
Synthesis of 2-Azetidinones via Cycloaddition Approaches: An Update



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
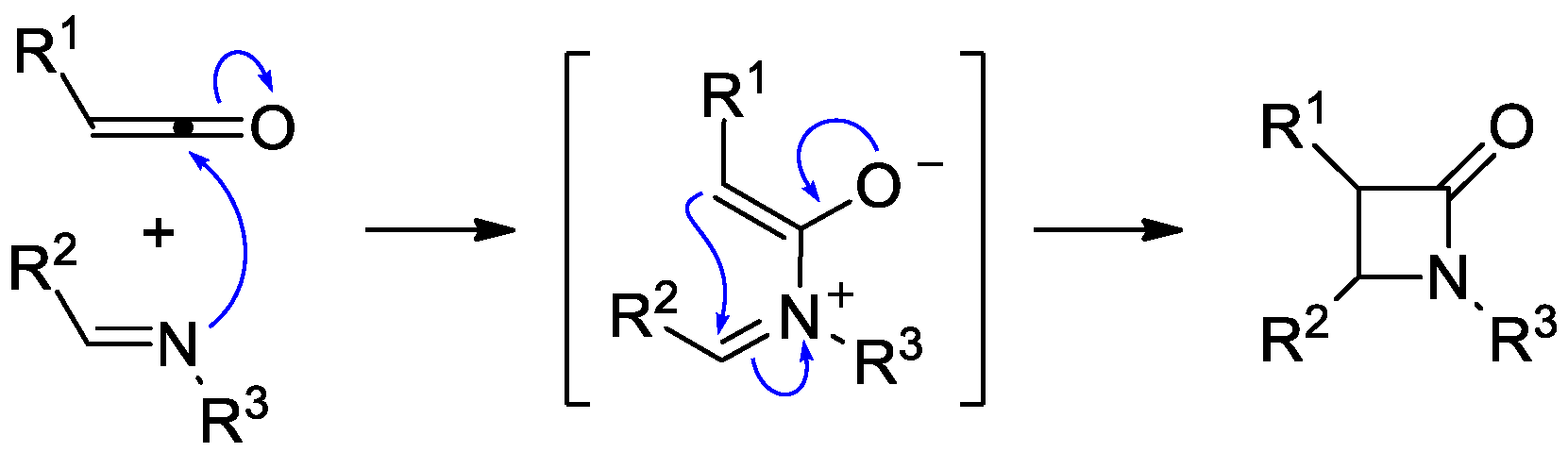
2. Ketene–Imine Cycloaddition (Staudinger Synthesis)
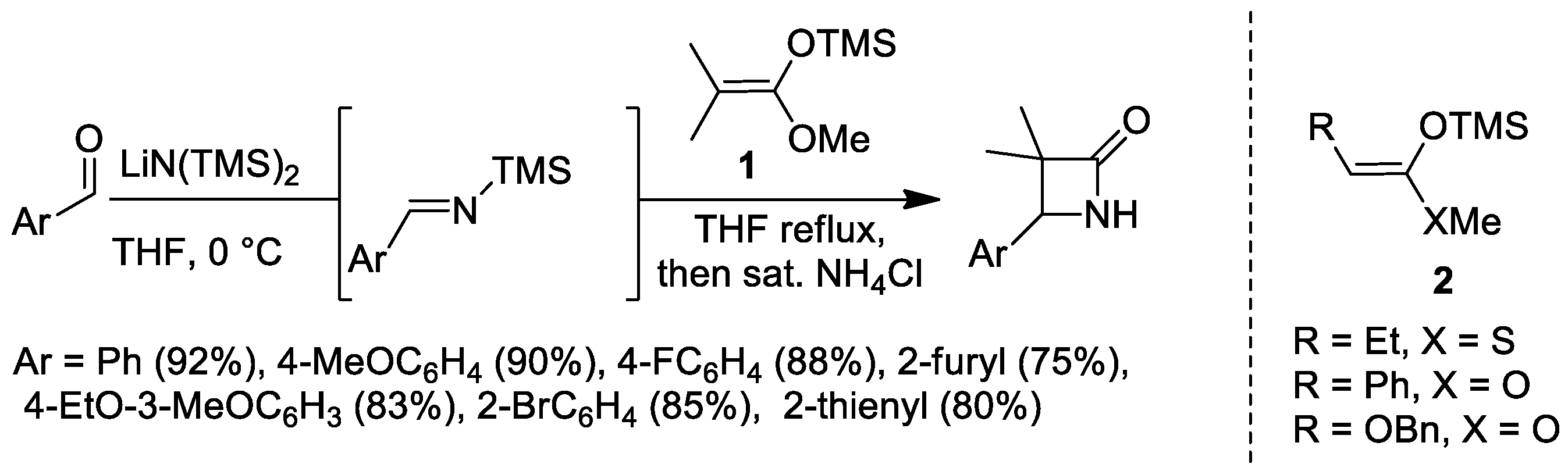
2.1. Ketene Generated In Situ from Ketene Acetals
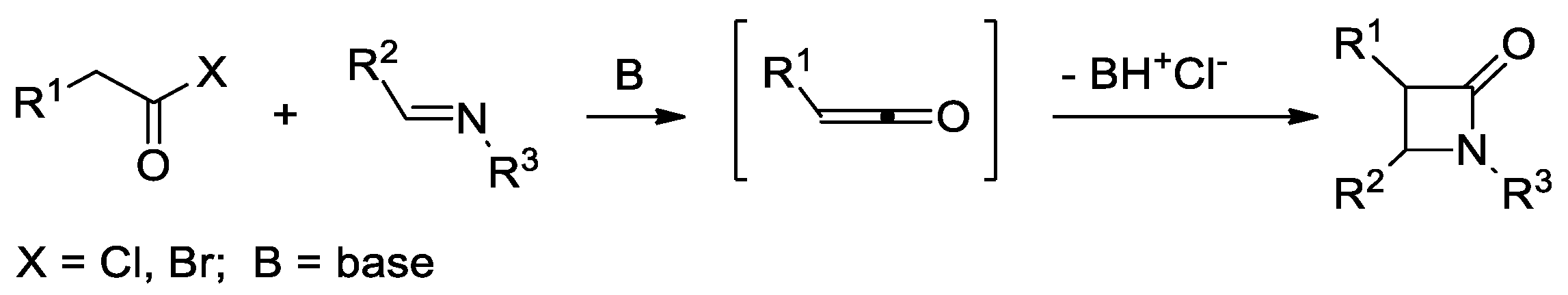
2.2. Ketene Generated In Situ from Acyl Chlorides and Acyl Bromides
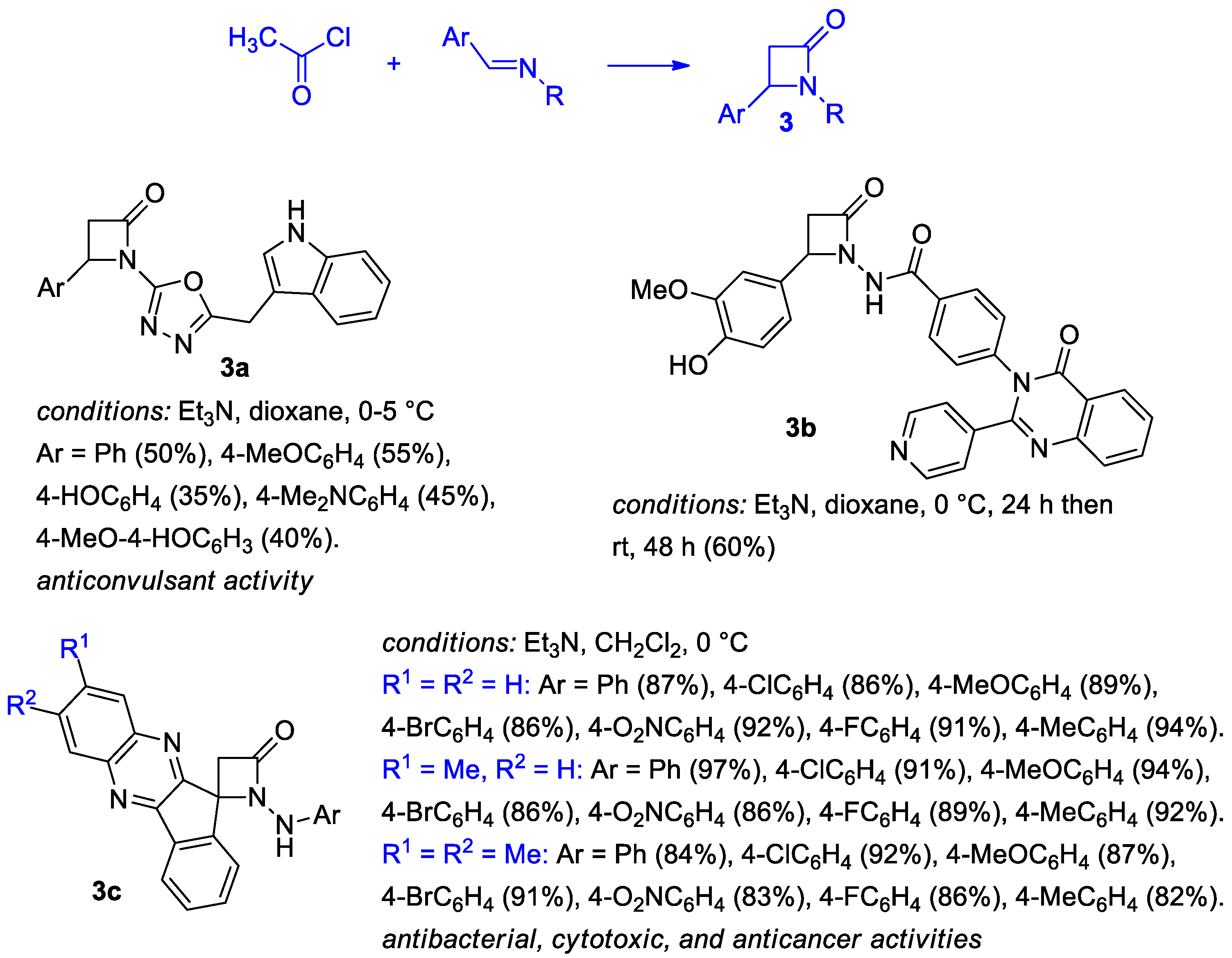
2.2.1. Ketene Generated In Situ from Acetyl Chloride
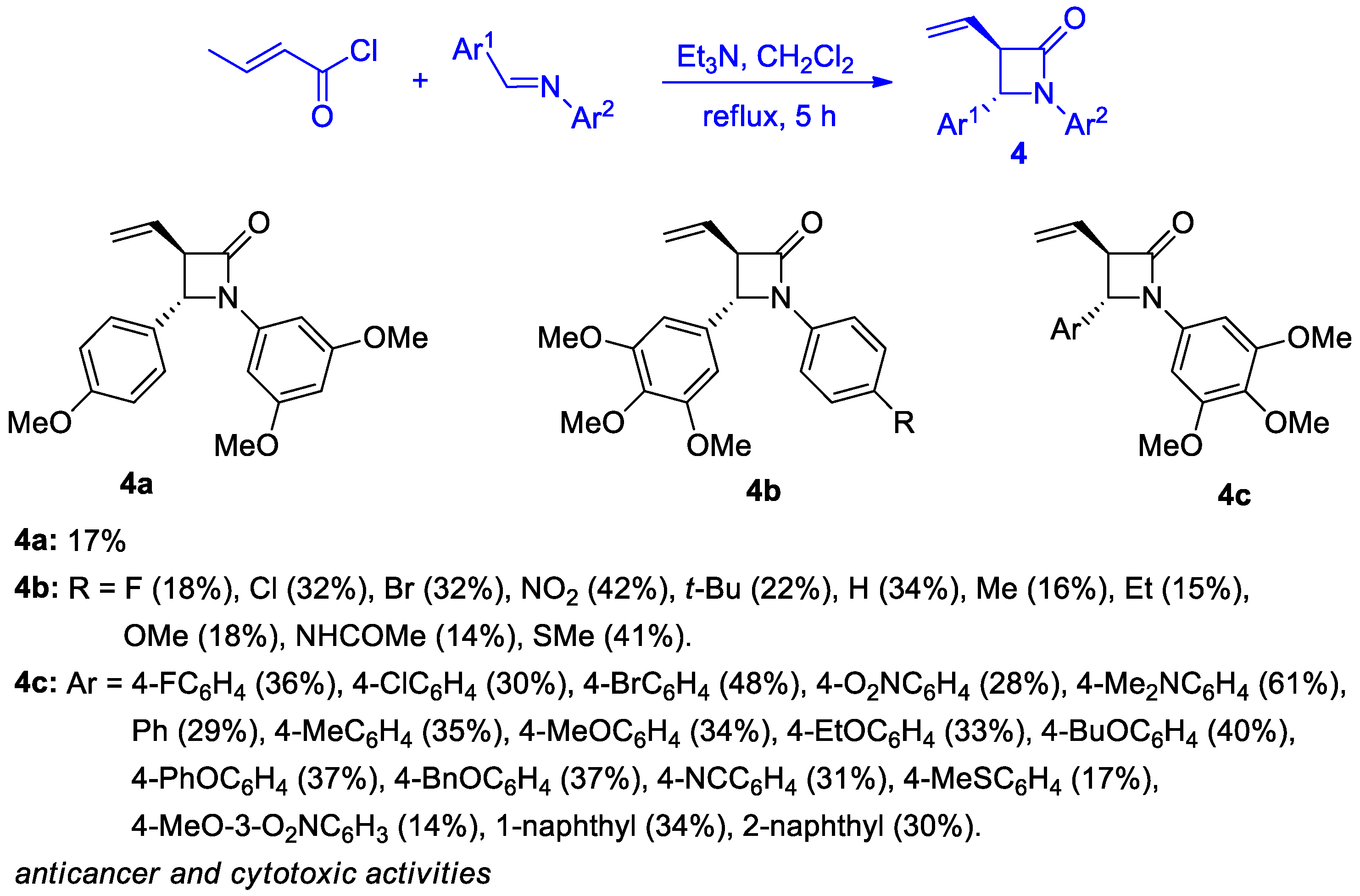
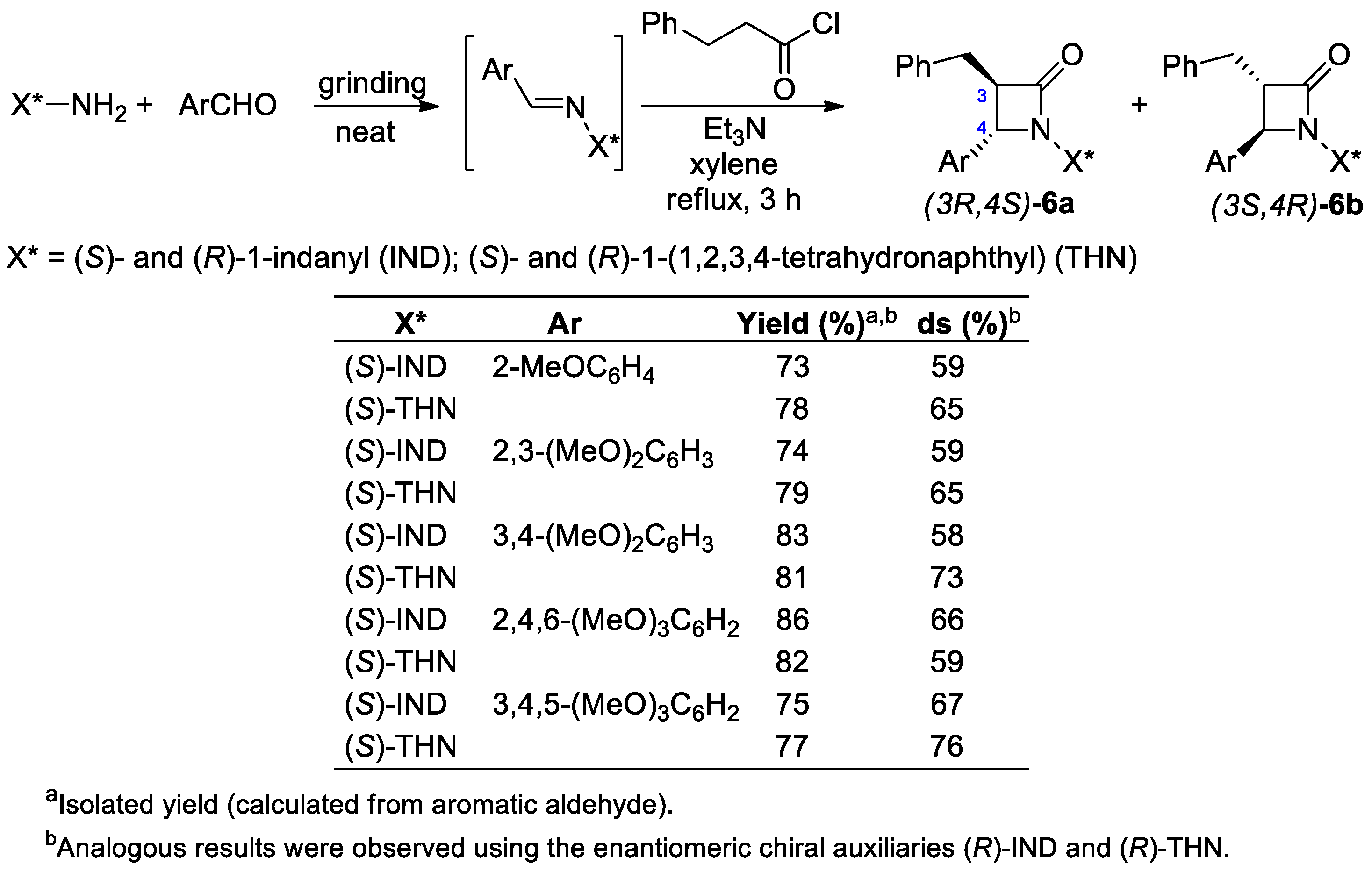
2.2.2. Ketene Generated In Situ from 2-Alkyl-, 2-Vinyl-, and 2-Arylacetyl Chloride
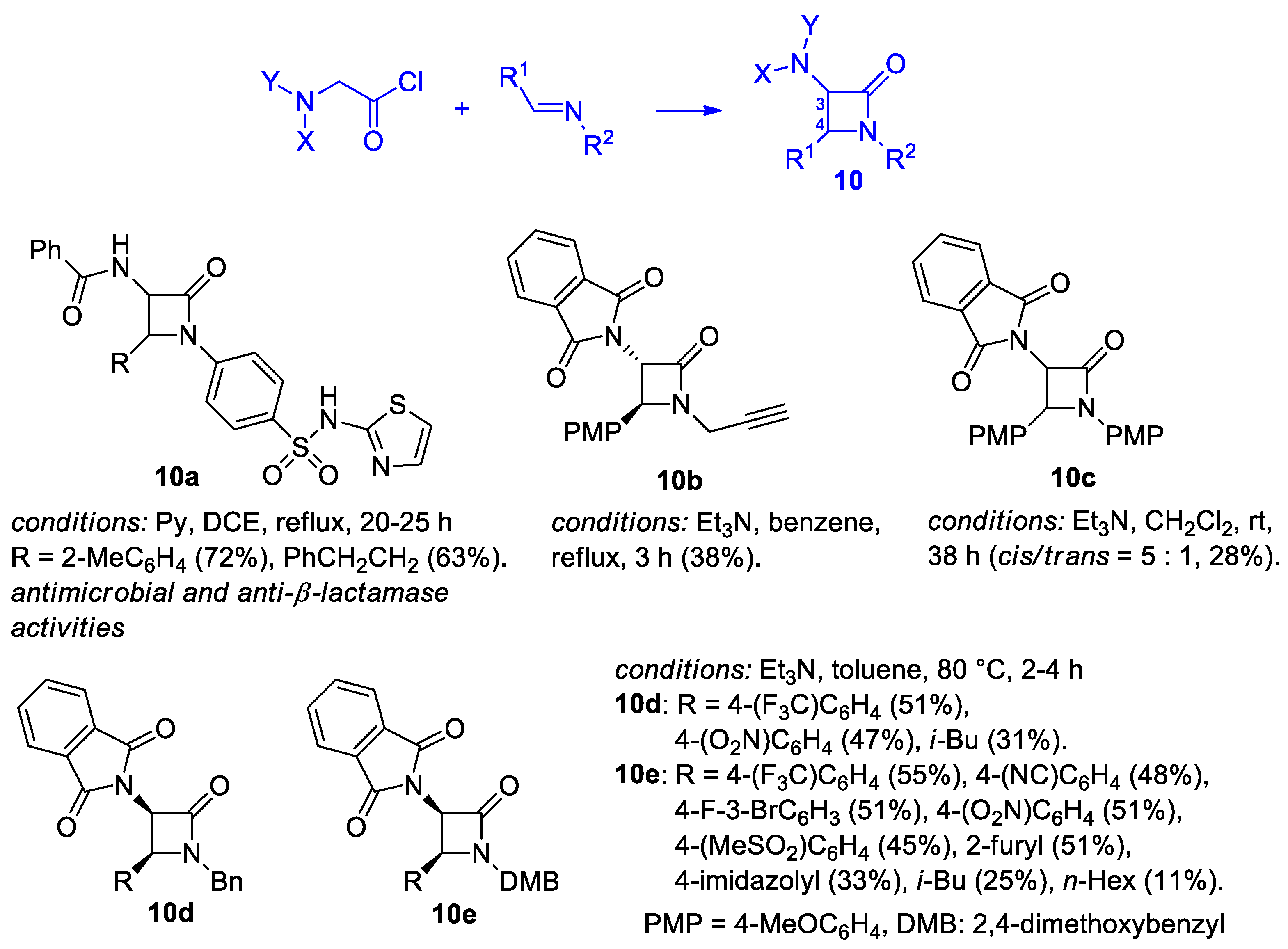
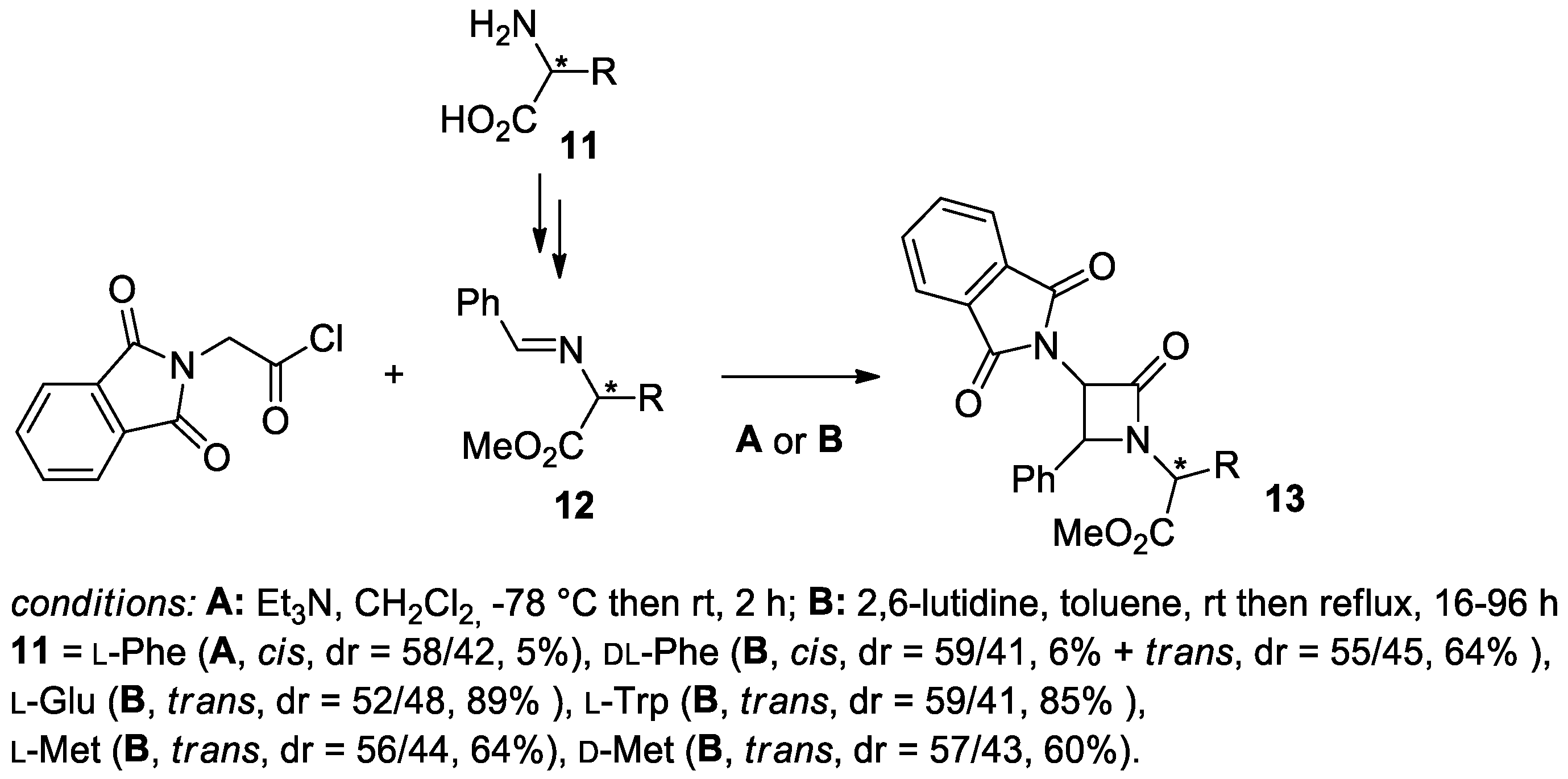
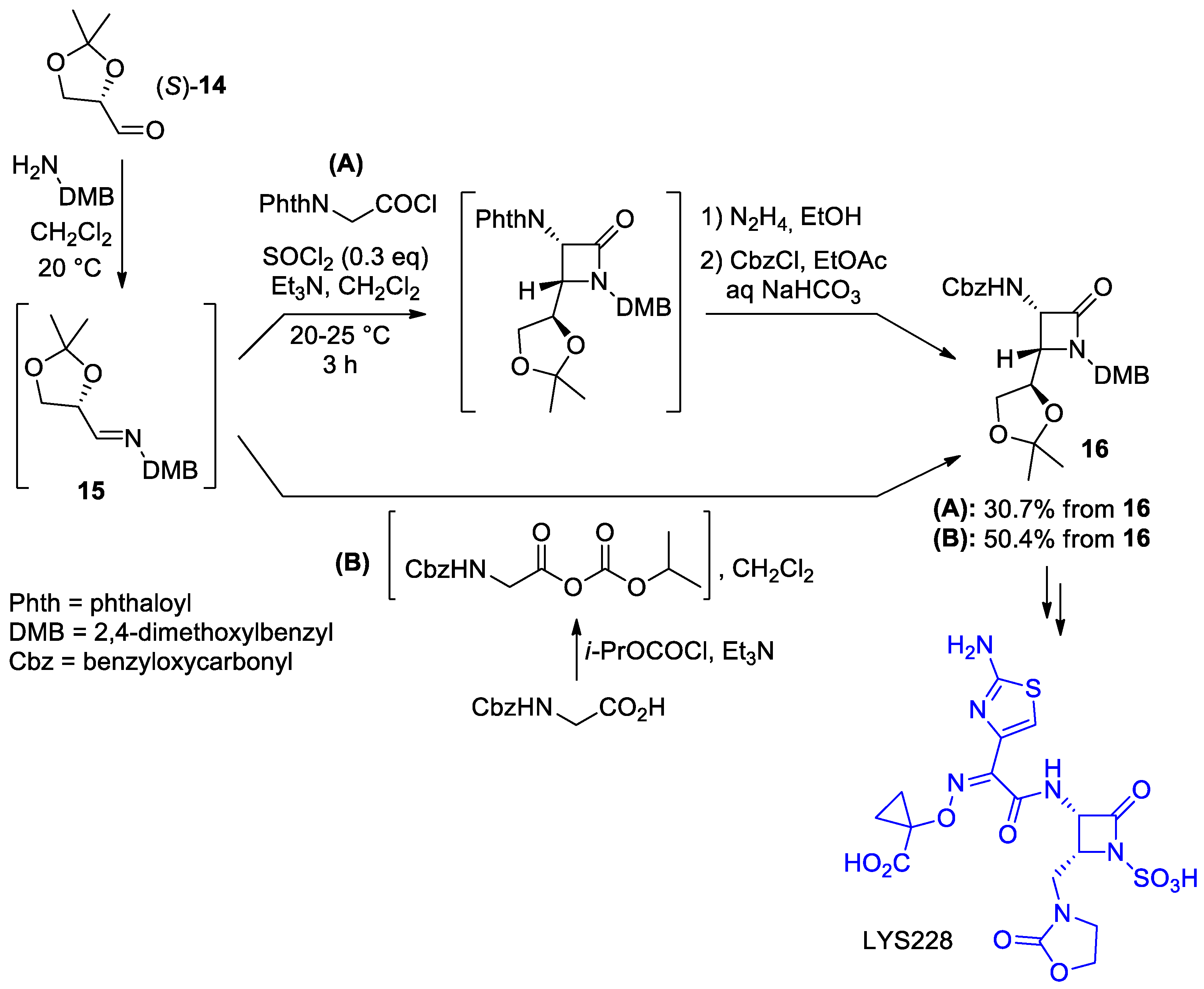
2.2.3. Ketene Generated In Situ from 2-Amidoacetyl Chloride
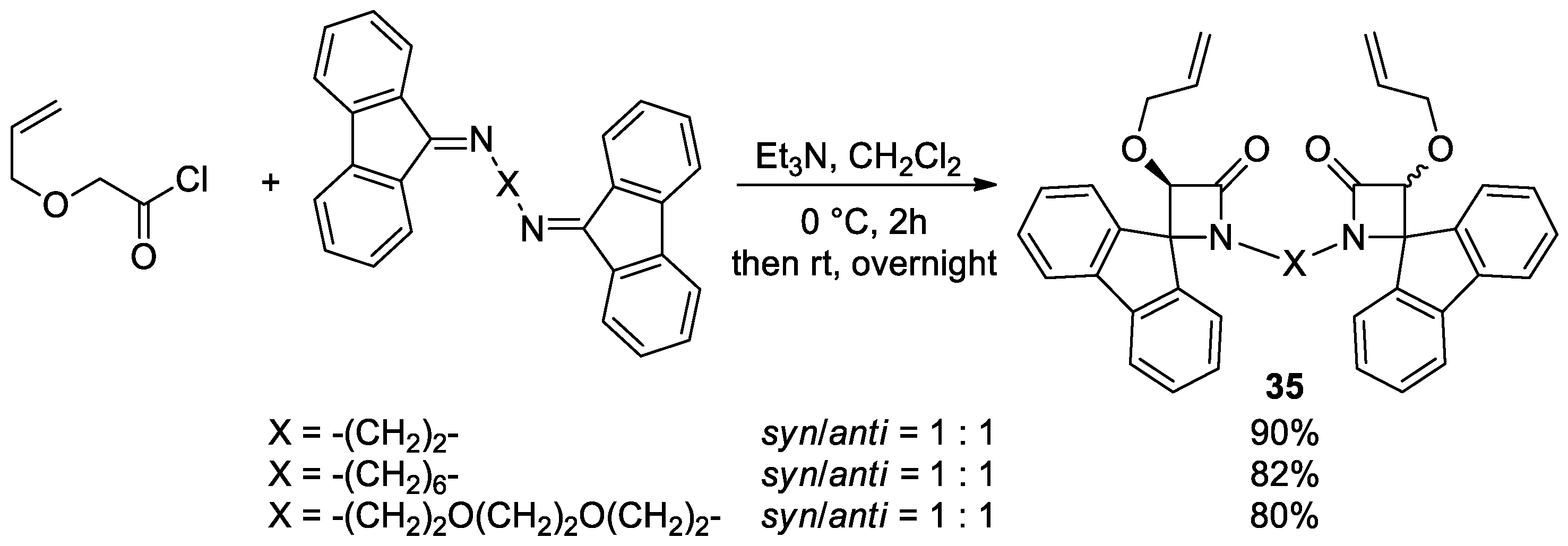
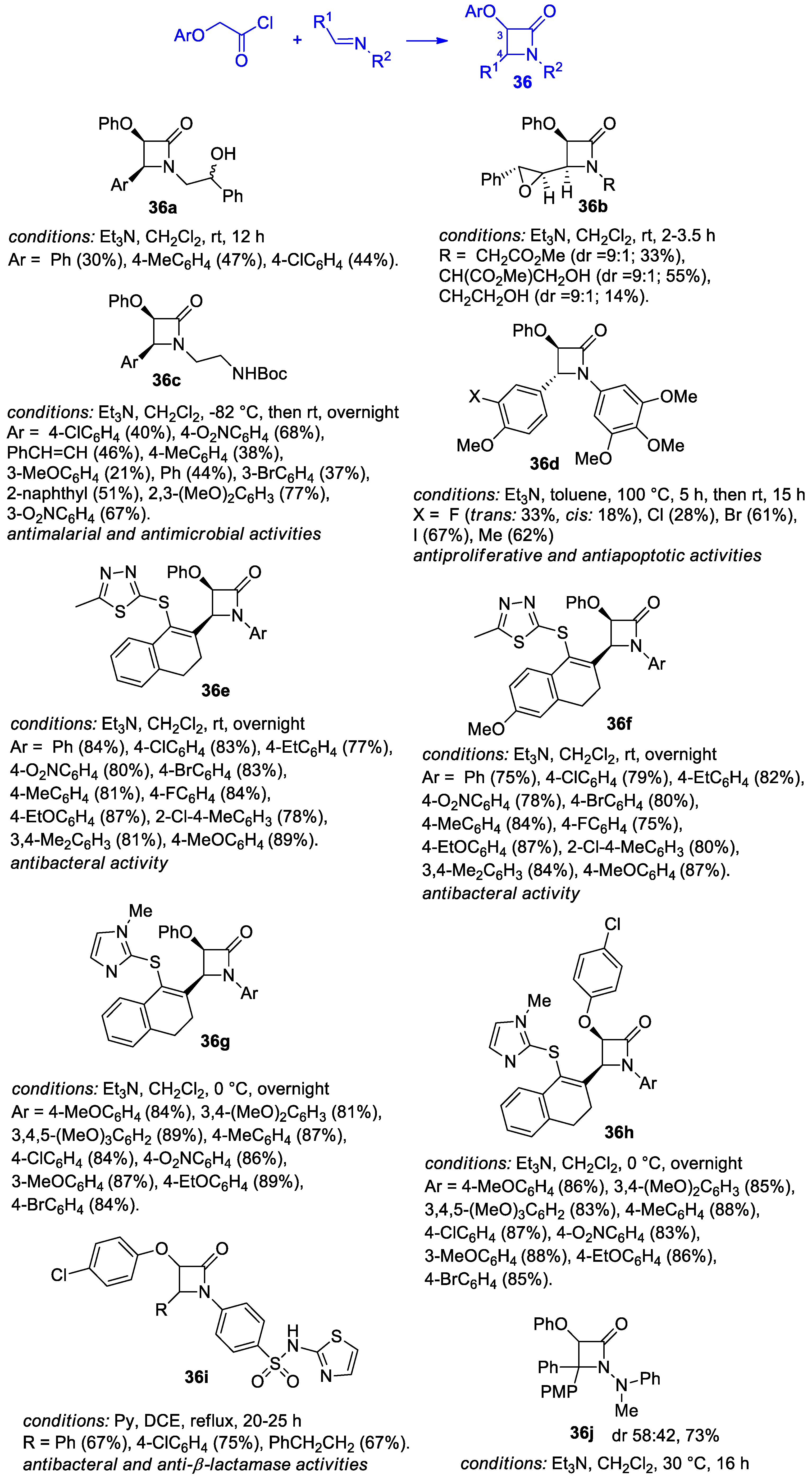
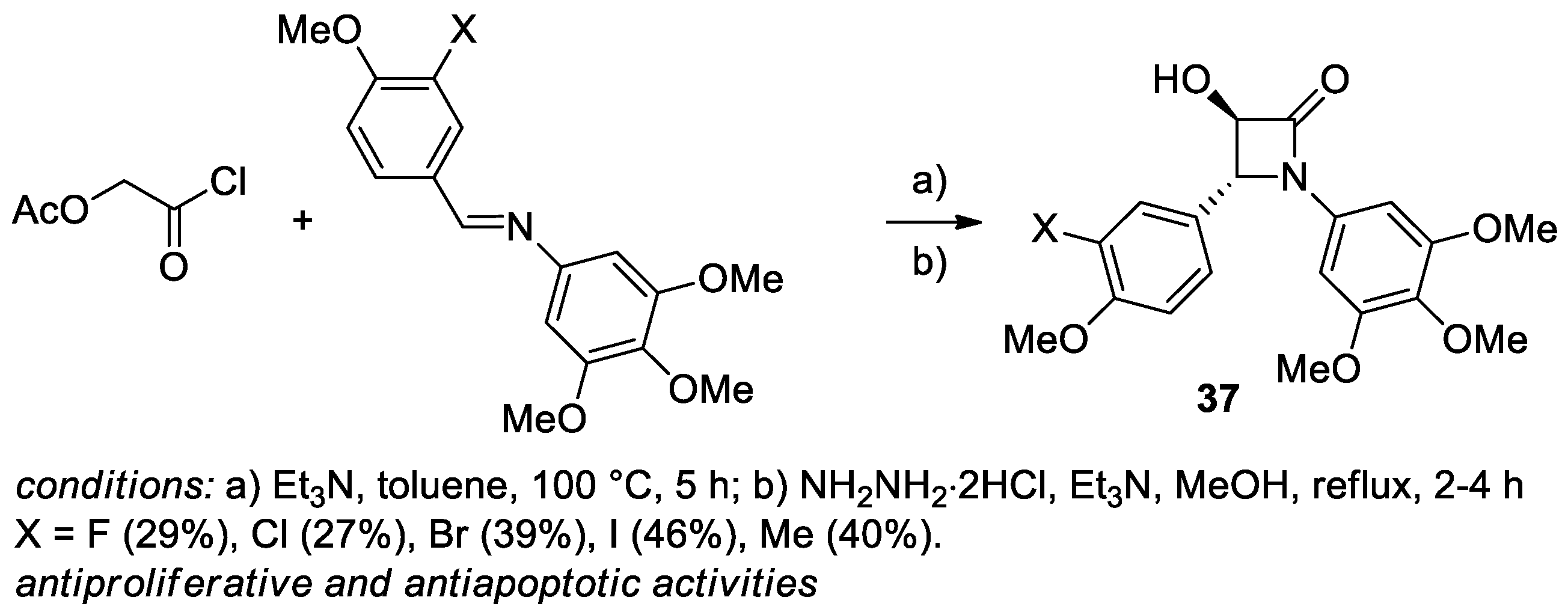
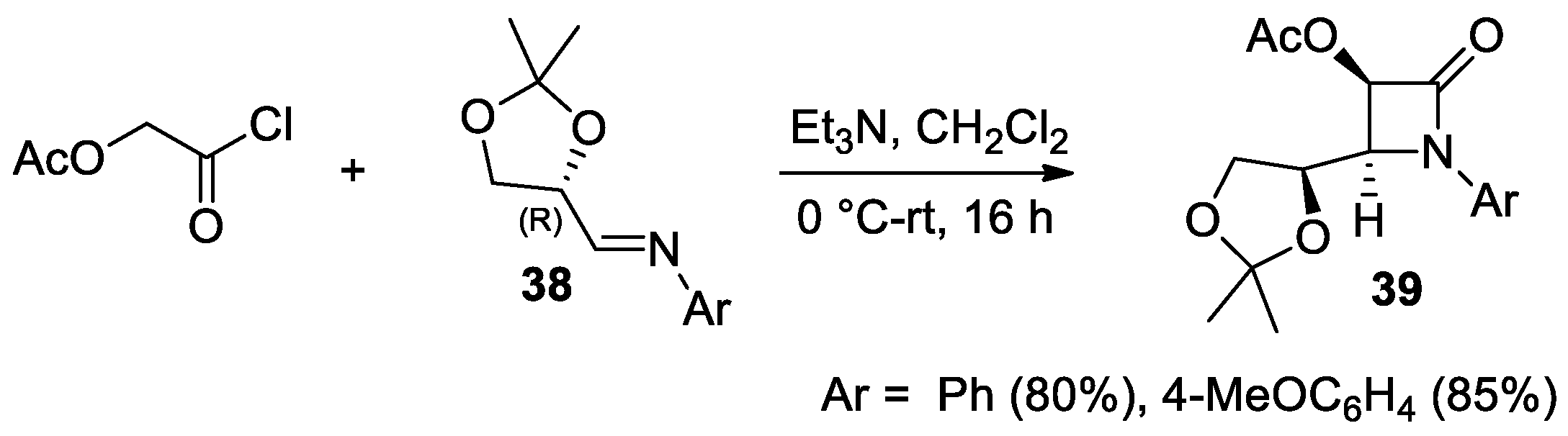
2.2.4. Ketene Generated In Situ from 2-Alkoxy-, 2-Aryloxy-, and 2-Acetoxyacetyl Chloride
2.2.5. Ketene Generated In Situ from 2-Phenylthioacetyl Chloride
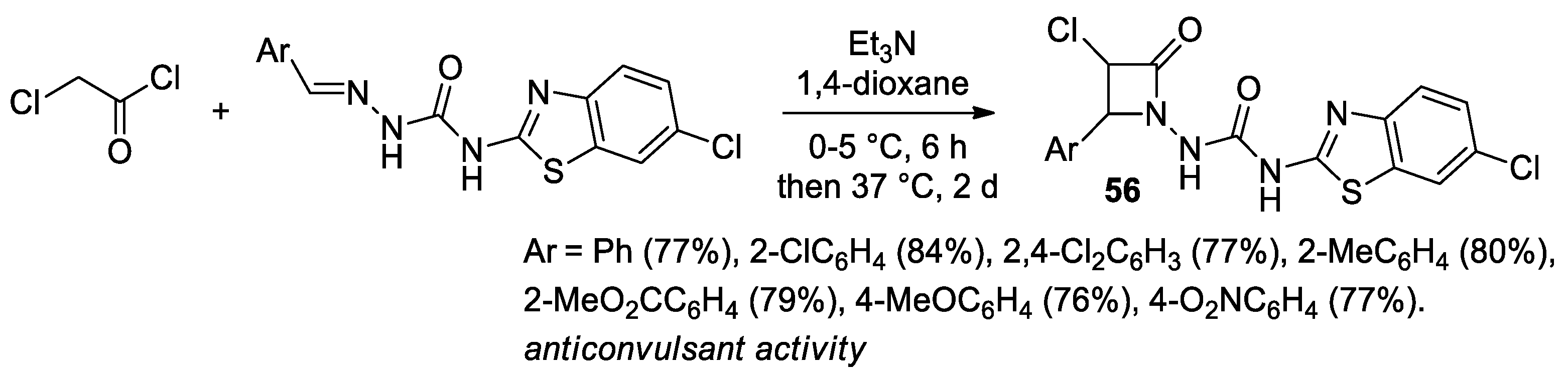
2.2.6. Ketene Generated In Situ from 2-Chloroacetyl Chloride
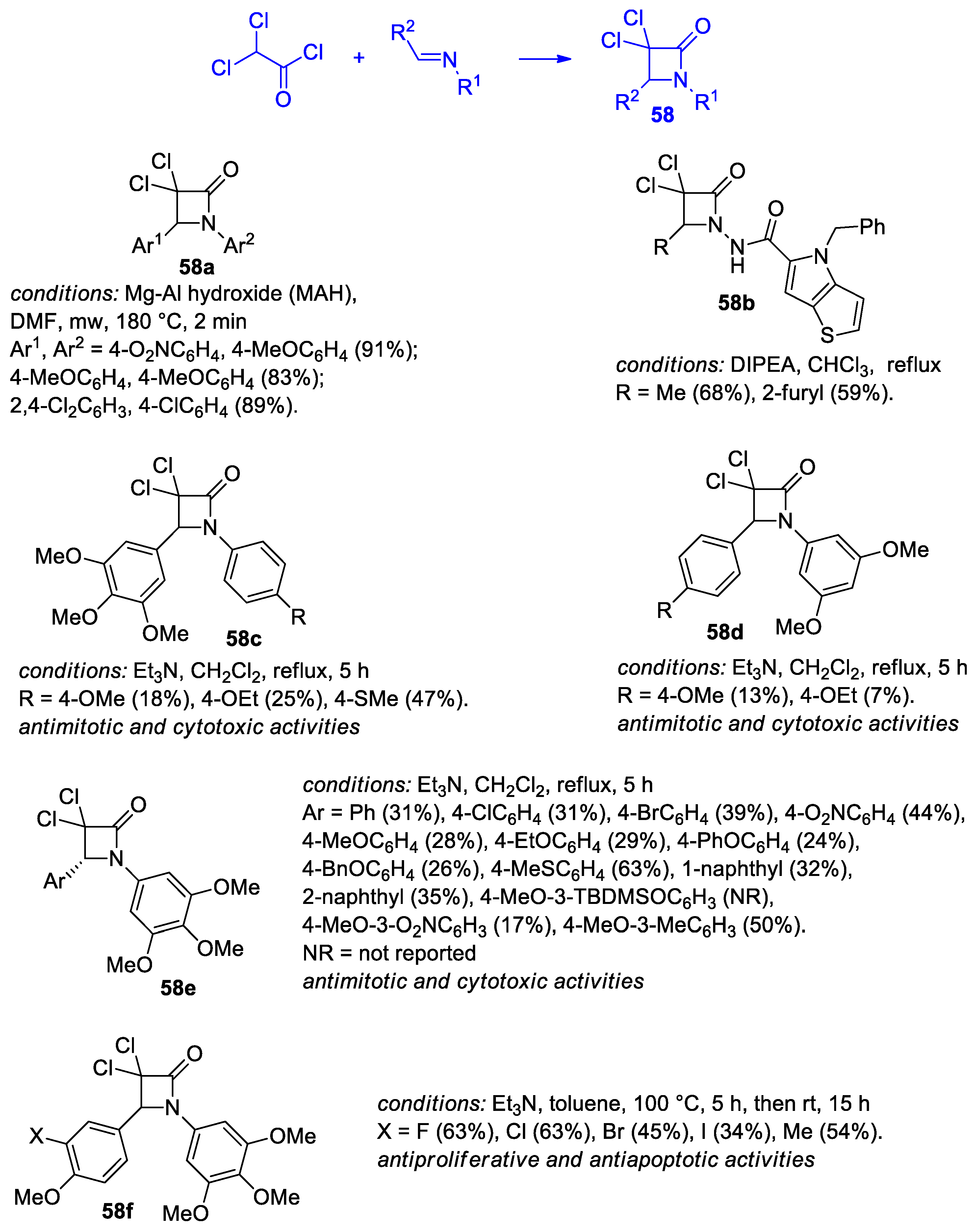
2.2.7. Ketene Generated In Situ from 2,2-Dichloroacetyl Chloride
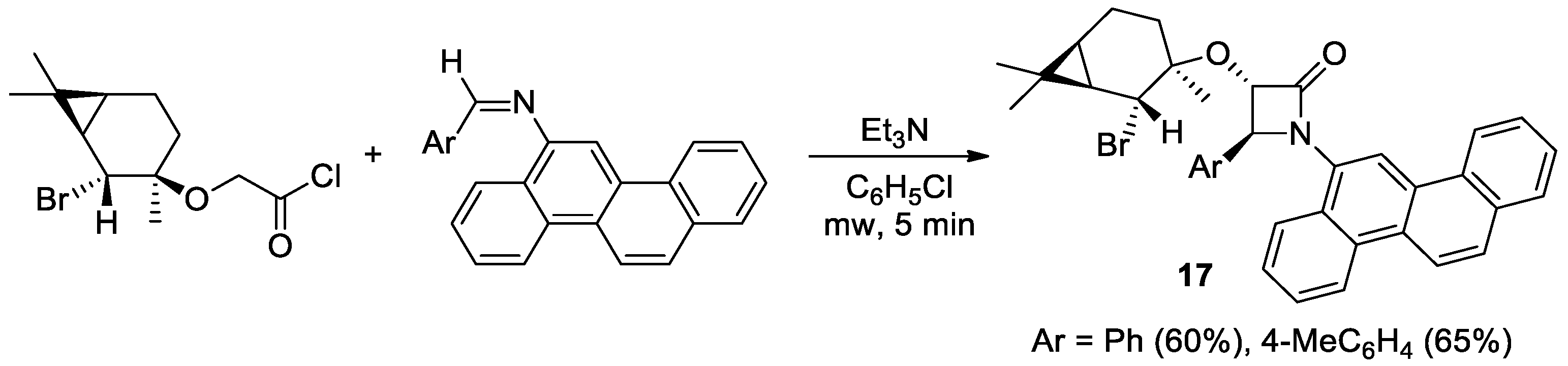
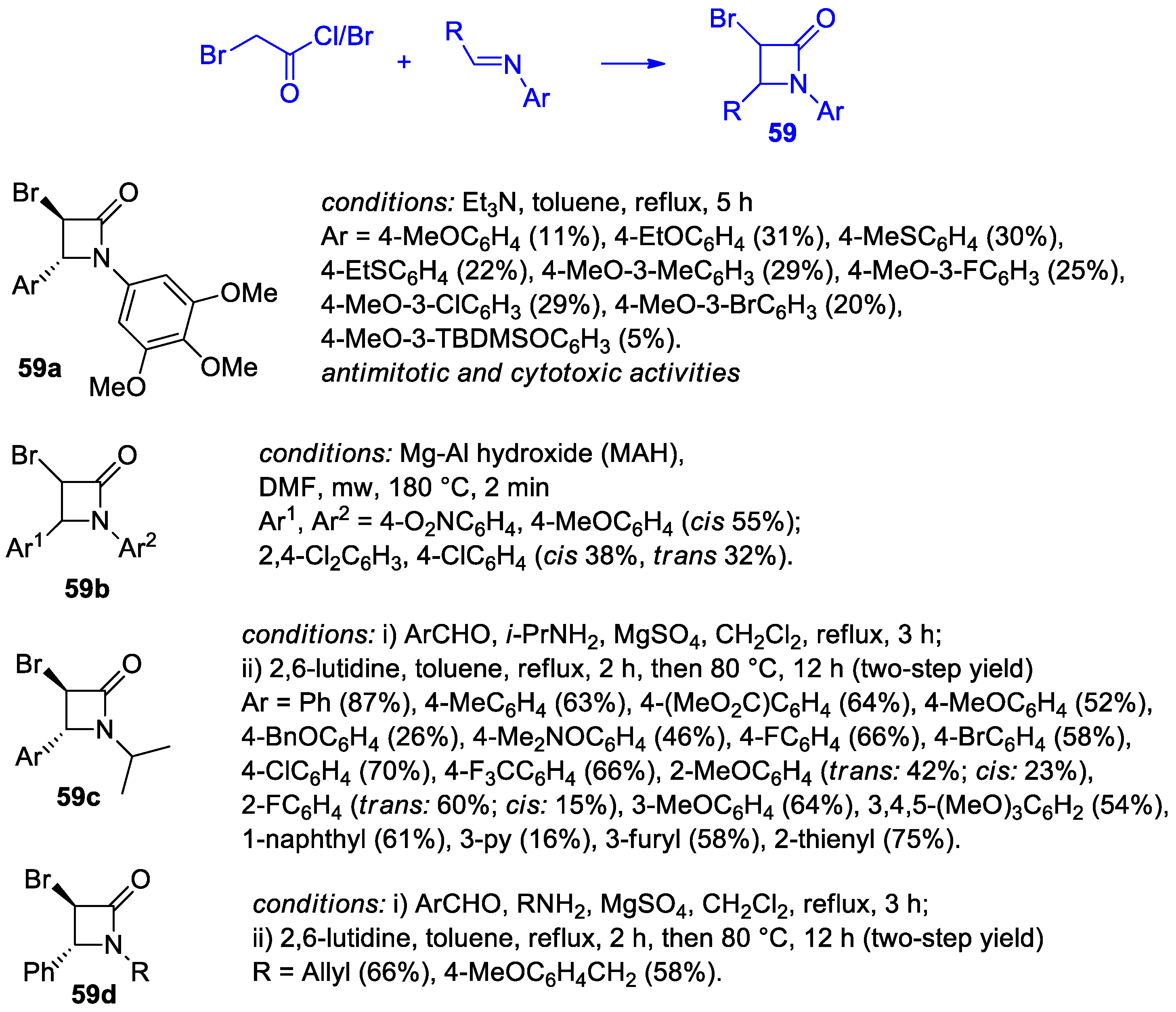
2.2.8. Ketene Generated In Situ from 2-Bromoacetyl Chloride/Bromide
2.3. Ketene Generated In Situ from Carboxylic Acids
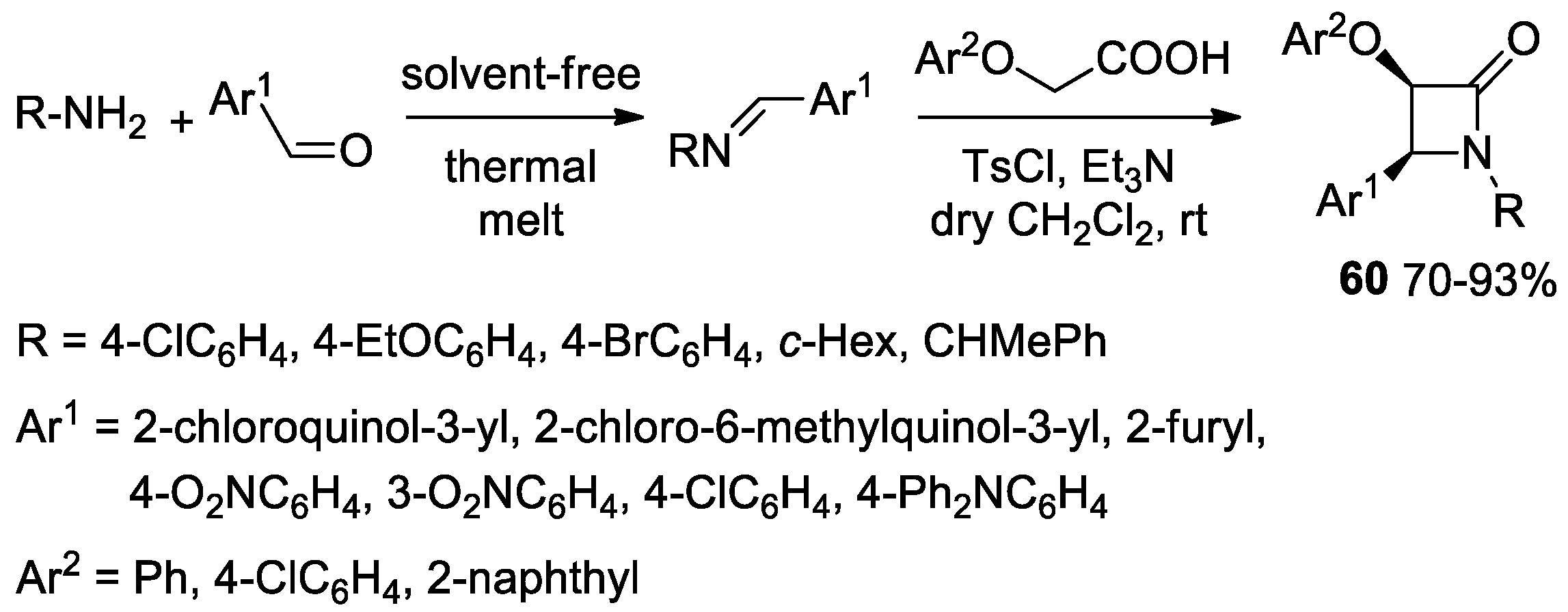
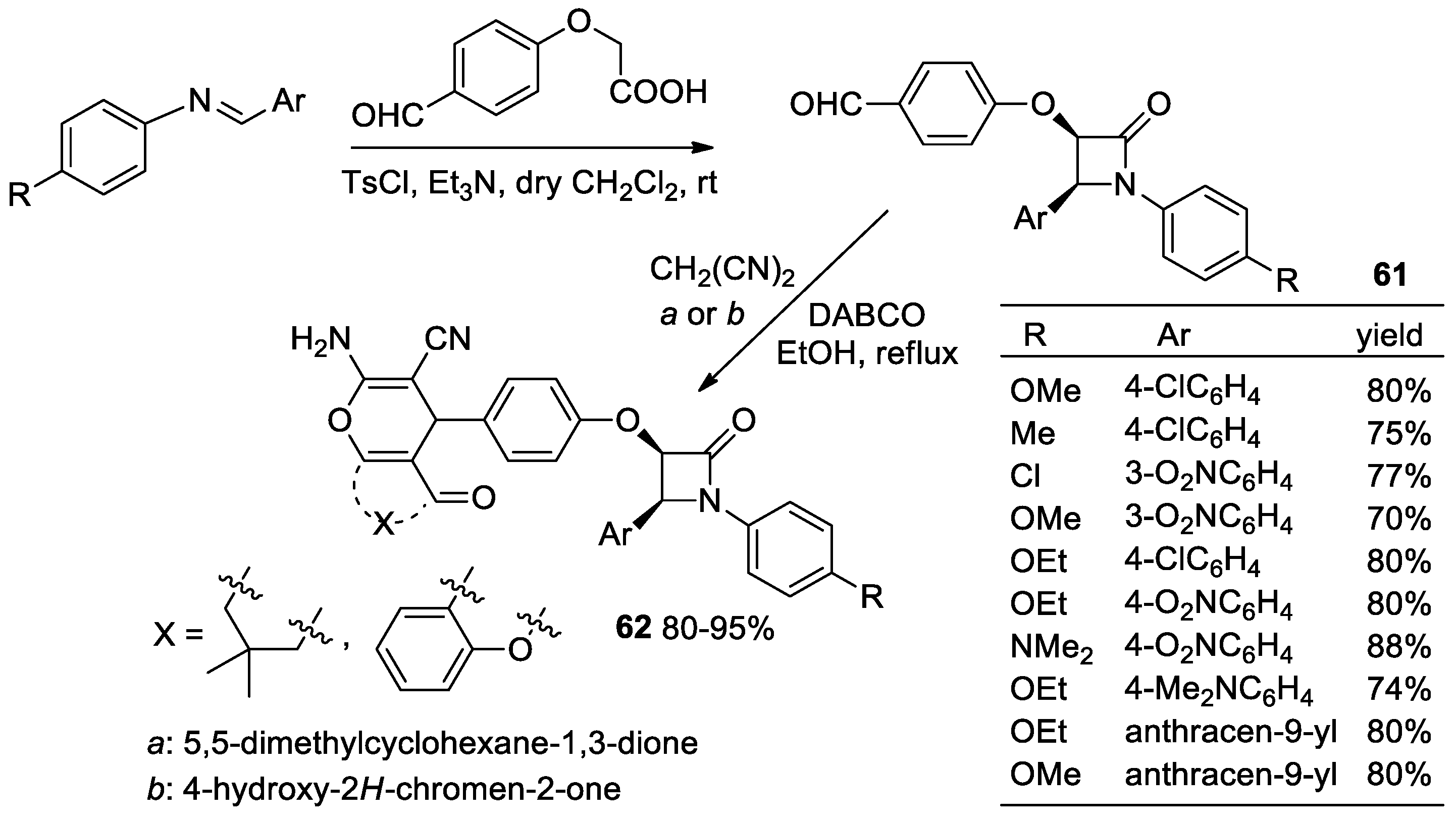
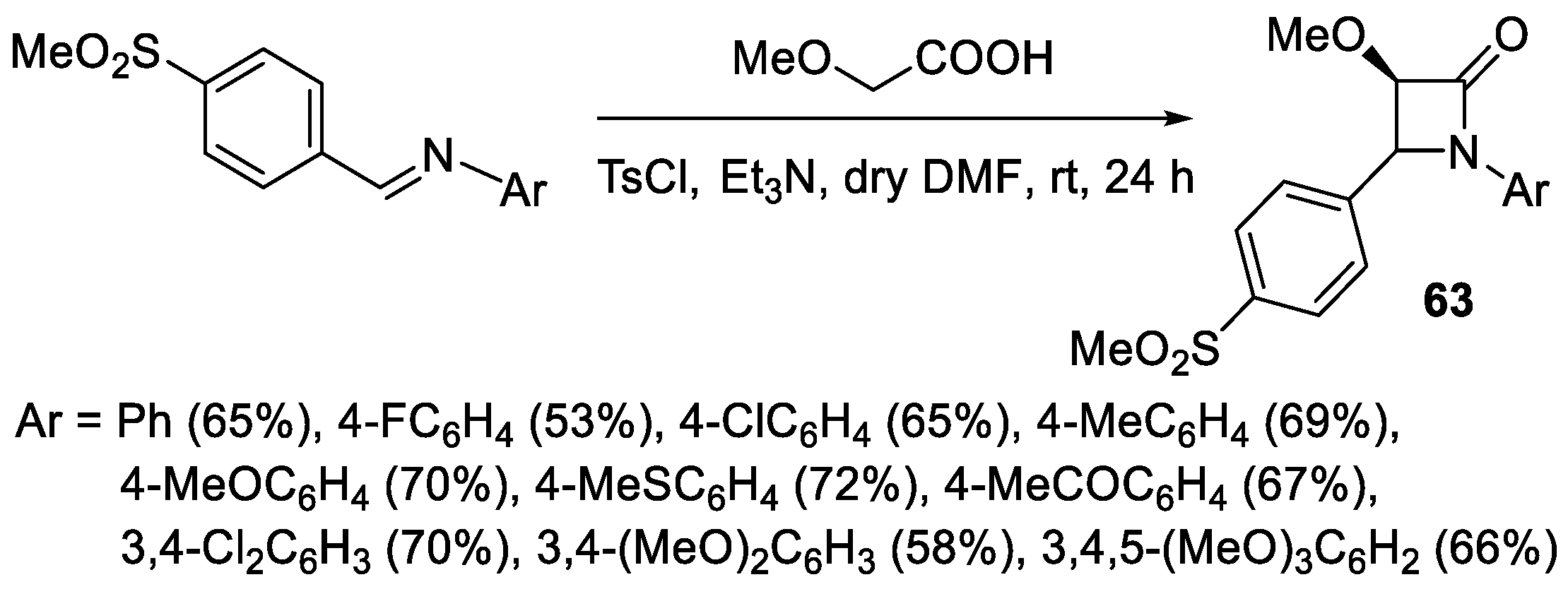
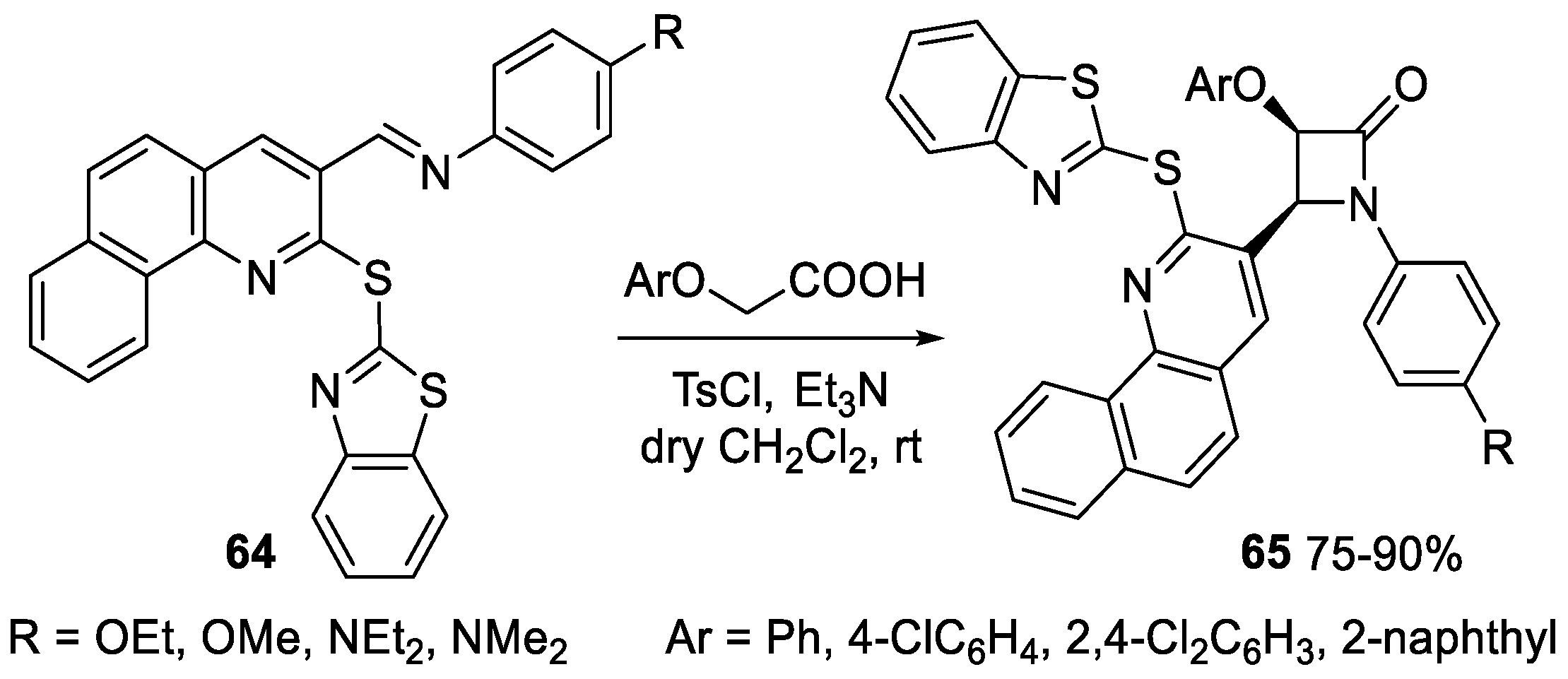
2.3.1. Ketene Generated In Situ from Carboxylic Acids and p-TsCl/Base
2.3.2. Ketene Generated In Situ from Carboxylic Acids and POCl3/Base
2.3.3. Ketene Generated In Situ from Carboxylic Acids and SOCl2/Base
2.3.4. Ketene Generated In Situ from Carboxylic Acids and Acyl Chlorides/Base or Trifosgene/Base
2.3.5. Ketene Generated In Situ from Carboxylic Acids and Vilsmeier Reagent/Base
2.3.6. Ketene Generated In Situ from Carboxylic Acids and Mukaiyama Reagent/Base
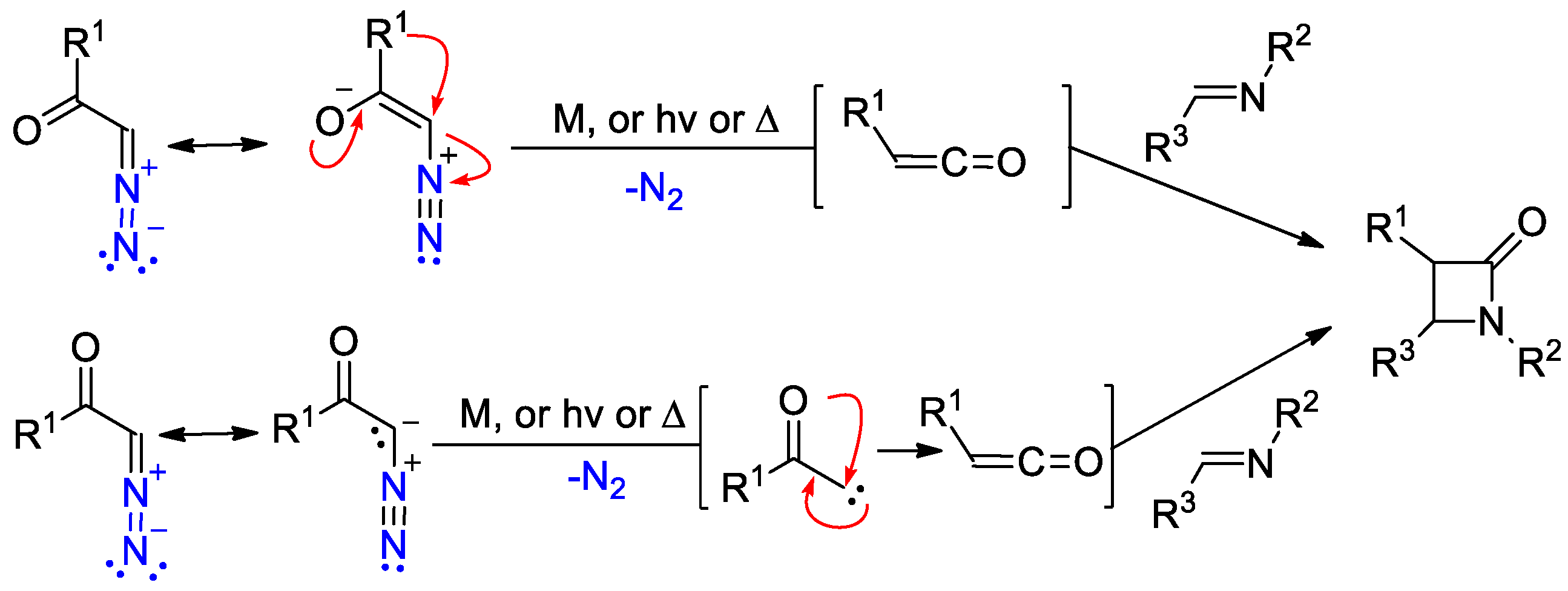
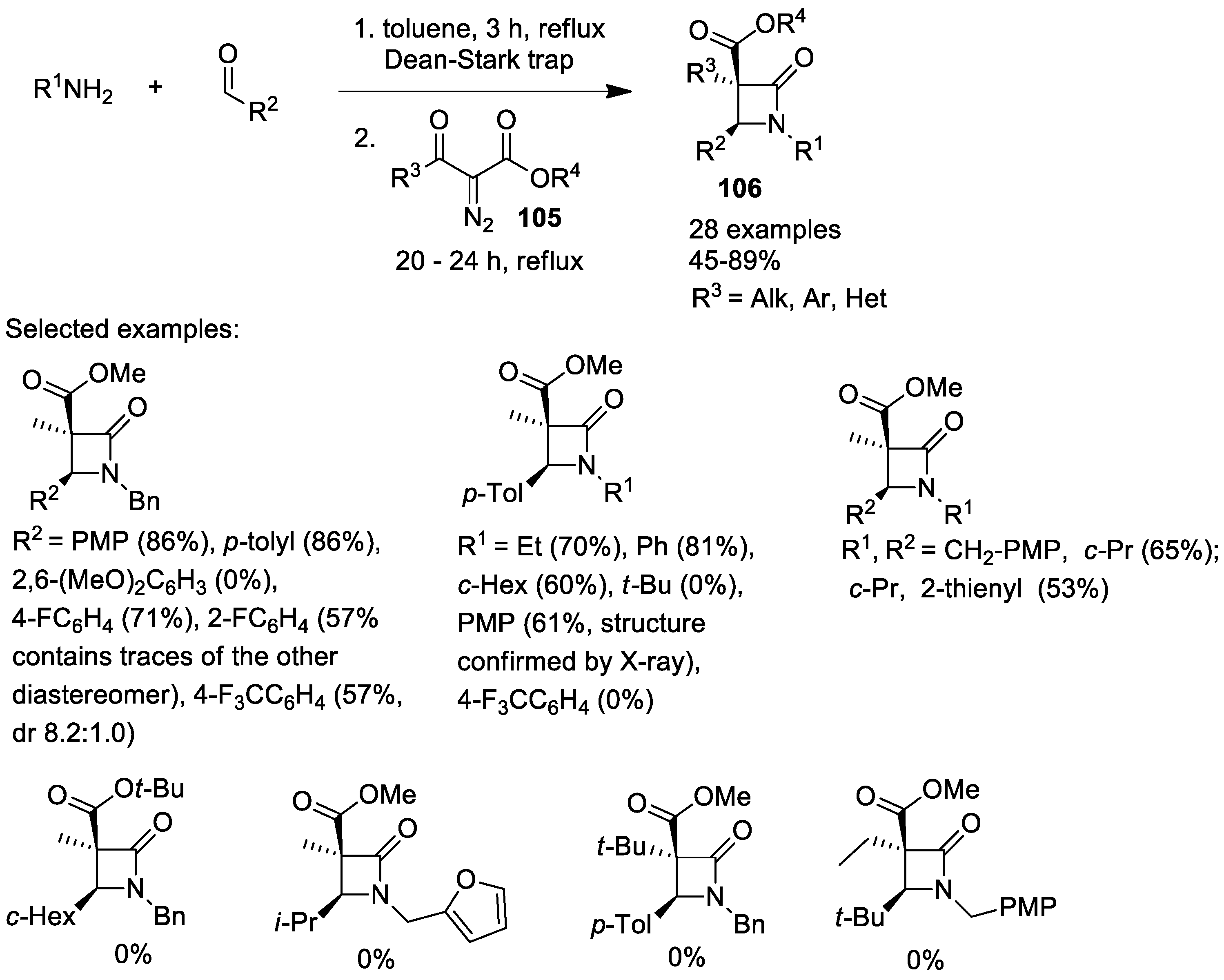
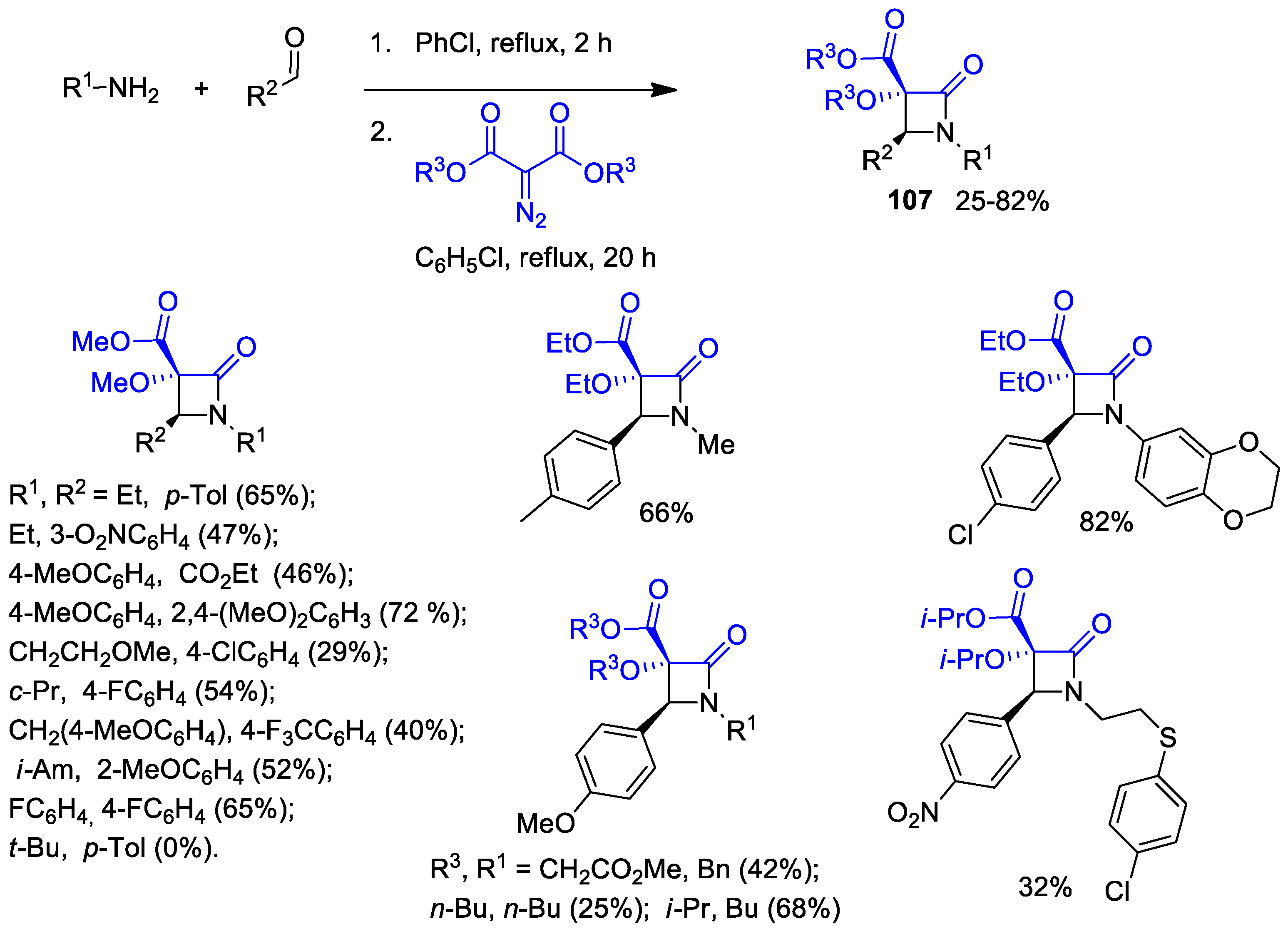
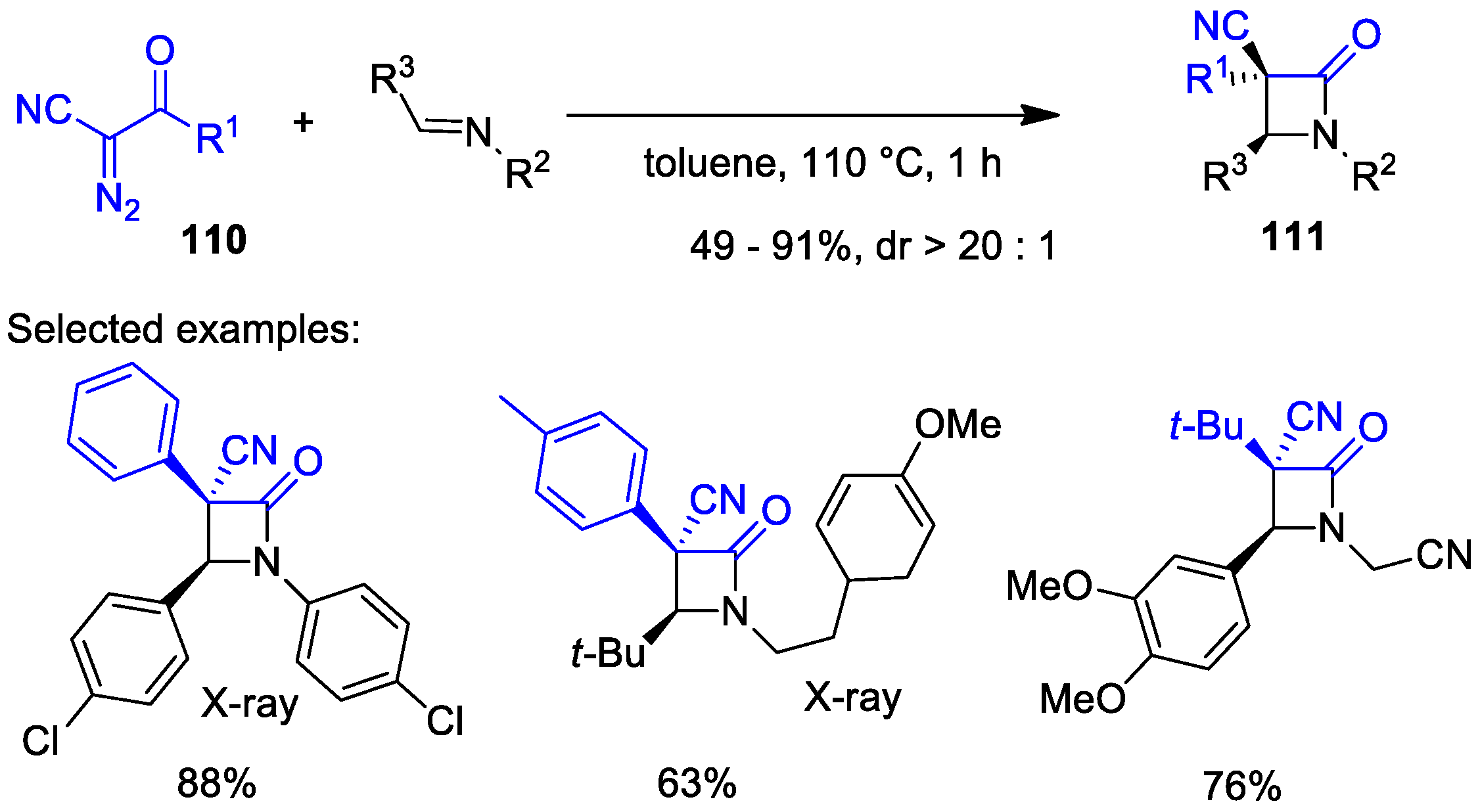
2.4. Ketene Generated In Situ from Diazo Compounds
2.5. Ketene Generated In Situ from α-Haloesters (Reformatsky-Type Reaction)
2.6. Ketene Generated In Situ from Ester (or Amido) Enolates
2.7. Ketene Generated In Situ from Cyclobutenones
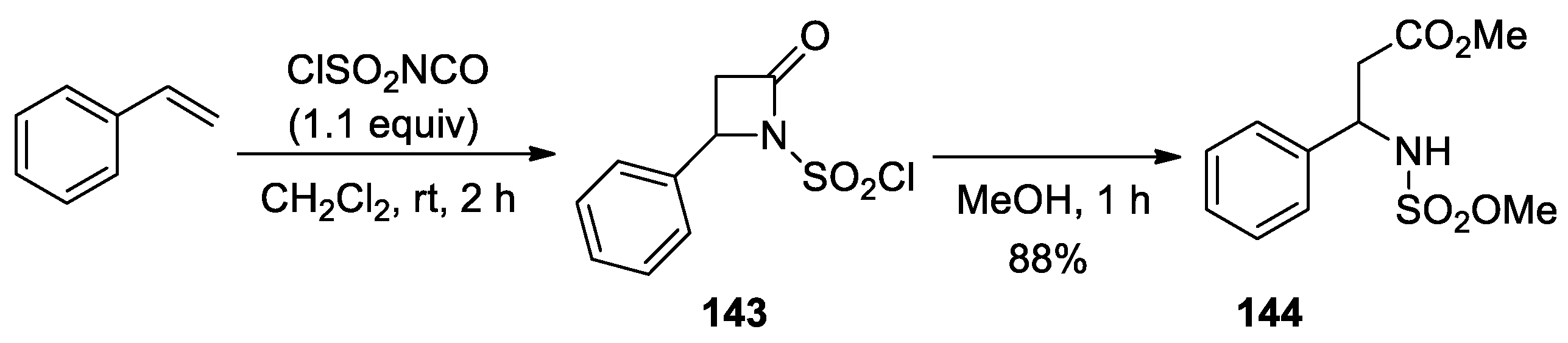
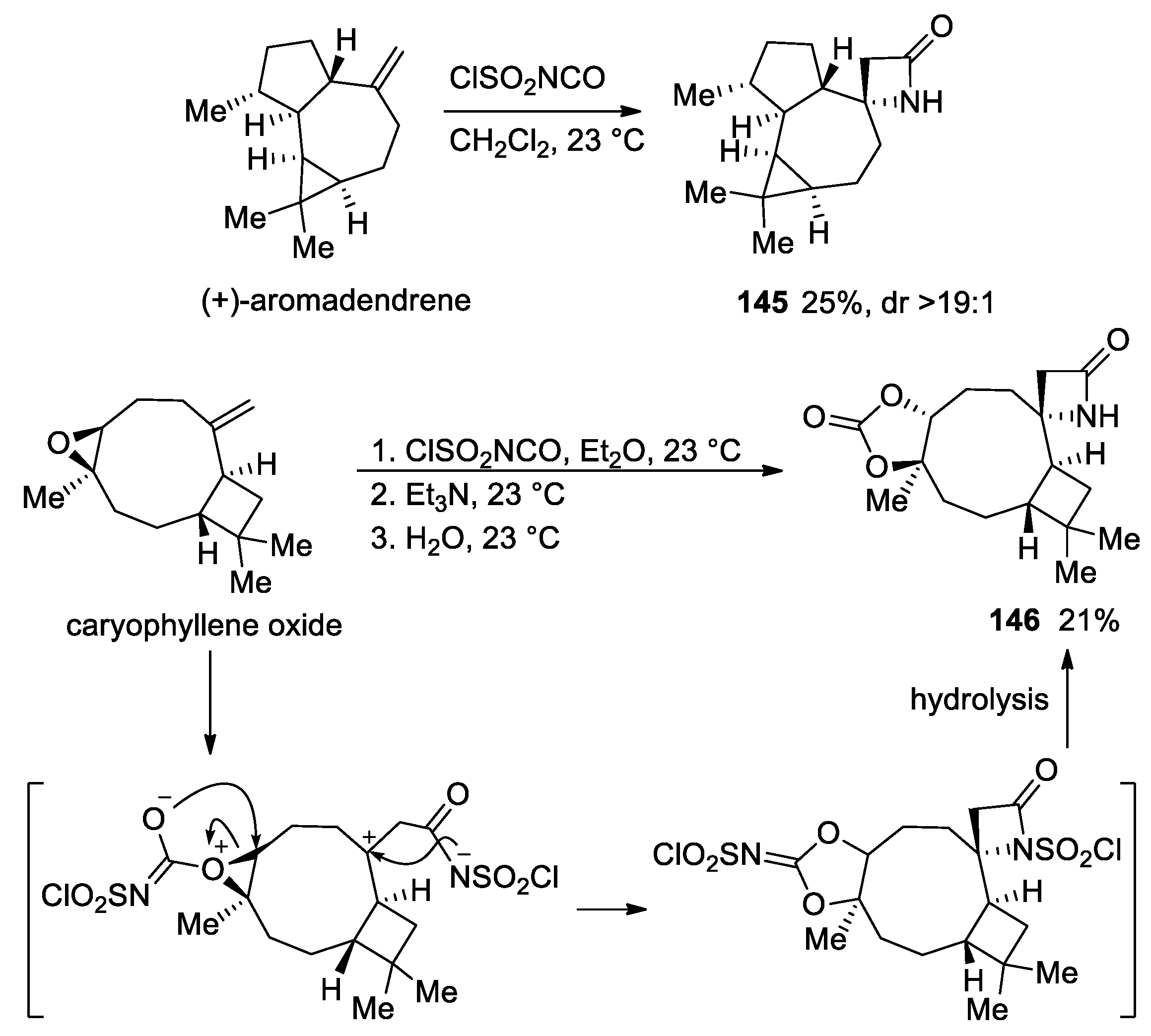
3. Alkene–Isocyanate Cycloaddition
4. Azetidin-2-Ones from Nitrones
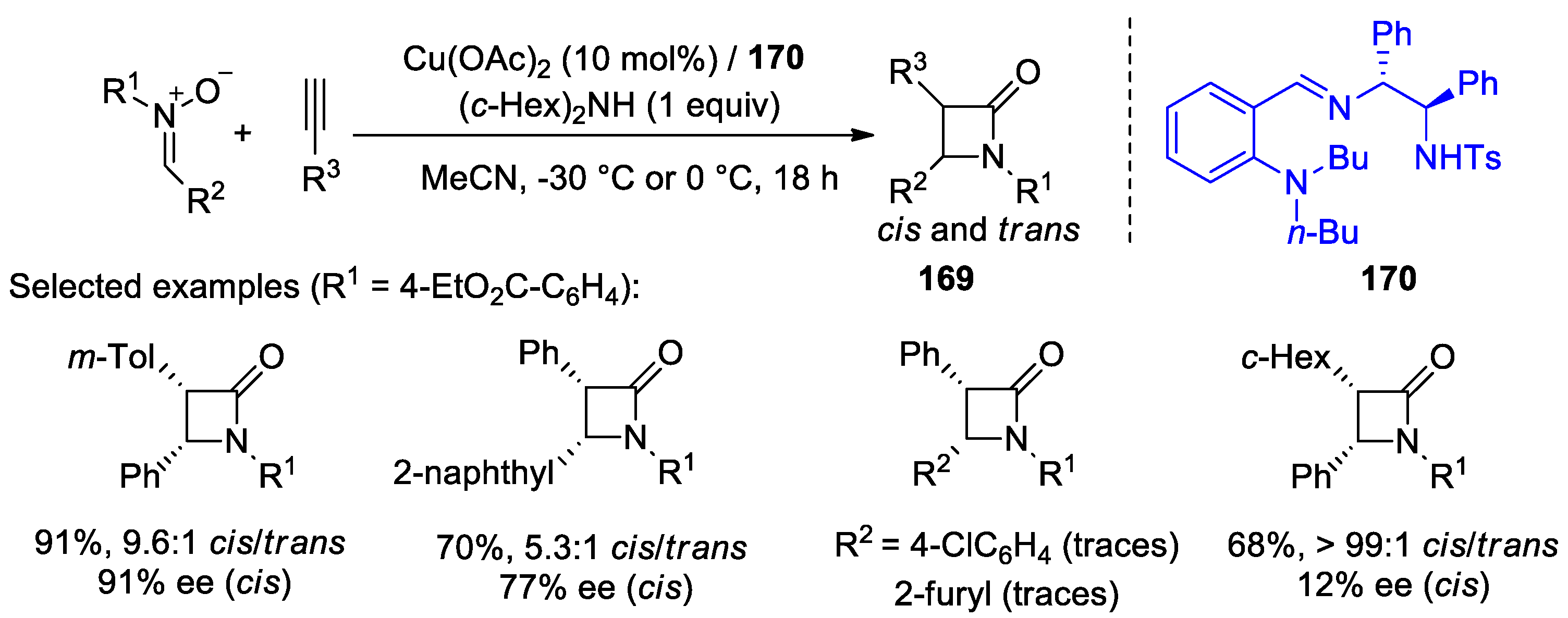
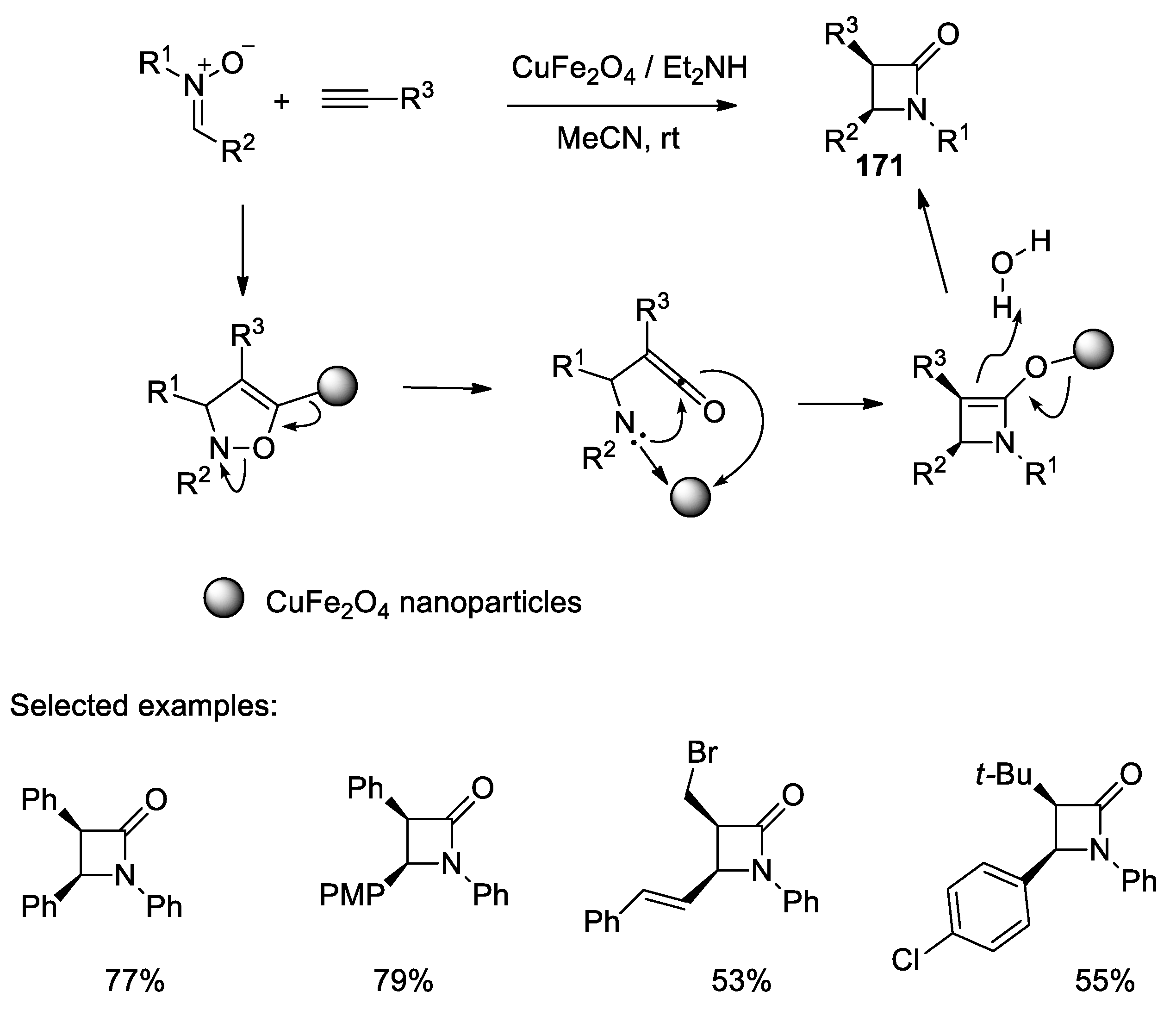
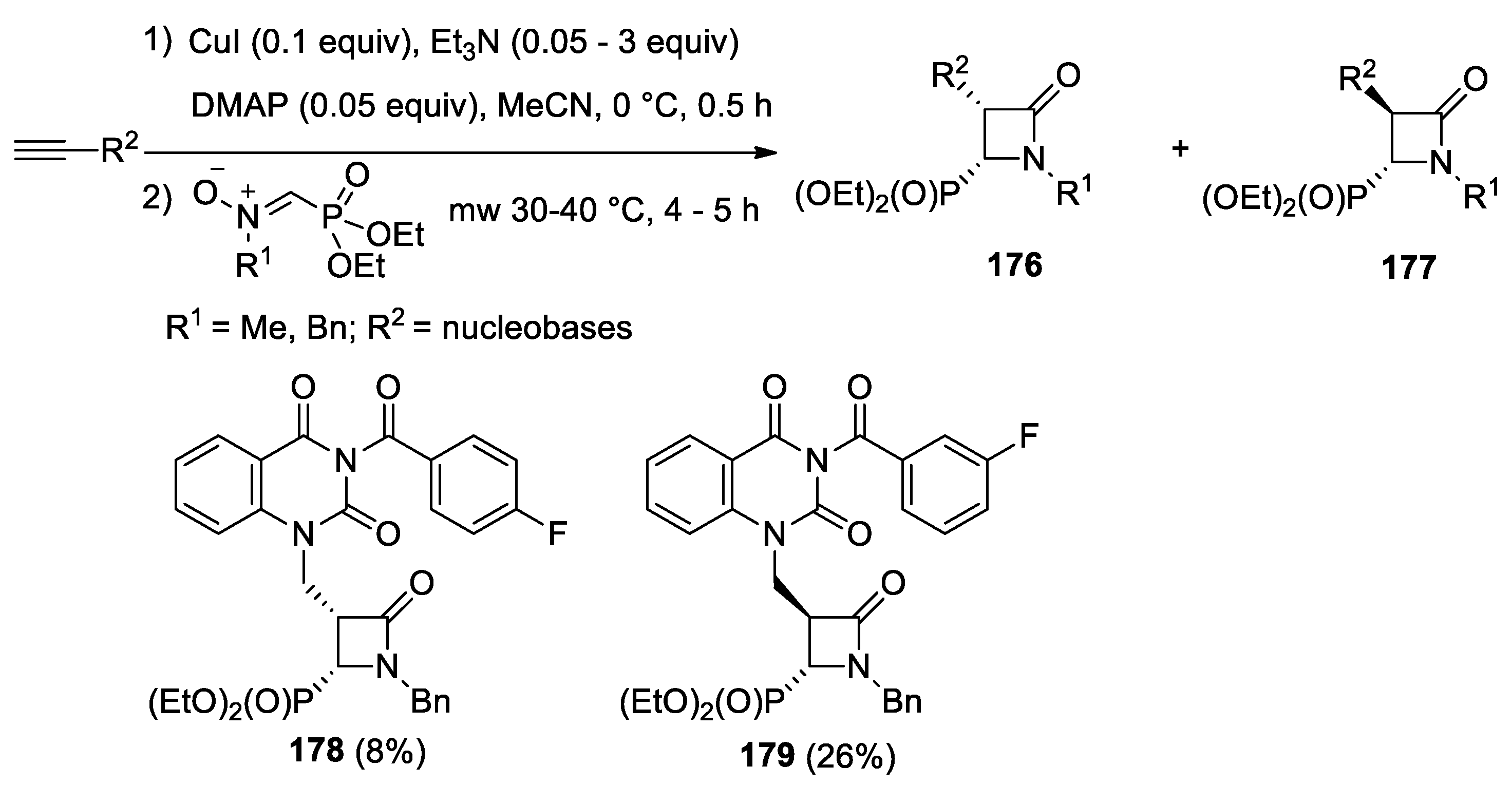
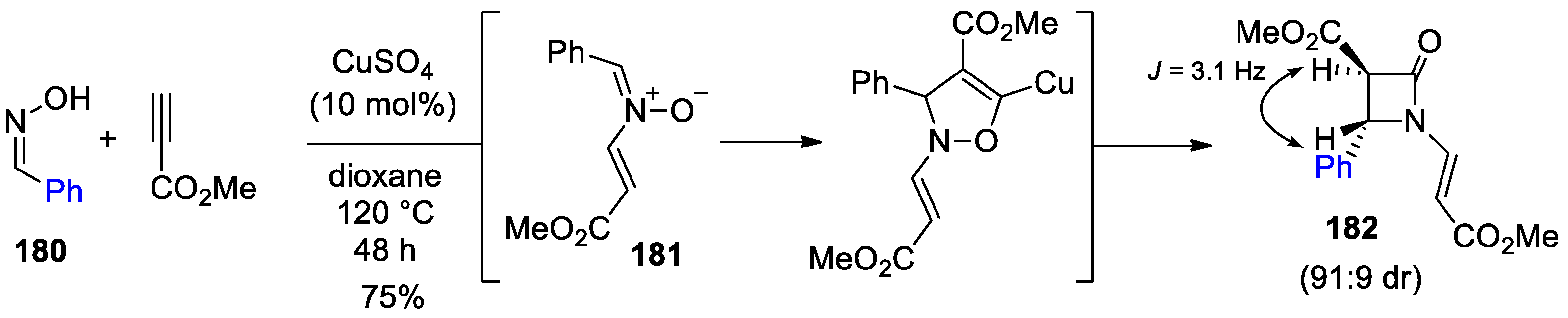
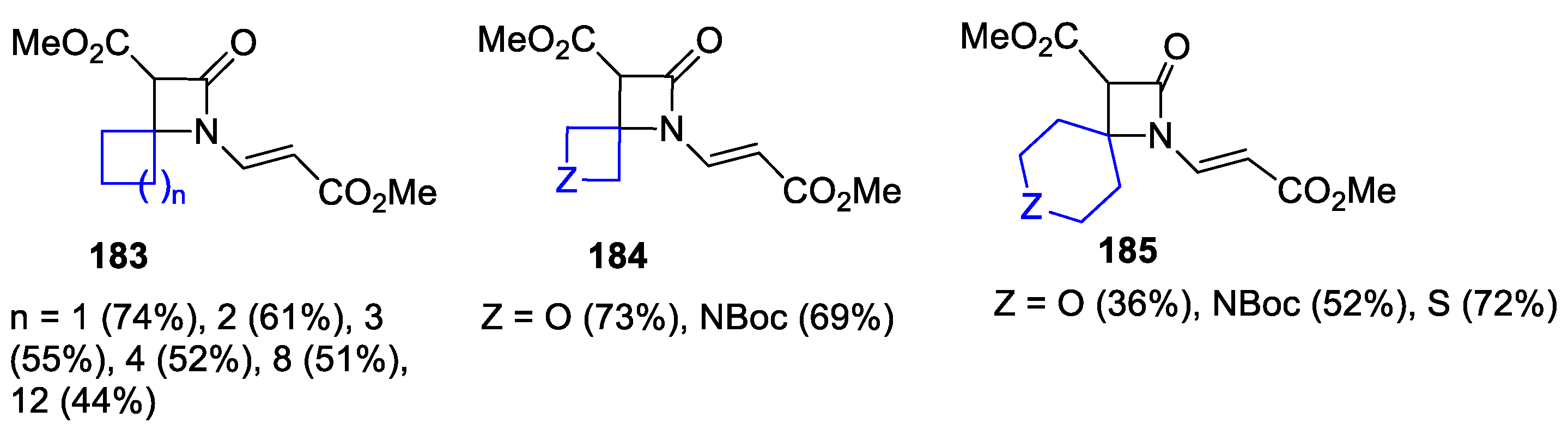
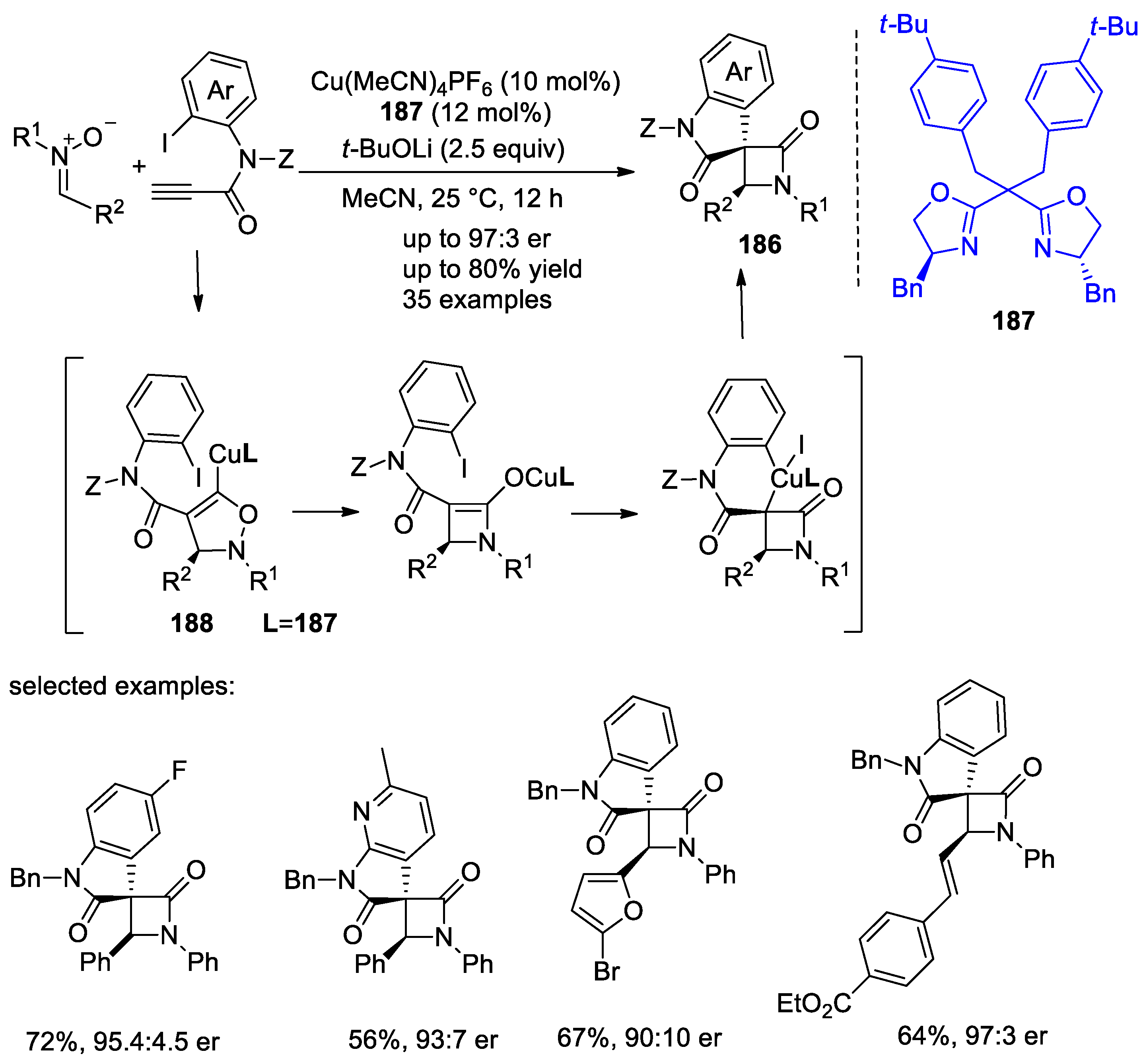
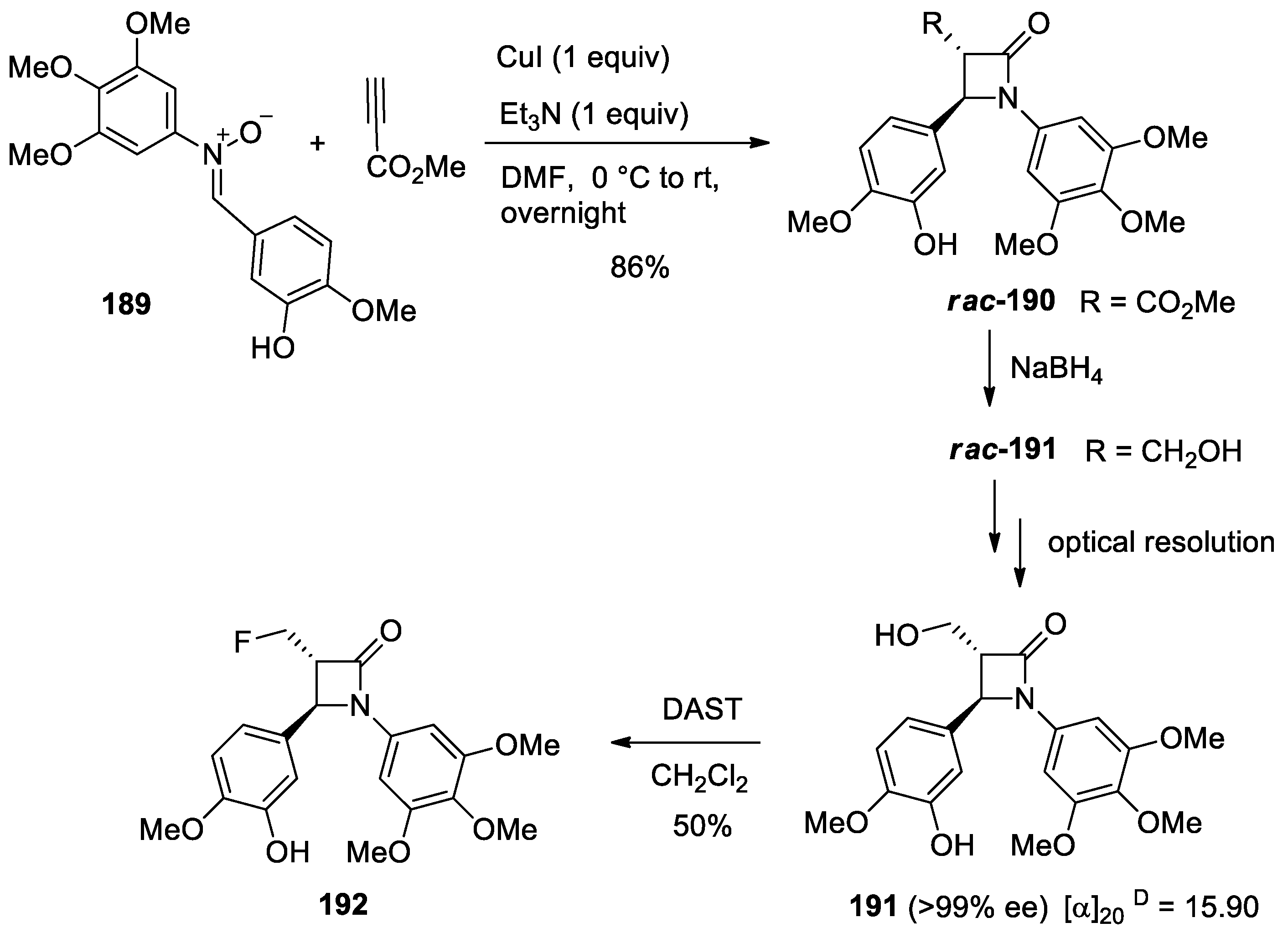
4.1. Nitrones and Alkynes (Kinugasa Reaction)
4.2. Nitrones and Polifluoropropenes
4.3. Nitrones and Methylenecyclopropanes
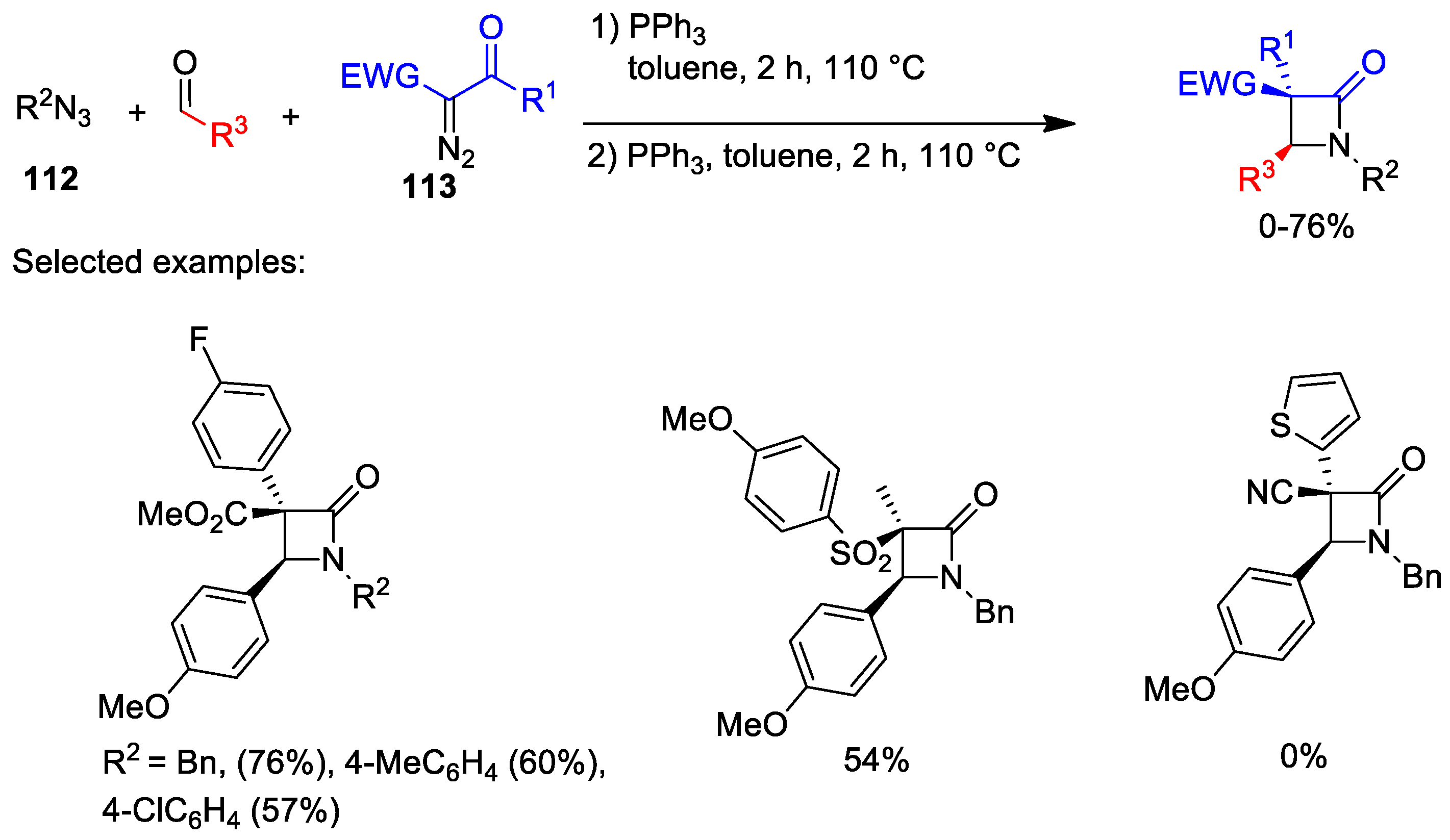
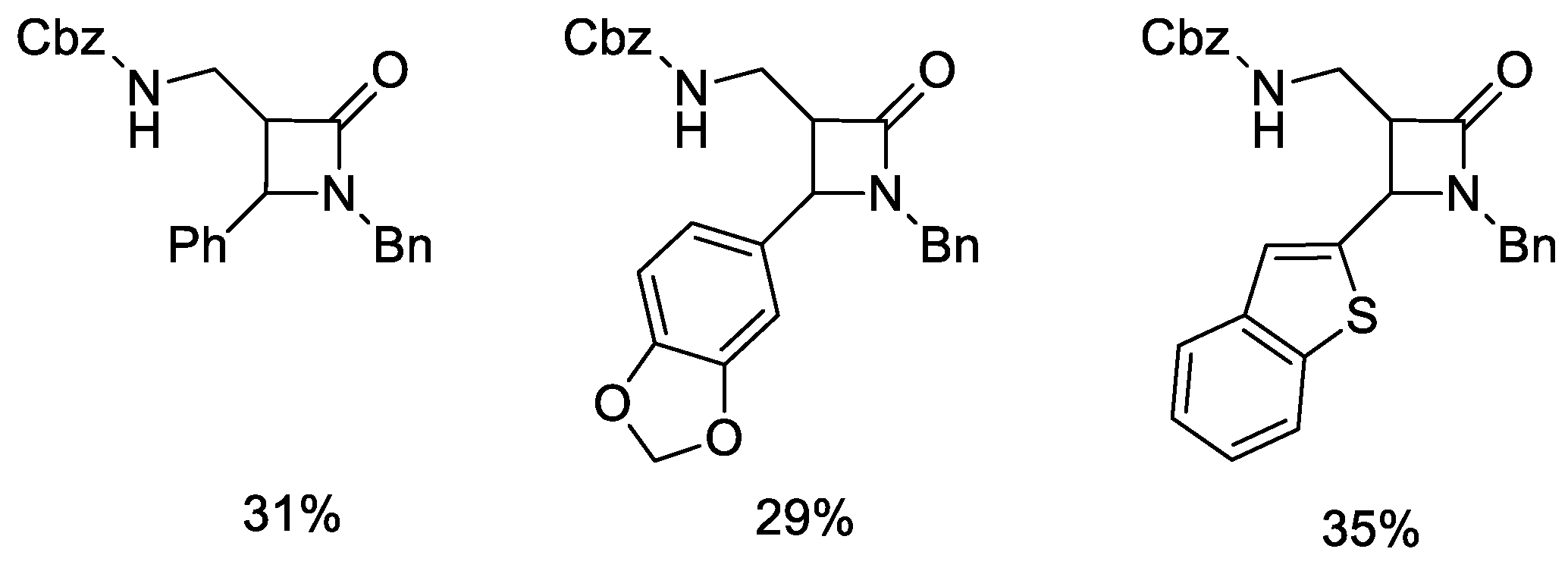
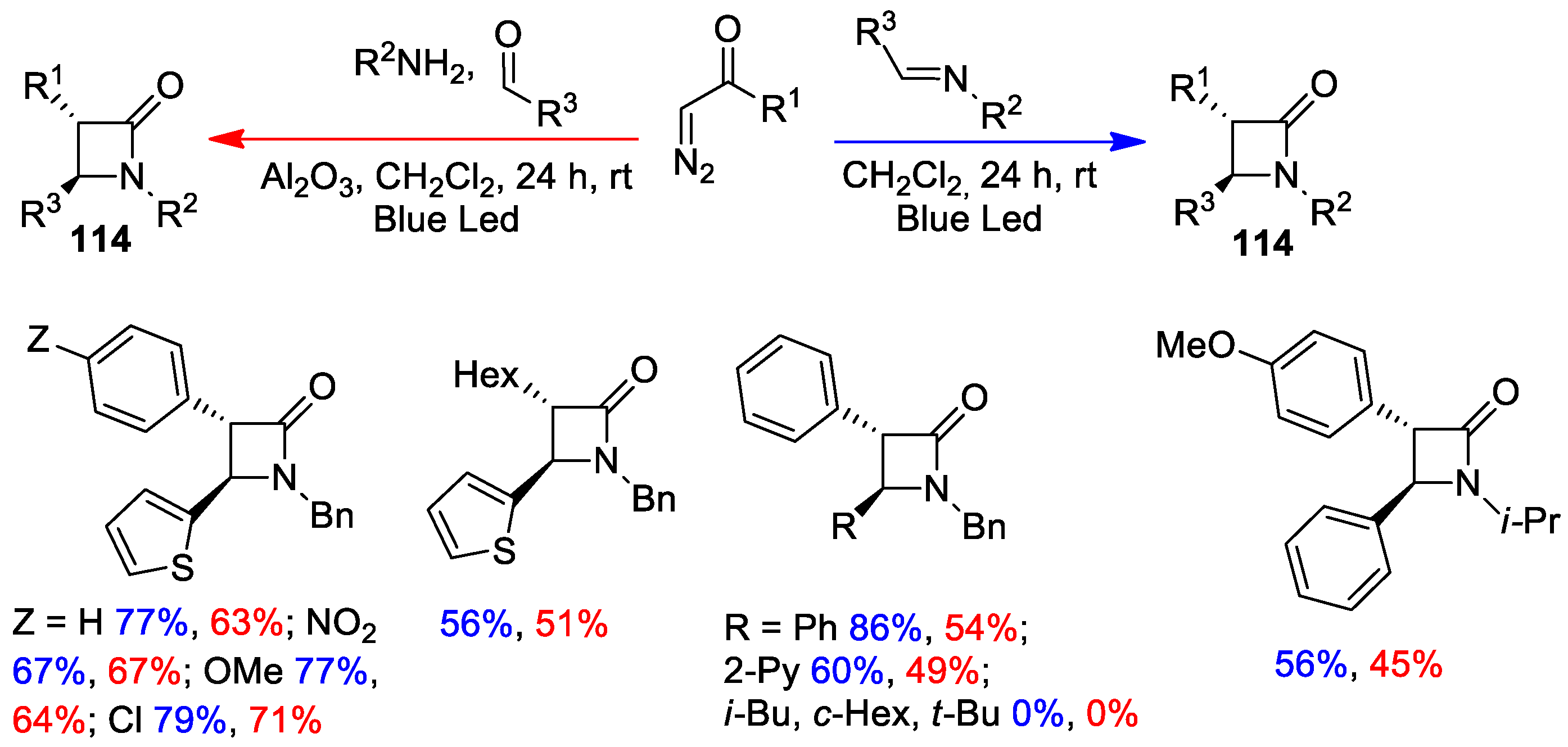
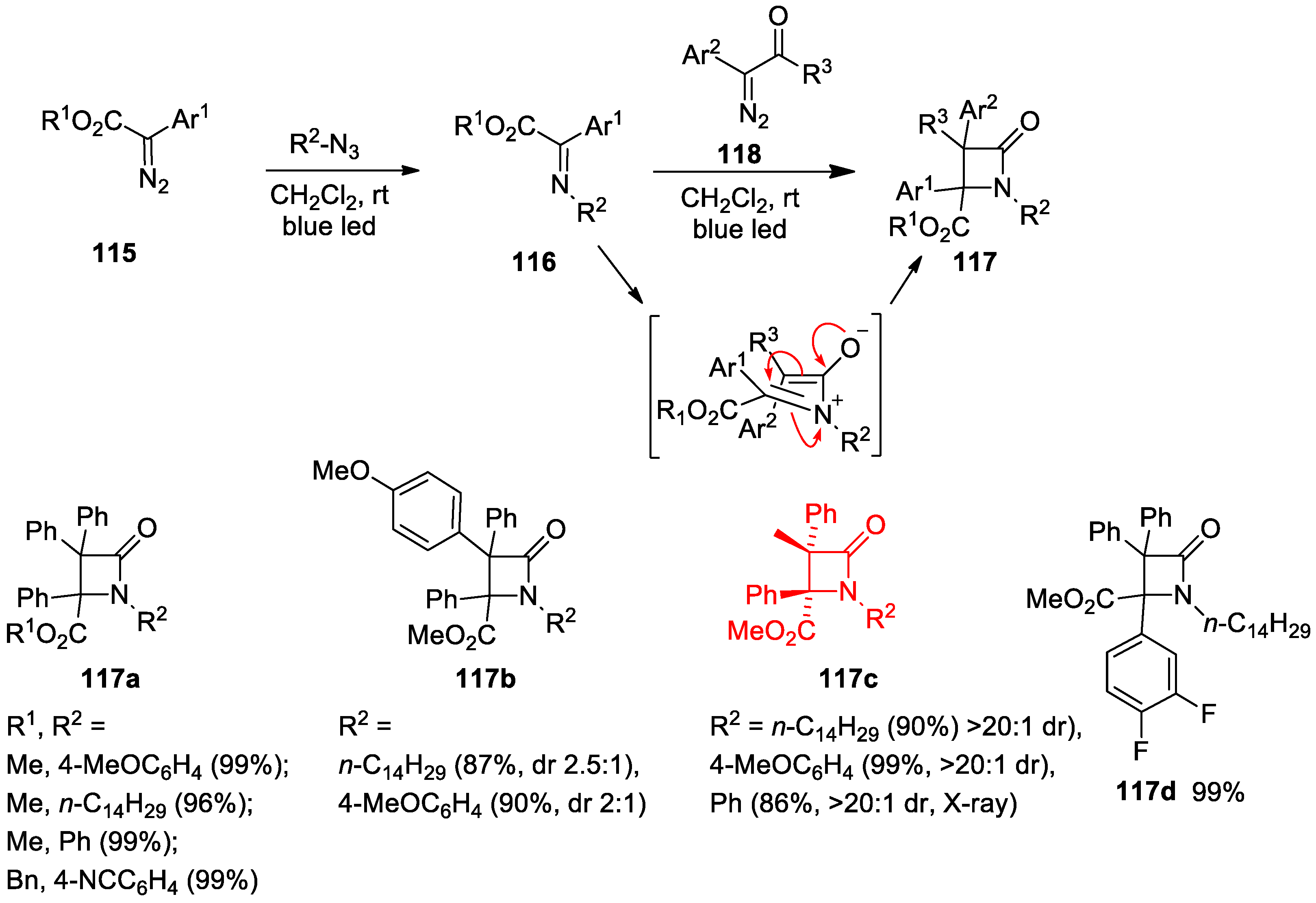
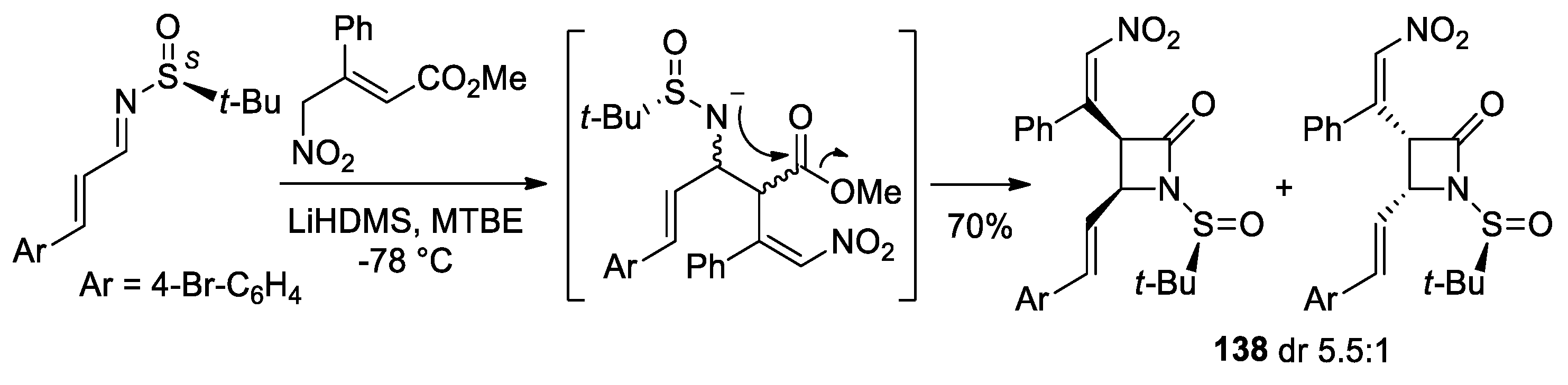
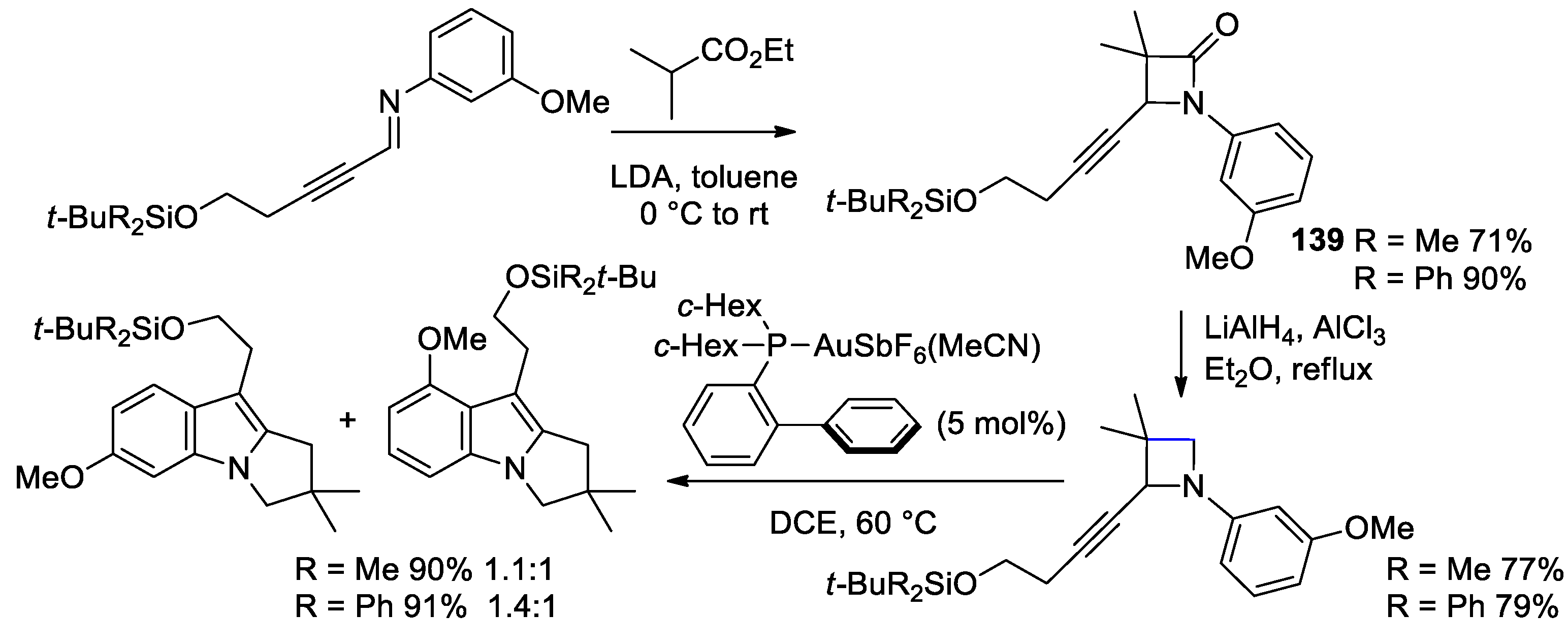
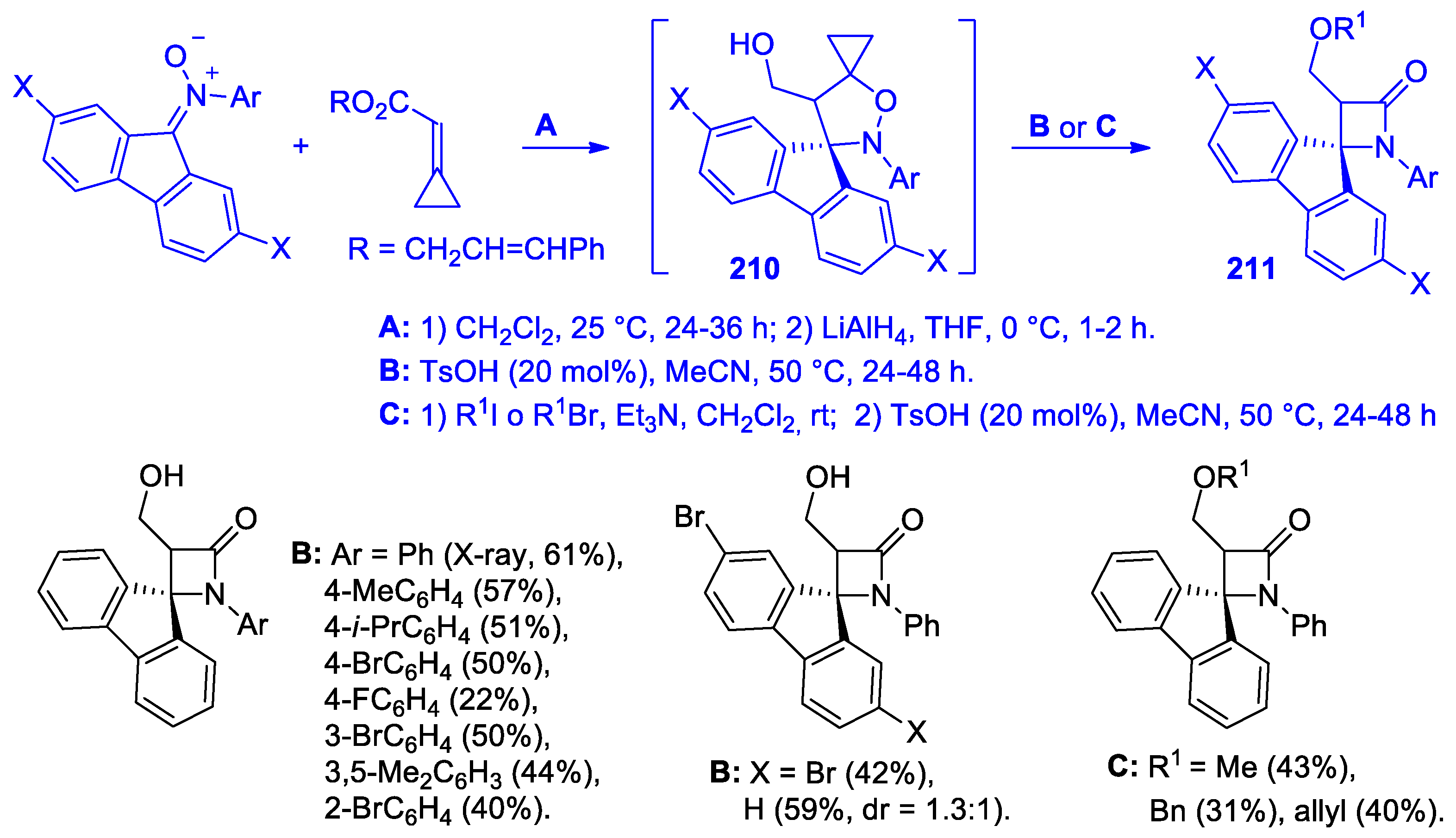
5. Miscellanea
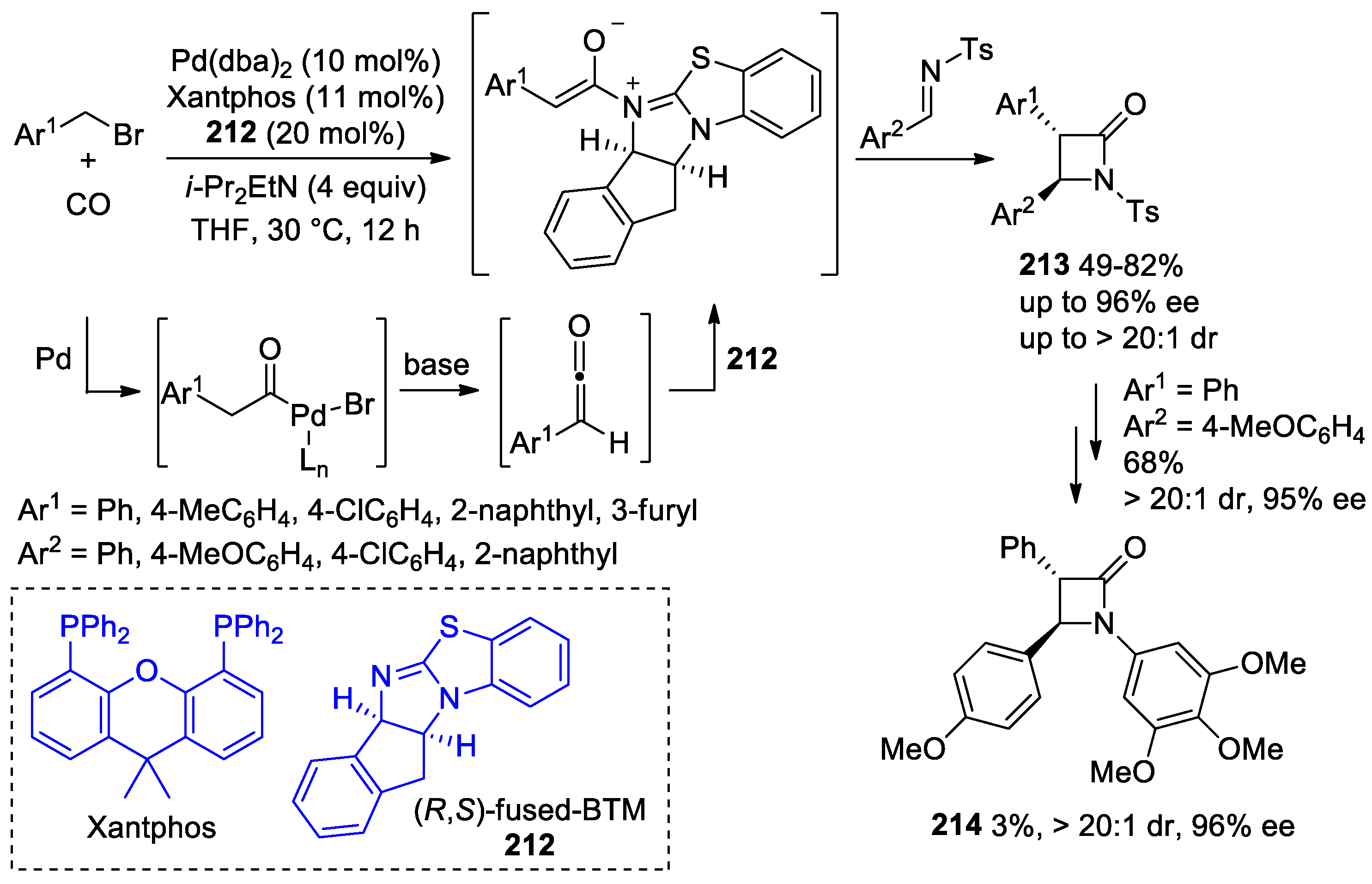
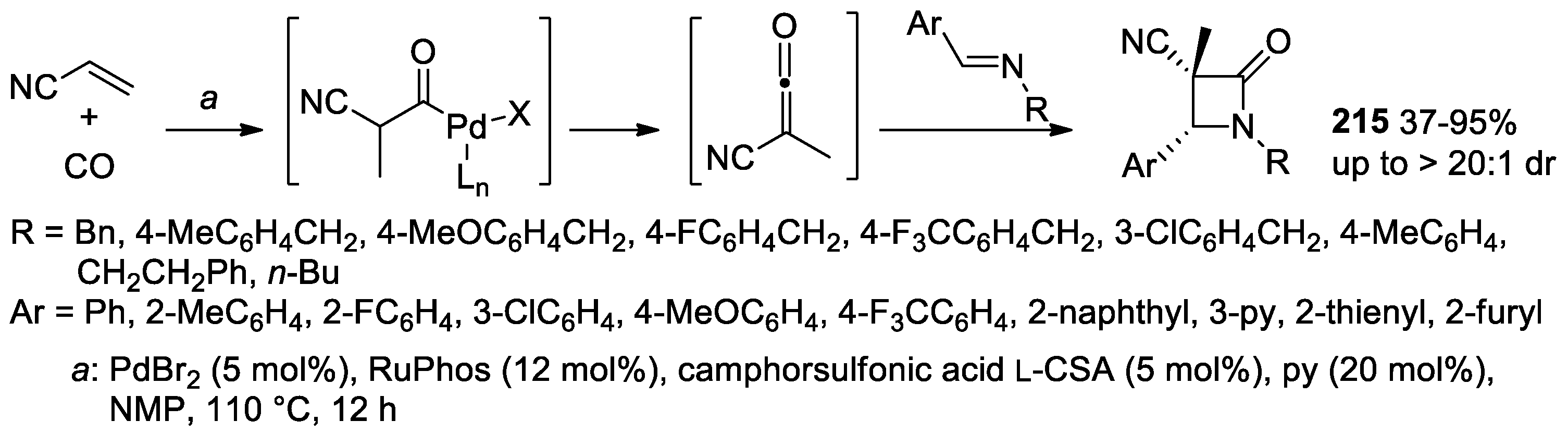
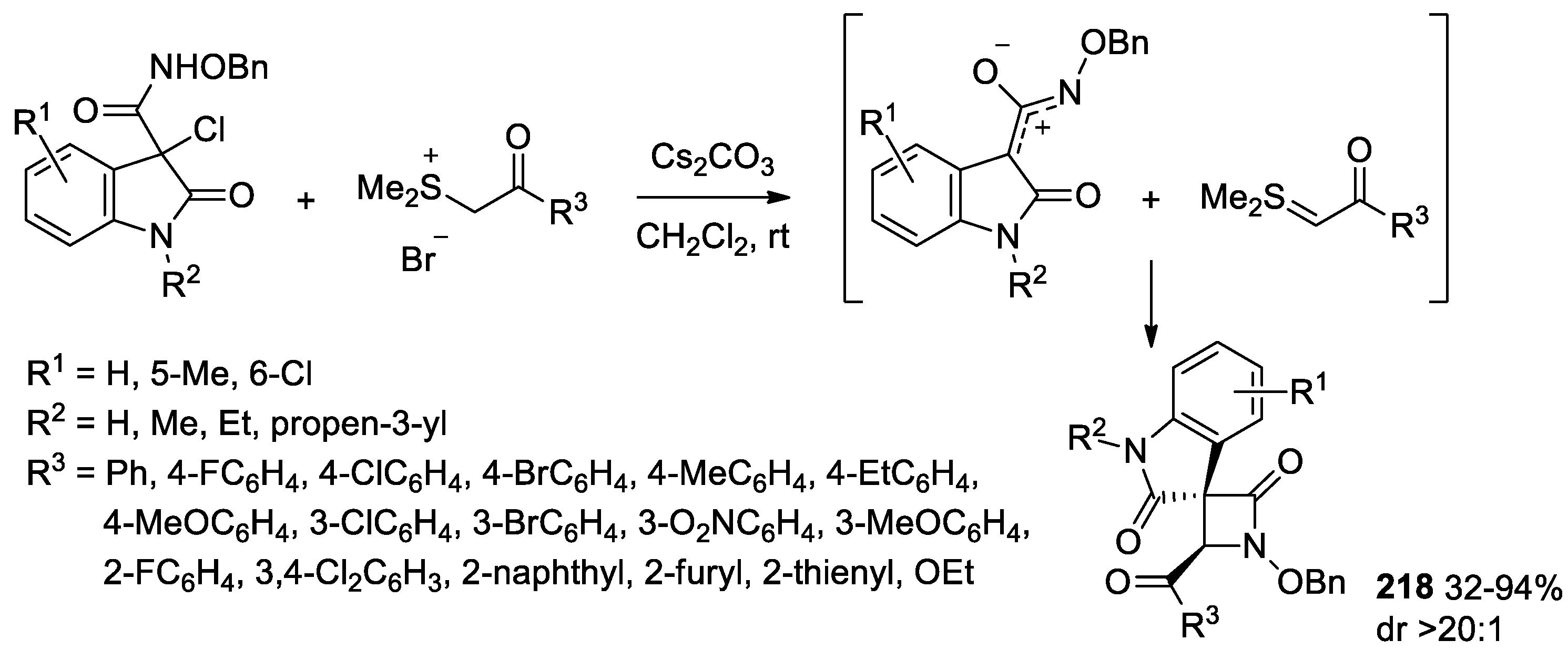
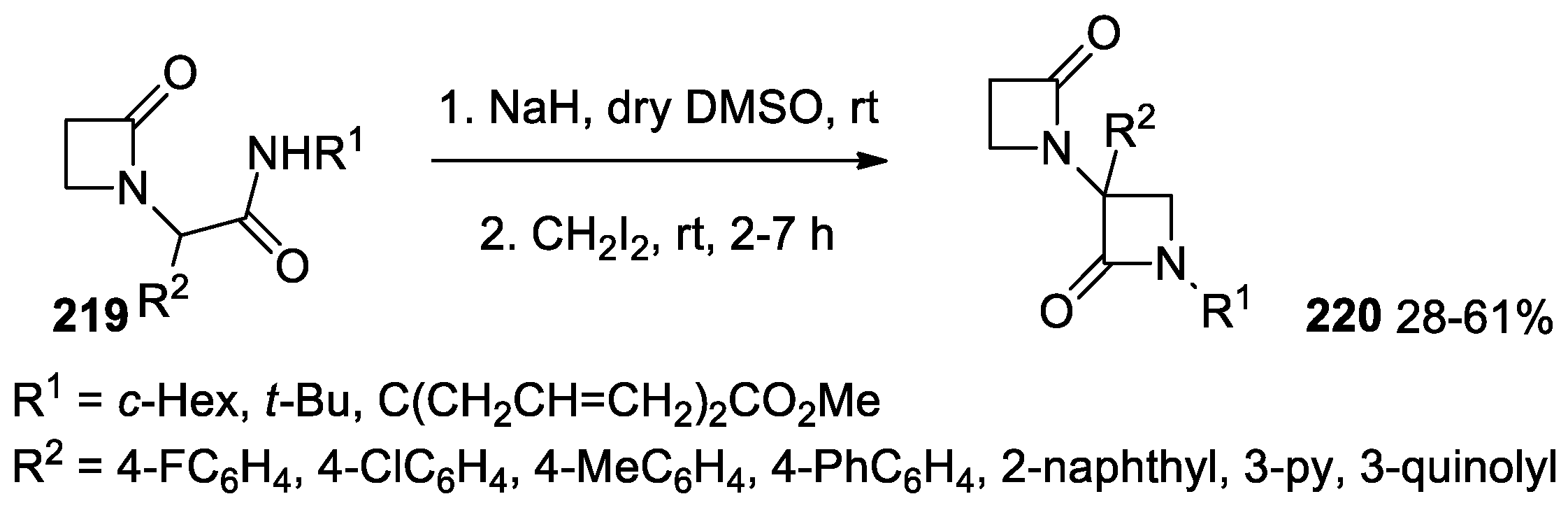
5.1. Formal [1+1+2] Cycloadditions
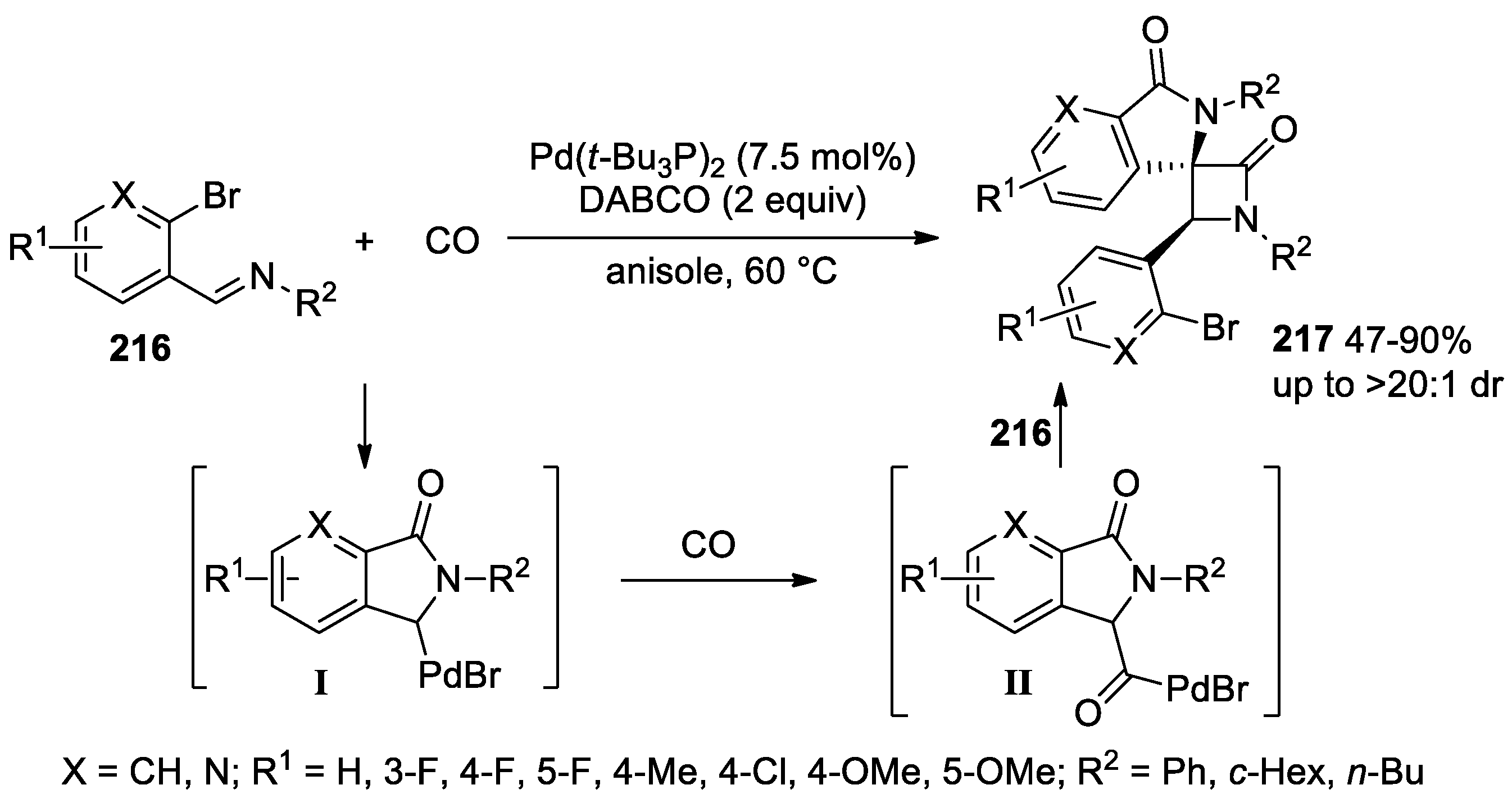
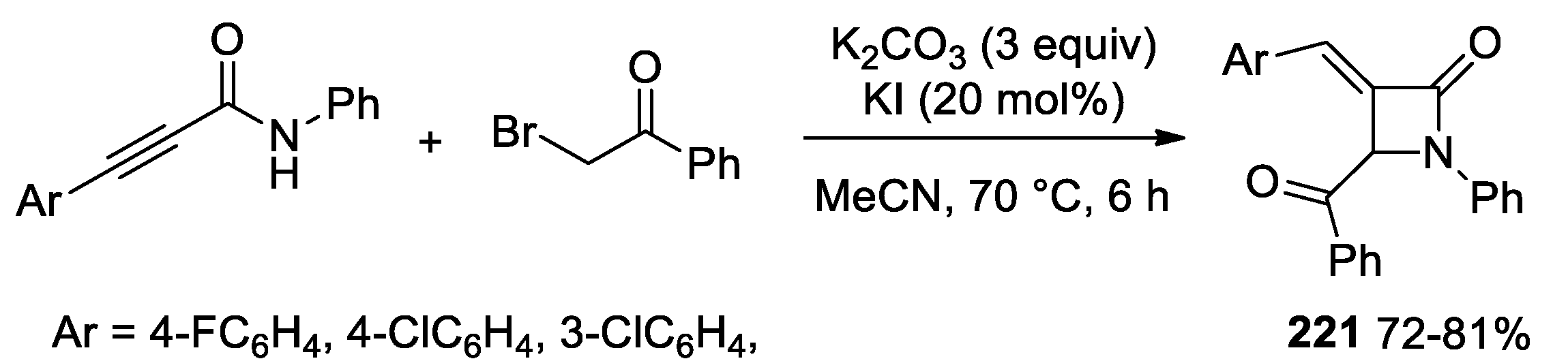
5.2. Formal [3+1] Cycloadditions
6. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Andresini, M.; Degennaro, L.; Luisi, R. Azetidines, Azetines and Azetes: Monocyclic. In Comprehensive Heterocyclic Chemistry IV; Elsevier: Amsterdam, The Netherlands, 2022; pp. 1–115. ISBN 978-0-12-818656-5. [Google Scholar]
- Singh, G.S.; D’hooghe, M.; De Kimpe, N. Azetidines, Azetines and Azetes: Monocyclic. In Comprehensive Heterocyclic Chemistry III; Elsevier: Amsterdam, The Netherlands, 2008; pp. 1–110. ISBN 978-0-08-044992-0. [Google Scholar]
- Pitts, C.R.; Lectka, T. Chemical Synthesis of β-Lactams: Asymmetric Catalysis and Other Recent Advances. Chem. Rev. 2014, 114, 7930–7953. [Google Scholar] [CrossRef]
- Brandi, A.; Cicchi, S.; Cordero, F.M. Novel Syntheses of Azetidines and Azetidinones. Chem. Rev. 2008, 108, 3988–4035. [Google Scholar] [CrossRef]
- Deketelaere, S.; Van Nguyen, T.; Stevens, C.V.; D’hooghe, M. Synthetic Approaches toward Monocyclic 3-Amino-β-Lactams. Chem. Open 2017, 6, 301–319. [Google Scholar] [CrossRef]
- Mehta, P.D.; Sengar, N.P.S.; Pathak, A.K. 2-Azetidinone—A New Profile of Various Pharmacological Activities. Eur. J. Med. Chem. 2010, 45, 5541–5560. [Google Scholar] [CrossRef]
- Gupta, A.; Halve, A.K. β-Lactams: A Mini Review of Their Biological Activity. Int. J. Pharm. Sci. Res. 2015, 6, 978–987. [Google Scholar]
- Galletti, P.; Giacomini, D. Monocyclic β-Lactams: New Structures for New Biological Activities. Curr. Med. Chem. 2011, 18, 4265–4283. [Google Scholar] [CrossRef]
- Leite, T.H.O.; Saraiva, M.F.; Pinheiro, A.C.; De Souza, M.V.N. Monocyclic β-Lactam: A Review on Synthesis and Potential Biological Activities of a Multitarget Core. Mini-Rev. Med. Chem. 2020, 20, 1653–1682. [Google Scholar] [CrossRef]
- Grabrijan, K.; Strašek, N.; Gobec, S. Monocyclic Beta–Lactams for Therapeutic Uses: A Patent Overview (2010–2020). Expert Opin. Ther. Pat. 2021, 31, 247–266. [Google Scholar] [CrossRef]
- Palomo, C.; Oiarbide, M. β-Lactam Ring Opening: A Useful Entry to Amino Acids and Relevant Nitrogen-Containing Compounds. In Heterocyclic Scaffolds I; Banik, B.K., Ed.; Topics in Heterocyclic Chemistry; Springer: Berlin/Heidelberg, Germany, 2010; Volume 22, pp. 211–259. ISBN 978-3-642-12844-8. [Google Scholar]
- Alcaide, B.; Almendros, P.; Aragoncillo, C. β-Lactams: Versatile Building Blocks for the Stereoselective Synthesis of Non-β-Lactam Products. Chem. Rev. 2007, 107, 4437–4492. [Google Scholar] [CrossRef]
- Muller, P. Glossary of Terms Used in Physical Organic Chemistry (IUPAC Recommendations 1994). Pure Appl. Chem. 1994, 66, 1077–1184. [Google Scholar] [CrossRef]
- Landa, A.; Mielgo, A.; Oiarbide, M.; Palomo, C. Asymmetric Synthesis of β-Lactams by the Staudinger Reaction. Org. React. 2018, 95, 423–594. [Google Scholar]
- Nelson, S.G.; Dura, R.D.; Peelen, T.J. Catalytic Asymmetric Ketene [2+2] and [4+2] Cycloadditions. Org. React. 2013, 82, 471–621. [Google Scholar]
