
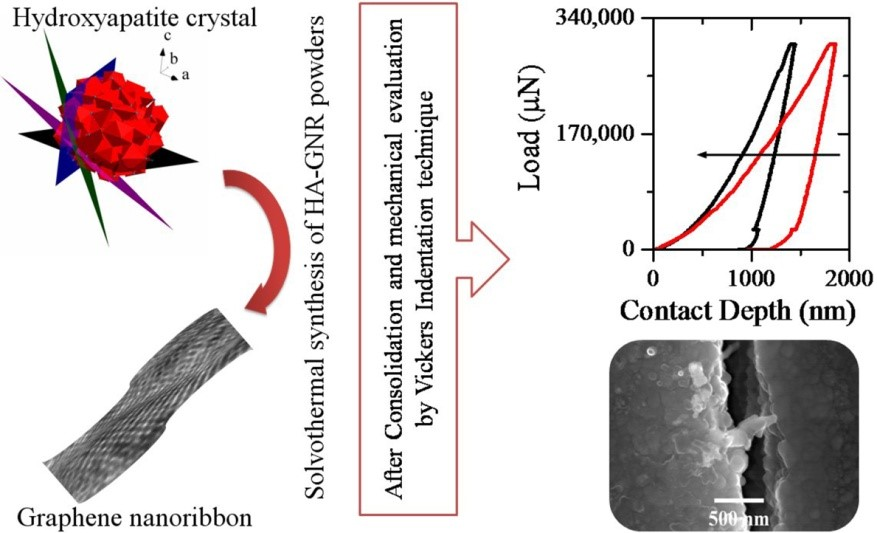
Synthesis of Graphene Nanoribbons–Hydroxyapatite Nanocomposite Applicable in Biomedicine and Theranostics



Abstract
:
1. Introduction
2. Materials and Methods
2.1. Evaluation of the SinteredSamples
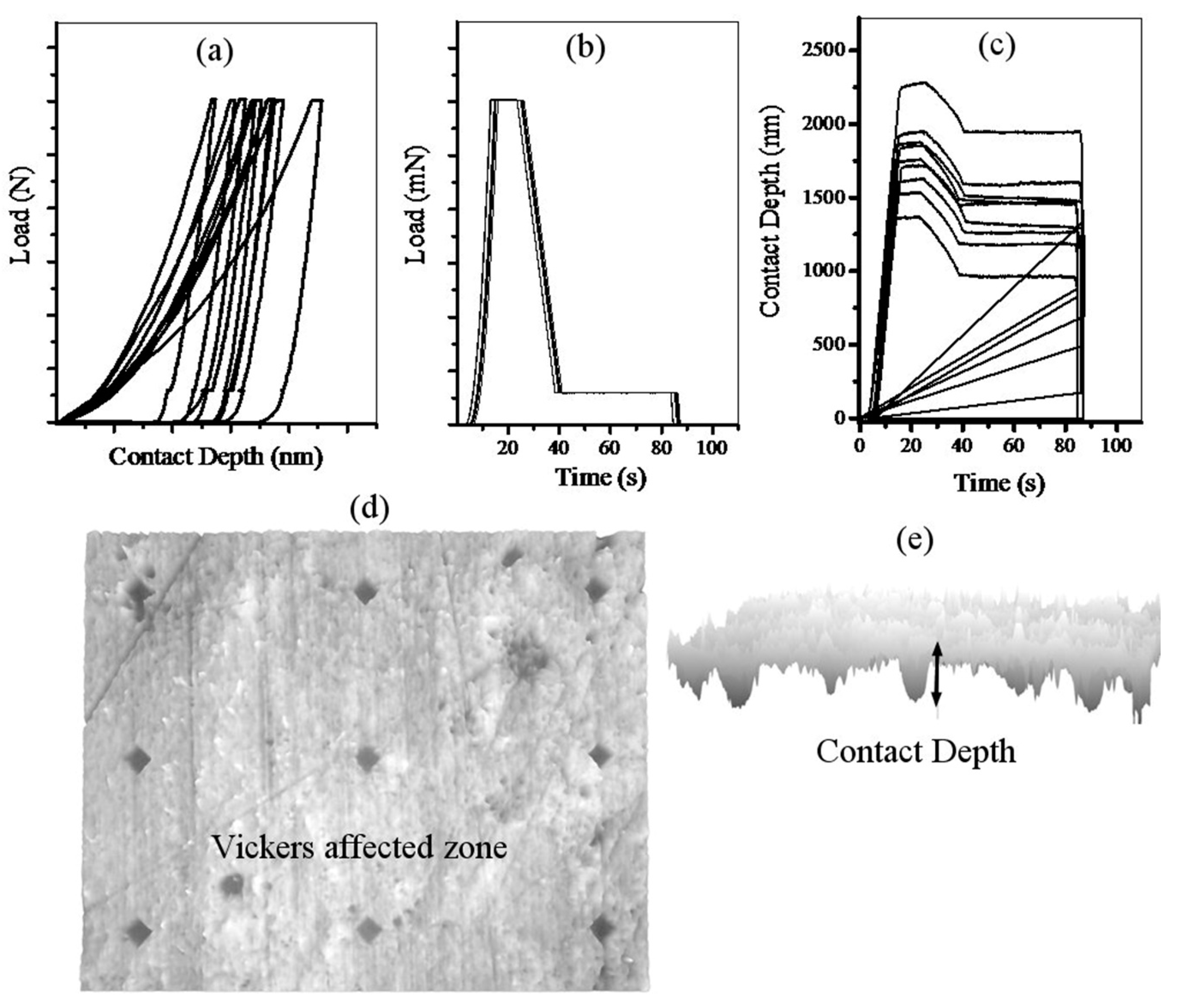
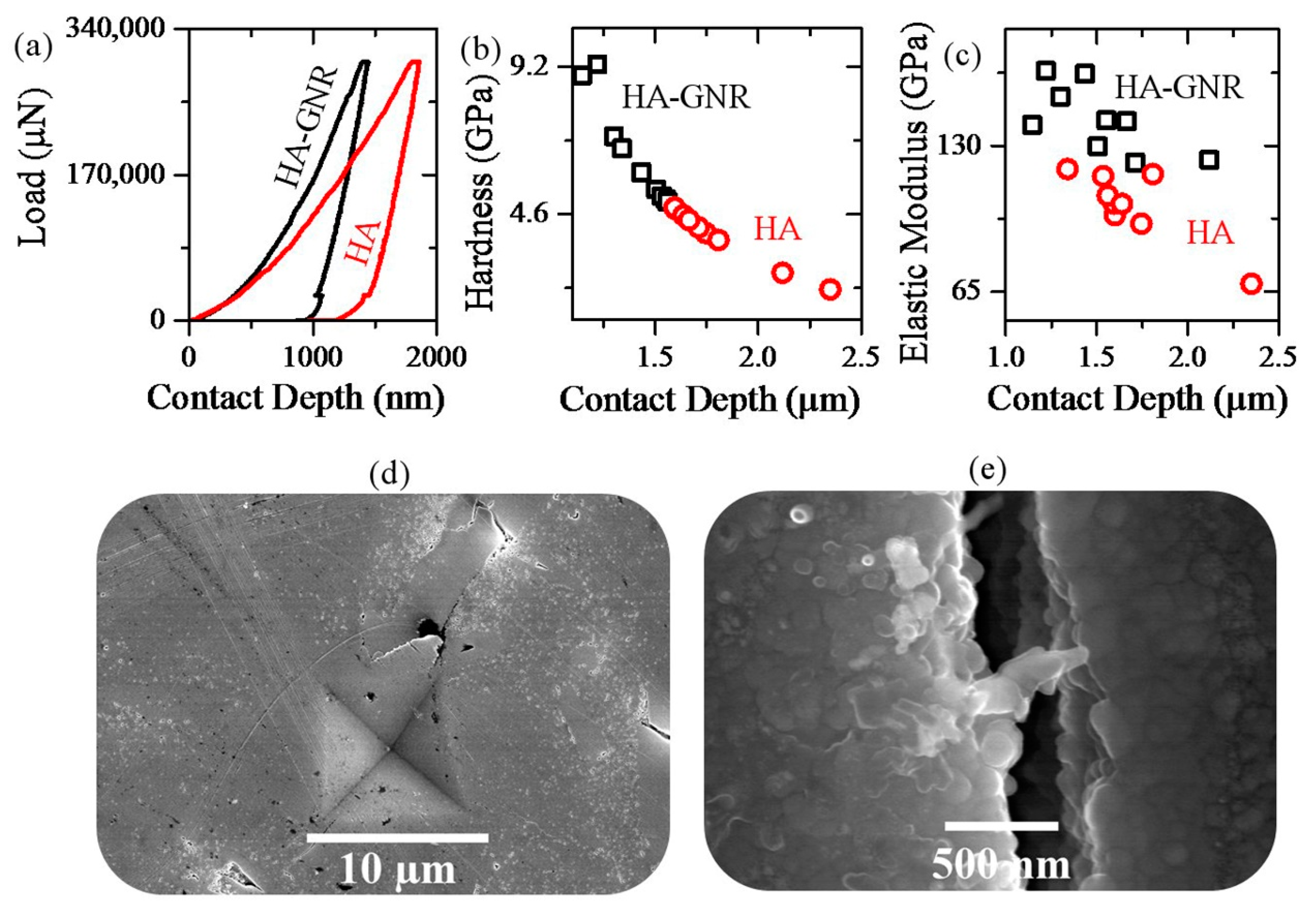
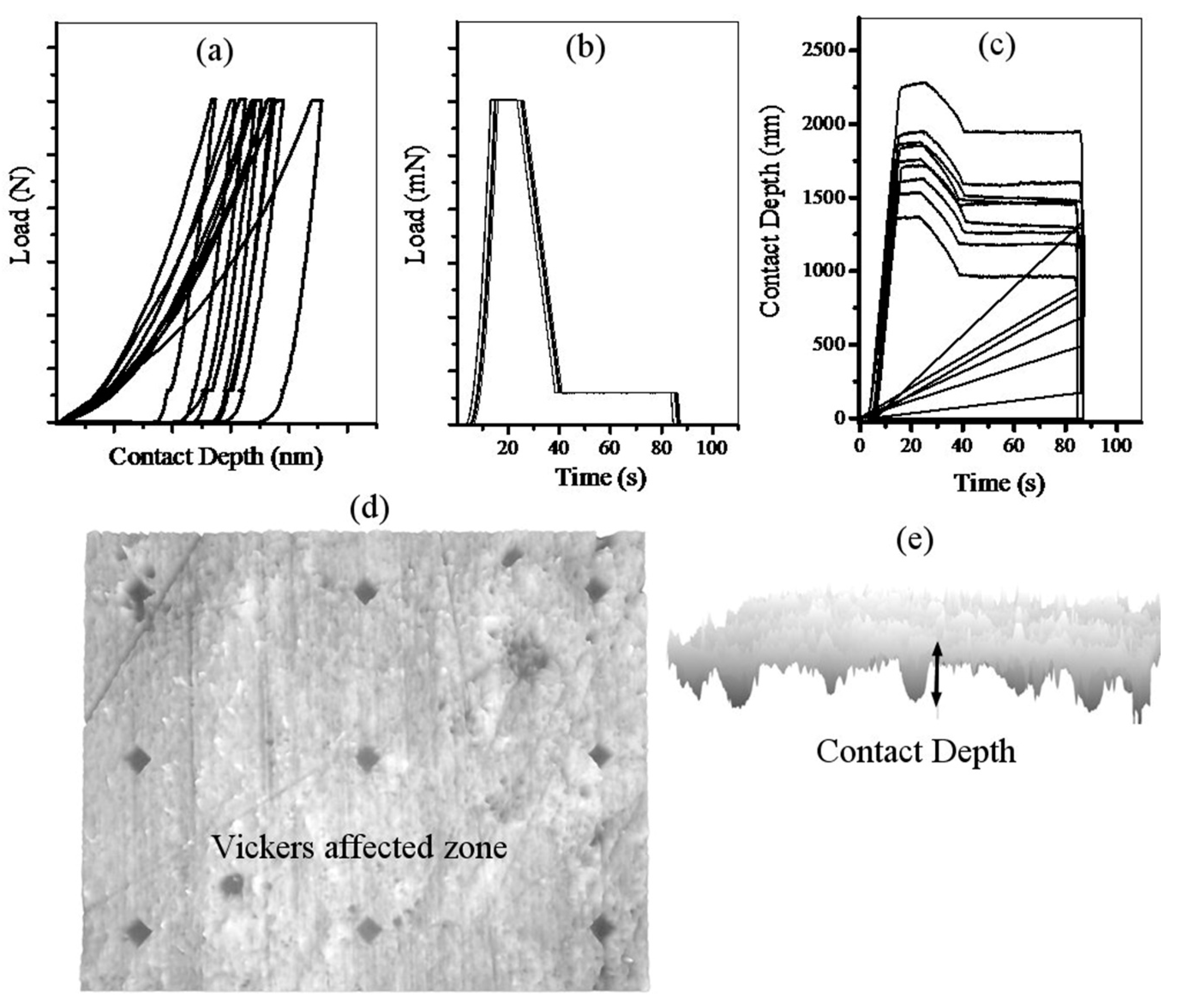
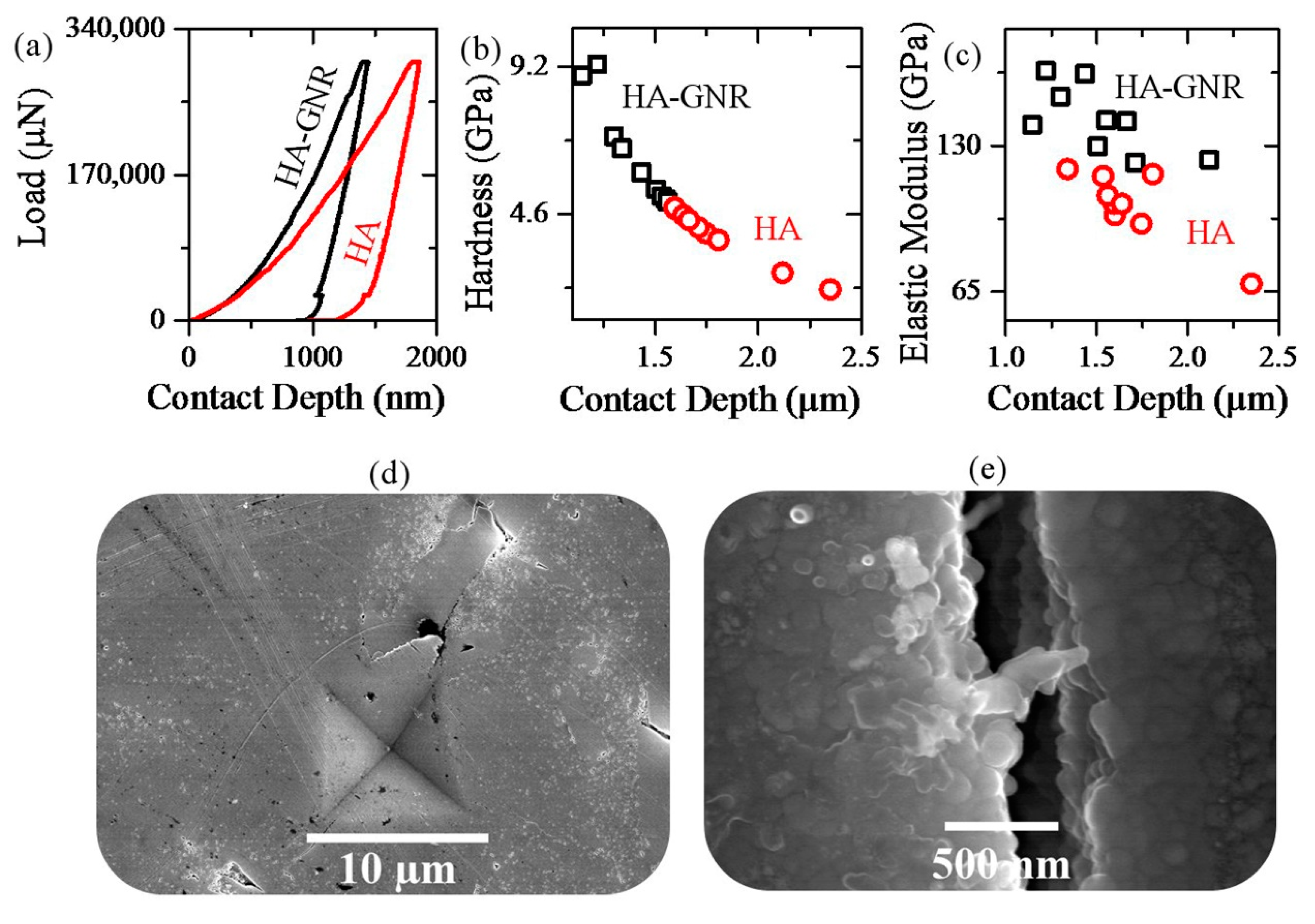
2.2. Vickers Indentation
2.3. Characterization Techniques
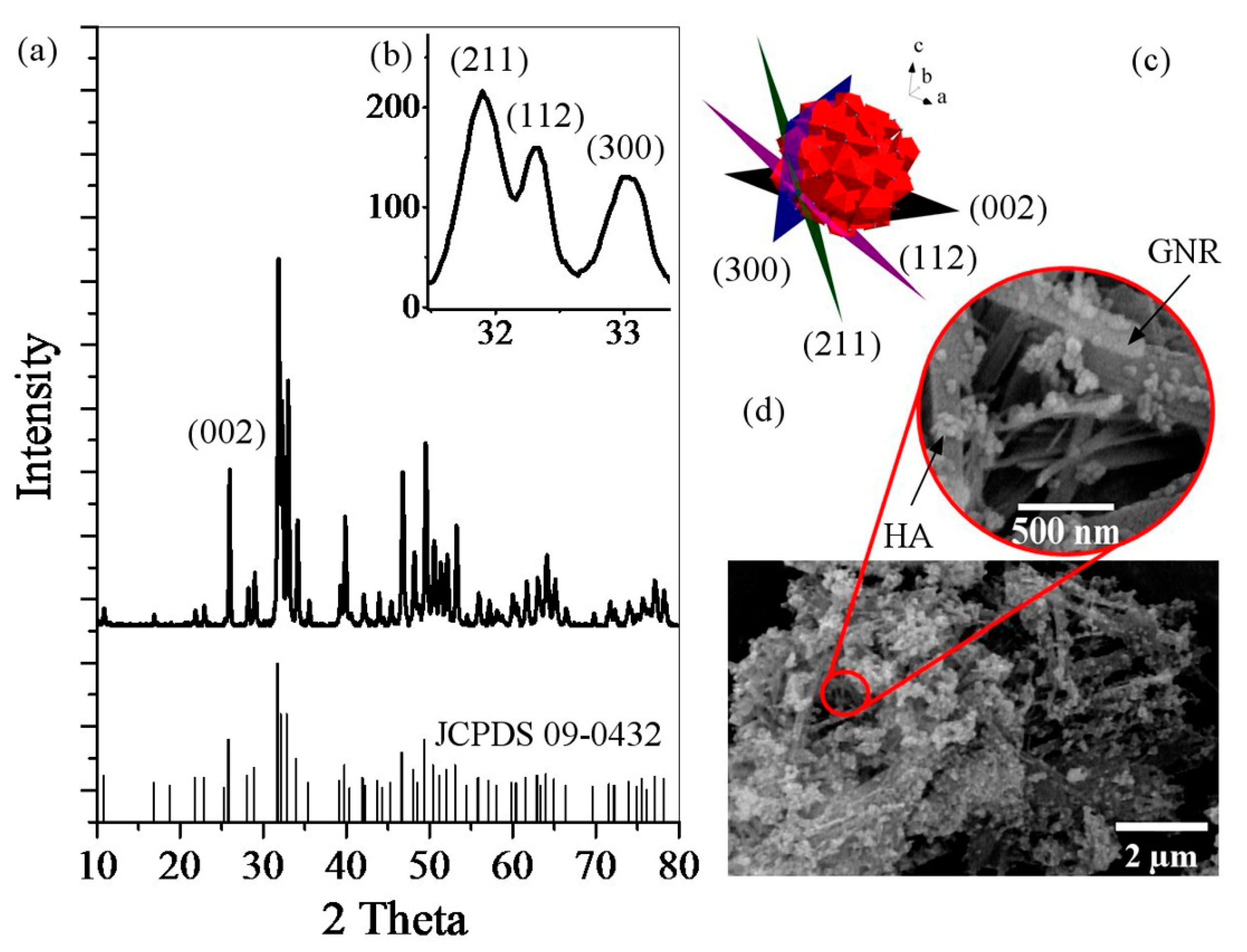
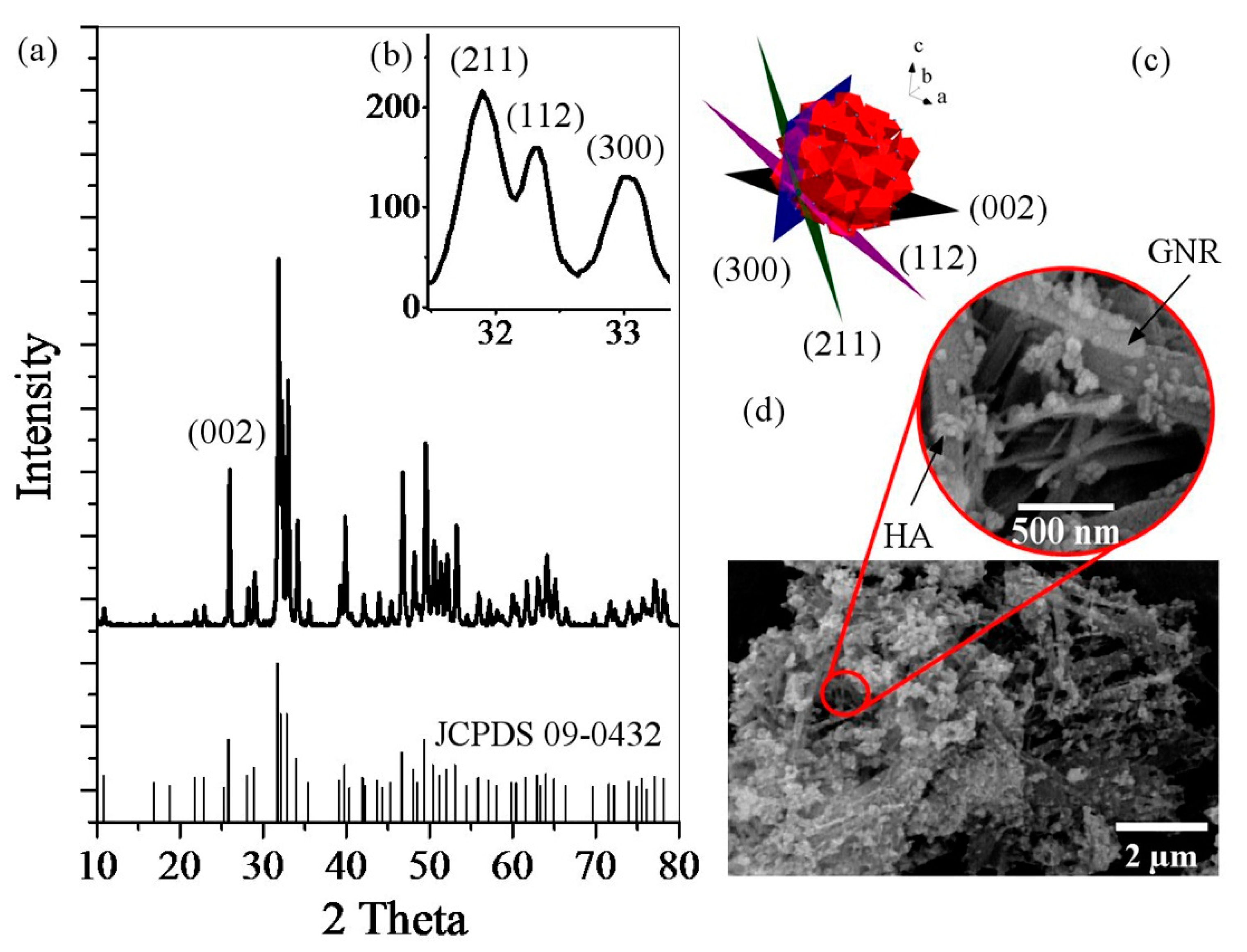
2.3.1. X-Ray Diffraction
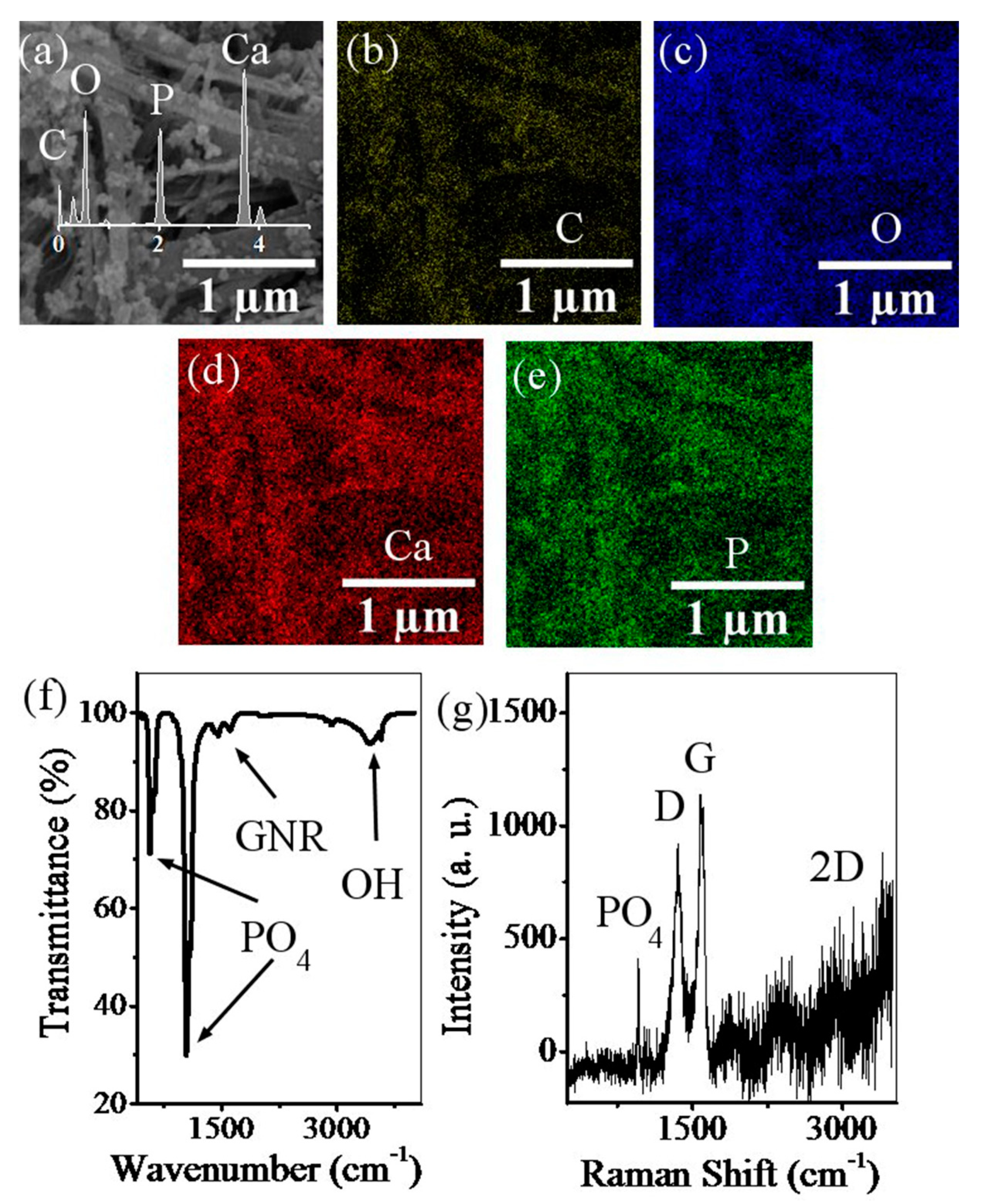
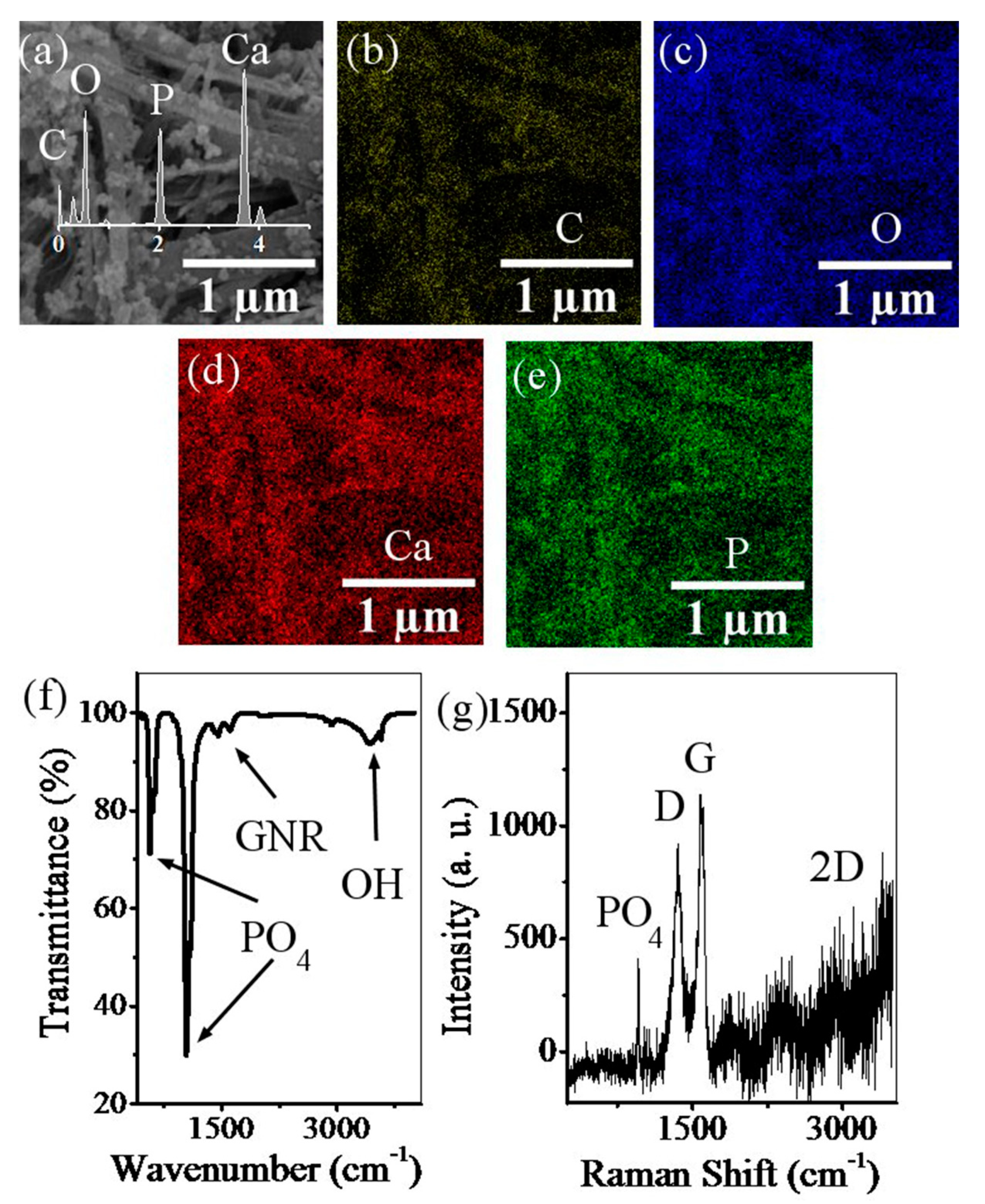
2.3.2. Field Emission Scanning Electron Microscopy
2.3.3. Fourier Transform Infrared Spectroscopy
2.3.4. Raman Spectroscopy
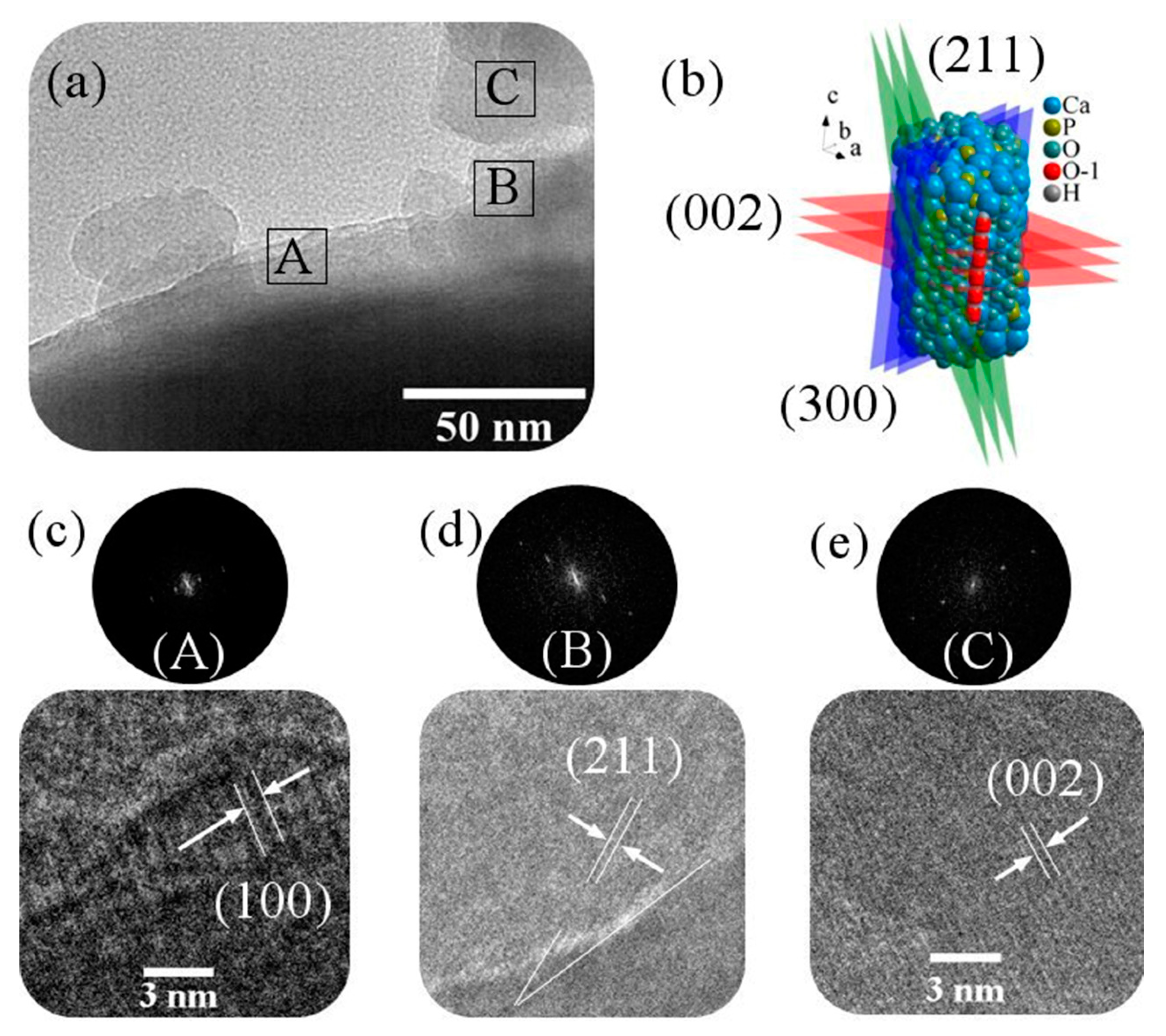
2.3.5. High-Resolution Transmission Electron Microscopy
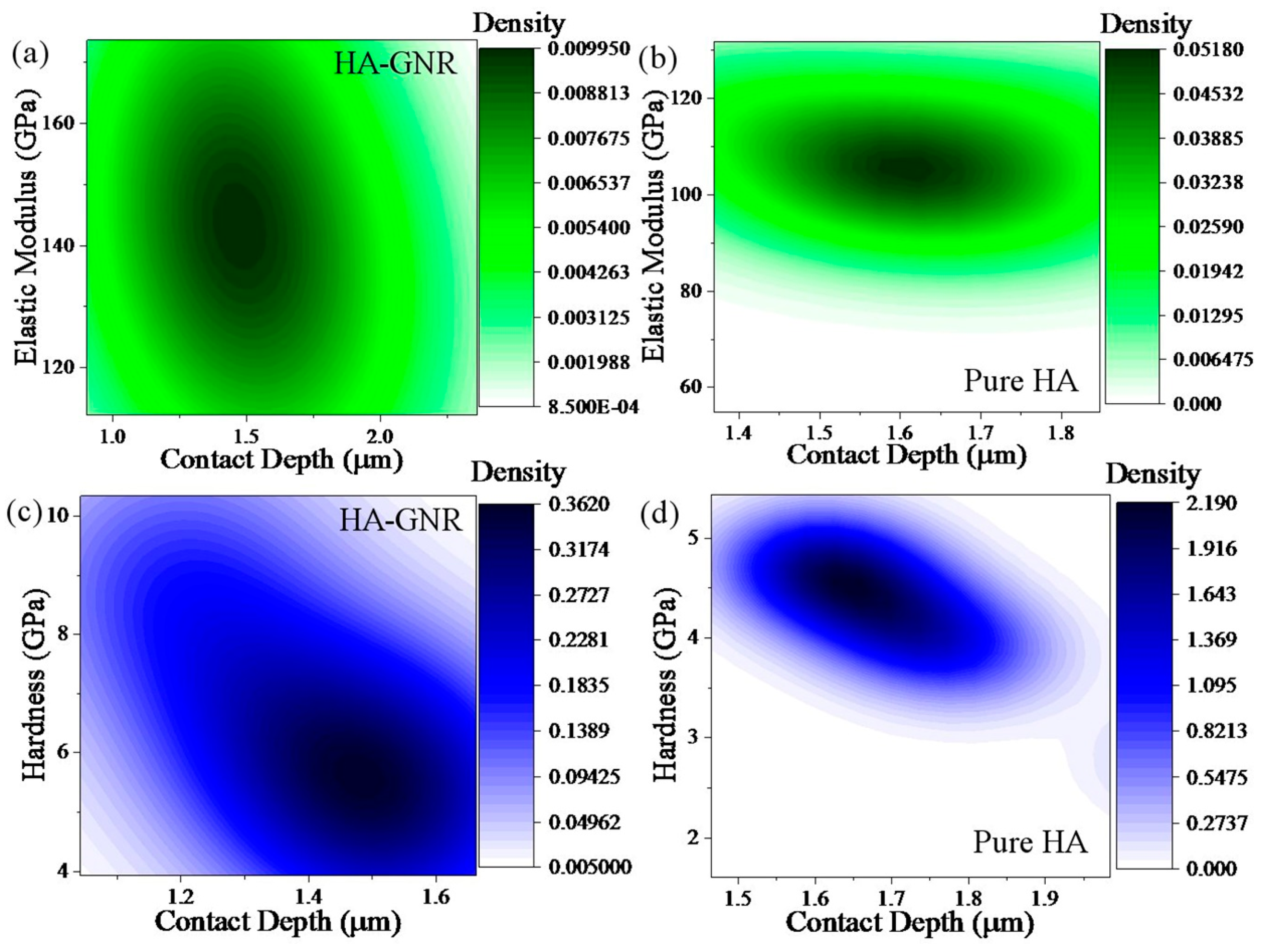
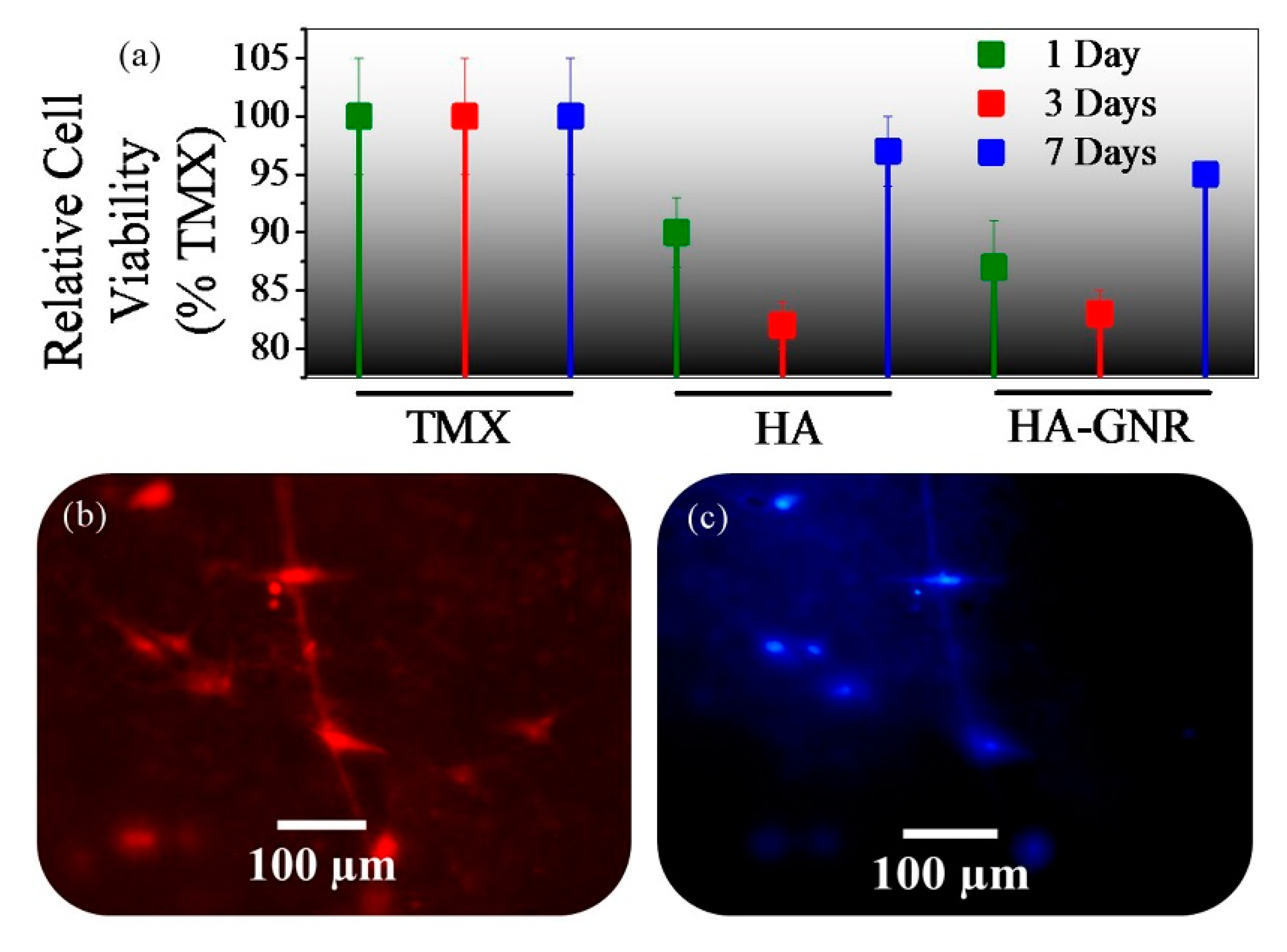
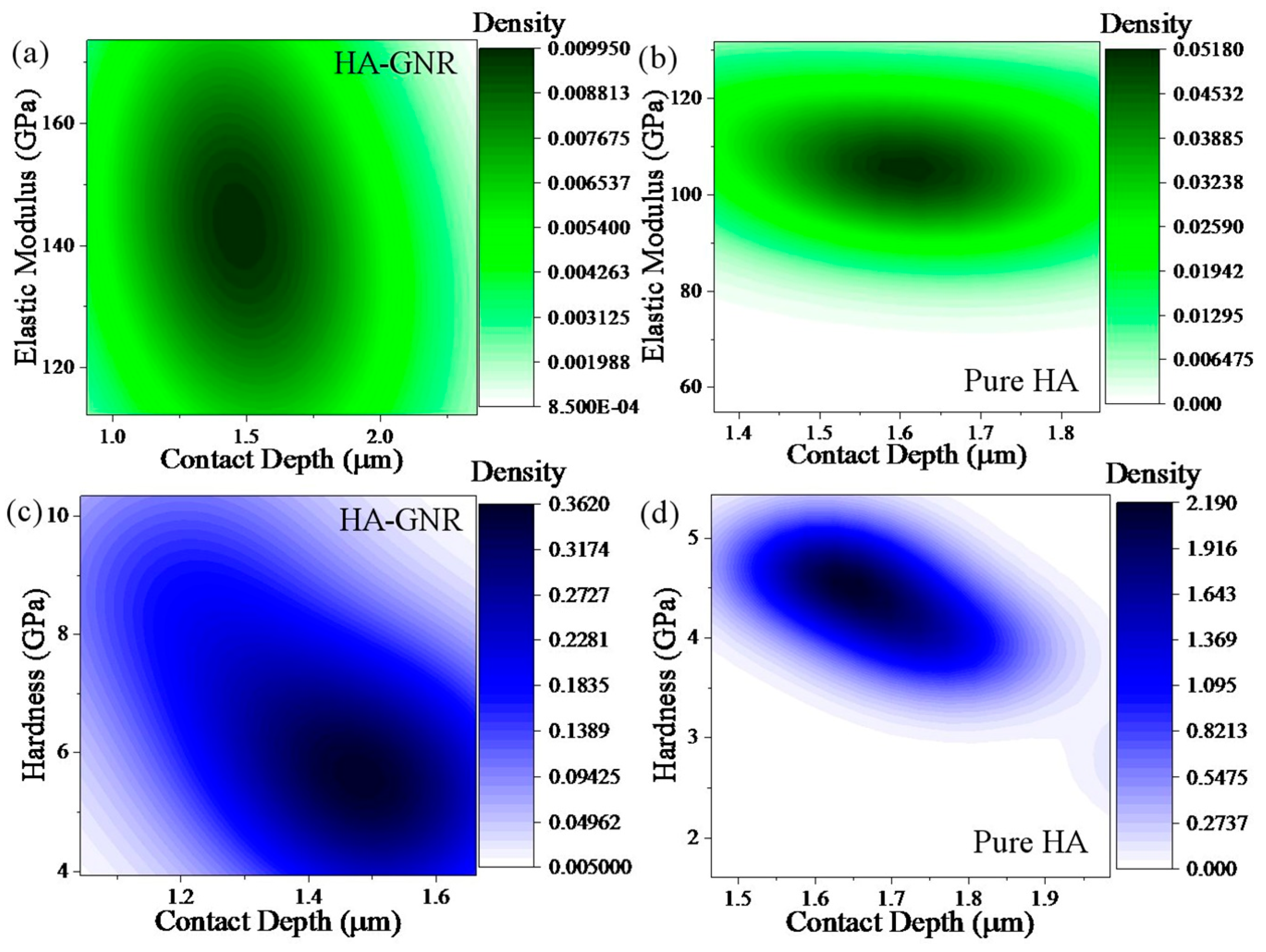
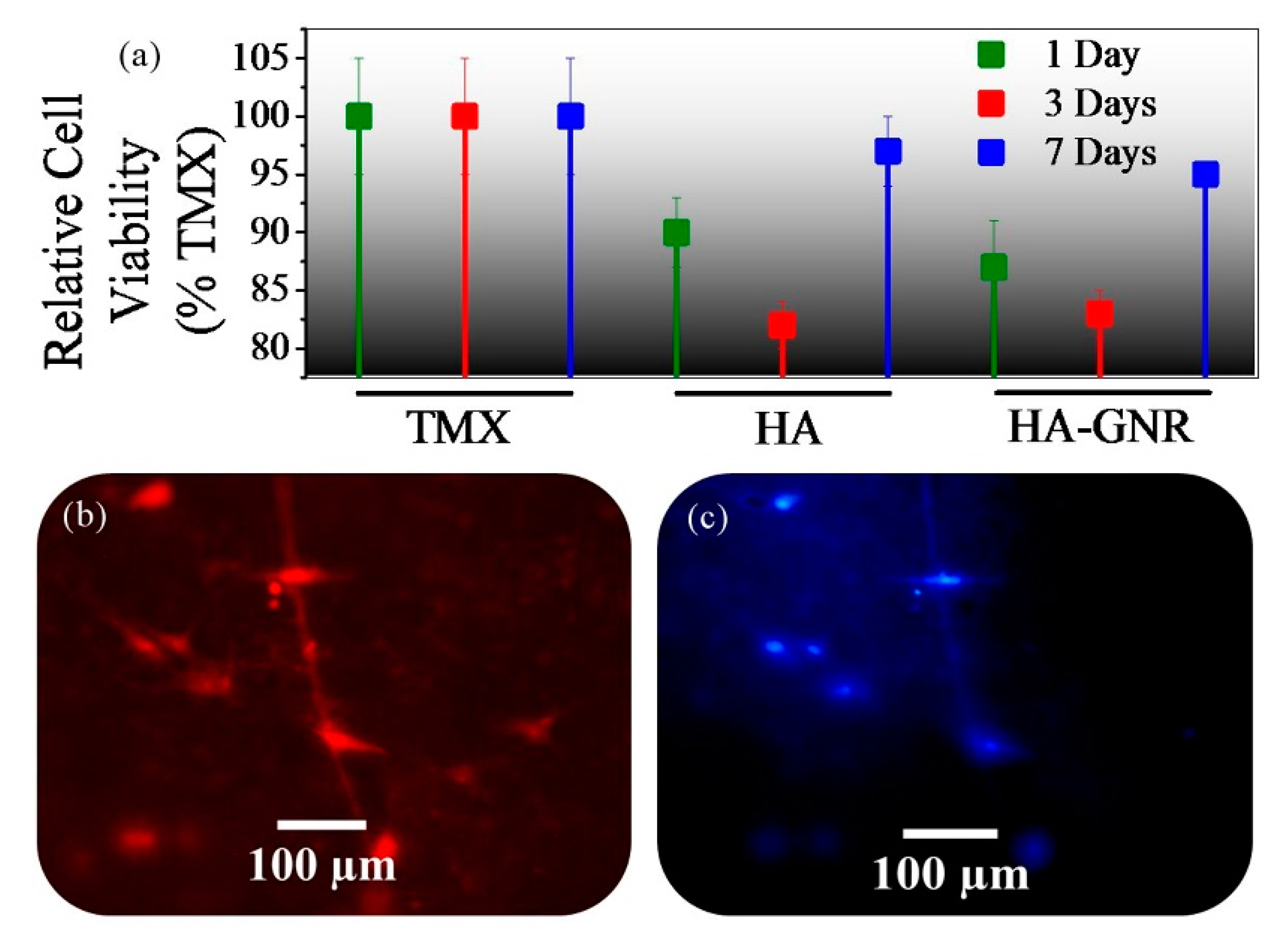
3. Results and Discussion
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Li, M.; Xiong, P.; Yan, F.; Li, S.; Ren, C.; Yin, Z.; Li, A.; Li, H.; Ji, X.; Zheng, Y.; et al. An overview of graphene-based hydroxyapatite composites for orthopedic applications. Bioact. Mater. 2018, 3, 1–18. [Google Scholar] [CrossRef] [PubMed]
- Gao, C.; Feng, P.; Peng, S.; Shuai, C. Carbon nanotube, graphene and boron nitride nanotube reinforced bioactive ceramics for bone repair. Acta Biomater. 2017, 61, 1–20. [Google Scholar] [CrossRef] [PubMed]
- Ahmadi, A.H.; Nosrati, H.; Sarraf-Mamoory, R. Decreasing β- three calcium phosphate particle size using graphite as nucleation sites and diethylene glycol as a chemical additive. J. Bioeng. Res. 2019, 1, 50–59. [Google Scholar]
- Nosrati, H.; Le, D.Q.S.; Emameh, R.Z.; Bunger, C.E. Characterization of the precipitated Dicalcium Phosphate Dehydrate on the Graphene Oxide surface as a bone cement reinforcement. J. Tissues Mater. 2019, 2, 33–46. [Google Scholar]
