Abstract
A proof of concept for a novel approach towards enantiomerically highly enriched acyclic secondary amines and β-aminothiols as non-cyclic target molecules when starting from 3-thiazolines as heterocycles is presented. Starting from 2,2,4,5,5-pentamethyl-3-thiazoline, we demonstrated this chemoenzymatic pathway to both of these types of amine molecules, which were isolated as urea derivatives with a non-optimized yield of up to 20%. As a substrate, 2,2,4,5,5-pentamethyl-3-thiazolidine, which was obtained with an enantiomeric excess (ee) of 99% in a biotransformation from the corresponding 3-thiazoline according to a recently developed protocol, was used. For the reductive desulfurization of this substrate leading to a sulfur-free secondary amine, in situ formed Ni2B turned out to be a suitable reducing reagent. However, when using lithium aluminum hydride as a reducing agent, β-aminothiol was obtained.
1. Introduction
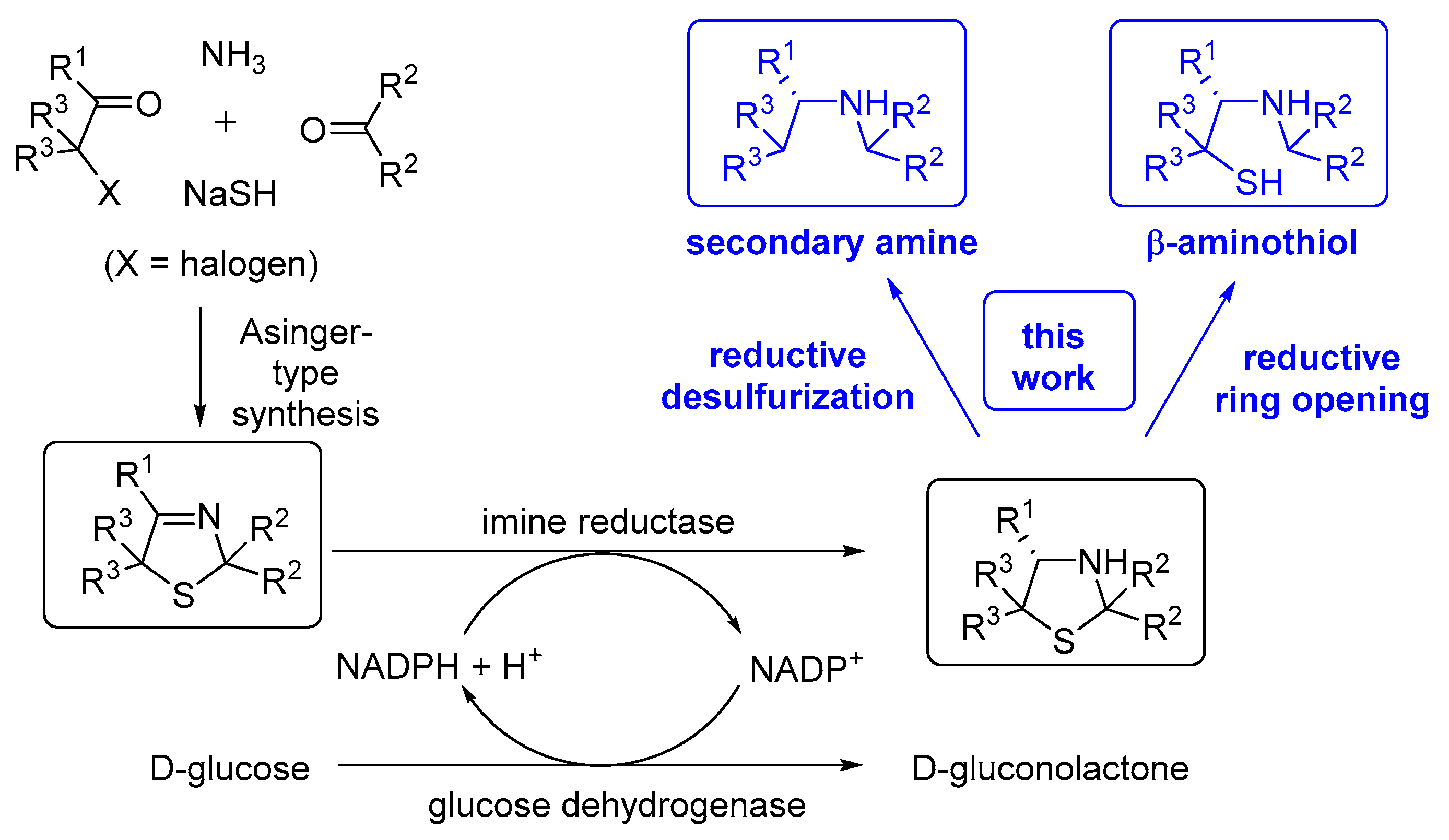
Friedrich Asinger, a professor of Technical Chemistry at RWTH Aachen, is the discoverer of a sulfur-based multi-component condensation reaction leading in an elegant fashion to 3-thiazoline heterocycles [1,2,3]. This one-pot process enabling the synthesis of a sulfur-containing heterocycle bearing an imine bond, which was named after him, turned out to have a broad substrate scope as well as industrial potential [2,4]. In the original form, 2 equivalents of an α-C-H-acidic carbonyl compound were condensed with ammonia and sulfur under formation of the corresponding 3-thiazoline [1,2,3]. Later, a more flexible variant was developed based on the use of a preformed α-thiolated carbonyl or α-halogenated carbonyl moiety, which enabled a broader substitution pattern of the resulting products (Scheme 1) [1,4]. The Asinger group members, Offermanns and Scherberich, introduced the Asinger synthesis at Degussa AG after joining this company and at Degussa AG then, this synthetic route was expanded towards an industrial process for the production of d-pencillamine [4], thus underlining the power of heterocyclic chemistry also for the preparation of non-cyclic industrial chemicals on a large scale. Inspired by this success in the chemical industry, in the following decades, numerous further efficient transformations of the 3-thiazoline heterocycle by addition of suitable nucleophiles have been reported, e.g., the pioneer achievements by Martens at Degussa AG and later accomplishments by his group at Oldenburg University with, in part, collaboration partners on the use of malonates (leading to β-amino acid derivatives) [5], isocyanides (within the Ugi reaction leading to totally protected α-amino acids) [6,7] and phosphites (leading to α-amino phosphonates) [8,9,10,11]. Although most of these methods give racemic products [5,6,7], it is noteworthy that Martens, Shibasaki and their coworkers demonstrated that the latter transformation was able to be conducted even in an asymmetric fashion leading to high enantioselectivities for the formation of the resulting α-amino phosphonates [9,10,11]. As a long-lasting challenge for decades, the (enantioselective) reduction of this C–N double bonds of 3-thiazolines leading to the non-cleaved thiazolidine heterocycles (which are a subunit, e.g., in the pharmaceutically important penicillin-type antibiotics) in high conversion and excellent enantiomeric excess was recently realized by conducting the reaction by means of enzyme catalysis (Scheme 1) [12,13,14]. In detail, the reduction is catalyzed by means of an imine reductase and the reduced form of a cofactor (NADPH), which is regenerated in situ by consuming d-glucose as the stoichiometrically required (cheap) reducing agent. This biocatalytic transformation opens a perspective towards enantiomerically pure secondary aliphatic amines and β-aminothiols by derivatizing the thiazolidines obtained in the biocatalytic step according to the synthetic concepts given in Scheme 1.
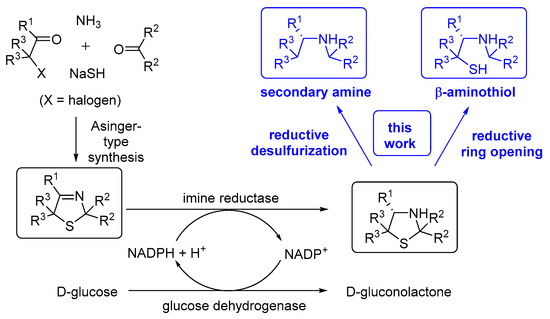
Scheme 1.
Conceptual overview about chemoenzymatic approaches towards enantiomerically secondary amines and β-aminothiols based on a combination of Asinger synthesis, biocatalytic reduction and chemical ring opening reactions.