- Kaur, N. Synthesis of Azetidines by Cycloaddition of Imines to Carbonyl Compounds. In Synthesis of Azetidines from Imines by Cycloaddition Reactions; Elsevier: Amsterdam, The Netherlands, 2023; pp. 161–201. ISBN 978-0-443-19204-3. [Google Scholar]
- Fu, N.; Tidwell, T.T. Preparation of β-Lactams by [2+2] Cycloaddition of Ketenes and Imines. Tetrahedron 2008, 64, 10465–10496. [Google Scholar] [CrossRef]
- Palomo, C.; Aizpurua, J.M.; Ganboa, I.; Oiarbide, M. Asymmetric Synthesis of β-Lactams by Staudinger Ketene-Imine Cycloaddition Reaction. Eur. J. Org. Chem. 1999, 1999, 3223–3235. [Google Scholar] [CrossRef]
- Cossío, F.P.; Arrieta, A.; Sierra, M.A. The Mechanism of the Ketene−Imine (Staudinger) Reaction in Its Centennial: Still an Unsolved Problem? Acc. Chem. Res. 2008, 41, 925–936. [Google Scholar] [CrossRef]
- Cossío, F.P.; de Cózar, A.; Sierra, M.A.; Casarrubios, L.; Muntaner, J.G.; Banik, B.K.; Bandyopadhyay, D. Role of Imine Isomerization in the Stereocontrol of the Staudinger Reaction between Ketenes and Imines. RSC Adv. 2022, 12, 104–117. [Google Scholar] [CrossRef]
- Cassú, D. Computational Insight into the Scope of the Staudinger Cycloaddition Reaction for the Preparation of β-Lactams. ChemistrySelect 2020, 5, 3278–3282. [Google Scholar] [CrossRef]
- Domingo, L.R.; Ríos-Gutiérrez, M.; Sáez, J.A. Unravelling the Mechanism of the Ketene-Imine Staudinger Reaction. An ELF Quantum Topological Analysis. RSC Adv. 2015, 5, 37119–37129. [Google Scholar] [CrossRef]
- Panagiotou, M.; Demos, V.; Magriotis, P.A. Direct and Practical Gilman-Speeter Synthesis of 3,4-Trisubstituted β-Lactams via the Thorpe-Ingold Effect. Tetrahedron Lett. 2020, 61, 152375. [Google Scholar] [CrossRef]
- Borsari, C.; Trader, D.J.; Tait, A.; Costi, M.P. Designing Chimeric Molecules for Drug Discovery by Leveraging Chemical Biology. J. Med. Chem. 2020, 63, 1908–1928. [Google Scholar] [CrossRef]
- Zeid, I.F.; Mohamed, N.A.; Khalifa, N.M.; Kassem, E.M.; Nossier, E.S.; Salman, A.A.; Mahmoud, K.; Al-Omar, M.A. PI3K Inhibitors of Novel Hydrazide Analogues Linked 2-Pyridinyl Quinazolone Scaffold as Anticancer Agents. J. Chem. 2019, 2019, 6321573. [Google Scholar] [CrossRef]
- Archana, A.; Saini, S. Synthesis and Anticonvulsant Studies of Thiazolidinone and Azetidinone Derivatives from Indole Moiety. Drug Res. 2019, 69, 445–450. [Google Scholar]
- Dabhi, R.A.; Dhaduk, M.P.; Bhatt, V.D.; Bhatt, B.S. Synthetic Approach toward Spiro Quinoxaline-β-lactam Based Heterocyclic Compounds: Spectral Characterization, SAR, Pharmacokinetic and Biomolecular Interaction Studies. J. Biomol. Struct. Dyn. 2023, 41, 5382–5398. [Google Scholar] [CrossRef]
- Wang, S.; Malebari, A.M.; Greene, T.F.; O’Boyle, N.M.; Fayne, D.; Nathwani, S.M.; Twamley, B.; McCabe, T.; Keely, N.O.; Zisterer, D.M.; et al. 3-Vinylazetidin-2-ones: Synthesis, Antiproliferative and Tubulin Destabilizing Activity in MCF-7 and MDA-MB-231 Breast Cancer Cells. Pharmaceuticals 2019, 12, 56. [Google Scholar] [CrossRef]
- Vigliotta, G.; Giordano, D.; Verdino, A.; Caputo, I.; Martucciello, S.; Soriente, A.; Marabotti, A.; De Rosa, M. New Compounds for a Good Old Class: Synthesis of Two β-Lactam Bearing Cephalosporins and Their Evaluation with a Multidisciplinary Approach. Bioorg. Med. Chem. 2020, 28, 115302. [Google Scholar] [CrossRef]
- Bendeddouche, S.; Bendeddouche, C.K.; Benhaoua, H. A Convenient Stereoselective Method for Synthesis of β-Lactams under Microwave Irradiation with [BmIm] OH as a Reusable Ionic Liquid. Lett. Org. Chem. 2021, 18, 929–935. [Google Scholar] [CrossRef]
- Malebari, A.M.; Fayne, D.; Nathwani, S.M.; O’Connell, F.; Noorani, S.; Twamley, B.; O’Boyle, N.M.; O’Sullivan, J.; Zisterer, D.M.; Meegan, M.J. β-Lactams with Antiproliferative and Antiapoptotic Activity in Breast and Chemoresistant Colon Cancer Cells. Eur. J. Med. Chem. 2020, 189, 112050. [Google Scholar] [CrossRef]
- Verma, V.A.; Saundane, A.R. Synthesis of Some Novel 5-(8-Substituted-11H-indolo[3,2-c]isoquinolin-5-ylthio)-1′,3′,4′-oxadiazol-2-amines Bearing Thiazolidinones and Azetidinones as Potential Antimicrobial, Antioxidant, Antituberculosis, and Anticancer Agents. Polycycl. Aromat. Compd. 2021, 41, 871–896. [Google Scholar] [CrossRef]
- Filatov, V.; Kukushkin, M.; Kuznetsova, J.; Skvortsov, D.; Tafeenko, V.; Zyk, N.; Majouga, A.; Beloglazkina, E. Synthesis of 1,3-Diaryl-spiro[azetidine-2,3′-indoline]-2′,4-diones via the Staudinger Reaction: Cis- or trans-Diastereoselectivity with Different Addition Modes. RSC Adv. 2020, 10, 14122–14133. [Google Scholar] [CrossRef]
- Bortolami, M.; Chiarotto, I.; Mattiello, L.; Petrucci, R.; Rocco, D.; Vetica, F.; Feroci, M. Organic Electrochemistry: Synthesis and Functionalization of β-Lactams in the Twenty-First Century. Heterocycl. Commun. 2021, 27, 32–44. [Google Scholar] [CrossRef]
- Kurteva, V.; Alexandrova, M. Constrained 1-Phenylethyl Amine Analogues as Chiral Auxiliaries in Stereoselective trans-β-Lactam Formation via Staudinger Cycloaddition. J. Heterocycl. Chem. 2019, 56, 930–937. [Google Scholar] [CrossRef]
- Avello, M.G.; Moreno-Latorre, M.; de la Torre, M.C.; Casarrubios, L.; Gornitzka, H.; Hemmert, C.; Sierra, M.A. β-Lactam and Penicillin Substituted Mesoionic Metal Carbene Complexes. Org. Biomol. Chem. 2022, 20, 2651–2660. [Google Scholar] [CrossRef] [PubMed]
- Mahmood, A.A.J.; Al-Iraqi, M.A.; Abachi, F.T. Design, Synthesis, and Evaluation the Anti-β-lactamase Activity of New Sulphathiazole-Derived Monobactam Compounds. Iraqi J. Pharm. 2020, 17, 19–36. [Google Scholar] [CrossRef]
- Asahina, Y.; Wurtz, N.R.; Arakawa, K.; Carson, N.; Fujii, K.; Fukuchi, K.; Garcia, R.; Hsu, M.-Y.; Ishiyama, J.; Ito, B.; et al. Discovery of BMS-986235/LAR-1219: A Potent Formyl Peptide Receptor 2 (FPR2) Selective Agonist for the Prevention of Heart Failure. J. Med. Chem. 2020, 63, 9003–9019. [Google Scholar] [CrossRef] [PubMed]
- Grabrijan, K.; Strašek, N.; Gobec, S. Synthesis of 3-Amino-4-substituted Monocyclic β-Lactams—Important Structural Motifs in Medicinal Chemistry. Int. J. Mol. Sci. 2022, 23, 360. [Google Scholar] [CrossRef] [PubMed]
- Decuyper, L.; Jukič, M.; Sosič, I.; Amoroso, A.M.; Verlaine, O.; Joris, B.; Gobec, S.; D’hooghe, M. Synthesis and Penicillin-Binding Protein Inhibitory Assessment of Dipeptidic 4-Phenyl-β-lactams from α-Amino Acid-derived Imines. Chem. Asian J. 2020, 15, 51–55. [Google Scholar] [CrossRef]
- Fei, Z.; Wu, Q.; Gong, W.; Fu, P.; Li, C.; Wang, X.; Han, Y.; Li, B.; Li, L.; Wu, B.; et al. Process Development for the Synthesis of a Monobactam Antibiotic-LYS228. Org. Process Res. Dev. 2020, 24, 363–370. [Google Scholar] [CrossRef]