- Gholibegloo, E.; Karbasi, A.; Pourhajibagher, M.; Chiniforush, N.; Ramazani, A.; Akbari, T.; Bahador, A.; Khoobi, M. Carnosine-graphene oxide conjugates decorated with hydroxyapatite as promising nanocarrier for ICG loading with enhanced antibacterial effects in photodynamic therapy against Streptococcus mutans. J. Photochem. Photobiol. B: Boil. 2018, 181, 14–22. [Google Scholar] [CrossRef]
- Iafisco, M.; Di Foggia, M.; Bonora, S.; Prat, M.; Roveri, N. Adsorption and spectroscopic characterization of lactoferrin on hydroxyapatite nanocrystals. Dalton Trans. 2011, 40, 820–827. [Google Scholar] [CrossRef]
- Tavafoghi, M.; Brodusch, N.; Gauvin, R.; Cerruti, M. Hydroxyapatite formation on graphene oxide modified with amino acids: Arginine versus glutamic acid. J. R. Soc. Interface 2016, 13, 20150986. [Google Scholar] [CrossRef]
- Zhang, R.; Metoki, N.; Sharabani-Yosef, O.; Zhu, H.; Eliaz, N. Hydroxyapatite/Mesoporous Graphene/Single-Walled Carbon Nanotubes Freestanding Flexible Hybrid Membranes for Regenerative Medicine. Adv. Funct. Mater. 2016, 26, 7965–7974. [Google Scholar] [CrossRef]
- Nosrati, H.; Sarraf-Mamoory, R.; Le, D.Q.S.; Bünger, C.E. Enhanced fracture toughness of three dimensional graphene-hydroxyapatite nanocomposites by employing the Taguchi method. Compos. B Eng. 2020, 190, 107928. [Google Scholar] [CrossRef]
- Moldovan, M.; Prodan, D.; Sarosi, C.; Carpa, R.; Socaci, C.; Rosu, M.-C.; Pruneanu, S.M. Synthesis, morpho-structural properties and antibacterial effect of silicate-based composites containing graphene oxide/hydroxyapatite. Mater. Chem. Phys. 2018, 217, 48–53. [Google Scholar] [CrossRef]
- Canillas, M.; Rivero, R.; García-Carrodeguas, R.; Barba, F.; Rivas-Mercury, J.; Garcia-Carrodeguas, R.; Rodriguez, M.A. Processing of hydroxyapatite obtained by combustion synthesis. Boletín de la Sociedad Española de Cerámica y Vidrio 2017, 56, 237–242. [Google Scholar] [CrossRef]
- Guo, X.; Yan, H.; Zhao, S.; Li, Z.; Li, Y.; Liang, X. Effect of calcining temperature on particle size of hydroxyapatite synthesized by solid-state reaction at room temperature. Adv. Powder Technol. 2013, 24, 1034–1038. [Google Scholar] [CrossRef]
- Shirkhanzadeh, M. Direct formation of nanophase hydroxyapatite on cathodically polarized electrodes. J. Mater. Sci. Mater. Electron. 1998, 9, 67–72. [Google Scholar] [CrossRef] [PubMed]