The development of novel synthetic approaches enabling a more facile synthesis towards such products is still a field of high interest for both academia and industry. For example, even simple representatives of (enantiomerically pure) β-aminothiols are hardly commercially available (such as the thiol analogue of alaninol), thus indicating the tediousness of current approaches. A representative chemical approach towards β-aminothiols has been described by Otto et al. converting at first l-amino acids to β-aminoethanols, which are then brominated and subsequently transformed with thiourea to the desired β-aminothiols in yields of up to 40% [15].
Addressing these challenges, in the following, we present a novel conceptual approach to such non-cyclic target molecules by starting from heterocycles. In detail, we report the proof of concept for a highly enantioselective chemoenzymatic pathway to both types of molecules starting from a 3-thiazoline, consisting of an initial biocatalytic reduction of easily accessible 3-thiazoline followed by different chemical reductive technologies.
2. Experimental
2.1. General Information
The commercially available reagents (Acros Organics (Geel, Belgium), Alfa Aesar (Ward Hill, MA, USA), Carl Roth (Karslruhe, Germany), CHEMSOL (Indianapolis, IN, USA), Thermo Fisher Scientific (Waltham, MA, USA), J.T Baker(Phillipsburg, NJ, USA), Merck (Darmstadt, Germany), Sigma-Aldrich (St. Louis, MO, USA), VWR (Radnor, PA, USA) were used without further purification. Solvents were used in high-grade purity or purified prior to use. NMR spectra were recorded on Bruker Advance III 500 or Bruker Advance III 500HD at a frequency of 500 MHz (1H) or 126 MHz (13C). The chemical shift δ is given in ppm and referenced to the corresponding solvent signal (CDCl3). Coupling constants (J) are given in Hz. Nano-electrospray ionization (Nano-ESI) mass spectra were recorded using an Esquire 3000 ion trap mass spectrometer (Bruker Daltonik GmbH, Bremen, Germany) equipped with a standard nano-ESI source. Samples were introduced by static nano-ESI using in-house pulled-glass emitters. Nitrogen served as both the nebulizer gas and the dry gas. Nitrogen was generated by a Bruker nitrogen generator NGM 11. Helium served as cooling gas for the ion trap and collision gas for MSn experiments. HRMS-ESI mass spectra were recorded using an Agilent 6220 time-of-flight mass spectrometer (Agilent Technologies, Santa Clara, CA, USA) in extended dynamic range mode equipped with a dual ESI source, operating with a spray voltage of 2.5 kV. Nitrogen served as both the nebulizer gas and the dry gas. Nitrogen was generated by a nitrogen generator NGM 11. Samples were introduced with a 1200 HPLC system consisting of an autosampler, a degasser, a binary pump, a column oven and a diode array detector (Agilent Technologies, Santa Clara, CA, USA) using a C18 Hypersil Gold column (length: 50 mm, diameter: 2.1 mm, particle size: 1.9 μm) with a short gradient (increase from 0% B to 98% B in 4 min, back to 0% B in 0.2 min; total run time: 7.5 min) at a flow rate of 250 μL/min and a column oven temperature of 40 °C. HPLC solvent A consisted of 94.9% water, 5% acetonitrile and 0.1% formic acid; solvent B consisted of 5% water, 94.9% acetonitrile and 0.1% formic acid. The mass axis was externally calibrated with ESI-L Tuning Mix (Agilent Technologies, Santa Clara, CA, USA) as a calibration standard. Electron ionization (EI) mass spectra were recorded using an Autospec X magnetic sector mass spectrometer with EBE geometry (Vacuum Generators, Manchester, UK) equipped with a standard EI source. Samples were introduced by push rod in aluminum crucibles. Ions were accelerated by a potential difference of 8 kV in EI mode. Thin-layer chromatography (TLC)-ESI mass spectrometry was performed on the ZQ2000 single-quadrupole mass spectrometer (Waters, Manchester, UK) equipped with an ESI source, operating with a spray voltage of 3.5 kV. Nitrogen served as both the nebulizer gas and the dry gas and was generated by a nitrogen generator NGM 11. The mass axis was externally calibrated with ESI-L Tuning Mix (Agilent Technologies, Santa Clara, CA, USA) as a calibration standard. Samples were introduced using an in-house built TLC interface, operated with a Hitachi L-600 LC pump (2-Propanol, flow rate: 400 μL/min). Flash column chromatography was performed using the Biotage Isolera One flash chromatography system with cyclohexane/ethyl acetate mixtures.
2.2. Construction and Preparation of Whole-Cell Catalyst
The whole-cell catalyst for the biotransformation according to Scheme 2 was constructed and prepared according to Reference [12].
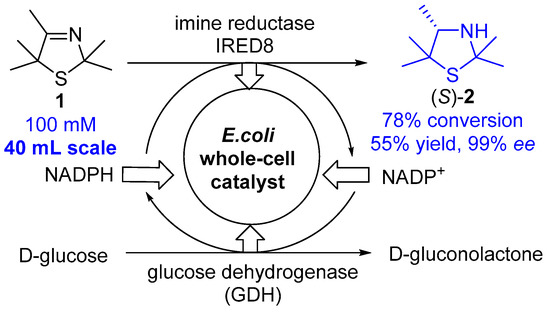
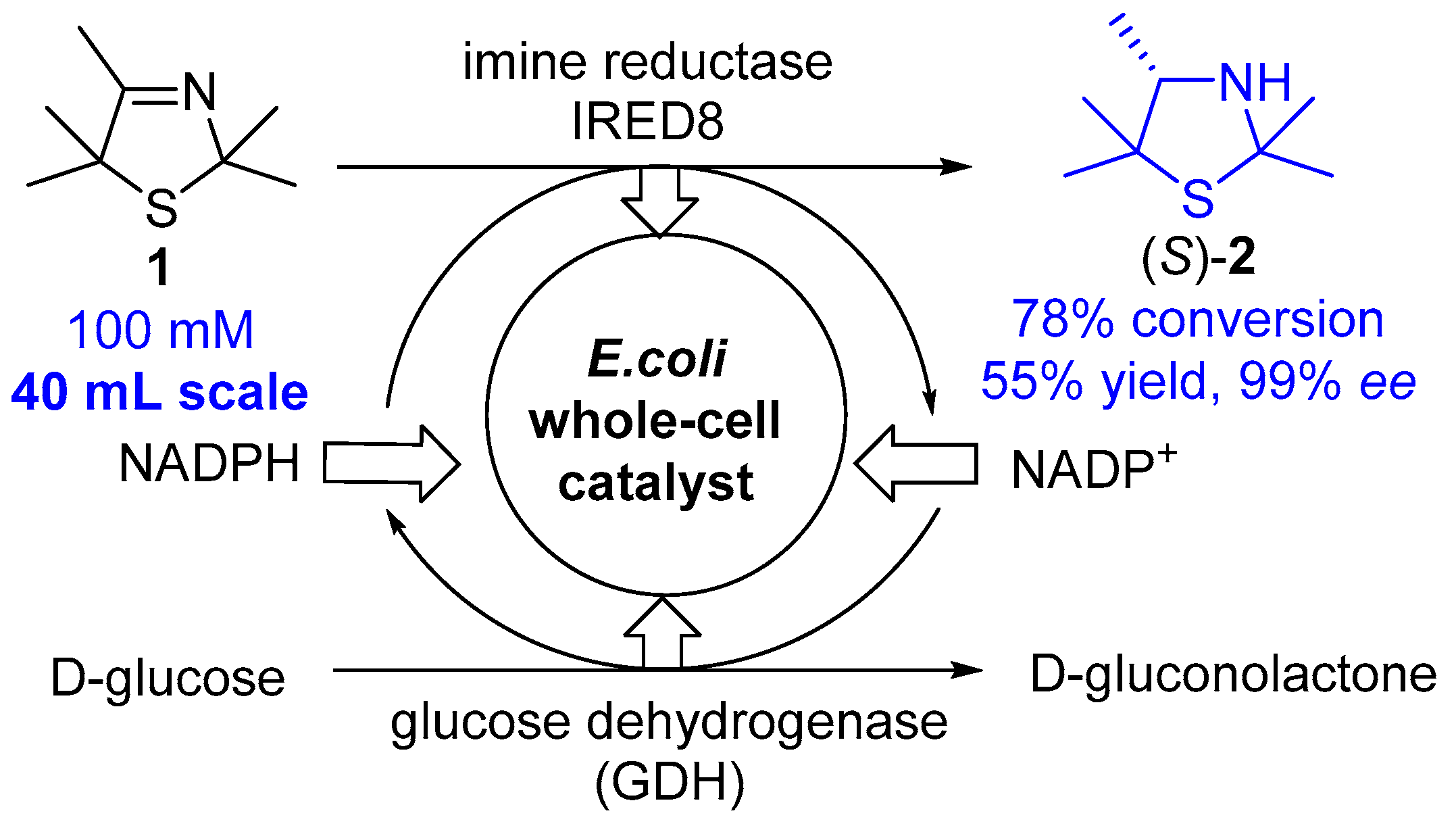
Scheme 2.