- Shaikh, A.L.; Yadav, R.N.; Banik, B.K. Microwave-Induced Enantiospecific Synthesis of trans-(3R,4R)-3-Acetoxy-4-aryl-1-(chrysen-6-yl)azetidin-2-ones via the Staudinger Cycloaddition Reaction of (+)-Car-3-ene with Polyaromatic Imines. Russ. J. Org. Chem. 2020, 56, 910–915. [Google Scholar] [CrossRef]
- Deketelaere, S.; Kaur, G.; Piens, N.; Deturck, D.; Depestel, R.; Van Hecke, K.; Stevens, C.V.; Kumar, V.; D’hooghe, M. Synthesis of 4-Imidoyl-, 4-Oxiranyl- and 4-Propargyloxyphenyl-Substituted β-Lactam Building Blocks. J. Heterocycl. Chem. 2022, 60, 423–430. [Google Scholar] [CrossRef]
- Sandmeier, T.; Goetzke, F.W.; Krautwald, S.; Carreira, E.M. Iridium-Catalyzed Enantioselective Allylic Substitution with Aqueous Solutions of Nucleophiles. J. Am. Chem. Soc. 2019, 141, 12212–12218. [Google Scholar] [CrossRef]
- Jarrahpour, A.; Rostami, M.; Sinou, V.; Latour, C.; Djouhri-Bouktab, L.; Brunel, J.M. Diastereoselective Synthesis of Potent Antimalarial cis-β-Lactam Agents. Iran. J. Pharm. Res. 2019, 18, 596–606. [Google Scholar]
- Yamamoto, Y.; Kodama, S.; Nishimura, R.; Nomoto, A.; Ueshima, M.; Ogawa, A. One-Pot Construction of Diverse β-Lactam Scaffolds via the Green Oxidation of Amines and Its Application to the Diastereoselective Synthesis of β-Amino Acids. J. Org. Chem. 2021, 86, 11571–11582. [Google Scholar] [CrossRef]
- Habib, O.M.; Mohamed, A.S.; Ibrahim, Y.A.; Al-Awadi, N.A. Sequential Diimination, Staudinger [2+2] Ketene-Imine Cycloaddition, and Ring-Closing Metathesis (RCM) Reactions: In Route to Bis(4-Spiro-Fused-β-Lactams)-Based Macrocycles. J. Org. Chem. 2021, 86, 14777–14785. [Google Scholar] [CrossRef] [PubMed]
- Mohamed, A.S.; Al-Awadhi, F.H.; Al-Awadi, N.A. Variable-Sized Bis(4-spiro-fused-β-lactam)-Based Unsaturated Macrocycles: Synthesis and Characterization. ACS Omega 2022, 7, 36795–36803. [Google Scholar] [CrossRef]
- Xie, W.; Sun, S.; Xu, J. Experimental Evidence on the Formation of Highly Strained 6,7-Dihydroazeto[2,1-b]oxazol-3-ium Derivatives as Reactive Intermediates. Helv. Chim. Acta 2022, 105, e202100187. [Google Scholar] [CrossRef]
- Mishra, M.K.; Sharma, S.; Ahmad, K.; Singh, V.N. Stereoselective Synthesis of Novel Monocyclic cis-β-Lactams Bearing 1,3,4-Thiadiazole Nucleus: Bioactive Agents and Potential Synthons. ChemistrySelect 2020, 5, 3784–3788. [Google Scholar] [CrossRef]
- Singh, V.N.; Sharma, S. The Facile and Efficient Synthesis of Novel Monocyclic cis-β-Lactam Conjugates with a 1-Methyl-1H-imidazole-2-thiol Nucleus. New J. Chem. 2021, 45, 19347–19357. [Google Scholar] [CrossRef]
- Livingstone, K.; Bertrand, S.; Kennedy, A.R.; Jamieson, C. Transition-Metal-Free Coupling of 1,3-Dipoles and Boronic Acids as a Sustainable Approach to C-C Bond Formation. Chem. Eur. J. 2020, 26, 10591–10597. [Google Scholar] [CrossRef]
- Yadav, R.N.; Paniagua, A.; Banik, B.K. An Intramolecular Oxa-Michael Addition on Prebuilt β-Lactam Tethered α, β-Unsaturated Ester: A Remarkable Synthesis of a Unique Scaffold of 2,3-Fused β-Lactam-1,4-dioxepane. J. Indian Chem. Soc. 2021, 98, 100010. [Google Scholar] [CrossRef]
- Galván, A.; de la Cruz, F.N.; Cruz, F.; Martínez, M.; Gomez, C.V.; Alcaraz, Y.; Domínguez, J.M.; Delgado, F.; Vázquez, M.A. Heterogeneous Catalysis with Basic Compounds to Achieve the Synthesis and C-N Cleavage of Azetidin-2-ones under Microwave Irradiation. Synthesis 2019, 51, 3625–3637. [Google Scholar] [CrossRef]
- Saini, P.; Bari, S.S.; Sahoo, S.C.; Khullar, S.; Mandal, S.K.; Bhalla, A. Stereoselective Synthesis and Characterization of Novel trans-4-(Thiophenyl)pyrazolyl-β-lactams and their C-3 Functionalization. Tetrahedron 2019, 75, 4591–4601. [Google Scholar] [CrossRef]
- Shaikh, A.L.; Banik, B. Krishna. Microwave-Induced Stereoselective Synthesis of β-Lactams Containing Aromatic Carboxylic Acids. Heterocycl. Lett. 2022, 12, 253–256. [Google Scholar]
- Berry, S.; Bari, S.S.; Yadav, P.; Garg, A.; Khullar, S.; Mandal, S.K.; Bhalla, A. Stereoselective Synthesis of trans-3-Functionalized-4-pyrazolo[5,1-b]thiazole-3-carboxylate Substituted β-Lactams: Potential Synthons for Diverse Biologically Active Agents. Synth. Commun. 2020, 50, 2969–2980. [Google Scholar] [CrossRef]
- Wang, F.; Tan, X.; Wu, T.; Zheng, L.-S.; Chen, G.-Q.; Zhang, X. Ni-Catalyzed Asymmetric Reduction of α-Keto-β-lactams via DKR Enabled by Proton Shuttling. Chem. Commun. 2020, 56, 15557–15560. [Google Scholar] [CrossRef] [PubMed]
- King, T.A.; Stewart, H.L.; Mortensen, K.T.; North, A.J.P.; Sore, H.F.; Spring, D.R. Cycloaddition Strategies for the Synthesis of Diverse Heterocyclic Spirocycles for Fragment-Based Drug Discovery. Eur. J. Org. Chem. 2019, 2019, 5219–5229. [Google Scholar] [CrossRef] [PubMed]
- Yadav, R.N.; Banik, I.; Banik, B.K. Microwave-induced New Synthesis of trans and cis 3-Phenylthio-4-Carboethoxy β-Lactams. J. Indian Chem. Soc. 2019, 96, 1355–1358. [Google Scholar]
- Yadav, R.N.; Singh, A.K.; Banik, I.; Banik, B.K. Microwave-induced Stereospecific Synthesis of trans 3-Phenylthio β-Lactams. J. Indian Chem. Soc. 2019, 96, 1359–1363. [Google Scholar]
- Koch, V.; Lorion, M.M.; Barde, E.; Bräse, S.; Cossy, J. Cobalt-Catalyzed α-Arylation of Substituted α-Halogeno β-Lactams. Org. Lett. 2019, 21, 6241–6244. [Google Scholar] [CrossRef]
- Sahni, T.; Sharma, S.; Verma, D.; Kaur, H.; Kaur, A. Synthesis and in Vitro Fungitoxic Evaluation of Syringaldehyde Schiff Bases and β-Lactams. Org. Prep. Proced. Int. 2022, 54, 370–379. [Google Scholar] [CrossRef]
- Malebari, A.M.; Wang, S.; Greene, T.F.; O’Boyle, N.M.; Fayne, D.; Khan, M.F.; Nathwani, S.M.; Twamley, B.; McCabe, T.; Zisterer, D.M.; et al. Synthesis and Antiproliferative Evaluation of 3-Chloroazetidin-2-ones with Antimitotic Activity: Heterocyclic Bridged Analogues of Combretastatin A-4. Pharmaceuticals 2021, 14, 1119. [Google Scholar] [CrossRef]
- Das, R.; Mehta, D.K. Evaluation and Docking Study of Pyrazine Containing 1, 3, 4-Oxadiazoles Clubbed with Substituted Azetidin-2-one: A New Class of Potential Antimicrobial and Antitubercular. Drug Res. 2021, 71, 26–35. [Google Scholar] [CrossRef] [PubMed]
- Aziz, D.M.; Azeez, H.J. Synthesis of New β-Lactam-N-(thiazol-2-yl)benzene Sulfonamide Hybrids: Their in Vitro Antimicrobial and in Silico Molecular Docking Studies. J. Mol. Struct. 2020, 1222, 128904. [Google Scholar] [CrossRef]
- Kumbar, S.S.; Shettar, A.; Joshi, S.D.; Patil, S.A. Design, Synthesis, Molecular Docking and Biological Activity Studies of Novel Coumarino-Azetidinones. J. Mol. Struct. 2021, 1231, 130016. [Google Scholar] [CrossRef]