- Jillavenkatesa, A.; Sr, R.A.C. Sol–gel processing of hydroxyapatite. J. Mater. Sci. 1998, 33, 4111–4119. [Google Scholar] [CrossRef]
- Kuriakose, T.; Kalkura, S.N.; Palanichamy, M.; Arivuoli, D.; Dierks, K.; Bocelli, G.; Betzel, C. Synthesis of stoichiometric nano crystalline hydroxyapatite by ethanol-based sol–gel technique at low temperature. J. Cryst. Growth 2004, 263, 517–523. [Google Scholar] [CrossRef]
- Sinitsyna, O.V.; Veresov, A.G.; Kovaleva, E.S.; Kolen’Ko, Y.V.; Putlyaev, V.I.; Tretyakov, Y.D. Synthesis of hydroxyapatite by hydrolysis of α-Ca3(PO4)2. Russ. Chem. Bull. 2005, 54, 79–86. [Google Scholar] [CrossRef]
- Liu, C.; Huang, Y.; Shen, W.; Cui, J. Kinetics of hydroxyapatite precipitation at pH 10 to 11. Biomaterials 2001, 22, 301–306. [Google Scholar] [CrossRef]
- Manuel, C.M.; Ferraz, M.P.; Monteiro, F.J. Synthesis of hydroxyapatite and tri calcium phosphate nanoparticles Preliminary Studies. Key Eng. Mater. 2003, 240–242, 555–558. [Google Scholar] [CrossRef]
- Yamashita, K.; Arashi, T.; Kitagaki, K.; Yamada, S.; Umegaki, T.; Ogawa, K. Preparation of Apatite Thin Films through rf-Sputtering from Calcium Phosphate Glasses. J. Am. Ceram. Soc. 1994, 77, 2401–2407. [Google Scholar] [CrossRef]
- Kimura, I. Synthesis of Hydroxyapatite by Interfacial Reaction in a Multiple Emulsion. Res. Lett. Mater. Sci. 2007, 2007, 1–4. [Google Scholar] [CrossRef] [Green Version]
- Tas, A.C. Synthesis of biomimetic Ca-hydroxyapatite powders at 37 °C in synthetic body fluids. Biomaterials 2000, 21, 1429–1438. [Google Scholar] [PubMed]
- Qi, C.; Zhu, Y.-J.; Ding, G.-J.; Wu, J.; Chen, F. Solvothermal synthesis of hydroxyapatite nanostructures with various morphologies using adenosine 5′-monophosphate sodium salt as an organic phosphorus source. RSC Adv. 2015, 5, 3792–3798. [Google Scholar] [CrossRef]
- Neira, I.S.; Kolen’Ko, Y.V.; Lebedev, O.I.; Van Tendeloo, G.; Gupta, H.S.; Guitián, F.; Yoshimura, M. An Effective Morphology Control of Hydroxyapatite Crystals via Hydrothermal Synthesis. Cryst. Growth Des. 2009, 9, 466–474. [Google Scholar] [CrossRef]
- Peng, F.; Yu, X.; Wei, M. In vitro cell performance on hydroxyapatite particles/poly(l-lactic acid) nanofibrous scaffolds with an excellent particle along nanofiber orientation. Acta Biomater. 2011, 7, 2585–2592. [Google Scholar] [CrossRef] [PubMed]