Biocatalytic enantioselective reduction of 3-thiazoline 1 to (S)-2 by means of an E. coli whole-cell catalyst bearing the imine reductase IRED8 from Mycobacterium smegmatis and glucose dehydrogenase from Bacillus subtilis.
2.3. 2,2,4,5,5-Pentamethyl-3-thiazoline (1)
According to reference [12] sodium hydrogen sulfide monohydrate (0.10 mol), ammonia-solution (13.3 M, 15 mL), and acetone (0.10 mol) were mixed and cooled to 4 °C. 3-Chloro-3-methyl-2-butanone (0.11 mol) was dissolved in dichloromethane (33 mL) and added to the yellow mixture, keeping the temperature under 10 °C. The reaction mixture was stirred overnight at room temperature, phases were separated, and the aqueous phase was extracted with dichloromethane (3 × 40 mL). The combined organic phases were dried over magnesium sulfate, the solvent was evaporated in vacuo, yielding 1 (7.68 g, 0.06 mol, 56%) as yellow oil via fractional distillation in vacuo (100 mbar, boiling point: 73–75 °C). 1H-NMR (500 MHz, CDCl3): δ [ppm] = 2.00 (s, 3 H, C4-CH3), 1.61 (s, 6 H, C2-(CH3)2) and 1.55 (s, 6 H, C5-(CH3)2). The analytical data corresponds with the literature data [12].
2.4. Achiral Gas Chromatography Analysis
According to reference [12] the determination of the conversion for the biotransformation of 3-thiazoline 1 to the corresponding thiazolidine (S)-2 was carried out by using the gas chromatography system GC-2010 Plus (Shimadzu, Kyoto, Japan) equipped with ZB-5MSi column (Phenomenex, Torrance, California, CA, USA, column dimensions 30 m × 0.25 mm × 0.25 μm; N2; linear velocity: 46.9 cm/s; split mode: 1:10; total flow rate: 28.8 mL/min; purge flow rate: 3.0 mL/min; column flow rate: 2.34 mL/min; pressure: 140.4 kPa) and coupled to an AOC-20i/s auto injector/auto sampler. Temperature range: 40–200 °C, temperature increase: 10 °C/min, observed retention times: tR (1): 4.6 min; tR (2): 5.1 min. The analytical data corresponds with the literature data [12].
2.5. (S)-2,2,4,5,5-Pentamethyl-3-thiazolidine (2)
According to reference [12], the biotransformation of 3-thiazoline 1 was performed on a 40 mL scale at 30 °C in 100 mM KPi buffer (pH: 7), with 2% methanol containing 240 mM d-glucose, 100 mM 3-thiazoline 1, 10 mg mL−1 lyophilized whole-cell catalyst and 0.5 mM NADP+. After 30 h, the reaction was stopped by adding 2 mL of 10 M NaOH solution and 40 mL dichloromethane. Phase separation was promoted by centrifugation and the conversion of 78% was determined by analyzing the organic phase by means of achiral GC. Flash column chromatography (ethyl acetate (5%–50% gradient) in cyclohexane) was performed yielding (S)-2,2,4,5,5-Pentamethyl-3-thiazolidine ((S)-2) (266 mg, 1.67 mmol, 55%). 1H-NMR (500 MHz, CDCl3): δ [ppm] = 3.27 (q, 3J = 6.6 Hz, 1H, C4-H), 1.60 (s, 3H, C2-CH3), 1.56 (s, 3H, C2-CH3), 1.41 (s, 3H, C5-CH3), 1.20 (s, 3H, C5-CH3), 1.11 (d, 3J = 6.49 Hz, 3H, C4-CH3). The analytical date corresponds with the literature data [12].
2.6. N-Isopropyl-3-methylbutan-2-amine (rac-3)
Acetone (80 mL, 1.08 mol), molecular sieve (0.4 nm) and 3-Methylbutan-2-amine (330 μL, 2.80 mmol) were mixed at room temperature. Concentrated sulfuric acid was added and the mixture was stirred for 20 h; the solvent was evaporated in vacuo. Sodium borohydride (219 mg, 5.80 mmol) dissolved in tetrahydrofuran (5 mL) was added and the mixture was stirred for 48 h at 30 °C. The reaction was stopped by adding water (50 mL) and 2 M NaOH solution (30 mL) and extracted with MTBE (3 × 40 mL). The combined organic phases were dried over magnesium sulfate, and the solvent was evaporated in vacuo and flash column chromatography (ethyl acetate (5%–100%) in cyclohexane) yielding rac-3 (36.0 mg, 0.27 mmol, 10%) as colorless oil. 1H-NMR (500 MHz, CDCl3): δ [ppm] = 4.56 (m, 1H, NCH-(CH3)2), 2.77 (m, 1H, NCH(CH3)), 1.92 (m, 1H, NC(CH3)CH-(CH3)2), 1.70 (d, 3J = 11.5 Hz, 3H, NCH(CH3)), 1.27–1.17 (m, 6H, NCH-(CH3)2, 0.95 (d, 3J = 6.9 Hz, 3H, NCH(CH3)CH-(CH3)2), 0.92 (d, 3J = 6.8 Hz, 3H, NCH(CH3)CH-(CH3)2). TLC-MS (ESI) m/z value calculated for C8H19N [M + H]+: 130.2, experimental value 130.2.
2.7. General Procedure 1 for Derivatization Using Phenylisocyanate
According to references [12,16], thiazolidine was dissolved in diethyl ether (5 mL), phenylisocyanate (1.3 equivalents (eq). or 2.6 eq.) was added, and the reaction mixture was stirred overnight while the solvent was evaporated in the meantime. The colorless solid was dried in vacuo.
rac-1-Isopropyl-1-(3-methylbutan-2-yl)-3-phenylurea (rac-4)
Preparation according to general procedure 1 using rac-3 (crude product) yielded rac-4 (72.6 mg, 0.31 mmol, 11%) as a colorless solid. 1H-NMR (500 MHz, CDCl3): δ [ppm] = 7.44–7.26 (m, 4H, Ar-H), 7.01 (m, 1H, Ar-H), 6.22 (s, 1H, NH), 3.94 (q, 3J = 6.9 Hz, 1H, NCH(CH3)), 3.36 (m, 1H, NCH-(CH3)2), 1.40–1.28 (m, 9H, NCH-(CH3)2, NCH(CH3)), 1.90 (m, 1H, NCH(CH3)-CH-(CH3)2), 0.99 (d, 3J = 7.0 Hz, 3H, NCH(CH3)-CH-(CH3)2), 0.97 (d, 3J = 7.6 Hz, 3H, NCH(CH3)-CH-(CH3)2). HRMS (ESI) m/z value calculated for C15H24N2O [M + Na]+: 271.1781,experimental value: 271.1779. IR (neat) [cm−1]: 3280, 2966, 2932, 2671, 1698, 1630, 1593, 1520, 1499, 1440, 1417, 1327, 1242, 1145, 1081, 743 and 692.
2.8. Attempts for the Synthesis of (S)-3 According to Ni/Al Alloy Routes a–c
2.8.1. Attempt a
According to references [17,18], thiazolidine (S)-2 (92.6 mg, 0.58 mmol) was dissolved in H2O (10 mL), methanol (2% v/v) and Ni/Al alloy (1.06 g; 50 wt% Ni, 50 wt% Al) are added. The mixture was heated to 90 °C and 1% aqueous (aq.) KOH (10 mL) was added. The suspension was stirred for 1 h at 90 °C. After filtration, the mixture was extracted with dichloromethane (3 × 20 mL). The combined organic phases were dried over magnesium sulfate, and the solvent was evaporated in vacuo. No formation of (S)-3 was observed.