- Kaur, B.; Mishra, S.; Kaur, R.; Kalotra, S.; Singh, P. Rationally Designed TNF-α Inhibitors: Identification of Promising Cytotoxic Agents. Bioorg. Med. Chem. Lett. 2021, 41, 127982. [Google Scholar] [CrossRef]
- Afreen; Manturthi, S.; nath Velidandi, A. Thiazole- and Coumarin-Conjugated (β-Lactam Scaffold) Azetidinones Synthesis and Their Substitution Effect in In Silico, and In Vitro Cell Viability Studies. Russ. J. Bioorg. Chem. 2022, 48, 1273–1281. [Google Scholar] [CrossRef]
- Verma, D.; Sharma, S.; Sahni, T.; Kaur, H.; Kaur, S. Designing, Antifungal and Structure Activity Relationship Studies of Azomethines and β-Lactam Derivatives of Aza Heterocyclic Amines. J. Indian Chem. Soc. 2022, 99, 100587. [Google Scholar] [CrossRef]
- Cebeci, Y.U.; Bayrak, H.; Şirin, Y. Synthesis of Novel Schiff Bases and Azol-β-lactam Derivatives Starting from Morpholine and Thiomorpholine and Investigation of their Antitubercular, Antiurease Activity, Acethylcolinesterase Inhibition Effect and Antioxidant Capacity. Bioorg. Chem. 2019, 88, 102928. [Google Scholar] [CrossRef]
- Mermer, A.; Bayrak, H.; Şirin, Y.; Emirik, M.; Demirbaş, N. Synthesis of Novel Azol-β-Lactam Derivatives Starting from Phenyl Piperazine and Investigation of Their Antiurease Activity and Antioxidant Capacity Comparing with Their Molecular Docking Studies. J. Mol. Struct. 2019, 1189, 279–287. [Google Scholar] [CrossRef]
- Mermer, A.; Bayrak, H.; Alyar, S.; Alagumuthu, M. Synthesis, DFT Calculations, Biological Investigation, Molecular Docking Studies of β-Lactam Derivatives. J. Mol. Struct. 2020, 1208, 127891. [Google Scholar] [CrossRef]
- Fahim, A.M.; Farag, A.M.; Mermer, A.; Bayrak, H.; Şirin, Y. Synthesis of Novel β-Lactams: Antioxidant Activity, Acetylcholinesterase Inhibition and Computational Studies. J. Mol. Struct. 2021, 1233, 130092. [Google Scholar] [CrossRef]
- Aldujaili, R.A.B.; Alhasan, A.A.Y. Preparation and Characterization of Some New Benzothiazole-Heterocyclic Derivatives. Egypt. J. Chem. 2021, 64, 2845–2855. [Google Scholar] [CrossRef]
- Gandhi, D.; Sethiya, A.; Agarwal, D.; Prajapat, P.; Agarwal, S. Design, Synthesis and Antimicrobial Study of Novel 1-(1,3-Benzothiazol-2-yl)-3-chloro-4H-spiro[azetidine-2,3′-indole]-2′,4(1′H)-diones through Ketene-Imine Cycloaddition Reaction. Lett. Org. Chem. 2020, 17, 141–148. [Google Scholar] [CrossRef]
- Anwer, K.E.; Sayed, G.H.; Hassan, H.H.; Azab, M.E. Conventional and Microwave Synthesis of Some New Pyridine Derivatives and Evaluation Their Antimicrobial and Cytotoxic Activities. Egypt. J. Chem. 2019, 62, 707–726. [Google Scholar] [CrossRef]
- Bakr, R.B.; Elkanzi, N.A.A. Preparation of Some Novel Thiazolidinones, Imidazolinones, and Azetidinone Bearing Pyridine and Pyrimidine Moieties with Antimicrobial Activity. J. Heterocycl. Chem. 2020, 57, 2977–2989. [Google Scholar] [CrossRef]
- Adem, K.; Boda, S.; Sirgamalla, R.; Macha, R. Synthesis, Antimicrobial Activities of Novel 3-Chloro-4-(2,4-difluorophenyl)-1-(3-aryl-1,8-naphthyridin-2-yl)azetidin-2-ones and 2-(2,4-Difluorophenyl)-3-(3-aryl-1,8-naphthyridin-2-yl)thiazolidin-4-ones. Synth. Commun. 2022, 52, 667–677. [Google Scholar] [CrossRef]
- Rekha, T.; Nagarjuna, U.; Padmaja, A.; Padmavathi, V. Synthesis, Molecular Properties Prediction and Antimicrobial Activity of Imidazolyl Schiff Bases, Triazoles and Azetidinones. Chem. Biodivers. 2019, 16, e1900073. [Google Scholar] [CrossRef] [PubMed]
- Rathod, A.S.; Biradar, J.S. Green Approach to the Synthesis of New Indole and Benzimidazole Analogs and Their Biological Evaluation. Russ. J. Org. Chem. 2021, 57, 1540–1551. [Google Scholar] [CrossRef]
- El Azab, I.H.; Gobouri, A.A.; Altalhi, T.A. 4-Chlorothiazole-5-carbaldehydes as Potent Precursors for Synthesis of Some New Pendant N-Heterocycles Endowed with Anti-Tumor Activity. J. Heterocycl. Chem. 2019, 56, 281–295. [Google Scholar] [CrossRef]
- Mahmoud, N.F.H.; Ghareeb, E.A. Synthesis of Novel Substituted Tetrahydropyrimidine Derivatives and Evaluation of Their Pharmacological and Antimicrobial Activities. J. Heterocycl. Chem. 2019, 56, 81–91. [Google Scholar] [CrossRef]
- Jori, A.; Dixit, S.R.; Pujar, G.V. Synthesis and in Vitro Cytotoxicity Study of Novel 4-Substituted Quinazolines Encompassed with Thiazolidinone and Azetidinone. Asian J. Chem. 2020, 32, 2617–2623. [Google Scholar] [CrossRef]
- Kusurkar, R.V.; Rayani, R.H.; Parmar, D.R.; Patel, D.R.; Patel, M.J.; Pandey, N.O.; Zunjar, V.; Soni, J.Y. Phenyl Substituted 3-Chloro 2-Azetidinones: Design, Green Synthesis, Antimicrobial Activity, and Molecular Docking Studies. J. Mol. Struct. 2023, 1272, 134185. [Google Scholar] [CrossRef]
- Gaba, J.; Sharma, S.; Kaur, P.; Joshi, S. Synthesis and Biological Evaluation of Thymol Functionalized Oxadiazole Thiol, Triazole Thione and β-Lactam Derivatives. Lett. Org. Chem. 2021, 18, 453–464. [Google Scholar] [CrossRef]
- Abdu-Rahem, L.R.; Ahmad, A.K.; Abachi, F.T. Docking and Synthesis of Some 2-Aminothiazole Derivatives as Antimicrobial Agents. Egypt. J. Chem. 2021, 64, 7269–7276. [Google Scholar] [CrossRef]
- Mohi, A.T.; Al-Rubaye, H.I.; Askar, F.W.; Abboud, H.J. Synthesis, Theoretical and Antimicrobial Activity Study of New Benzimidazole Derivatives. Egypt. J. Chem. 2020, 63, 2877–2886. [Google Scholar] [CrossRef]
- Abdalhassan, H.; Jabbar, S.; Khalf, A.J.; Ibrahim, R.; Mutanabbi, A. Synthesis, Antimicrobial, Antioxidant and Docking Study of Novel 2H-1,4-Benzoxazin-3(4H)-one Derivatives. Egypt. J. Chem. 2020, 63, 225–238. [Google Scholar]
- Ayyash, A.N. Synthesis and Antimicrobial Screening of Novel Azetidin-2-ones Derived from Pyromellitic Diimide via [2+2]-Cycloaddition Reaction. Russ. J. Org. Chem. 2019, 55, 1961–1966. [Google Scholar] [CrossRef]
- Alghamdi, S.; Almehmadi, M.M.; Asif, M. Antimicrobial Activity of N-(3-Chloro-2-aryl-4-oxoazetidin-1-yl)-4-nitro Benzamide Derivatives. Indian J. Heterocycl. Chem. 2022, 32, 91–95. [Google Scholar]
- Giri, T.; Shashikala, K.; Ramesh, D.; Siddhartha, M.; Laxminarayana, E. Microwave-Assisted Synthesis of Thieno(3,2-d) Pyrimidine Amino Derivatives and Their Molecular Docking Studies. Indian J. Heterocycl. Chem. 2021, 31, 397–404. [Google Scholar]
- Idrees, M.; Bodkhe, Y.G.; Siddiqui, N.J.; Kola, S.S. Synthesis, Characterization and in Vitro Antimicrobial Screening of Some Novel Series of 2-Azetidinone Derivatives Integrated with Quinoline, Pyrazole and Benzofuran Moieties. Asian J. Chem. 2020, 32, 896–900. [Google Scholar] [CrossRef]
- Torosyan, S.A.; Nuriakhmetova, Z.F.; Vostrikov, N.S.; Gimalova, F.A. Synthesis of 4-Benzylthieno[3,2-b]pyrrole Derivatives Containing 1,3,4-Oxadiazole and Azetidinone Fragments. Russ. J. Org. Chem. 2021, 57, 1455–1460. [Google Scholar] [CrossRef]