- Jevtić, M.; Mitrić, M.; Škapin, S.; Jančar, B.; Ignjatović, N.L.; Uskoković, D.P. Crystal Structure of Hydroxyapatite Nanorods Synthesized by Sonochemical Homogeneous Precipitation. Cryst. Growth Des. 2008, 8, 2217–2222. [Google Scholar]
- Costa, D.O.; Dixon, S.J.; Rizkalla, A. One- and Three-Dimensional Growth of Hydroxyapatite Nanowires during Sol–Gel–Hydrothermal Synthesis. ACS Appl. Mater. Interfaces 2012, 4, 1490–1499. [Google Scholar] [CrossRef]
- Zhang, Y.; Lu, J.; Wang, J.; Yang, S.; Chen, Y. Synthesis of nanorod and needle-like hydroxyapatite crystal and role of pH adjustment. J. Cryst. Growth 2009, 311, 4740–4746. [Google Scholar] [CrossRef]
- Chandanshive, B.B.; Rai, P.; Rossi, A.; Ersen, O.; Khushalani, D. Synthesis of hydroxyapatite nanotubes for biomedical applications. Mater. Sci. Eng. C 2013, 33, 2981–2986. [Google Scholar]
- Sun, Q.; Lou, J.-T.; Kang, F.; Chen, J.-F.; Wang, J.-X. Facile preparation of hydroxyapatite nanotubes assisted by needle-like calcium carbonate. Powder Technol. 2014, 261, 49–54. [Google Scholar] [CrossRef]
- Medeiros, J.S.; Oliveira, A.M.; De Carvalho, J.O.; Ricci, R.; Martins, M.D.C.C.; Rodrigues, B.V.M.; Webster, T.J.; Viana, B.C.; De Vasconcellos, L.M.R.; Canevari, R.A.; et al. Nanohydroxyapatite/Graphene Nanoribbons Nanocomposites Induce in Vitro Osteogenesis and Promote in Vivo Bone Neoformation. ACS Biomater. Sci. Eng. 2018, 4, 1580–1590. [Google Scholar] [CrossRef]
- Li, H.; Song, X.; Li, B.; Kang, J.; Liang, C.; Wang, H.; Yu, Z.; Qiao, Z. Carbon nanotube-reinforced mesoporous hydroxyapatite composites with excellent mechanical and biological properties for bone replacement material application. Mater. Sci. Eng. C 2017, 77, 1078–1087. [Google Scholar]
- Abden, M.; Afroze, J.; Alam, M.; Bahadur, N. Pressureless sintering and mechanical properties of hydroxyapatite/functionalized multi-walled carbon nanotube composite. Mater. Sci. Eng. C 2016, 67, 418–424. [Google Scholar]
- Qu, Y.; He, F.; Yu, C.; Liang, X.; Liang, N.; Ma, L.; Zhang, Q.; Lv, J.; Wu, J. Advances on graphene-based nanomaterials for biomedical applications. Mater. Sci. Eng. C 2018, 90, 764–780. [Google Scholar]