2.8.2. Attempt b
According to reference [17], thiazolidine (S)-2 (107 mg, 0.67 mmol) was dissolved in 10% aq. NaOH (50 mL), ethanol (5 mL) heated to 90 °C and Ni/Al alloy (5.01 g, 50% Ni, 50% Al) were added carefully and the mixture was stirred for 2 h at 90 °C. After filtration, the mixture was washed with H2O (40 mL) and extracted with dichloromethane (3 × 30 mL). The combined organic phases were dried over magnesium sulfate, and the solvent was evaporated in vacuo. No formation of (S)-3 was observed.
2.8.3. Attempt c
In conformity with the work reported in reference [17], thiazolidine (S)-2 (104 mg, 0.67 mmol) was dissolved in 10% aq. NaOH (50 mL), toluene (5 mL) heated at 90 °C and Ni/Al alloy (5.02 g, 50% Ni, 50% Al) were added carefully and the mixture was refluxed for 5 h. No formation of (S)-3 was observed.
2.9. Attempts for the Synthesis of (S)-3 by Using Raney Ni (W4)
2.9.1. Attempt a
In conformity with the works reported in references [19,20] and according to reference [21], thiazolidine (S)-2 (100 mg, 0.63 mmol) was dissolved in ethanol (10 mL) and heated to 78 °C. Raney Ni (approx. 1 g) was added carefully and the mixture was refluxed for 2 h. After filtration over Celite®, the mixture was washed with dichloromethane (3 × 20 mL). The combined organic phases were dried over magnesium sulfate, and the solvent was evaporated in vacuo yielding yellow oil. Formation of a complex product mixture was confirmed by thin-layer chromatography.
2.9.2. Attempt b
In conformity with the works reported in references [19,20] and considering reference [22], thiazolidine (S)-2 (217 mg, 1.34 mmol) was dissolved in ethanol (10 mL) and heated to 78 °C under H2 atmosphere. An excess of Raney Ni (approx. 10 g) was added carefully and the mixture was refluxed for 4 h. The cooled suspension was filtrated over Celite® and the mixture was washed with dichloromethane (30 mL). A pH value of 10 was established using 1 M aq. NaOH, and the suspension was filtered and extracted with dichloromethane (3 × 30 mL). The combined organic phases were dried over magnesium sulfate, and the solvent was evaporated in vacuo yielding yellow oil. TLC-MS (ESI) m/z value calculated for C8H19N [M + H]+: 130.2, experimental value: 130.2.
2.10. (S)-N-Isopropyl-3-methylbutan-2-amine (S-3)
According to references [23,24], thiazolidine (S)-2 (110 mg, 0.69 mmol) was dissolved in ethanol (5 mL) and cooled to 4 °C. Nickel(II) chloride hexahydrate (480 mg, 2.31 mmol, 3 eq.) was added and the mixture was stirred for 1.5 h at room temperature. After filtration through Celite®, the mixture was washed with dichloromethane (3 × 30 mL). The combined organic phases were dried over magnesium sulfate, and the solvent was evaporated in vacuo yielding yellow oil.
2.11. (S)-1-Isopropyl-1-(3-methylbutan-2-yl)-3-phenylurea (S-4)
The preparation of (S)-4 was carried out according to general procedure 1 using (S)-N-isopropyl-3-methylbutan-2-amine (S-3) as yellow crude oil (from Section 2.10: Ni2B-route). Flash column chromatography (ethyl acetate (5%–40% gradient) in cyclohexane) gives (S)-4 (15.5 mg, 0.06 mmol, 10%, 99% ee) as a colorless solid. 1H-NMR (500 MHz, CDCl3): δ [ppm] = 7.39–7.27 (m, 2H, Ar-H), 7.30–7.23 (m, 2H, Ar-H), 7.1 (m, 1H, Ar-H), 6.23 (s, 1H, NH), 3.92 (m, 1H, NCH(CH3)), 3.36 (m, 1H, NCH-(CH3)2), 1.95 (m, 1H, NCH(CH3)-CH-(CH3)2), 1.37–1.29 (m, 9H, NCH(CH3)2, NCH(CH3)), 0.99 (d, 3J = 6.6 Hz, 3H, NCH(CH3)CH(CH3)2), 0.97 (d, 3J = 6.7 Hz, 3H, NCH(CH3)CH(CH3)2). 13C-NMR (127 MHz, CDCl3): δ [ppm] = 154.9 (CO), 139.4 (Ar-C), 128.9 (Ar-C), 122.6 (Ar-C), 119.7 (Ar-C), 57.2 (NCH(CH3)), 46.1 (NCH-(CH3)2), 32.9 (NCHCH-(CH3)2), 21.9 (NCH-(CH3)2), 21.7 (NCH-(CH3)2), 20.9 (NCHCH-(CH3)2), 20.8 (NCHCH-(CH3)2) and 18.4 (NCH-CH3). HRMS (ESI) m/z value calculated for C15H24N2O [M + Na]+: 271.1781, experimental value: 271.1776. IR (neat) [cm−1]: 3271, 2965 2925, 2870, 1626, 1593, 1521, 1499, 1440, 1417, 1374, 1319, 1243, 1146, 1054, 743 and 692.
2.12. General Procedure 2 for Synthesis of β-aminothiols
According to reference [2], lithium aluminium hydride powder (2 eq.) was dissolved in diethyl ether (5 mL) and cooled to 4 °C. 3-Thiazoline 1 or 3-thiazolidine (S)-2 were dissolved in diethyl ether (5 mL) and added to the reaction mixture. The suspension was stirred for 2 h at room temperature and was stopped by addition of water (10 mL), and the mixture was extracted with diethyl ether (3 × 30 mL). The combined organic phases were dried over magnesium sulfate and the solvent was evaporated in vacuo, yielding yellow oil.
2.12.1. S-(3-(1-Isopropyl-3-phenylureido)-2-methylbutan-2-yl)phenylcarbamothioate (rac-6)
The compound rac-6 was prepared according to general procedure 2 using 3-thiazoline 1 (200 mg, 1.27 mmol) and lithium aluminium hydride (98.3 mg, 2.54 mmol, 2 eq.), yielding a yellow oil. Without further purification, the derivatization was conducted according to general procedure 1 using phenylisocyanate (357 μL, 3.30 mmol, 2.6 eq.), yielding a colorless solid. Flash column chromatography (ethyl acetate (5%–40% gradient) in cyclohexane) gives rac-6 (149 mg, 0.37 mmol, 29%) as a colorless solid. 1H-NMR (500 MHz, CDCl3): δ [ppm] = 7.63 (d, 3J = 8.6, 2H, Ar-H), 7.44 (d, 3J = 7.77 Hz, 2H, Ar-H), 7.37–7.30 (m, 2H, Ar-H), 7.28–7.26 (m, 2H, Ar-H), 7.15 (t, 3J = 7.4 Hz, 1H, Ar-H), 6.97 (t, 3J = 7.4 Hz, 1H, Ar-H), 4.79 (m, 1H, NCH(CH3)), 3.41 (m, 1 H, NCH-(CH3)2), 1.81 (s, 3 H, SC-(CAH3)), 1.55 (d, 3J = 6.7 Hz, 3H, NCH(CH3)), 1.50 (d, 3J = 6.6 Hz, 6H, NCH-(CH3)2), 1.39 (s, 3H, SC-(CBH3)). HRMS (ESI) m/z value calculated for C22H29N3O2S [M + Na]+: 422.1873, experimental value: 422.1870. IR (neat) [cm−1]: 3342, 3232, 2973, 2924, 2848, 2361, 1634, 1541, 1497, 1442, 1330, 1245, 1108, 884, 745, 690 and 503.