- Gilani, S.J.; Hassan, M.Z.; Imam, S.S.; Kala, C.; Dixit, S.P. Novel Benzothiazole Hydrazine Carboxamide Hybrid Scaffolds as Potential in Vitro GABA AT Enzyme Inhibitors: Synthesis, Molecular Docking and Antiepileptic Evaluation. Bioorg. Med. Chem. Lett. 2019, 29, 1825–1830. [Google Scholar] [CrossRef] [PubMed]
- Mandal, M.K.; Ghosh, S.; Bhat, H.R.; Naesens, L.; Singh, U.P. Synthesis and Biological Evaluation of Substituted Phenyl Azetidine-2-one Sulphonyl Derivatives as Potential Antimicrobial and Antiviral Agents. Bioorg. Chem. 2020, 104, 104320. [Google Scholar] [CrossRef]
- Deketelaere, S.; Van Den Broeck, E.; Cools, L.; Deturck, D.; Naeyaert, H.; Van Hecke, K.; Stevens, C.V.; Van Speybroeck, V.; D’hooghe, M. Unexpected Formation of 2,2-Dichloro-N-(chloromethyl) acetamides during Attempted Staudinger 2,2-Dichloro-β-lactam Synthesis. Eur. J. Org. Chem. 2021, 2021, 5823–5830. [Google Scholar] [CrossRef]
- Lorion, M.M.; Koch, V.; Nieger, M.; Chen, H.-Y.; Lei, A.; Bräse, S.; Cossy, J. Cobalt-Catalyzed α-Arylation of Substituted α-Bromo α-Fluoro β-Lactams with Diaryl Zinc Reagents: Generalization to Functionalized Bromo Derivatives. Chem. Eur. J. 2020, 26, 13163–13169. [Google Scholar] [CrossRef] [PubMed]
- Mozaffari, A.; Jarrahpour, A.; Alborz, M.; Turos, E. One-Pot Multicomponent Synthesis of β-Lactams via in situ Generated Imines. ChemistrySelect 2019, 4, 5950–5953. [Google Scholar] [CrossRef]
- Borazjani, N.; Sepehri, S.; Behzadi, M.; Jarrahpour, A.; Rad, J.A.; Sasanipour, M.; Mohkam, M.; Ghasemi, Y.; Akbarizadeh, A.R.; Digiorgio, C.; et al. Three-Component Synthesis of Chromeno β-Lactam Hybrids for Inflammation and Cancer Screening. Eur. J. Med. Chem. 2019, 179, 389–403. [Google Scholar] [CrossRef]
- Arefi, H.; Naderi, N.; Shemirani, A.B.I.; Kiani Falavarjani, M.; Azami Movahed, M.; Zarghi, A. Design, Synthesis, and Biological Evaluation of New 1,4-Diarylazetidin-2-one Derivatives (β-Lactams) as Selective Cyclooxygenase-2 Inhibitors. Arch. Pharm. 2020, 353, e1900293. [Google Scholar] [CrossRef] [PubMed]
- Borazjani, N.; Jarrahpour, A.; Rad, J.A.; Mohkam, M.; Behzadi, M.; Ghasemi, Y.; Mirzaeinia, S.; Karbalaei-Heidari, H.R.; Ghanbari, M.M.; Batta, G.; et al. Design, Synthesis and Biological Evaluation of Some Novel Diastereoselective β-Lactams Bearing 2-Mercaptobenzothiazole and Benzoquinoline. Med. Chem. Res. 2019, 28, 329–339. [Google Scholar] [CrossRef]
- Borazjani, N.; Behzadi, M.; Aseman, M.D.; Jarrahpour, A.; Rad, J.A.; Kianpour, S.; Iraji, A.; Nabavizadeh, S.M.; Ghanbari, M.M.; Batta, G.; et al. Cytotoxicity, Anticancer, and Antioxidant Properties of Mono and bis-Naphthalimido β-Lactam Conjugates. Med. Chem. Res. 2020, 29, 1355–1375. [Google Scholar] [CrossRef]
- Bashiri, M.; Jarrahpour, A.; Rastegari, B.; Iraji, A.; Irajie, C.; Amirghofran, Z.; Malek-Hosseini, S.; Motamedifar, M.; Haddadi, M.; Zomorodian, K.; et al. Synthesis and Evaluation of Biological Activities of Tripodal Imines and β-Lactams Attached to the 1,3,5-Triazine Nucleus. Monatsh. Chem. 2020, 151, 821–835. [Google Scholar] [CrossRef]
- Ranjbari, S.; Behzadi, M.; Sepehri, S.; Aseman, M.D.; Jarrahpour, A.; Mohkam, M.; Ghasemi, Y.; Akbarizadeh, A.R.; Kianpour, S.; Atioglu, Z.; et al. Investigations of Antiproliferative and Antioxidant Activity of β-Lactam Morpholino-1,3,5-triazine Hybrids. Bioorg. Med. Chem. 2020, 28, 115408. [Google Scholar] [CrossRef]
- Alborz, M.; Pournejati, R.; Ameri Rad, J.; Jarrahpour, A.; Reza Karbalaei-Heidari, H.; Michel Brunel, J.; Turos, E. Design and Preparation of β-Lactam Derivatives Bearing Phenanthrenimidazole as Cytotoxic Agents. ChemistrySelect 2022, 7, e202202306. [Google Scholar] [CrossRef]
- Heiran, R.; Jarrahpour, A.; Riazimontazer, E.; Gholami, A.; Troudi, A.; Digiorgio, C.; Brunel, J.M.; Turos, E. Sulfonamide-β-Lactam Hybrids Incorporating the Piperazine Moiety as Potential Antiinflammatory Agent with Promising Antibacterial Activity. ChemistrySelect 2021, 6, 5313–5319. [Google Scholar] [CrossRef]
- Heiran, R.; Sepehri, S.; Jarrahpour, A.; Digiorgio, C.; Douafer, H.; Brunel, J.M.; Gholami, A.; Riazimontazer, E.; Turos, E. Synthesis, Docking and Evaluation of in Vitro Anti-Inflammatory Activity of Novel Morpholine Capped β-Lactam Derivatives. Bioorg. Chem. 2020, 102, 104091. [Google Scholar] [CrossRef] [PubMed]
- Gummidi, L.; Kerru, N.; Awolade, P.; Ibeji, C.U.; Karpoormath, R.; Singh, P. N-Phenyl Substituent Controlled Diastereoselective Synthesis of β-Lactam-Isatin Conjugates. Tetrahedron Lett. 2020, 61, 151602. [Google Scholar] [CrossRef]
- Jarrahpour, A.; Jowkar, Z.; Haghighijoo, Z.; Heiran, R.; Rad, J.A.; Sinou, V.; Rouvier, F.; Latour, C.; Brunel, J.M.; Özdemir, N. Synthesis, in-Vitro Biological Evaluation, and Molecular Docking Study of Novel Spiro-β-lactam-Isatin Hybrids. Med. Chem. Res. 2022, 31, 1026–1034. [Google Scholar] [CrossRef]
- Bashiri, M.; Jarrahpour, A.; Nabavizadeh, S.M.; Karimian, S.; Rastegari, B.; Haddadi, E.; Turos, E. Potent Antiproliferative Active Agents: Novel Bis Schiff Bases and Bis Spiro β-Lactams Bearing Isatin Tethered with Butylene and Phenylene as Spacer and DNA/BSA Binding Behavior as Well as Studying Molecular Docking. Med. Chem. Res. 2021, 30, 258–284. [Google Scholar] [CrossRef]
- Filatov, V.E.; Kuznetsova, J.; Petrovskaya, L.; Yuzabchuk, D.; Tafeenko, V.A.; Zyk, N.V.; Beloglazkina, E.K. cis-Diastereoselective Synthesis of Spirooxindolo-β-lactams by Staudinger Cycloaddition with TsCl as Activating Co-Reagent. ACS Omega 2021, 6, 22740–22751. [Google Scholar] [CrossRef]
- Filatov, V.E.; Iuzabchuk, D.A.; Tafeenko, V.A.; Grishin, Y.K.; Roznyatovsky, V.A.; Lukianov, D.A.; Fedotova, Y.A.; Sukonnikov, M.A.; Skvortsov, D.A.; Zyk, N.V.; et al. Dispirooxindole-β-lactams: Synthesis via Staudinger Ketene-Imine Cycloaddition and Biological Evaluation. Int. J. Mol. Sci. 2022, 23, 6666. [Google Scholar] [CrossRef] [PubMed]
- Pandey, S.; Thakur, A.; Reshma; Bari, S.S.; Thapar, R. Studies towards Synthesis and Lewis Acid Catalysed Functionalization of 3-(4′-Substitutedphenylthio)-azetidin-2-ones. J. Chem. Sci. 2020, 132, 129. [Google Scholar] [CrossRef]
- Pandey, S.; Nagpal, R.; Thakur, A.; Bari, S.S.; Thapar, R. A Highly Stereoselective Oxidation and an Easy One Pot Elimination Methodology for 3-Allyl-3-phenylthio-β-lactams. J. Sulfur Chem. 2022, 43, 275–289. [Google Scholar] [CrossRef]
- Al-Khazragie, Z.K.; Al-Fartosy, A.J.M.; Al-Salami, B.K. Synthesis, Characterization and Biological Activity of β-Lactam and Thiazolidinone Derivatives Based on Sulfonamide. Egypt. J. Chem. 2022, 65, 621–645. [Google Scholar]