- Nie, C.; Ma, L.; Li, S.; Fan, X.; Yang, Y.; Cheng, C.; Zhao, W.; Zhao, C. Recent progresses in graphene based biofunctional nanostructures for advanced biological and cellular interfaces. Nano Today 2019, 26, 57–97. [Google Scholar] [CrossRef]
- Gadipelli, S.; Guo, Z.X.; Srinivas, G. Graphene-based materials: Synthesis and gas sorption, storage and separation. Prog. Mater. Sci. 2015, 69, 1–60. [Google Scholar] [CrossRef] [Green Version]
- Bai, R.G.; Ninan, N.; Muthoosamy, K.; Manickam, S. Graphene: A versatile platform for nanotheranostics and tissue engineering. Prog. Mater. Sci. 2018, 91, 24–69. [Google Scholar]
- Shin, S.R.; Li, Y.-C.; Jang, H.L.; Khoshakhlagh, P.; Akbari, M.; Nasajpour, A.; Zhang, Y.S.; Tamayol, A.; Khademhosseini, A. Graphene-based materials for tissue engineering. Adv. Drug Deliv. Rev. 2016, 105, 255–274. [Google Scholar] [CrossRef] [Green Version]
- Stoller, M.D.; Park, S.; Zhu, Y.; An, J.; Ruoff, R.S. Graphene-Based Ultracapacitors. Nano Lett. 2008, 8, 3498–3502. [Google Scholar] [CrossRef]
- Wang, S.; Zhang, S.; Wang, Y.; Sun, X.; Sun, K. Reduced graphene oxide/carbon nanotubes reinforced calcium phosphate cement. Ceram. Int. 2017, 43, 13083–13088. [Google Scholar] [CrossRef]
- Baradaran, S.; Moghaddam, E.; Basirun, W.; Mehrali, M.; Sookhakian, M.; Hamdi, M.; Moghaddam, M.N.; Alias, Y. Mechanical properties and biomedical applications of a nanotube hydroxyapatite-reduced graphene oxide composite. Carbon 2014, 69, 32–45. [Google Scholar] [CrossRef]
- Nosrati, H.; Mamoory, R.S.; Le, D.Q.S.; Bünger, C.E. Preparation of reduced graphene oxide/hydroxyapatite nanocomposite and evaluation of graphene sheets/hydroxyapatite interface. Diam. Relat. Mater. 2019, 100, 107561. [Google Scholar] [CrossRef]
- Meng, X.; Cui, X.; Rager, M.; Zhang, S.; Wang, Z.; Yu, J.; Harn, Y.W.; Kang, Z.; Wagner, B.K.; Liu, Y.; et al. Cascade charge transfer enabled by incorporating edge-enriched graphene nanoribbons for mesostructured perovskite solar cells with enhanced performance. Nano Energy 2018, 52, 123–133. [Google Scholar] [CrossRef]
- Nosrati, H.; Mamoory, R.S.; Dabir, F.; Le, D.Q.S.; Bünger, C.E.; Perez, M.C.; Rivas-Mercury, J. Effects of hydrothermal pressure on in situ synthesis of 3D graphene- hydroxyapatite nano structured powders. Ceram. Int. 2019, 45, 1761–1769. [Google Scholar] [CrossRef]
