chelate co-ordination. J. Soc. Chem. Ind. Lond. Rev. Sect. 1923, 42, 847–850. Available online: https://archive.org/details/ost-chemistry-chemistryindustr01soci/page/n871 (accessed on 12 August 2019). [CrossRef]
- Lowry, T.M. Co-ordination and the electron. J. Soc. Chem. Ind. Lond. Rev. Sect. 1923, 42, 1004–1007. Available online: https://archive.org/details/ost-chemistry-chemistryindustr01soci/page/n1029 (accessed on 12 August 2019). [CrossRef]
- McKenney, R.E.B. Mono- and di-basic phosphates. Science 1910, 32, 836–837. [Google Scholar] [CrossRef]
- Ransom, J.H. Inorganic nomenclature. Science 1911, 33, 63–64. [Google Scholar] [CrossRef]
- International Association of Chemical Societies. Nature 1912, 89, 245–246. [CrossRef] [Green Version]
- K., C.G. International Association of Chemical Societies. J. Chem. Soc. 1912, 86–88. [Google Scholar]
- Van Tiggelen, B.; Fauque, D. The Formation of the International Association of Chemical Societies. Chem. Int. 2012, 34, 8–12. [Google Scholar] [CrossRef]
- Fennell, R.W. History of IUPAC 1919–1987; Blackwell Science: Oxford, UK, 1994. [Google Scholar]
- Marshall, A.G. The Interallied Conference of Scientific Academies, in London. Publ. Astron. Soc. Pac. 1918, 30, 331–335. Available online: https://iopscience.iop.org/article/10.1086/122768/pdf (accessed on 4 August 2019). [CrossRef]
- Walker, J. Presidential address delivered at the annual general meeting, March 22nd, 1923. Symbols and formulae. J. Chem. Soc. Trans. 1923, 123, 939–946. [Google Scholar] [CrossRef]
- Chemical nomenclature. Science 1923, 57, 474. [CrossRef]
- Comptes Rendus de la Deuxieme Conference Internationale de la Chimie. Brussels, Belgium, 1921.
- Revision of inorganic nomenclature. Analyst 1926, 51, 192–195. [CrossRef]
- Delépine, M. Réforme de la nomenclature chimique. Bull. Soc. Encour. Ind. Natl. 1929, 775–811. Available online: http://cnum.cnam.fr/CGI/fpage.cgi?BSPI.143/774/100/906/20/860 (accessed on 5 August 2019).
- Meyer, R.J.; Rosenheim, A. Die Vorschläge der deutschen Nomenklaturkommission für anorganische Chemie. Z. Angew. Chem. 1925, 38, 713–715. [Google Scholar] [CrossRef]
- Meyer, R.J. Zur Nomenklatur der anorganischen Chemie. Z. Angew. Chem. 1929, 42, 1059–1062. [Google Scholar] [CrossRef]
- Meyer, R.J. Rapport sur la nomenclature des combinations inorganiques. Helv. Chim. Acta 1937, 20, 159–175. [Google Scholar] [CrossRef]
- Meyer, R.J. Chem. Weekblad. 1936, 33, 722–729.
- Leigh, G.J. A History of CNIC. Chem. Int. 2019, 41, 39–43. [Google Scholar] [CrossRef]
- Jorissen, W.P.; Bassett, H.; Damiens, A.; Fichter, F.; Remy, H. International Union of Chemistry. Rules for Naming Inorganic Compounds. Analyst 1940, 65, 509–511. [Google Scholar]
- Jorissen, W.P.; Bassett, H.; Damiens, A.; Fichter, F.; Remy, H. Internationale Union für Chemie, Kommission für die Reform der Nomenklatur der anorganischen Chemie: Richtsätze für die Benennung anorganischer Verbindungen. Ber. Dtsch. Chem. Ges. A 1940, 73A, 53–70. [Google Scholar] [CrossRef]
- Jorissen, W.P.; Bassett, H.; Damiens, A.; Fichter, F.; Remy, H. Internationale Chemische Union. Kommission für die Reform der Nomenklatur der anorganischen Chemie: Richtsätze für die Benennung anorganischer Verbindungen. Helv. Chim. Acta 1940, 23, 997–1011. [Google Scholar]
- Jorissen, W.P.; Bassett, H.; Damiens, A.; Fichter, F.; Remy, H. Union Internationale de Chimie. Regles relatives a la nomenclature des composes mineraux. Rapport de la commission de reforme de la nomenclature de chimie inorganique 1940. Helv. Chim. Acta 1940, 23, 1012–1024. [Google Scholar]
- Jorissen, W.P.; Bassett, H.; Damiens, A.; Fichter, F.; Remy, H. Report of the Committee for the Reform of Inorganic Chemical Nomenclature, 1940. J. Chem. Soc. 1940, 1404–1415. [Google Scholar] [CrossRef]
- Jorissen, W.P.; Bassett, H.; Damiens, A.; Fichter, F.; Remy, H. Rules for Naming Inorganic Compounds* Report of the Committee of the International Union of Chemistry for the Reform of Inorganic Chemical Nomenclature, 1940. J. Am. Chem. Soc. 1941, 63, 889–897. [Google Scholar] [CrossRef]
- Scott, J.D. The need for reform in inorganic chemical nomenclature. Chem. Rev. 1943, 32, 73–97. [Google Scholar] [CrossRef]
- Chemical Nomenclature; ACS Series No 8. American Chemical Society: Washington, DC, USA, 1953.