- Chopde, H.N.; Pandhurnekar, C.P.; Yadao, B.G.; Bhattacharya, D.M.; Mungole, A.J. Synthesis, Characterization and Antibacterial Activity of 1-([6-Bromo-2-hydroxynaphthalen-1-yl]arylphenyl)methyl)-3-chloro-4-(arylphenyl)-azetidin-2-one. J. Heterocycl. Chem. 2020, 57, 3499–3504. [Google Scholar] [CrossRef]
- Bhalla, J.; Bari, S.S.; Chaudhary, G.R.; Kumar, A.; Rathee, A.; Sharma, R.; Bhalla, A. Stereoselective Synthesis and in-silico Evaluation of C4-Benzimidazolyloxyphenyl Substituted trans-β-Lactam Derivatives as Promising Novel PPARgamma Activators. Synth. Commun. 2021, 51, 3758–3767. [Google Scholar] [CrossRef]
- Saini, P.; Bari, S.S.; Thakur, S.; Garg, A.; Kumar, S.; Mandal, S.K.; Bhalla, A. Stereoselective Synthesis, Characterization and Mechanistic Insights of ortho-/meta-/para-(2-Benzo[d]oxazolyl) phenyl Substituted trans-β-Lactams: Potential Synthons for Variegated Heterocyclic Molecules. Synth. Commun. 2022, 52, 1742–1755. [Google Scholar] [CrossRef]
- Baruah, S.; Aier, M.; Puzari, A. Fluorescent Probe Sensor Based on (R)-(-)-4-Phenyl-2-oxazolidone for Effective Detection of Divalent Cations. Luminescence 2020, 35, 1206–1216. [Google Scholar] [CrossRef] [PubMed]
- Baruah, S.; Aier, M.; Puzari, A. (S)-4-(4-Aminobenzyl)-2-oxazolidinone Based 2-Azetidinones for Antimicrobial Application and Luminescent Sensing of Divalent Metal Cations. J. Heterocycl. Chem. 2020, 57, 2498–2511. [Google Scholar] [CrossRef]
- Jin, J.-H.; Zhao, J.; Yang, W.-L.; Deng, W.-P. Asymmetric Synthesis of Spirooxindole β-Lactams via Isothiourea-catalyzed Mannich/lactamization Reaction of Aryl Acetic Acids with Isatin-derived Ketimines. Adv. Synth. Catal. 2019, 361, 1592–1596. [Google Scholar] [CrossRef]
- Mishra, M.K.; Singh, V.N.; Muhammad, S.; Aloui, Z.; Sangeeta, S.; Noorussabah, N.; Ahmad, K.; Choudhary, M.; Sharma, S. An Efficient and Eco-Friendly Synthesis, Computational Assay and Antimicrobial Evaluation of Some Novel Diastereoselective Monocyclic cis-β-Lactams. J. Mol. Struct. 2020, 1219, 128638. [Google Scholar] [CrossRef]
- Kocz, R.; Roestamadji, J.; Mobashery, S. A Convenient Triphosgene-Mediated Synthesis of Symmetric Carboxylic Acid Anhydrides. J. Org. Chem. 1994, 59, 2913–2914. [Google Scholar] [CrossRef]
- Moslehi, A.; Zarei, M. Application of Magnetic Fe3O4 Nanoparticles as a Reusable Heterogeneous Catalyst in the Synthesis of ß-Lactams Containing Amino Groups. New J. Chem. 2019, 43, 12690–12697. [Google Scholar] [CrossRef]
- Mohamadzadeh, M.; Zarei, M.; Vessal, M. Synthesis, in vitro Biological Evaluation and in silico Molecular Docking Studies of Novel β-Lactam-Anthraquinone Hybrids. Bioorg. Chem. 2020, 95, 103515. [Google Scholar] [CrossRef] [PubMed]
- Mohamadzadeh, M.; Zarei, M. Anticancer Activity and Evaluation of Apoptotic Genes Expression of 2-Azetidinones Containing Anthraquinone Moiety. Mol. Divers. 2021, 25, 2429–2439. [Google Scholar] [CrossRef] [PubMed]
- Salarinejad, S.; Islami, M.R.; Abbasnejad, M.; Zigheimat, F.; Kooshki, R.; Pouramiri, B.; Hosseini, F.S. Access to the Naproxen Ring System, a Crowded β-Lactam, through in situ Generated Ketenes: Synthesis, Molecular Docking, and Evaluation of Anticonvulsant Activity. ChemistrySelect 2020, 5, 14190–14197. [Google Scholar] [CrossRef]
- Amiri, M.; Islami, M.R.; Mortazavi, Z.F. al-Sadat Stereoselective Synthesis of New β-Lactams from the Main Functional Group of Indomethacin. J. Iran. Chem. Soc. 2022, 19, 2475–2480. [Google Scholar] [CrossRef]
- Synofzik, J.; Dar’in, D.; Novikov, M.S.; Kantin, G.; Bakulina, O.; Krasavin, M. α-Acyl-α-diazoacetates in Transition-Metal-Free β-Lactam Synthesis. J. Org. Chem. 2019, 84, 12101–12110. [Google Scholar] [CrossRef] [PubMed]
- Synofzik, J.; Bakulina, O.; Dar’in, D.; Kantin, G.; Krasavin, M. Dialkyl Diazomalonates in Transition-Metal-Free, Thermally Promoted, Diastereoselective Wolff β-Lactam Synthesis. Synlett 2020, 31, 1273–1276. [Google Scholar]
- Synofzik, J.; Bakulina, O.; Balabas, O.; Dar’in, D.; Krasavin, M. Catalyst-Free Synthesis of Diastereomerically Pure 3-Sulfonylazetidin-2-ones via Microwave-Assisted Tandem Wolff Rearrangement-Staudinger Cycloaddition. Synthesis 2020, 52, 3029–3035. [Google Scholar]
- Levashova, E.; Bakulina, O.; Dar’in, D.; Krasavin, M. Catalyst-Free Synthesis of Diastereomerically Pure 3-Cyanoazetidin-2-Ones via Thermally Promoted Tandem Wolff Rearrangement-Staudinger [2+2] Cycloaddition. ChemistrySelect 2021, 6, 13582–13588. [Google Scholar] [CrossRef]
- Paramonova, P.; Lebedev, R.; Bakulina, O.; Dar’in, D.; Krasavin, M. In Situ Generation of Imines by the Staudinger/Aza-Wittig Tandem Reaction Combined with Thermally Induced Wolff Rearrangement for One-Pot Three-Component β-Lactam Synthesis. Org. Biomol. Chem. 2022, 20, 9679–9683. [Google Scholar] [CrossRef]
- Minuto, F.; Lambruschini, C.; Basso, A. Ketene 3-Component Staudinger Reaction (K-3CSR) to β-Lactams: A New Entry in the Class of Photoinduced Multicomponent Reactions. Eur. J. Org. Chem. 2021, 2021, 3270–3273. [Google Scholar] [CrossRef]
- Munaretto, L.S.; dos Santos, C.Y.; Gallo, R.D.C.; Okada, C.Y., Jr.; Deflon, V.M.; Jurberg, I.D. Visible-Light-Mediated Strategies to Assemble Alkyl 2-Carboxylate-2,3,3-Trisubstituted β-Lactams and 5-Alkoxy-2,2,4-Trisubstituted Furan-3(2H)-ones Using Aryldiazoacetates and Aryldiazoketones. Org. Lett. 2021, 23, 9292–9296. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Wang, K.; Shao, Y.; Sun, J. Stereoselective Synthesis of Fully Substituted β-Lactams via Metal-Organo Relay Catalysis. Org. Lett. 2019, 21, 3804–3807. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Zhang, L.; Shao, Y.; Xu, G.; Zhang, X.; Tang, S.; Sun, J. Rhodium-Catalyzed C=N Bond Formation through a Rebound Hydrolysis Mechanism and Application in β-Lactam Synthesis. Org. Lett. 2019, 21, 4124–4127. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Xu, R.; Xu, Z.; Xu, X. Rh2(Esp)2-Catalyzed Redox/Cycloaddition Cascade of Diazoacetoacetate Enones with N-Methyl Nitrones: Diastereoselective Synthesis of β-Lactams with Two Adjacent Chiral Centers. Synthesis 2022, 54, 2433–2446. [Google Scholar]
- Koothradan, F.F.; Jayarani, A.; Sivasankar, C. Synthesis of β-Lactams through Carbonylation of Diazo Compounds Followed by the [2 + 2] Cycloaddition Reaction. J. Org. Chem. 2024, 89, 4294–4308. [Google Scholar] [CrossRef] [PubMed]
- Dar’in, D.; Kantin, G.; Krasavin, M. A “Sulfonyl-Azide-Free” (SAFE) Aqueous-Phase Diazo Transfer Reaction for Parallel and Diversity-Oriented Synthesis. Chem. Commun. 2019, 55, 5239–5242. [Google Scholar] [CrossRef] [PubMed]
- Golubev, A.A.; Smetanin, I.A.; Agafonova, A.V.; Rostovskii, N.V.; Khlebnikov, A.F.; Starova, G.L.; Novikov, M.S. [2+1+1] Assembly of Spiro β-Lactams by Rh(II)-Catalyzed Reaction of Diazocarbonyl Compounds with Azirines/Isoxazoles. Org. Biomol. Chem. 2019, 17, 6821–6830. [Google Scholar] [CrossRef]