2.12.2. (S)-S-(3-(1-Isopropyl-3-phenylureido)-2-methylbutan-2-yl)phenylcarbamothioate ((S)-6)
The compound (S)-6 was prepared according to general procedure 2 using thiazolidine (S)-2 (133 mg, 0.84 mmol) and lithium aluminium hydride (64.6 mg, 1.67 mmol, 2.3 eq.), yielding a yellow oil. Without further purification, the derivatization was conducted according to general procedure 1 using phenylisocyanate (177 μL, 1.64 mmol, 2.5 eq.), yielding a colorless solid. Flash column chromatography (ethyl acetate (5%–40% gradient) in cyclohexane) yielded (S)-6 (50.4 mg, 0.13 mmol, 20%, 99% ee) as a colorless solid. 1H-NMR (500 MHz, CDCl3): δ [ppm] = 7.63 (d, 3J = 8.2 Hz, 2 H, Ar-H), 7.46–7.41 (m, 2H, Ar-H), 7.34 (d, 3J = 8.5 Hz, 2H, Ar-H), 7.28–2.23 (m, 2H, Ar-H), 7.15 (m, 1H, Ar-H), 6.97 (t, 3J = 7.3 Hz, 1H, Ar-H), 4.79 (m, 1H, NCH(CH3)), 3.41 (m, 1H, NCH-(CH3)2), 1.81 (s, 3 H, SC-(CAH3)2), 1.55 (d, 3J = 6.8 Hz, 3H, NCH(CH3)), 1.50 (d, 3J = 6.6 Hz, 6H, NCH-(CH3)2), 1.39 (s, 3 H, SC-(CBH3)2). 13C-NMR (127 MHz, CDCl3): δ [ppm] = 179.9 (CO), 155.5 (CO), 129.2 (Ar-C), 128.6 (Ar-C), 122.0 (Ar-C), 119.6 (Ar-C), 55.9 (NCH(CH3)), 47.7 (NCH-(CH3)2), 26.9 (NCH(CH3)C-(CH3)2), 26.3 (NCH-(CH3)2), 24.3 (NCH-(CH3)2), 22.0 (NCH(CH3)C-(CH3)2), 20.4 (NCH(CH3)C-(CH3)2), 13.9 (NCH(CH3)). HRMS (ESI) m/z value calculated for C22H29N3O2S [M + Na]+: 422.1873, experimental value: 422.1872. IR (neat) [cm−1]: 3348, 3233, 2973, 2924, 2649, 2359, 1634, 1956, 1497, 1140, 1324, 1239, 1199, 1104, 881, 748, 688 and 502.
2.13. Chiral Supercritical Fluid Chromatography–High Performance Liquid Chromatography Analysis
Chiral SFC–HPLC analysis (for determination of the enantiomeric excess of (S)-2, rac-4, (S)-4, rac-6 and (S)-6) was performed using the LC2000 SFC-HPLC system from Jasco (Easton, USA) and the HPLC column Chiralpak IC from Daicel (Tokyo, Japan) (supercritical CO2:EtOH (Et2NH) = 10:90 (0.01) (v/v), 1 mL/min, 20 °C, 10 MPa, 210 nm): tR ((S)-2), 10.5 min [8a]; tR (rac-4), 10.6 min and 12.1 min; tR ((S)-4), 10.8 min (see Supplementary Materials Figures S1 and S2) or by the use of HPLC column Chiralpak OJ-H from Daicel (Tokyo, Japan) (supercritical CO2:2-Propanol (Et2NH) = 20:80 (0.01), 1 mL/min, 20 °C, 10 MPa, 210 nm): tR (rac-6), 23.7 min and 27.4 min; tR ((S)-6), 27.3 min (see Supplementary Materials Figures S3 and S4). Enantiomeric excess was determined based on area percentage of the enantiomers. The absolute configuration of the products 4 and 6 were assigned based on the assumption that the stereogenic center of the chiral substrate (S)-2 was not affected in the reactions leading to (S)-4 and (S)-6 (as the reactions did not take place at this stereogenic center).
3. Results and Discussion
3.1. Biocatalytic Synthesis of an Enantiomerically Pure Thiazolidine
At first, we chose the prochiral 2,2,4,5,5-pentamethyl-substituted 3-thiazoline 1 prepared via modified Asinger synthesis as the model substrate for our study. Following our recently developed protocol [12] for the biocatalytic reduction of 3-thiazolines in the presence of the recombinant imine reductase IRED8 from Mycobacterium smegmatis [25] and a glucose dehydrogenase from Bacillus subtilis [26] (utilized as our designed recombinant Escherichia coli (E. coli) whole-cell catalyst with a concentration of 2 mg/mL) [12] as well as a cofactor in catalytic amount and d-glucose as the reducing agent added in stoichiometric amount, we transformed this 3-thiazoline 1 into the thiazolidine (S)-2 on a 20 mM substrate concentration and on a 10 mL scale with a high conversion of >95% (see reference [12]: 82% conversion with 1.2 mg mL−1 IRED8 crude cell extract) and an excellent enantioselectivity of 99% ee. Since larger amounts of the thiazolidine (S)-2 were needed for the further studies on the planned synthesis of the envisaged desulfurization furnishing the secondary amine and the reduction to form the β-aminothiol, we carried out the biocatalytic reduction to 3-thiazolidine (S)-2 at an elevated lab scale of 40 mL using an increased substrate concentration of 100 mM utilizing a biocatalytic amount of 10 mg/mL of the E. coli whole-cell catalyst overexpressing IRED8 and GDH (Scheme 2). At this elevated lab scale and under these modified (although not optimized) conditions, a decrease of the conversion from 92% to 78% was observed. Additionally, under non-optimized conditions, a yield of 55% was obtained which was sufficient for the further chemical reductive trans-formations.
With this enantiomerically pure 3-thiazolidine (S)-2 in hand, we studied two different reductive approaches in order to cleave this 3-thiazolidine heterocycle (S)-2 with and without removal of sulfur, thus enabling an access to the corresponding desired enantiomerically pure secondary amine and β-aminothiol.
3.2. Reductive Desulfurization of Thiazolidine (S)-2
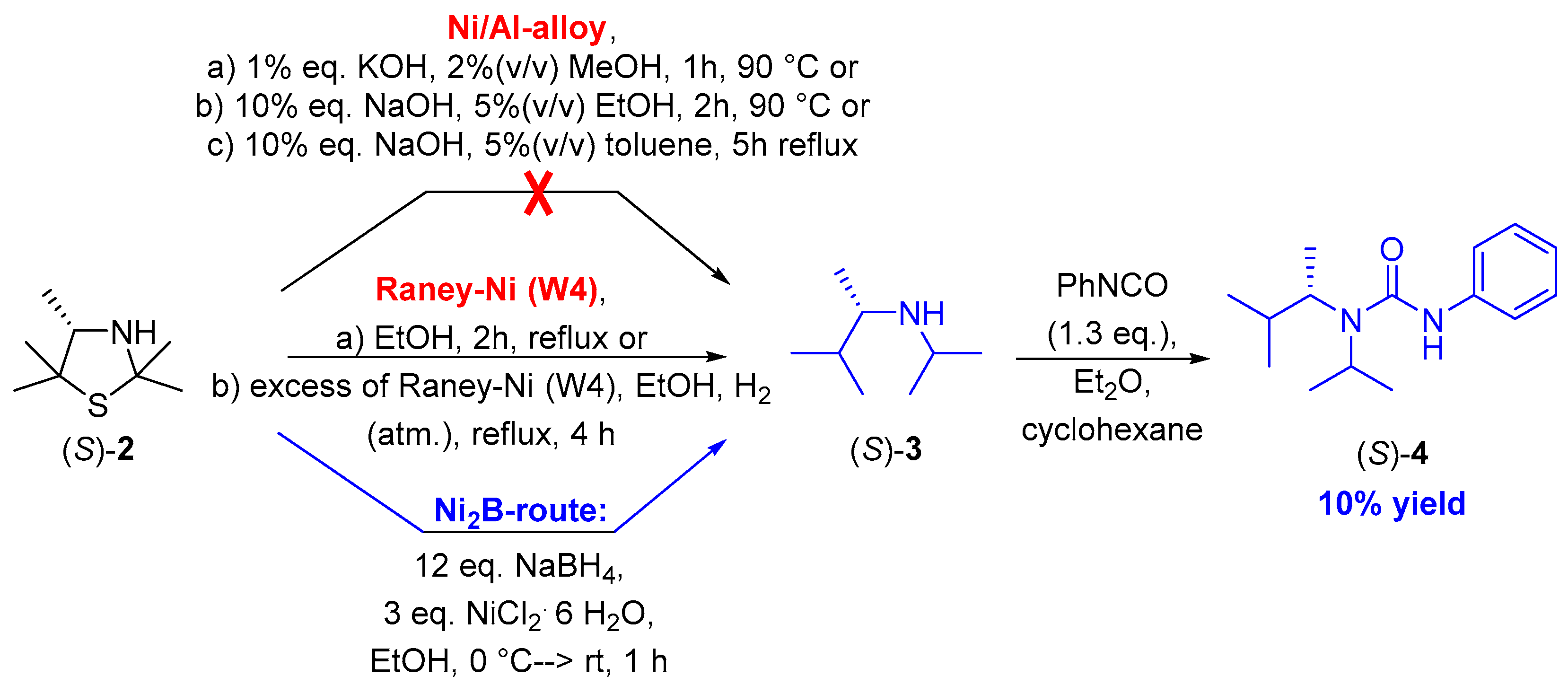
Therefore, we first conducted a screening of different methodologies which we considered to be suitable for the desired reductive desulfurization of this 3-thiazolidine (S)-2 using the racemic secondary amine rac-3, which was previously prepared, as a reference. The first method, which we applied in our experiments, was based on utilizing a Ni/Al-based alloy as a reducing agent following a protocol reported by Whitman et al. and Tsukinoki et al. (Scheme 3) [17,18]. Accordingly, a solution of (S)-2 in distilled water and methanol was treated with this Ni/Al-reducing agent, followed by adding a solution of KOH after increasing the temperature of this suspension to 90 °C. However, in these experiments, we did not observe any formation of the desired amine (S)-3. Further experiments in analogy to modified protocols developed by Whitman et al. [17] based on the use of a more concentrated base as well as modified solvents (ethanol and toluene) also did not give the desired product (S)-3 but showed a range of side products (Scheme 3). The formation of numerous side products, which are at least eight and seven for the latter two experiments (Scheme 3; Ni/Al alloys b and c, respectively), indicated a decomposition of the starting material. We were able to confirm this assumption by means of an experiment treating 3-thiazolidine (S)-2 in a solution of NaOH of the same concentration for two hours at 90 °C, showing a strong decomposition of this substrate according to 1H-NMR spectroscopy under such strong basic reaction conditions.