- Nosrati, H.; Mamoory, R.S.; Dabir, F.; Perez, M.C.; Rivas-Mercury, J.; Le, D.Q.S.; Bünger, C.E. In situ synthesis of three dimensional graphene-hydroxyapatite nano powders via hydrothermal process. Mater. Chem. Phys. 2019, 222, 251–255. [Google Scholar] [CrossRef]
- Nikolaev, A.; Gopin, A.; Severin, A.V.; Rudin, V.; Mironov, M.; Dezhkunov, N. Ultrasonic synthesis of hydroxyapatite in non-cavitation and cavitation modes. Ultrason. Sonochem. 2018, 44, 390–397. [Google Scholar] [CrossRef]
- Turon, P.; Del Valle, L.J.; Alemán, C.; Puiggali, J. Biodegradable and Biocompatible Systems Based on Hydroxyapatite Nanoparticles. Appl. Sci. 2017, 7, 60. [Google Scholar] [CrossRef] [Green Version]
- Chen, M.H.; Yoshioka, T.; Ikoma, T.; Hanagata, N.; Lin, F.H.; Tanaka, J. Photoluminescence and doping mechanism of theranostic Eu3+/Fe3+ dual-doped hydroxyapatite nanoparticles. Sci. Technol. Adv. Mater. 2014, 15, 055005. [Google Scholar] [CrossRef]
- Cipreste, M.F.; De Rezende, M.R.; Hneda, M.; Peres, A.M.; Cotta, A.A.C.; Teixeira, V.; Macedo, W.A.D.A.; De Sousa, E.M.B. Functionalized-radiolabeled hydroxyapatite/tenorite nanoparticles as theranostic agents for osteosarcoma. Ceram. Int. 2018, 44, 17800–17811. [Google Scholar] [CrossRef]
- Roy, S.; Jaiswal, A. Graphene-Based Nanomaterials for Theranostic Applications. Rep. Adv. Phys. Sci. 2017, 1, 1750011. [Google Scholar] [CrossRef] [Green Version]
- Liu, Z.; Liang, X.-J. Nano-Carbons as Theranostics. Theranostics 2012, 2, 235–237. [Google Scholar] [CrossRef]
- Orecchioni, M.; Cabizza, R.; Bianco, A.; Delogu, L.G. Graphene as Cancer Theranostic Tool: Progress and Future Challenges. Theranostics 2015, 5, 710–723. [Google Scholar] [CrossRef] [PubMed]
- Nosrati, H.; Mamoory, R.S.; Le, D.Q.S.; Bünger, C.E.; Emameh, R.Z.; Dabir, F. Gas injection approach for synthesis of hydroxyapatite nanorods via hydrothermal method. Mater. Charact. 2020, 159, 110071. [Google Scholar] [CrossRef]
- Oliver, W.; Pharr, G. An improved technique for determining hardness and elastic modulus using load and displacement sensing indentation experiments. J. Mater. Res. 1992, 7, 1564–1583. [Google Scholar] [CrossRef]