- Bassett, H. Some General Principles of Inorganic Chemical Nomenclature. ACS Ser. 1953, 8, 5–8. [Google Scholar]
- Fernelius, W.C. Nomenclature of Coordination Compounds and Its Relation to General Inorganic Nomenclature. ACS Ser. 1953, 8, 9–37. [Google Scholar]
- In Proceedings of the IUPAC Comptes Rendus of the 17th Conference; 1953; pp. 98–119.
- IUPAC. Nomenclature of Inorganic Chemistry. 1957 Rules, 2nd ed.; Butterworths: London, UK, 1959. [Google Scholar]
- IUPAC. 1957 Report of the Commission on the Nomenclature of Inorganic Chemistry. Nomenclature of Inorganic Chemistry Definitive Rules for Nomenclature of Inorganic Chemistry. J. Am. Chem. Soc. 1960, 82, 5523–5544. [Google Scholar]
- Cahn, R.S. An Introduction to Chemical Nomenclature; Butterworths: London, UK, 1964. [Google Scholar]
- IUPAC. Nomenclature of Inorganic Chemistry, 2nd ed.; Butterworths: London, UK, 1971. [Google Scholar]
- Kelly, D.P. Sulfur and its Doppelgänger. Arch. Microbiol. 1995, 163, 157–158. [Google Scholar] [CrossRef]
- So long sulphur. Nat. Chem. 2009, 1, 333. [CrossRef] [PubMed]
- Cohn, R.S. An Introduction to Chemical Nomenclature, 4th ed.; Butterworths: London, UK, 1974. [Google Scholar]
- Leigh, G.J. (Ed.) Nomenclature of Inorganic Chemistry. Recommendations Rules 1990; Blackwell: Oxford, UK, 1990. [Google Scholar]
- McCleverty, J.A.; Connelly, N.G. (Eds.) Nomenclature of Inorganic Chemistry II. Recommendations 2000; Royal Society of Chemistry: Cambridge, UK, 2001. [Google Scholar]
- Hartshorn, R.M.; Hellwich, K.-H.; Yerin, A.; Damhus, T.; Hutton, A.T. Brief guide to the nomenclature of inorganic chemistry. Pure Appl. Chem. 2015, 87, 1039–1049. [Google Scholar] [CrossRef]
- Brief Guide to the Nomenclature of Inorganic Chemistry. Available online: https://iupac.org/cms/wp-content/uploads/2018/05/Inorganic-Brief-Guide-V1-3.pdf (accessed on 25 July 2019).
- Housecroft, C.E.; Sharpe, A.G. Inorganic Chemistry, 5th ed.; Pearson: Harlow, UK, 2018. [Google Scholar]
- Brief Guide to the Nomenclature of Inorganic Chemistry. Available online: https://www.qmul.ac.uk/sbcs/iupac/BriefGuide/inorganic.html (accessed on 5 August 2019).
- Seidel, S.; Seppelt, K. Xenon as a Complex Ligand: The Tetra Xenono Gold(II) Cation in AuXe42+(Sb2F11−)2. Science 2000, 290, 117–118. [Google Scholar] [CrossRef] [PubMed]
- REACH Legislation. Available online: https://echa.europa.eu/regulations/reach/legislation (accessed on 25 July 2019).
- Streng, A.G.; Grosse, A.V. Acid of Krypton and Its Barium Salt. Science 1964, 143, 242–243. [Google Scholar] [CrossRef]








© 2019 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Constable, E.C. What’s in a Name?—A Short History of Coordination Chemistry from Then to Now. Chemistry 2019, 1, 126-163. https://doi.org/10.3390/chemistry1010010
Constable EC. What’s in a Name?—A Short History of Coordination Chemistry from Then to Now. Chemistry. 2019; 1(1):126-163. https://doi.org/10.3390/chemistry1010010
Chicago/Turabian StyleConstable, Edwin C. 2019. "What’s in a Name?—A Short History of Coordination Chemistry from Then to Now" Chemistry 1, no. 1: 126-163. https://doi.org/10.3390/chemistry1010010
APA StyleConstable, E. C. (2019). What’s in a Name?—A Short History of Coordination Chemistry from Then to Now. Chemistry, 1(1), 126-163. https://doi.org/10.3390/chemistry1010010