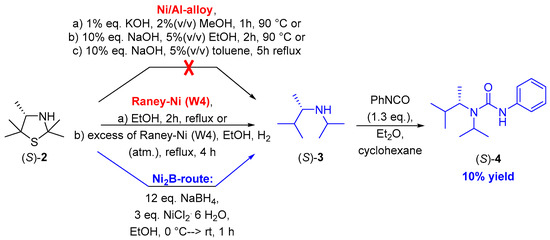
Scheme 3.
Screening of methodologies for the reductive desulfurization of 3-thiazolidine (S)-2 and prioritization of a preferred route leading to acyclic secondary amine (S)-3 and its urea derivative (S)-4.
In order to avoid such strong basic reaction conditions, next we evaluated a method disclosed by Mozingo et al., which is based on the utilization of Raney Ni as a reducing agent [19,20]. Accordingly, a Ni/Al-based alloy was added to a solution of NaOH at 25–50 °C, thus forming aluminate, molecular hydrogen and a sponge-like Raney nickel material. This “Ni sponge” allows molecular hydrogen to be attached to the surface, thus making an addition of external source of molecular hydrogen not necessary. The handling and storage of this in situ formed Raney Ni material turned out to be difficult (according to the literature showing one “tea spoon” of such a material corresponds to approximately 4 g of Raney Ni; Scheme 3) [20]. Although the first attempts of the desulfurization of thiazolidine (S)-2 by means of stoichiometric amount of such a Raney Ni furnished a complex product mixture as confirmed via thin-layer chromatography with at least seven compounds (see Reference [21]) and again no hint for the formation of the desired secondary amine, the subsequent use of an excess [19] of Raney Ni led to a product mixture containing the desired product (S)-3 as confirmed by 1H-NMR spectroscopy and mass spectrometrical analysis of the TLC spots. However, attempts for purification by chromatography or precipitation as either the amine or a derivative thereof (via formation of the urea derivative by conversion with phenylisocyanate) were not successful. It should be added that also in the literature such difficulties in product isolations for reductive desulfurizations with Raney Ni are mentioned [22].
As a third method for the reductive desulfurization we explored the in situ preparation of Ni2B and its use as a reducing agent following a procedure described by Sun et al. [23]. Toward this end, nickel(II) chloride was reduced with sodium borohydride, thus forming Ni2B and molecular hydrogen. In the literature [23,24], optimized results were found when operating at a molar ratio of NaBH4:substrate of 12:1 and at a molar ratio of NiCl2:substrate of 3:1. When conducting the reduction of 3-thiazolidine (S)-2 under such conditions, formation of the desired secondary amine (S)-3 was detected via mass spectrometric analysis of a corresponding TLC spot (m/z ratio of 130.2 corresponding to that of [M + H]+). As isolation again turned out to be difficult by column chromatography as well as short-path distillation, we decided to directly derivatize amine (S)-3 without isolation by transformation into its urea derivative (S)-4 through addition of phenylisocyanate (Scheme 3). We were pleased to find that, by means of subsequent automated column chromatography, the desired urea derivative (S)-4 could be isolated as a colorless solid, although the yield did not exceed 10% (Scheme 3). It is noteworthy that the enantiomeric purity remained excellent with an enantiomeric excess of 99% ee for the isolated product (S)-4, which was confirmed by chiral HPLC chromatography with the previously prepared racemic compound rac-4 as a reference. The deprotection of the urea compounds could be performed afterwards by means of the well-established solvolysis in neutral methanol as described by Hutchby et al. [27], leading towards the corresponding carbamate and the acyclic secondary amine (S)-3.
3.3. Reductive Ring Opening Toward Synthesis of β-aminothiols
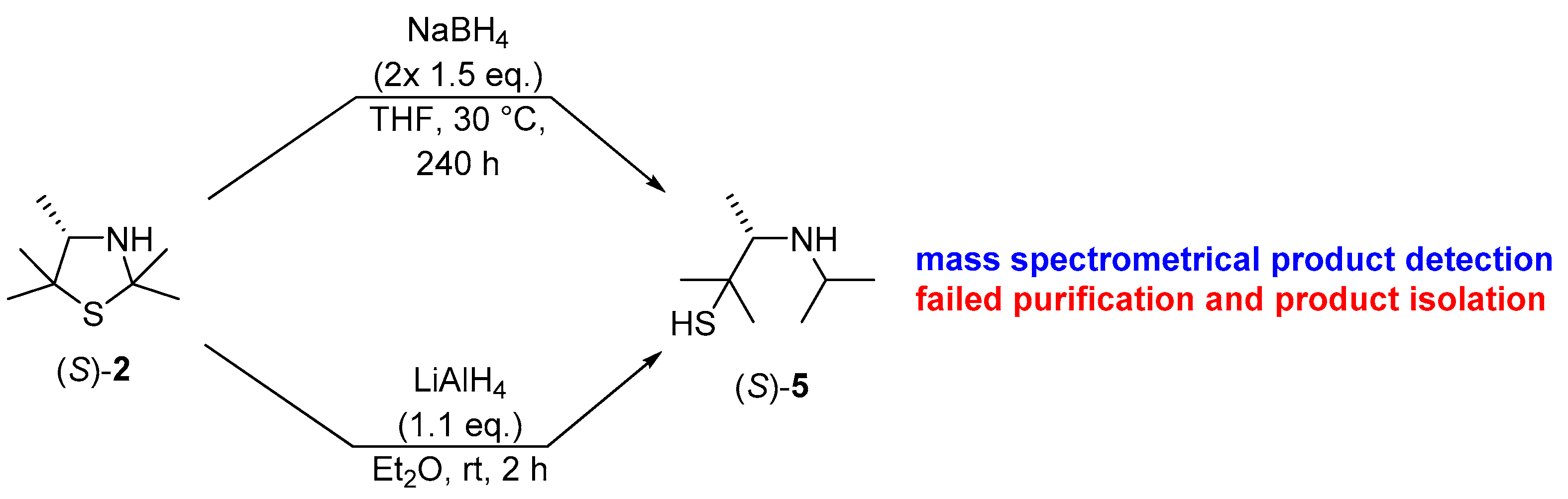
After this proof of concept for the conversion of an enantiomerically pure 3-thiazolidine into its secondary amine under retention of the excellent enantiomeric purity, we became interested if in analogy a reduction methodology could lead to reductive ring opening of the nitrogen, sulfur-acetal moiety, thus enabling the formation of the corresponding β-aminothiols with high enantiomeric excess when starting from the enzymatically prepared 3-thiazolidine. In this study, we utilized again the 3-thiazolidine (S)-2, showing an enantiomeric excess of 99% ee, as a model substrate. In modification of a previous method described by Martens et al. [16], a successful transformation was achieved when using directly non-modified sodium borohydride and expanding the reaction time to 10 days at a slightly increased reaction temperature of 30 °C (Scheme 4). Although formation of the desired product (S)-5 was detected by TLC spot-based mass spectrometry (m/z ratio of 162.3 for [M + H]+), again work-up, which was conducted by distillation in this case, turned out to be tedious.