- Hart, D.J.; Ha, D.-C. The Ester Enolate-Imine Condensation Route to β-Lactams. Chem. Rev. 1989, 89, 1447–1465. [Google Scholar] [CrossRef]
- Malebari, A.M.; Duffy Morales, G.; Twamley, B.; Fayne, D.; Khan, M.F.; McLoughlin, E.C.; O’Boyle, N.M.; Zisterer, D.M.; Meegan, M.J. Synthesis, Characterisation and Mechanism of Action of Anticancer 3-Fluoroazetidin-2-ones. Pharmaceuticals 2022, 15, 1044. [Google Scholar] [CrossRef]
- Braun, M. The Asymmetric Difluoro-Reformatsky Reaction. Eur. J. Org. Chem. 2021, 2021, 1825–1836. [Google Scholar] [CrossRef]
- Kapoor, A.; Rajput, J.K. Staudinger Ketene–Imine [2+2] Cycloaddition of Novel Azomethines to Synthesize Biologically Active Azetidinone Derivatives and Their in Vitro Antimicrobial Studies. J. Heterocycl. Chem. 2021, 58, 2304–2323. [Google Scholar] [CrossRef]
- Cao, Q.; Stark, R.T.; Fallis, I.A.; Browne, D.L. A Ball-Milling-Enabled Reformatsky Reaction. ChemSusChem 2019, 12, 2554–2557. [Google Scholar] [CrossRef]
- Kirillov, N.F.; Nikiforova, E.A.; Baibarodskikh, D.V.; Zakharova, T.A.; Govorushkin, L.S. Synthesis of New bis(spiro-β-Lactams) via Interaction of Methyl 1-Bromocycloalcanecarboxylates with Zinc and N,N’-bis(Arylmethylidene)benzidines. J. Chem. 2019, 2019, 7496512. [Google Scholar] [CrossRef]
- Nikiforova, E.A.; Baibarodskikh, D.V.; Kirillov, N.F.; Glavatskikh, L.A. Reformatsky Reaction of Methyl 1-Bromocyclohexanecarboxylate with N,N′-(1,4-Phenylene)bis(1-arylmethanimines). Russ. J. Org. Chem. 2020, 56, 1029–1033. [Google Scholar] [CrossRef]
- Sato, K.; Isoda, M.; Tarui, A.; Omote, M. Reductive Carbon-Carbon Bond Forming Reactions with Carbonyls Mediated by Rh-H Complexes. Eur. J. Org. Chem. 2020, 2020, 6503–6511. [Google Scholar] [CrossRef]
- Das, P.; Delost, M.D.; Qureshi, M.H.; Bao, J.; Fell, J.S.; Houk, K.N.; Njardarson, J.T. Dramatic Effect of γ-Heteroatom Dienolate Substituents on Counterion Assisted Asymmetric Anionic Amino-Cope Reaction Cascades. J. Am. Chem. Soc. 2021, 143, 5793–5804. [Google Scholar] [CrossRef] [PubMed]
- Miaskiewicz, S.; Weibel, J.-M.; Pale, P.; Blanc, A. A Gold(I)-Catalysed Approach towards Harmalidine an Elusive Alkaloid from Peganum Harmala. RSC Adv. 2022, 12, 26966–26974. [Google Scholar] [CrossRef]
- Ji, D.-S.; Liang, H.; Yang, K.-X.; Feng, Z.-T.; Luo, Y.-C.; Xu, G.-Q.; Gu, Y.; Xu, P.-F. Solvent Directed Chemically Divergent Synthesis of β-Lactams and α-Amino Acid Derivatives with Chiral Isothiourea. Chem. Sci. 2022, 13, 1801–1807. [Google Scholar] [CrossRef]
- Alcaide, B.; Almendros, P.; Lázaro-Milla, C. Convenient Access to 2,3-Disubstituted-cyclobut-2-en-1-ones under Suzuki Conditions and Their Synthetic Utility. Chem. Eur. J. 2019, 25, 7547–7552. [Google Scholar] [CrossRef] [PubMed]
- Cossío, F.P.; Lecea, B.; Lopez, X.; Roa, G.; Arrieta, A.; Ugalde, J.M. An Ab Initio Study on the Mechanism of the Alkene–Isocyanate Cycloaddition Reaction to Form β-Lactams. J. Chem. Soc. Chem. Commun. 1993, 1450–1452. [Google Scholar] [CrossRef]
- Atmaca, U.; Daryadel, S.; Taslimi, P.; Çelik, M.; Gülçin, I. Synthesis of β-Amino Acid Derivatives and Their Inhibitory Profiles against Some Metabolic Enzymes. Arch. Pharm. Weinh. Ger. 2019, 352, 1900200. [Google Scholar] [CrossRef] [PubMed]
- Jouanneau, M.; Vellalath, S.; Kang, G.; Romo, D. Natural Product Derivatization with β-Lactones, β-Lactams and Epoxides toward “infinite” Binders. Tetrahedron 2019, 75, 3348–3354. [Google Scholar] [CrossRef]
- Liu, L.; Courtney, K.C.; Huth, S.W.; Rank, L.A.; Weisblum, B.; Chapman, E.R.; Gellman, S.H. Beyond Amphiphilic Balance: Changing Subunit Stereochemistry Alters the Pore-Forming Activity of Nylon-3 Polymers. J. Am. Chem. Soc. 2021, 143, 3219–3230. [Google Scholar] [CrossRef] [PubMed]
- Du, X.; Xu, D.; Xu, G.; Yu, C.; Jiang, X. Synthesis of Imidized Cyclobutene Derivatives by Strain Release of [1.1.1]Propellane. Org. Lett. 2022, 24, 7323–7327. [Google Scholar] [CrossRef] [PubMed]
- Yu, S.; Noble, A.; Bedford, R.B.; Aggarwal, V.K. Methylenespiro[2.3]hexanes via Nickel-Catalyzed Cyclopropanations with [1.1.1]Propellane. J. Am. Chem. Soc. 2019, 141, 20325–20334. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Mayer, R.J.; Ofial, A.R.; Mayr, H. From Carbodiimides to Carbon Dioxide: Quantification of the Electrophilic Reactivities of Heteroallenes. J. Am. Chem. Soc. 2020, 142, 8383–8402. [Google Scholar] [CrossRef] [PubMed]
- Shellhamer, D.F.; Brady, D.L.; Flores, F.V.; Perry, M.C. Reaction of p-Toluenesulfonyl Isocyanate with Electron-Rich Alkenes and Monofluoroalkenes. J. Undergrad. Chem. Res. 2019, 19, 10–13. [Google Scholar]
- Allen, M.A.; Ivanovich, R.A.; Beauchemin, A.M. O-Isocyanates as Uncharged 1,3-Dipole Equivalents in [3+2] Cycloadditions. Angew. Chem. Int. Ed. 2020, 59, 23188–23197. [Google Scholar] [CrossRef]
- Balijepalli, A.S.; McNeely, J.H.; Hamoud, A.; Grinstaff, M.W. Guidelines for β-Lactam Synthesis: Glycal Protecting Groups Dictate Stereoelectronics and [2+2] Cycloaddition Kinetics. J. Org. Chem. 2020, 85, 12044–12057. [Google Scholar] [CrossRef]
- Brar, A.; Unruh, D.K.; Aquino, A.J.; Krempner, C. Lewis Acid Base Chemistry of Bestmann’s Ylide, Ph3PCCO, and Its Bulkier Analogue, (Cyclohexyl)3PCCO. Chem. Commun. 2019, 55, 3513–3516. [Google Scholar] [CrossRef] [PubMed]
- Mansour, S.T.; Abd-El-Maksoud, M.A.; El-Hussieny, M.; Awad, H.M.; Hashem, A.I. Efficient Synthesis and Antiproliferative Evaluation of New Bioactive N-, P-, and S-Heterocycles. Russ. J. Gen. Chem. 2022, 92, 1761–1774. [Google Scholar] [CrossRef]
- Kinugasa, M.; Hashimoto, S. The Reactions of Copper(I) Phenylacetylide with Nitrones. J. Chem. Soc. Chem. Commun. 1972, 466–467. [Google Scholar] [CrossRef]
- Lo, M.M.-C.; Fu, G.C. Cu(I)/Bis(Azaferrocene)-Catalyzed Enantioselective Synthesis of β-Lactams via Couplings of Alkynes with Nitrones. J. Am. Chem. Soc. 2002, 124, 4572–4573. [Google Scholar] [CrossRef] [PubMed]
- Santoro, S.; Himo, F. Mechanism of the Kinugasa Reaction Revisited. J. Org. Chem. 2021, 86, 10665–10671. [Google Scholar] [CrossRef]
- Qi, J.; Wei, F.; Huang, S.; Tung, C.-H.; Xu, Z. Copper(I)-Catalyzed Asymmetric Interrupted Kinugasa Reaction: Synthesis of α-Thiofunctional Chiral β-Lactams. Angew. Chem. Int. Ed. 2021, 60, 4561–4565. [Google Scholar] [CrossRef]
- Qi, J.; Wei, F.; Tung, C.-H.; Xu, Z. Modular Synthesis of α-Quaternary Chiral β-Lactams by a Synergistic Copper/Palladium-Catalyzed Multicomponent Reaction. Angew. Chem. Int. Ed. 2021, 60, 13814–13818. [Google Scholar] [CrossRef]