- Zhang, B.; Chen, G.; Guo, Q. Static Frame Model Validation with Small Samples Solution Using Improved Kernel Density Estimation and Confidence Level Method. Chin. J. Aeronaut. 2012, 25, 879–886. [Google Scholar] [CrossRef] [Green Version]
- O’Brien, T.; Kashinath, K.; Cavanaugh, N.; Collins, W.; O’Brien, J.P. A fast and objective multidimensional kernel density estimation method: FastKDE. Comput. Stat. Data Anal. 2016, 101, 148–160. [Google Scholar] [CrossRef] [Green Version]
- Wijesinghe, W.; Mantilaka, M.M.M.G.P.G.; Premalal, E.; Herath, H.; Mahalingam, S.; Edirisinghe, M.; Rajapakse, R. Facile synthesis of both needle-like and spherical hydroxyapatite nanoparticles: Effect of synthetic temperature and calcination on morphology, crystallite size and crystallinity. Mater. Sci. Eng. C 2014, 42, 83–90. [Google Scholar]
- Kaygili, O.; Keser, S.; Bulut, N.; Ates, T. Characterization of Mg-containing hydroxyapatites synthesized by combustion method. Phys. B Condens. Matter 2018, 537, 63–67. [Google Scholar] [CrossRef]
- Saleh, T.A. The influence of treatment temperature on the acidity of MWCNT oxidized by HNO3 or a mixture of HNO3/H2SO4. Appl. Surface Sci. 2011, 257, 7746–7751. [Google Scholar] [CrossRef]
- Saleh, T.A. Nanocomposite of carbon nanotubes/silica nanoparticles and their use for adsorption of Pb(II): From surface properties to sorption mechanism. Desalination Water Treat. 2015, 57, 1–15. [Google Scholar] [CrossRef]
- Fadillah, G.; Saleh, T.A.; Wahyuningsih, S.; Putri, E.N.K.; Febrianastuti, S. Electrochemical removal of methylene blue using alginate-modified graphene adsorbents. Chem. Eng. J. 2019, 378, 122140. [Google Scholar] [CrossRef]
- Saleh, T.A.; Fadillah, G. Recent trends in the design of chemical sensors based on graphene–metal oxide nanocomposites for the analysis of toxic species and biomolecules. TrAC Trends Anal. Chem. 2019, 120, 115660. [Google Scholar] [CrossRef]
- Saleh, T.A. Simultaneous adsorptive desulfurization of diesel fuel over bimetallic nanoparticles loaded on activated carbon. J. Clean. Prod. 2018, 172, 2123–2132. [Google Scholar] [CrossRef]