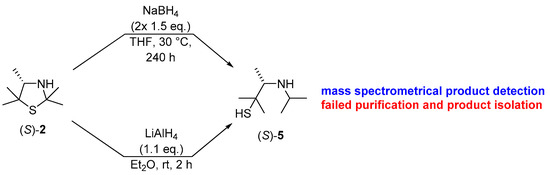
Scheme 4.
Initial studies on the reduction of the (S)-thiazolidine (S)-2 with metal hydrides and characterization of the resulting product (S)-5 via mass spectrometry.
Thus, as a further alternative, the use of lithium aluminum hydride has been explored in analogy to protocols reported by Asinger et al. [28], Gröger et al. [12] and Jeon et al. [29] (Scheme 4). The successful formation of β-aminothiol (S)-5 was again confirmed through mass spectrometry (m/z ratio of 162.2 for [M + H]+) and through 1H-NMR spectroscopy of the crude product.
In order to remove traces of impurities, preparative thin-layer separation was investigated but again led to non-satisfactory results. In this connection, it also appears that decomposition products were formed during the chosen procedure. Thus, in order to circumvent difficulties at the stage of the product isolation of the β-aminothiol (S)-5, we again decided to trap these products by addition of phenylisocyanate, thus forming the urea derivative (S)-6 (Scheme 5).
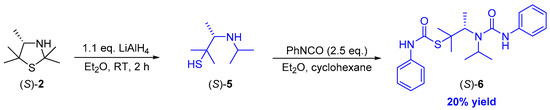
Scheme 5.
Reduction of 3-thiazolidine (S)-2 under formation of β-aminothiol (S)-5 and subsequent derivatization with phenylisocyanate towards the urea derivative (S)-6.
We were pleased to find that through column chromatographical purification the bis-derivatized β-aminothiol (S)-5 could be obtained in 20% yield as a colorless solid. Additionally, it is noteworthy that the enantiomeric excess of the bis-carbamate (S)-6 remained as high as 99% ee (according to chiral HPLC using the racemic compound rac-6 being prepared beforehand as a reference) and was not influenced by the reductive ring opening process of 3-thiazolidine (S)-2 or the subsequent double carbamoylation of β-aminothiol (S)-5. The removal of the protection groups could be performed afterwards by a two-step deprotection procedure including the well-established solvolysis in neutral methanol and a base-catalyzed degradation of thiocarbamates in an aqueous solution as described by Reisser et al. [27,30].
4. Conclusions
A proof of concept for a novel approach towards enantiomerically highly enriched acyclic secondary amines and β-aminothiols as non-cyclic target molecules when starting from 3-thiazolines as heterocycles has been presented. In detail, we exemplified this chemoenzymatic pathway to both of these types of molecules for the synthesis of (S)-3 and (S)-5, and both of these compounds were isolated as urea derivatives (S)-4 and (S)-6, respectively, with a non-optimized yield of up to 20%. As a substrate for the reductive ring-opening steps, 3-thiazolidine (S)-2, which was obtained with 99% ee in a biotransformation from 3-thiazoline 1 according to a recently developed protocol, was used. For the reductive desulfurization of (S)-2 leading to secondary amine (S)-3, in situ formed Ni2B turned out to be a suitable reagent. When using lithium aluminum hydride as a reducing agent, (S)-2 was converted into the desired β-aminothiol (S)-5. Based on this proof of concept work, the expansion of the substrate scope represents a task for the next current and future research steps.
Supplementary Materials
The following are available online at https://www.mdpi.com/2624-8549/1/1/12/s1.
Author Contributions
Conceptualization, M.H., N.Z., H.O. and H.G.; methodology, M.H., N.Z. and H.G.; validation, M.H.; formal analysis, M.H.; investigation, M.H.; resources, H.G.; data curation, M.H.; writing of original draft preparation, M.H., N.Z. and H.G.; writing of review and editing, M.H., N.Z., H.O. and H.G.; supervision, H.G.; project administration, H.G.; funding acquisition, H.G.
Funding
This research was funded by the German Federal Ministry of Education and Research (Bundesministerium für Bildung und Forschung, BMBF) within the project “Biotechnologie 2020+, Nächste Generation biotechnologischer Verfahren” (grant number: 031A184A).
Acknowledgments
We acknowledge support for the Article Processing Charge by the Deutsche Forschungsgemeinschaft (DFG) and the Open Access Publication Fund of Bielefeld University. The authors thank Hans Iding and Dennis Wetzl for providing us with the plasmid encoding for the imine reductase IRED8.
Conflicts of Interest
The authors declare no conflicts of interest
References
- Keim, W.; Offermanns, H. Friedrich Asinger (1907–1999): A mediator between basic and applied research. Angew. Chem. Int. Ed. 2007, 46, 6010–6013. [Google Scholar] [CrossRef] [PubMed]
- Asinger, F.; Offermanns, H. Syntheses with ketones, sulfur, and ammonia or amines at room temperature. Angew. Chem. Int. Ed. 1967, 6, 907–919. [Google Scholar] [CrossRef]
- Asinger, F. Über die gemeinsame Einwirkung von Schwefel und Ammoniak auf Ketone. Angew. Chem. 1956, 68, 413. [Google Scholar] [CrossRef]
- Weigert, W.M.; Offermanns, H.; Scherberich, P. d-Penicillamine--production and properties. Angew. Chem. Int. Ed. 1975, 14, 330–336. [Google Scholar] [CrossRef] [PubMed]
- Martens, J.; Kintscher, J.; Arnold, W. Synthese von 4-Thiazolidinessigsäuren und β-Homopenicillamin. Synthesis 1991, 1991, 497–498. [Google Scholar] [CrossRef]
- Kintscher, J.; Martens, J. Vereinfachte Peptidsynthese mit schutzgruppenfreien Aminosäure-Hydrochloriden nach dem Prinzip der Vierkomponenten-Kondensation. Synthesis 1992, 1992, 837–838. [Google Scholar] [CrossRef]
- Gröger, H.; Hatam, M.; Kintscher, J.; Martens, J. Synthesis of Glutathione Analogues, Peptide Nucleic Acids and Phosphonooligopeptides from Heterocyclic Imines. Synth. Commun. 1996, 26, 3383–3394. [Google Scholar] [CrossRef]
- Drauz, K.; Koban, H.G.; Martens, J.; Schwarze, W. Phosphonic and Phosphinic Acid Analogs of Penicillamine. Liebigs Ann. Chem. 1985, 1985, 448–452. [Google Scholar] [CrossRef]
- Gröger, H.; Saida, Y.; Arai, S.; Martens, J.; Sasai, H.; Shibasaki, M. First catalytic asymmetric hydrophosphonylation of cyclic imines: Highly efficient enantioselective approach to a 4-thiazolidinylphosphonate via chiral titanium and lanthanoid catalysts. Tetrahedron Lett. 1996, 37, 9291–9292. [Google Scholar] [CrossRef]
- Gröger, H.; Saida, Y.; Sasai, H.; Yamaguchi, K.; Martens, J.; Shibasaki, M. A New and Highly Efficient Asymmetric Route to Cyclic α-Amino Phosphonates: The First Catalytic Enantioselective Hydrophosphonylation of Cyclic Imines Catalyzed by Chiral Heterobimetallic Lanthanoid Complexes. J. Am. Chem. Soc. 1998, 120, 3089–3103. [Google Scholar] [CrossRef]
- Schlemminger, I.; Saida, Y.; Gröger, H.; Maison, W.; Durot, N.; Sasai, H.; Shibasaki, M.; Martens, J. Concept of improved rigidity: How to make enantioselective hydrophosphonylation of cyclic imines catalyzed by chiral heterobimetallic lanthanoid complexes almost perfect. J. Org. Chem. 2000, 65, 4818–4825. [Google Scholar] [CrossRef] [PubMed]
- Zumbrägel, N.; Merten, C.; Huber, S.M.; Gröger, H. Enantioselective reduction of sulfur-containing cyclic imines through biocatalysis. Nat. Commun. 2018, 9, 1949. [Google Scholar] [CrossRef] [PubMed]
- Zumbrägel, N.; Gröger, H. Merging Heterocyclic Chemistry and Biocatalysis in One-Pot Processes through Compartmentalization of the Reaction Steps. Bioengineering 2018, 5, 60. [Google Scholar] [CrossRef] [PubMed]
- Zumbrägel, N.; Gröger, H. One-pot synthesis of a 3-thiazolidine through combination of an Asinger-type multi-component-condensation reaction with an enzymatic imine reduction. J. Biotechnol. 2019, 291, 35–40. [Google Scholar] [CrossRef] [PubMed]
- Meinzer, A.; Breckel, A.; Thaher, B.A.; Manicone, N.; Otto, H.-H. Properties and Reactions of Substituted 1,2-Thiazetidine 1,1-Dioxides: Chiral Mono- and Bicyclic 1,2-Thiazetidine 1,1-Dioxides from α-Amino Acids. Helv. Chim. Acta 2004, 87, 90–105. [Google Scholar] [CrossRef]
- Reiners, I.; Gröger, H.; Martens, J. A New Enantioselective Synthetic Approach to β-Aminothio-Compoundsvia enantioselective reduction of N,S-heterocyclic imines. J. Prakt. Chem. 1997, 339, 541–546. [Google Scholar] [CrossRef]
- Papa, D.; Schwenk, E.; Whitman, B. Reductions with nickel-aluminium alloy and aqueous alkali. J. Org. Chem. 1942, 07, 587–590. [Google Scholar] [CrossRef]
- Tsukinoki, T.; Kanda, T.; Liu, G.-B.; Tsuzuki, H.; Tashiro, M. Organic reaction in water. Part 3: A facile method for reduction of aromatic rings using a raney Ni–Al alloy in dilute aqueous alkaline solution under mild conditions. Tetrahedron Lett. 2000, 41, 5865–5868. [Google Scholar] [CrossRef]
- Mozingo, R.; Wolf, D.E.; Harris, S.A.; Folkers, K. Hydrogenolysis of Sulfur Compounds by Raney Nickel Catalyst. J. Am. Chem. Soc. 1943, 65, 1013–1016. [Google Scholar] [CrossRef]
- Mozingo, R. Catalyst, Raney Nickel, W-2. Org. Synth. 1941, 21, 15. [Google Scholar] [CrossRef]
- Rentner, J.; Kljajic, M.; Offner, L.; Breinbauer, R. Recent advances and applications of reductive desulfurization in organic synthesis. Tetrahedron 2014, 70, 8983–9027. [Google Scholar] [CrossRef]
- Keefer, L.K.; Lunn, G. Nickel-aluminum alloy as a reducing agent. Chem. Rev. 1989, 89, 459–502. [Google Scholar] [CrossRef]
- Meng, X.; Li, L.; Li, K.; Zhou, P.; Zhang, H.; Jia, J.; Sun, T. Desulfurization of fuels with sodium borohydride under the catalysis of nickel salt in polyethylene glycol. J. Clean Prod. 2018, 176, 391–398. [Google Scholar] [CrossRef]
- Ohshima, T.; Xu, Y.; Takita, R.; Shimizu, S.; Zhong, D.; Shibasaki, M. Enantioselective total synthesis of (-)-strychnine using the catalytic asymmetric Michael reaction and tandem cyclization. J. Am. Chem. Soc. 2002, 124, 14546–14547. [Google Scholar] [CrossRef] [PubMed]
- Wetzl, D.; Berrera, M.; Sandon, N.; Fishlock, D.; Ebeling, M.; Müller, M.; Hanlon, S.; Wirz, B.; Iding, H. Expanding the Imine Reductase Toolbox by Exploring the Bacterial Protein-Sequence Space. ChemBioChem 2015, 16, 1749–1756. [Google Scholar] [CrossRef] [PubMed]
- Vázquez-Figueroa, E.; Chaparro-Riggers, J.; Bommarius, A.S. Development of a thermostable glucose dehydrogenase by a structure-guided consensus concept. ChemBioChem 2007, 8, 2295–2301. [Google Scholar] [CrossRef] [PubMed]
- Hutchby, M.; Houlden, C.E.; Ford, J.G.; Tyler, S.N.G.; Gangné, M.R.; Lloyd-Jones, G.C.; Booker-Milburn, K.I. Hindered Ureas as Masked Isocyanates: Facile Carbamolyation of Nucleophiles under Neutral Conditions. Angew. Chem. Int. Ed. 2009, 48, 8721–8724. [Google Scholar] [CrossRef]
- Thiel, M.; Asinger, F.; Häussler, K.; Körner, T. Über die gemeinsame Einwirkung von elementarem Schwefel und gasförmigem Ammoniak auf Ketone, XXII: Mercaptoamine durch Reduktion von Thiazolinen-Δ3 oder Dihydro-metathiazinen-Δ3 mit Lithiumalanat. Liebigs Ann. Chem. 1959, 622, 107–116. [Google Scholar] [CrossRef]
- Jeon, H.B.; Jang, Y. Reversible inactivation of bovine plasma amine oxidase by cysteamine and related analogs. Biochem. Biophys. Res. Commun. 2010, 403, 442–446. [Google Scholar] [CrossRef]
- Reisser, M.; Schmidt, B.F.; Brown, W. Synthesis, Characterization, and Solvolysis of Mono- and Bis-S-(glutathionyl) Adducts of Methylene-bis-(phenylisocyate) (MDI). Chem. Res. Toxicol. 2002, 15, 1235–1241. [Google Scholar] [CrossRef]
Sample Availability: Samples of the compounds are not available from the authors. |
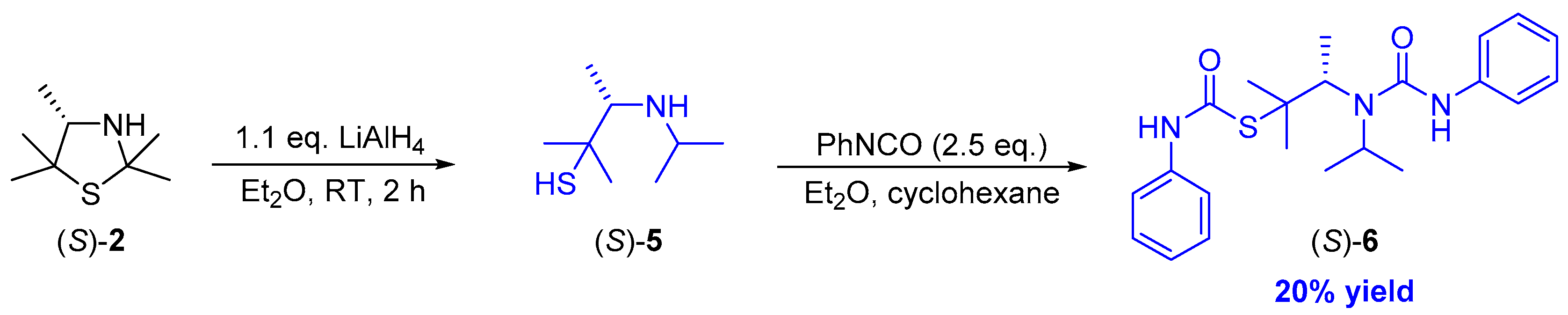
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).