- Nosrati, H.; Sarraf-Mamoory, R.; Dabir, F. Crystallographic study of hydrothermal synthesis of hydroxyapatite nano-rods using Brushite precursors. J. Tissues Mater. 2019, 2, 1–8. [Google Scholar]
- Nosrati, H.; Mamoory, R.S.; Le, D.Q.S.; Bünger, C.E. Fabrication of gelatin/hydroxyapatite/3D-graphene scaffolds by a hydrogel 3D-printing method. Mater. Chem. Phys. 2020, 239, 122305. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Chemical | Company | Purity | Formulation |
|---|---|---|---|
| Graphene oxide nanoribbons (GONRs) | GONRs were synthesized by chemical unzipping of CNTs under oxidative condition [42] | ||
| Calcium nitrate tetrahydrate | Merck (Kenilworth, NJ, USA) | >99% | Ca(NO3)2·4H2O |
| Diammonium Hydrogenphosphate | (NH4)2HPO4 | ||
| Ammonium solution | 25% | NH4OH | |
| Anhydrous ethanol | Sigma Aldrich (St. Louis, MO, USA) | >99% | CH3CH2OH |
| Dimethyl formamide (DMF) | (CH3)2NC(O)H | ||
| Dyethylene glycol (DEG) | (HOCH2CHc)2O | ||
| 2θ (°) | (hkl), d-spacing (Å) |
|---|---|
| 26 | (002), 3.43 |
| 32 | (211), 2.81 |
| 32.3 | (112), 2.77 |
| 33 | (300), 2.71 |
| 35.6 | (202), 2.52 |
| 39.9 | (310), 2.25 |
| 49.6 | (213), 1.84 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nosrati, H.; Sarraf-Mamoory, R.; Ahmadi, A.H.; Canillas Perez, M. Synthesis of Graphene Nanoribbons–Hydroxyapatite Nanocomposite Applicable in Biomedicine and Theranostics. J. Nanotheranostics 2020, 1, 6-18. https://doi.org/10.3390/jnt1010002
Nosrati H, Sarraf-Mamoory R, Ahmadi AH, Canillas Perez M. Synthesis of Graphene Nanoribbons–Hydroxyapatite Nanocomposite Applicable in Biomedicine and Theranostics. Journal of Nanotheranostics. 2020; 1(1):6-18. https://doi.org/10.3390/jnt1010002
Chicago/Turabian StyleNosrati, Hassan, Rasoul Sarraf-Mamoory, Amir Hossein Ahmadi, and Maria Canillas Perez. 2020. "Synthesis of Graphene Nanoribbons–Hydroxyapatite Nanocomposite Applicable in Biomedicine and Theranostics" Journal of Nanotheranostics 1, no. 1: 6-18. https://doi.org/10.3390/jnt1010002
APA StyleNosrati, H., Sarraf-Mamoory, R., Ahmadi, A. H., & Canillas Perez, M. (2020). Synthesis of Graphene Nanoribbons–Hydroxyapatite Nanocomposite Applicable in Biomedicine and Theranostics. Journal of Nanotheranostics, 1(1), 6-18. https://doi.org/10.3390/jnt1010002