- Xu, C.; Yang, Y.; Wu, Y.; He, F.; He, H.; Deng, P.; Zhou, H. Development of TsDPEN Based Imine-Containing Ligands for the Copper-Catalysed Asymmetric Kinugasa Reaction. RSC Adv. 2020, 10, 18107–18114. [Google Scholar] [CrossRef]
- Zarei, M. CuFe2O4 Nanoparticles Catalyze the Reaction of Alkynes and Nitrones for the Synthesis of 2-Azetidinones. New J. Chem. 2020, 44, 17341–17345. [Google Scholar] [CrossRef]
- Popik, O.; Grzeszczyk, B.; Staszewska-Krajewska, O.; Furman, B.; Chmielewski, M. Synthesis of β-Lactams via Diastereoselective, Intramolecular Kinugasa Reactions. Org. Biomol. Chem. 2020, 18, 2852–2860. [Google Scholar] [CrossRef]
- Glowacka, I.E.; Grabkowska-Druzyc, M.; Andrei, G.; Schols, D.; Snoeck, R.; Witek, K.; Podlewska, S.; Handzlik, J.; Piotrowska, D.G. Novel N-Substituted 3-Aryl-4-(diethoxyphosphoryl)azetidin-2-ones as Antibiotic Enhancers and Antiviral Agents in Search for a Successful Treatment of Complex Infections. Int. J. Mol. Sci. 2021, 22, 8032. [Google Scholar] [CrossRef]
- Głowacka, I.E.; Grabkowska-Druzyc, M.; Lebelt, L.; Andrei, G.; Schols, D.; Snoeck, R.; Piotrowska, D.G. β-Lactam Analogs of Oxetanocins—Synthesis and Biological Activity. Acta Pol. Pharm. 2022, 79, 167–196. [Google Scholar] [CrossRef]
- Qi, Z.; Wang, S. Copper-Catalyzed β-Lactam Formation Initiated by 1,3-Azaprotio Transfer of Oximes and Methyl Propiolate. Org. Lett. 2021, 23, 5777–5781. [Google Scholar] [CrossRef]
- Zhong, X.; Huang, M.; Xiong, H.; Liang, Y.; Zhou, W.; Cai, Q. Asymmetric Synthesis of spiro[Azetidine-3,3′-indoline]-2,2′-diones via Copper(I)-Catalyzed Kinugasa/C-C Coupling Cascade Reaction. Angew. Chem. Int. Ed. 2022, 61, e202208323. [Google Scholar] [CrossRef] [PubMed]
- Peng, Y.; Shi, Z.; Liang, Y.; Ding, K.; Wang, Y. Targeting the Tumor Microenvironment by an Enzyme-Responsive Prodrug of Tubulin Destabilizer for Triple-Negative Breast Cancer Therapy with High Safety. Eur. J. Med. Chem. 2022, 236, 114344. [Google Scholar] [CrossRef]
- Kutaszewicz, R.; Grzeszczyk, B.; Górecki, M.; Staszewska-Krajewska, O.; Furman, B.; Chmielewski, M. Bypassing the Stereoselectivity Issue: Transformations of Kinugasa Adducts from Chiral Alkynes and Achiral Open-Chain Nitrones. Org. Biomol. Chem. 2019, 17, 6251–6268. [Google Scholar] [CrossRef] [PubMed]
- Bilodeau, D.A.; Margison, K.D.; Ahmed, N.; Strmiskova, M.; Sherratt, A.R.; Pezacki, J.P. Optimized Aqueous Kinugasa Reactions for Bioorthogonal Chemistry Applications. Chem. Commun. 2020, 56, 1988–1991. [Google Scholar] [CrossRef]
- Imai, K.; Takayama, Y.; Murayama, H.; Ohmiya, H.; Shimizu, Y.; Sawamura, M. Asymmetric Synthesis of α-Alkylidene-β-lactams through Copper Catalysis with a Prolinol-Phosphine Chiral Ligand. Org. Lett. 2019, 21, 1717–1721. [Google Scholar] [CrossRef] [PubMed]
- Hosseini, A.; Schreiner, P.R. Synthesis of Exclusively 4-Substituted β-Lactams through the Kinugasa Reaction Utilizing Calcium Carbide. Org. Lett. 2019, 21, 3746–3749. [Google Scholar] [CrossRef]
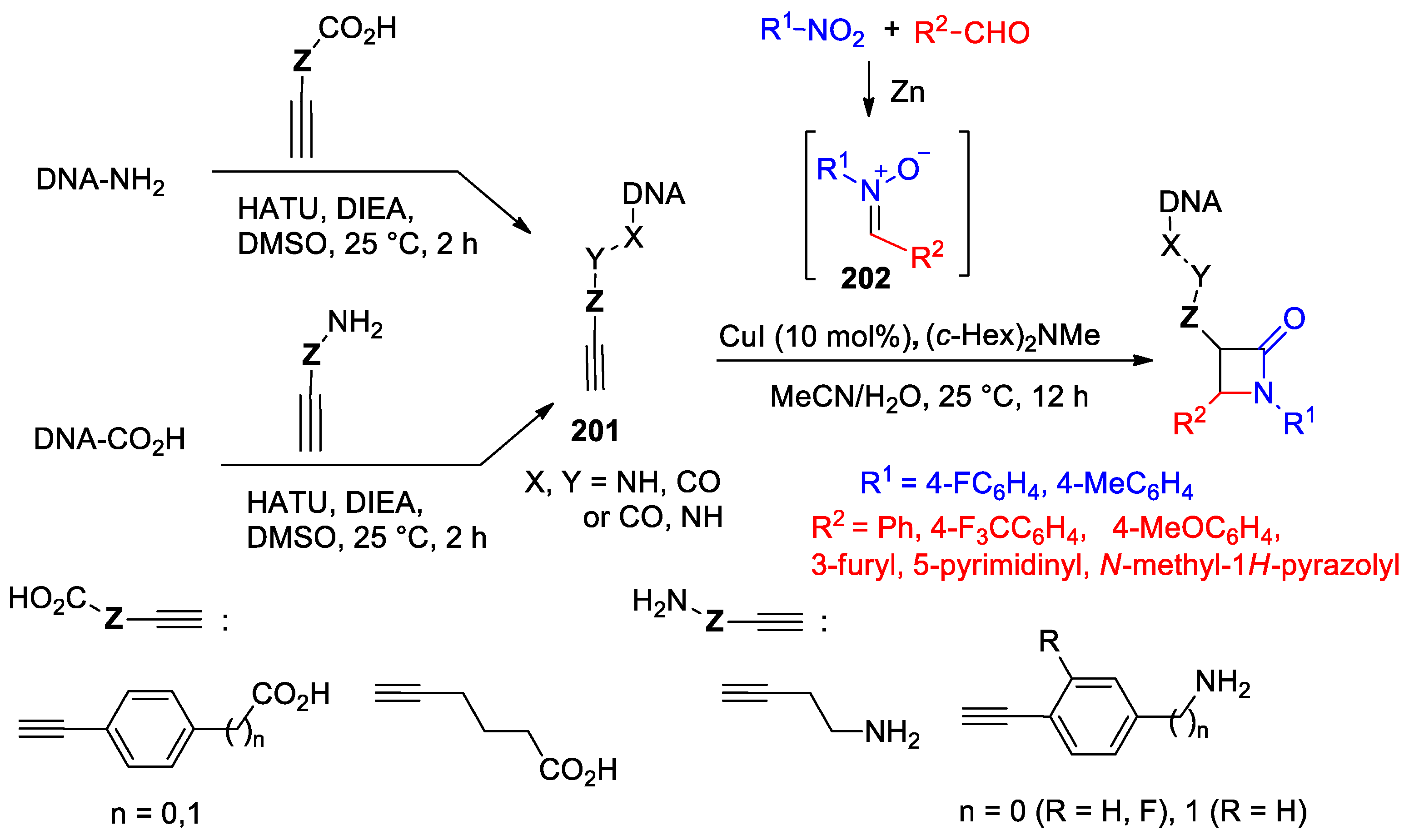
- Luo, A.; Zhang, Z.; Zeng, F.; Wang, X.; Zhao, X.; Yang, K.; Hu, Y.J. Kinugasa Reaction for DNA-Encoded β-Lactam Library Synthesis. Org. Lett. 2022, 24, 5756–5761. [Google Scholar] [CrossRef]
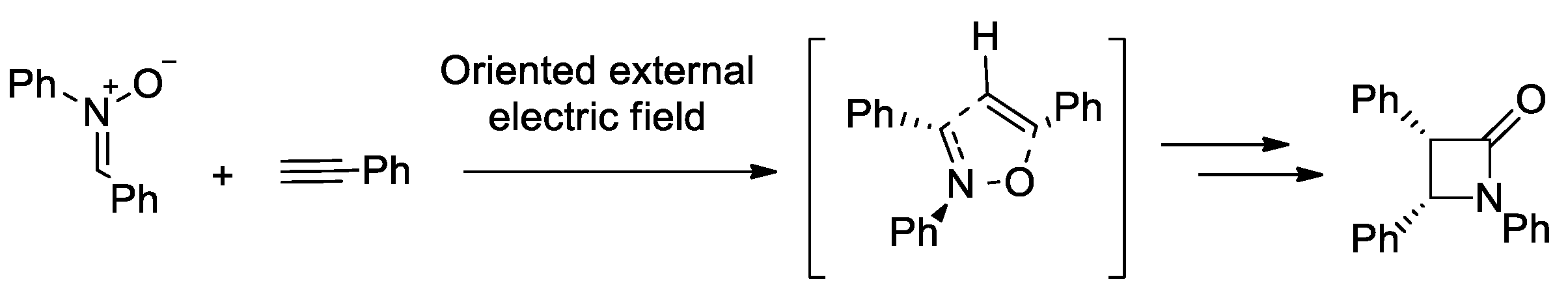
- Mandal, S.; Datta, A. Metal-free Kinugasa Reaction Catalyzed by External Electric Field. J. Phys. Org. Chem. 2022, 35, e4327. [Google Scholar] [CrossRef]
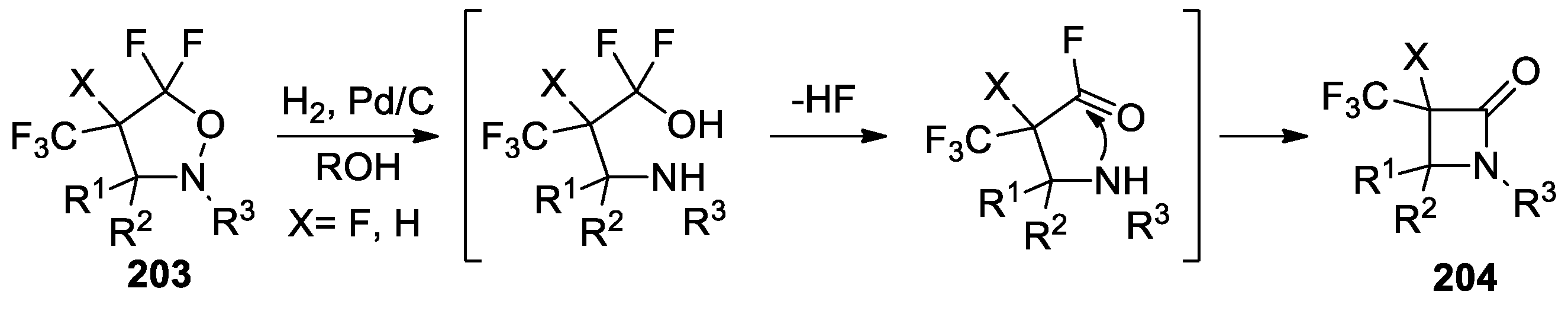
- Jakowiecki, J.; Loska, R.; Makosza, M. Synthesis of α-Trifluoromethyl-β-Lactams and Esters of β-Amino Acids via 1,3-Dipolar Cycloaddition of Nitrones to Fluoroalkenes. J. Org. Chem. 2008, 73, 5436–5441. [Google Scholar] [CrossRef] [PubMed]
- Brześkiewicz, J.; Loska, R. Palladium-Catalyzed Access to Benzocyclobutenone-Derived Ketonitrones via C(sp2)–H Functionalization. Org. Lett. 2022, 24, 3960–3964. [Google Scholar] [CrossRef] [PubMed]
- Cordero, F.M.; Brandi, A. Synthesis of β-Lactams and β-Homoprolines by Fragmentative Rearrangement of 5-Spirocyclopropaneisoxazolidines Mediated by Acids. Chem. Rec. 2021, 21, 284–294. [Google Scholar] [CrossRef] [PubMed]
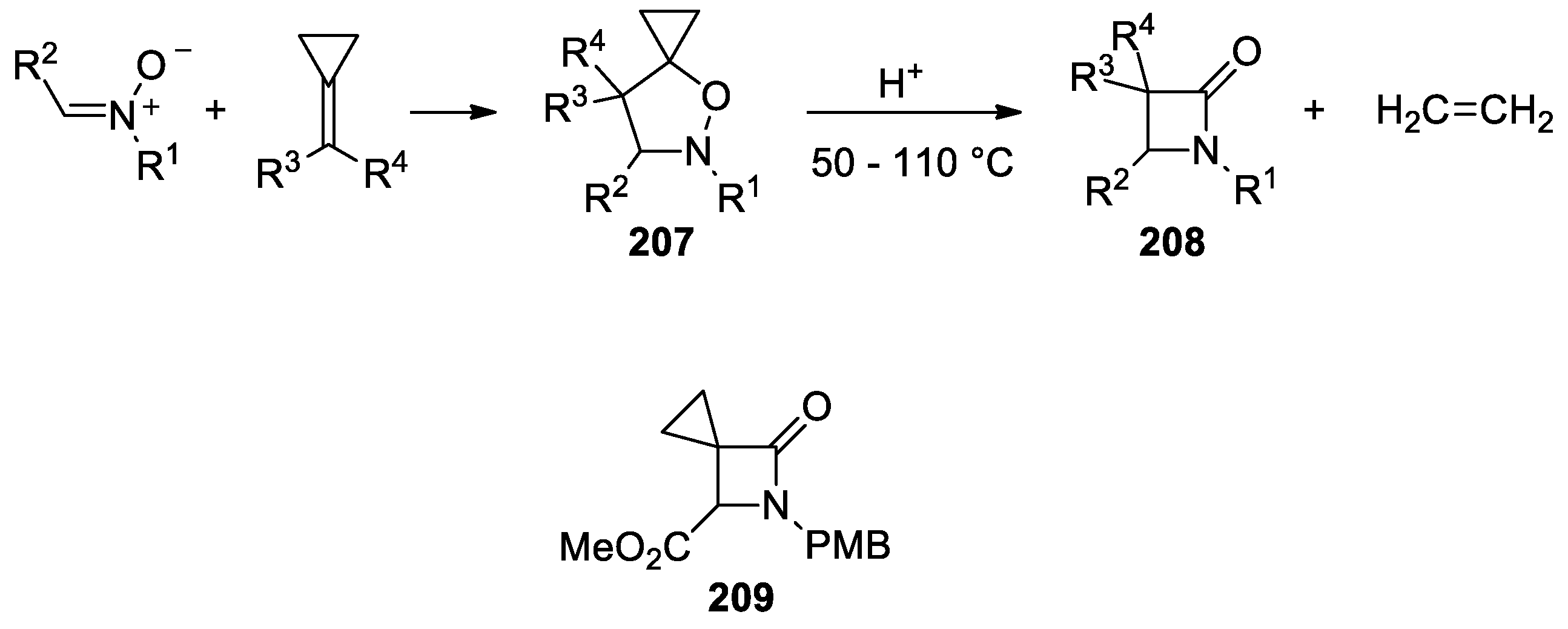
- Wei, C.; Zhu, J.-F.; Zhang, J.-Q.; Deng, Q.; Mo, D.-L. Synthesis of Spirofluorenyl-β-lactams through Cycloaddition and Ring Contraction from N-Aryl Fluorenone Nitrones and Methylenecyclopropanes. Adv. Synth. Catal. 2019, 361, 3965–3973. [Google Scholar] [CrossRef]
- Li, L.-L.; Ding, D.; Song, J.; Han, Z.-Y.; Gong, L.-Z. Catalytic Generation of C1 Ammonium Enolates from Halides and CO for Asymmetric Cascade Reactions. Angew. Chem. Int. Ed. 2019, 58, 7647–7651. [Google Scholar] [CrossRef] [PubMed]
- Ding, Y.; Huang, H. Carbonylative Cycloaddition of Alkenes and Imines to β-Lactams Enabled by Resolving the Acid-Base Paradox. Chem Catal. 2022, 2, 1467–1479. [Google Scholar] [CrossRef]
- Yan, J.; Ding, Y.; Huang, H. Palladium-Catalyzed Chemodivergent Carbonylation of ortho-Bromoarylimine to Biisoindolinones and Spiroisoindolinones. J. Org. Chem. 2023, 88, 5194–5204. [Google Scholar] [CrossRef] [PubMed]
- Leng, H.-J.; Li, Q.-Z.; Xiang, P.; Qi, T.; Dai, Q.-S.; Jia, Z.-Q.; Gou, C.; Zhang, X.; Li, J.-L. Diastereoselective [3 + 1] Cyclization Reaction of Oxindolyl Azaoxyallyl Cations with Sulfur Ylides: Assembly of 3,3′-spiro[β-Lactam]-oxindoles. Org. Lett. 2021, 23, 1451–1456. [Google Scholar] [CrossRef]
- Deeksha; Singh, R. Aza-oxyallyl Cations and their Applications in (3+m) Cycloaddition Reactions. Eur. J. Org. Chem. 2022, 2022, e202201043. [Google Scholar] [CrossRef]
- Cheibas, C.; Cordier, M.; Li, Y.; El Kaïm, L. A Ugi Straightforward Access to bis-β-Lactam Derivatives. Eur. J. Org. Chem. 2019, 2019, 4457–4463. [Google Scholar] [CrossRef]
- Cho, S.; Lee, Y.; Lee, K.; Lee, H.; Lee, Y.; Jung, B. Synthesis of Alkynamides through Reaction of Alkyl- or Aryl-Substituted Alkynylaluminums with Isocyanates. Org. Biomol. Chem. 2022, 20, 139–151. [Google Scholar] [CrossRef] [PubMed]


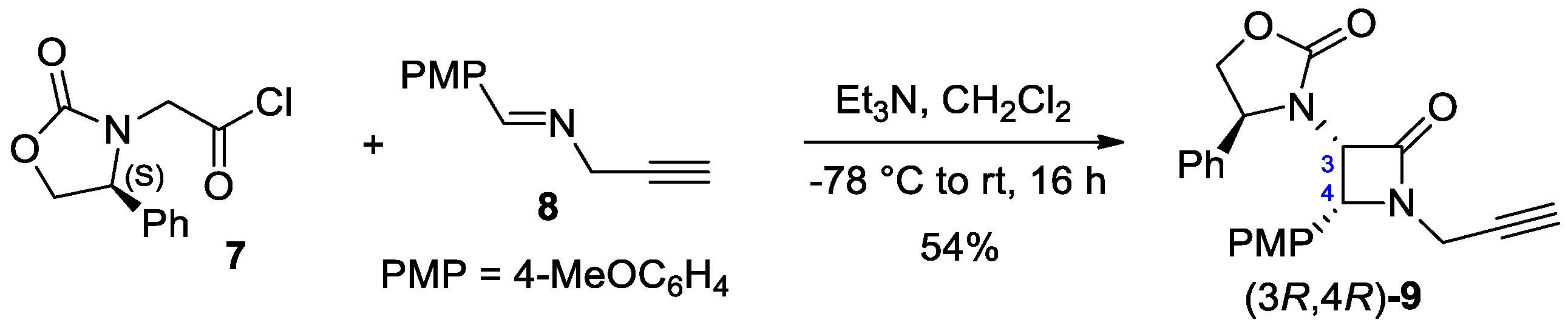
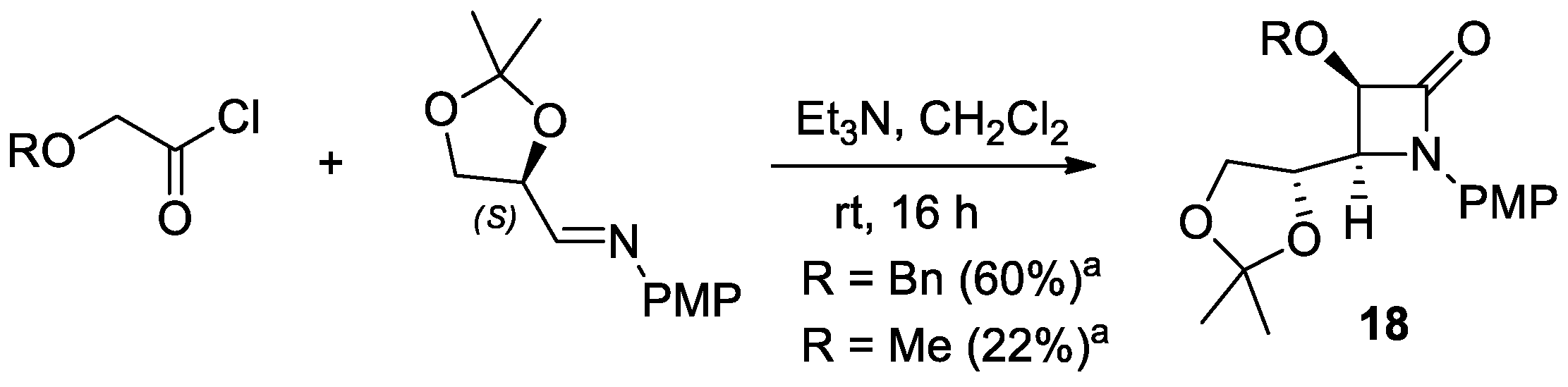
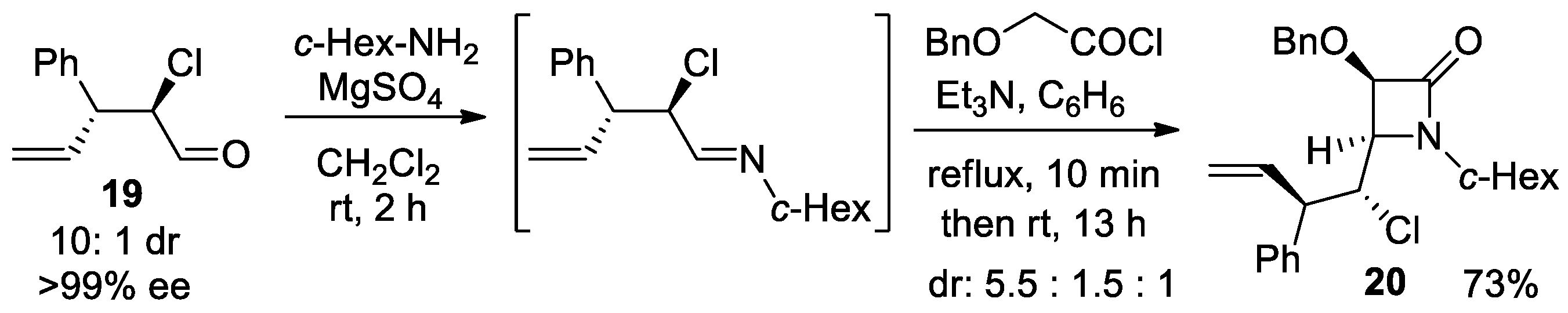
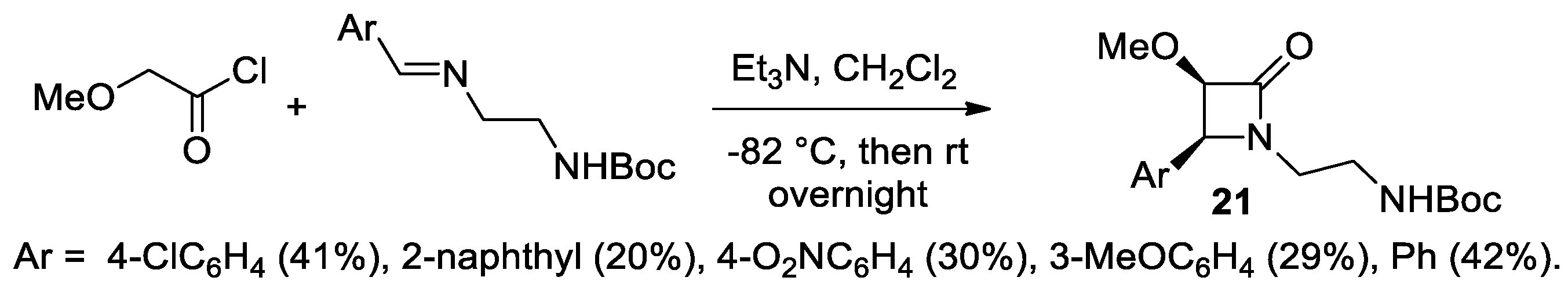
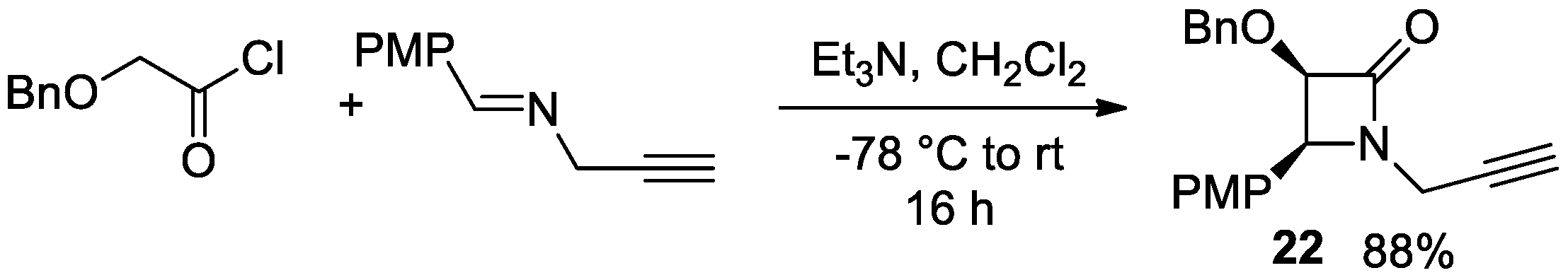
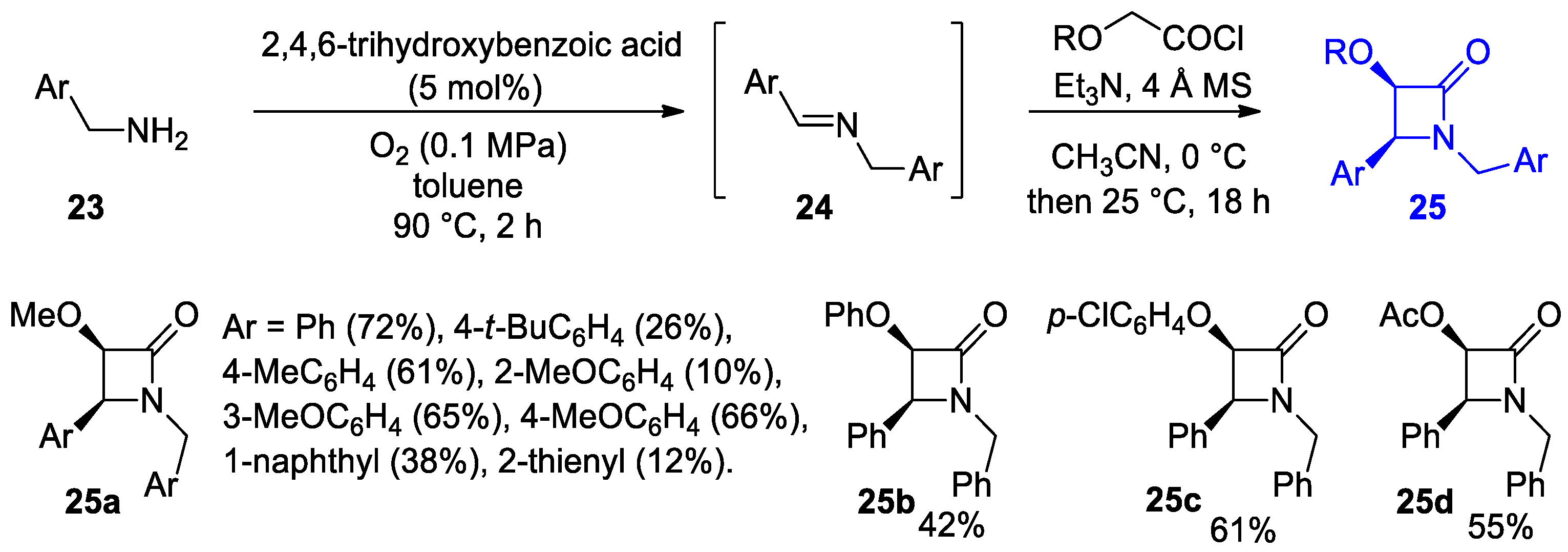
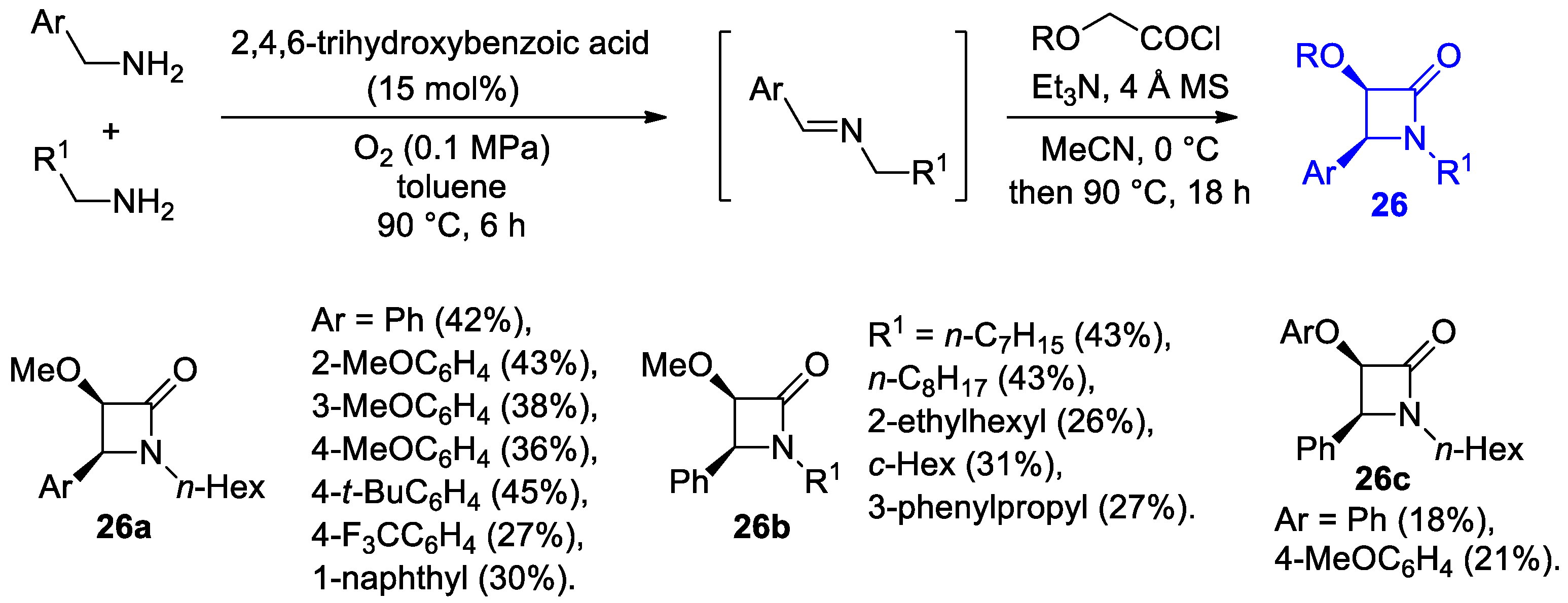

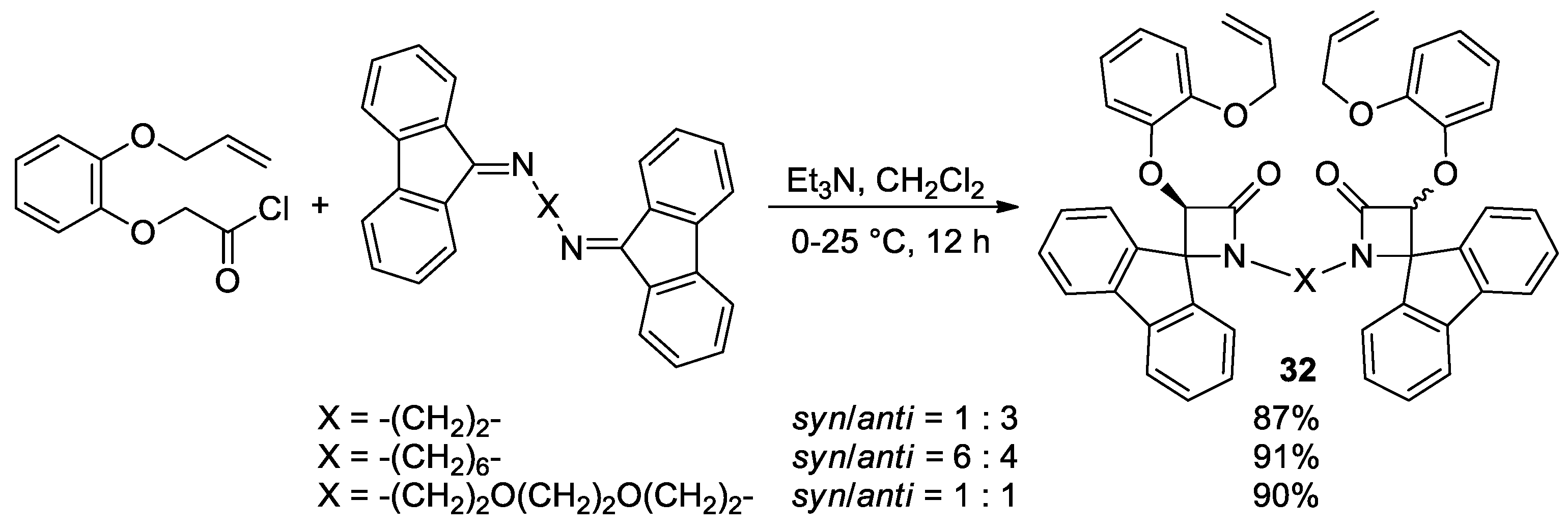
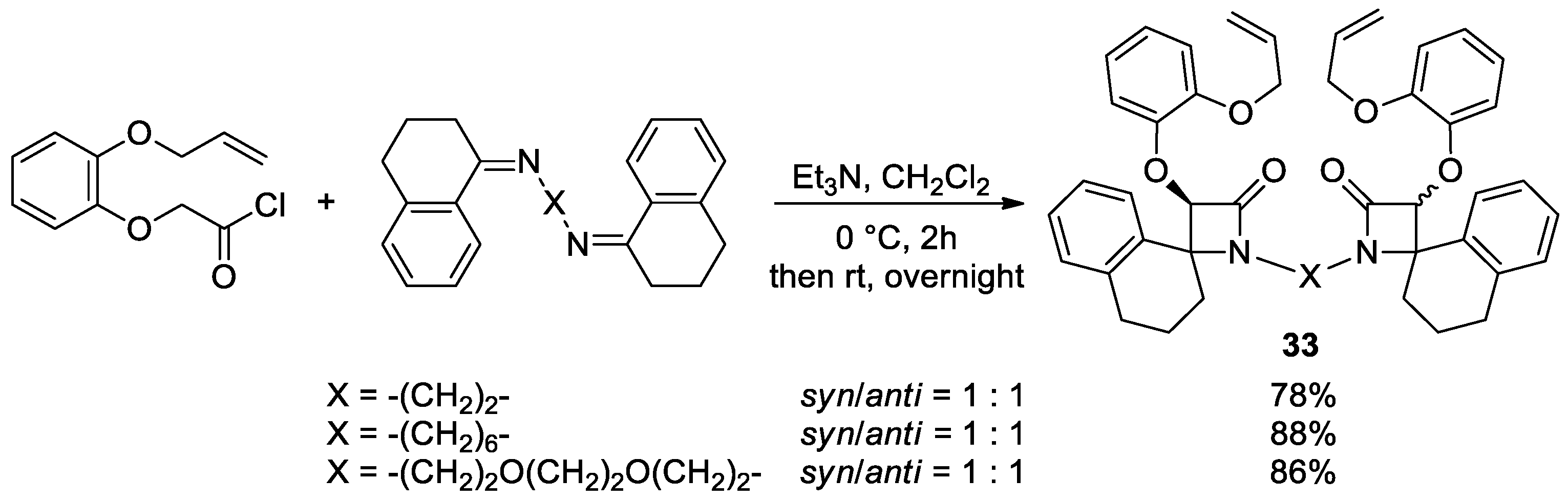
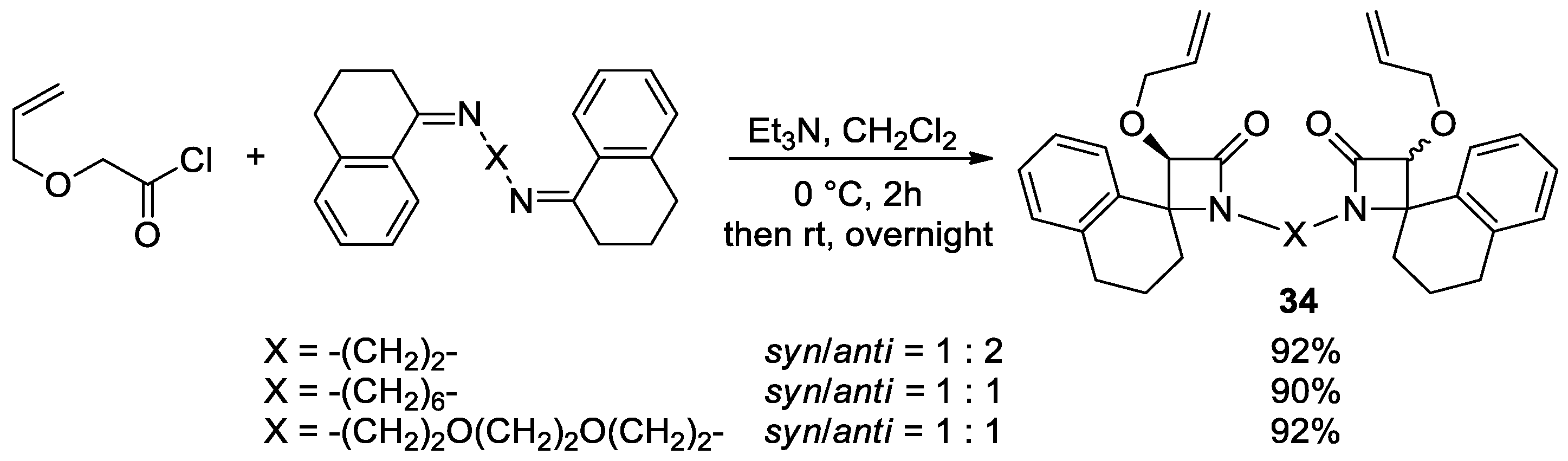
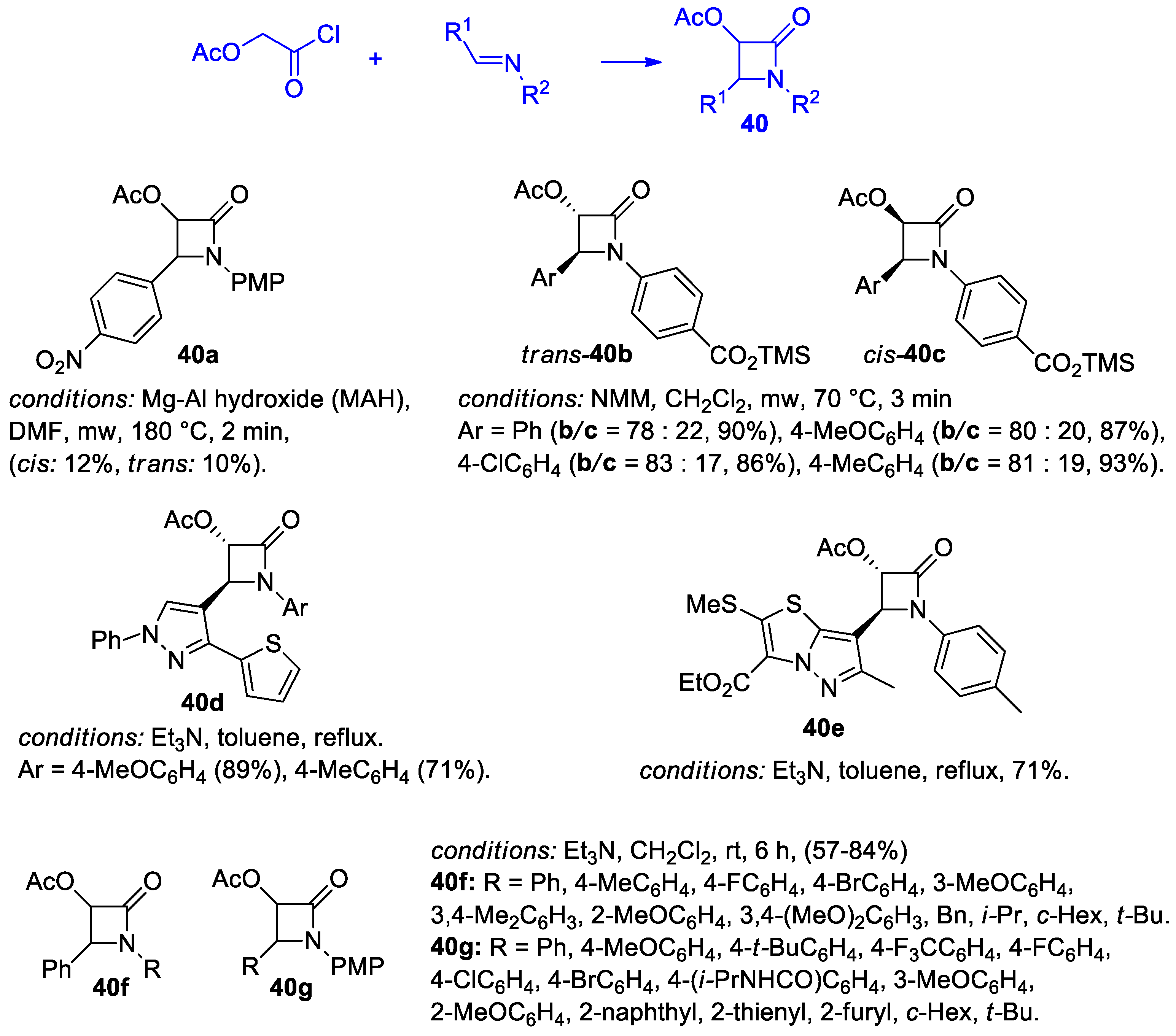
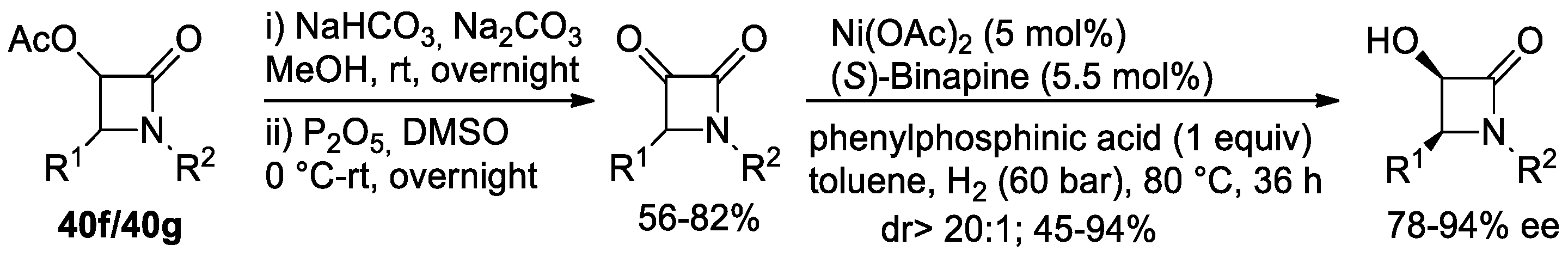
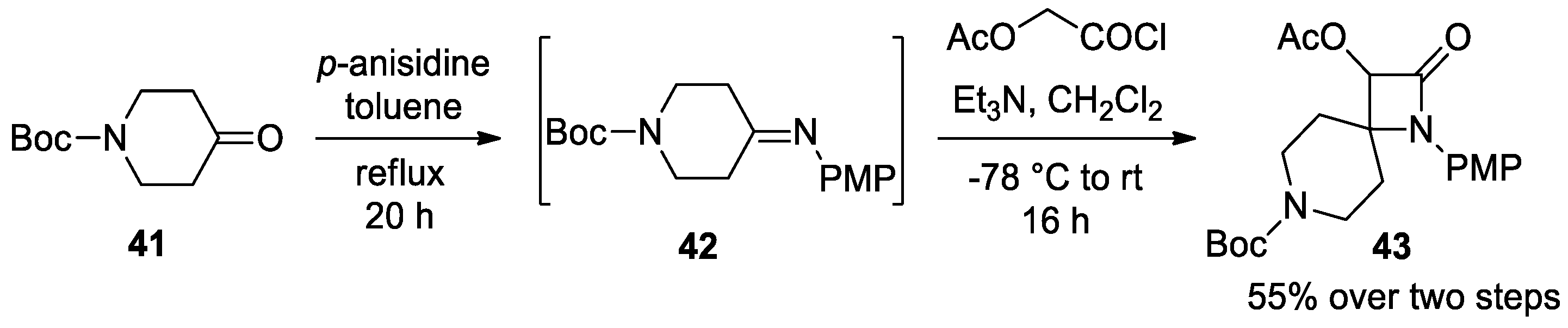
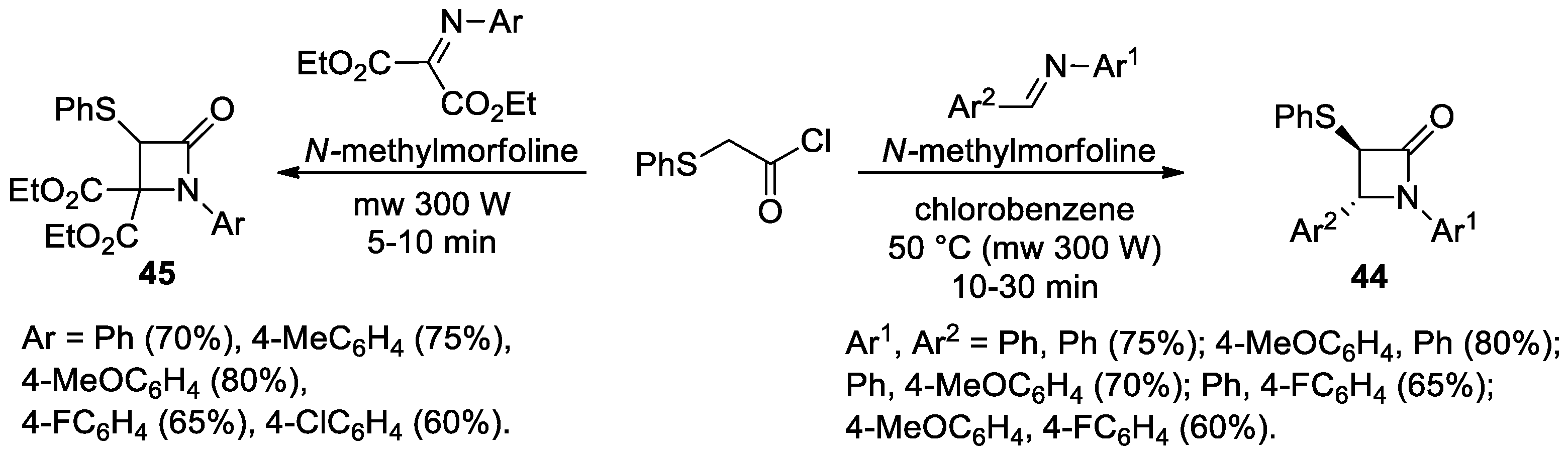






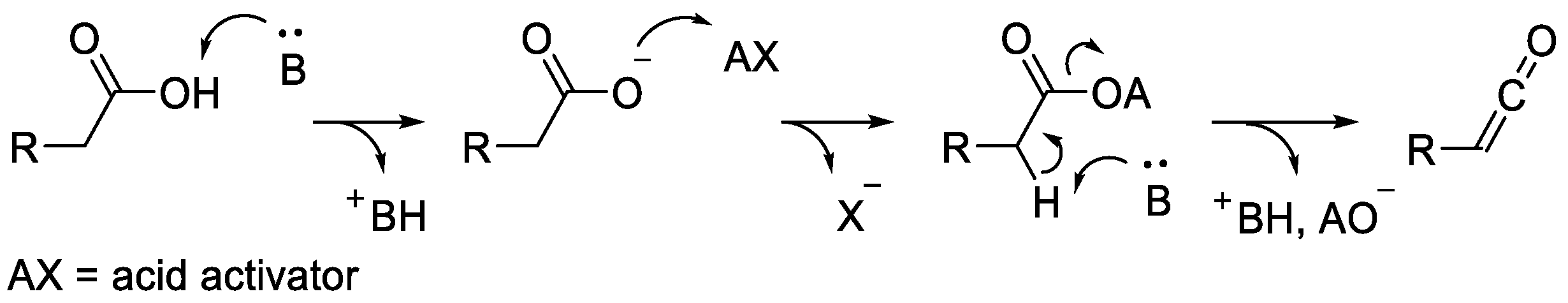






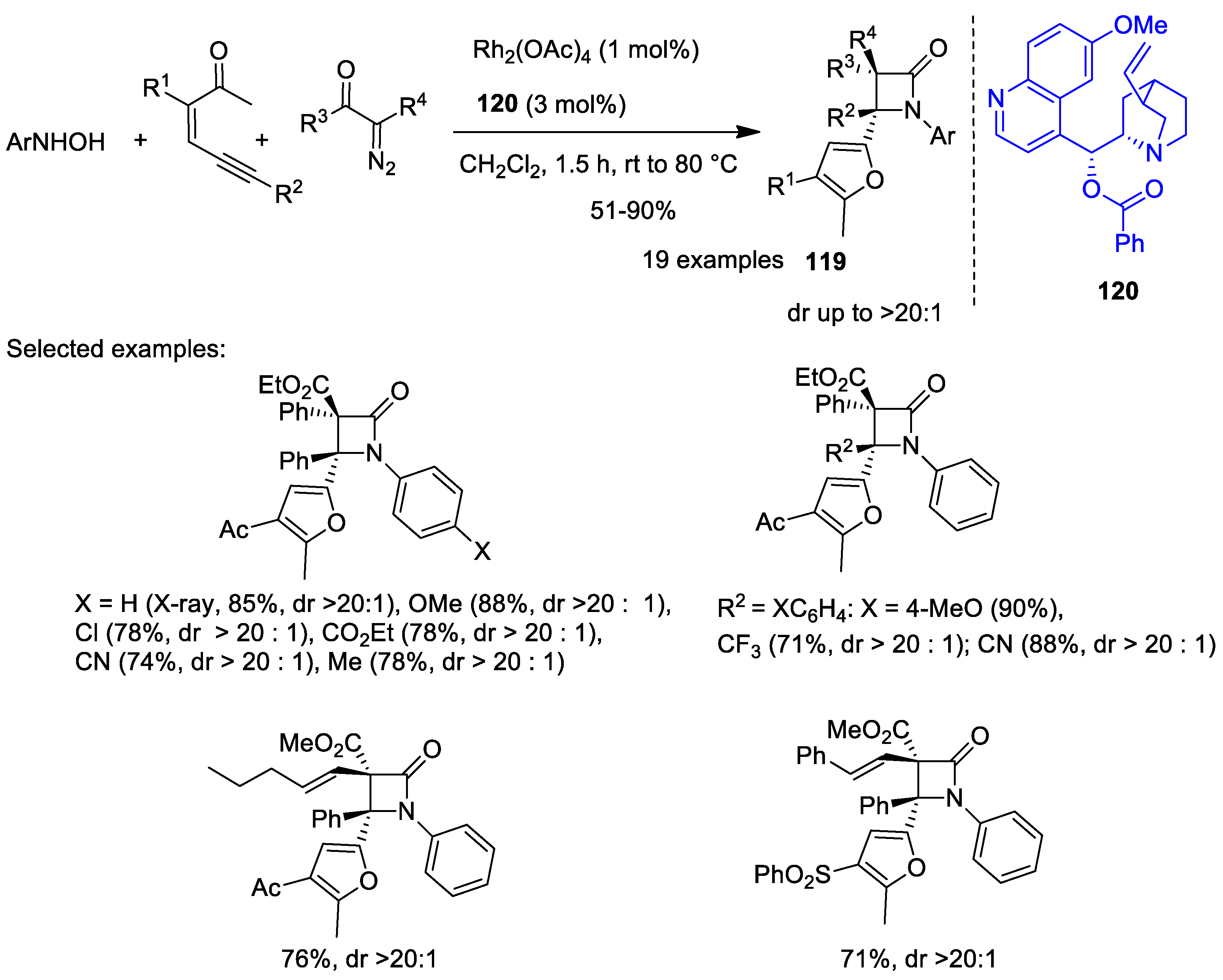
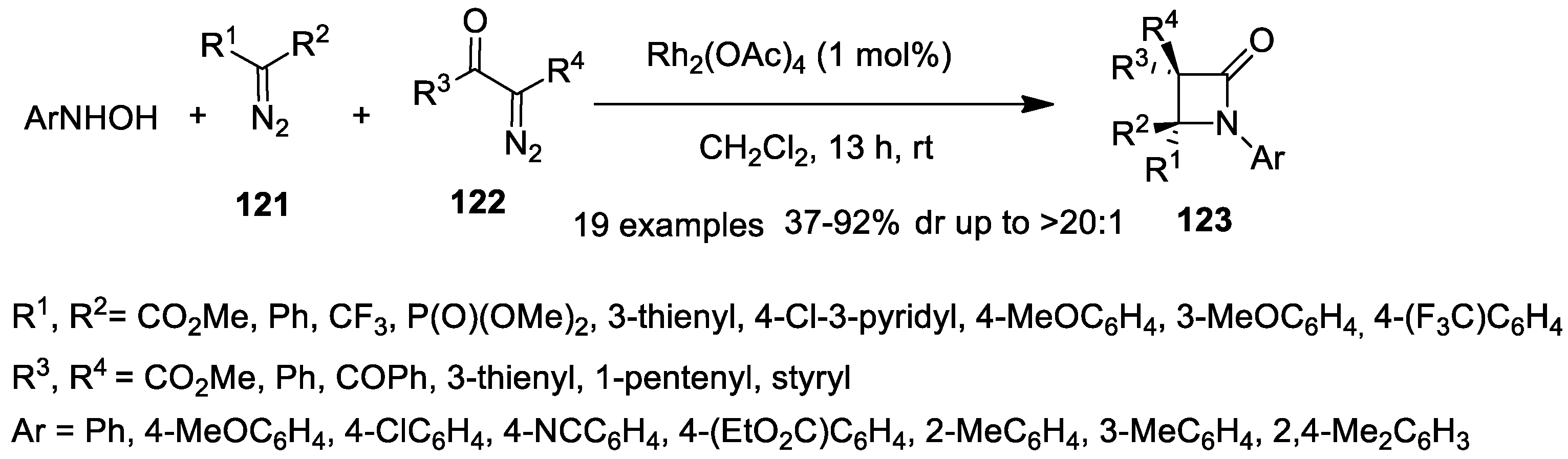
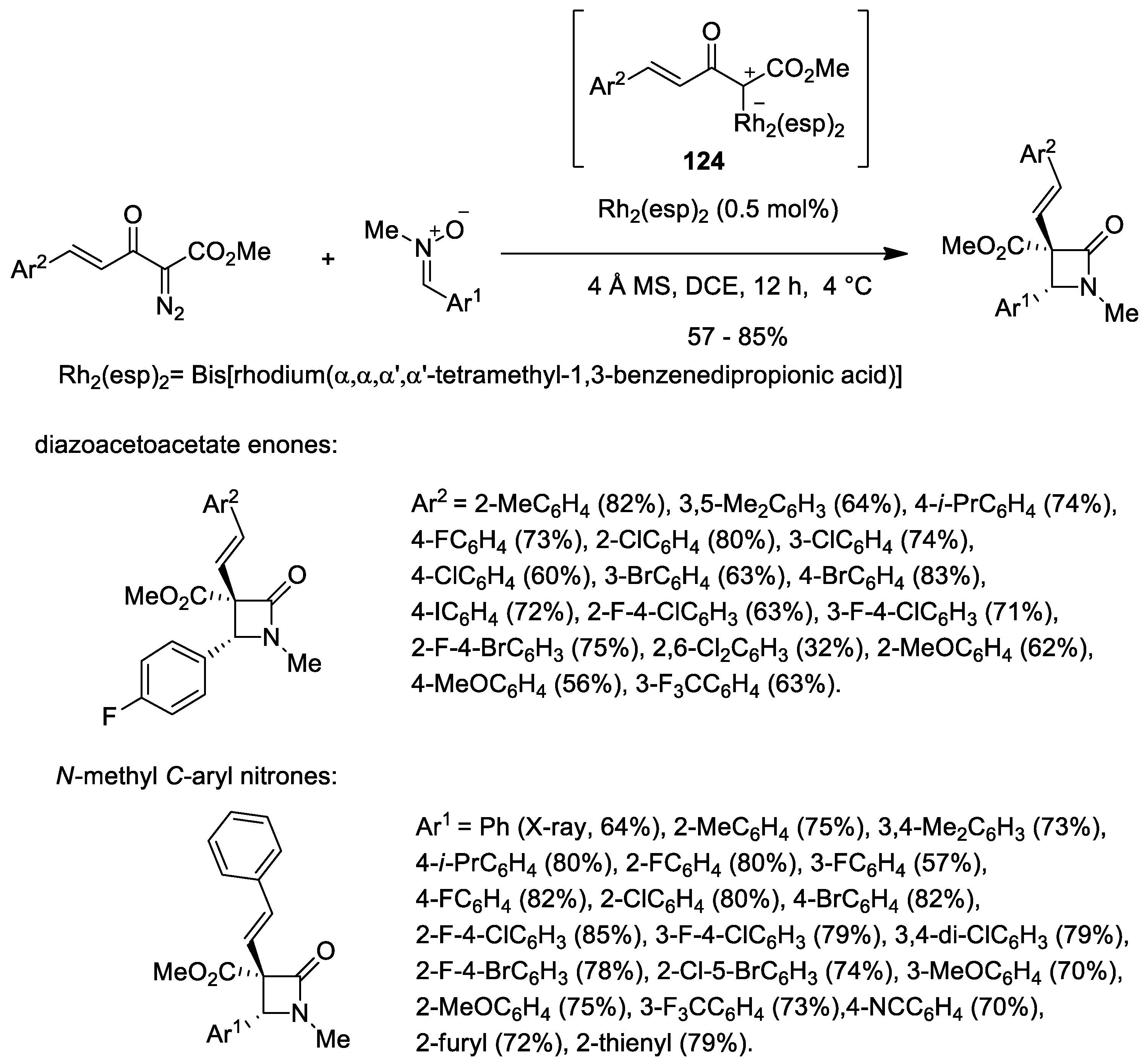
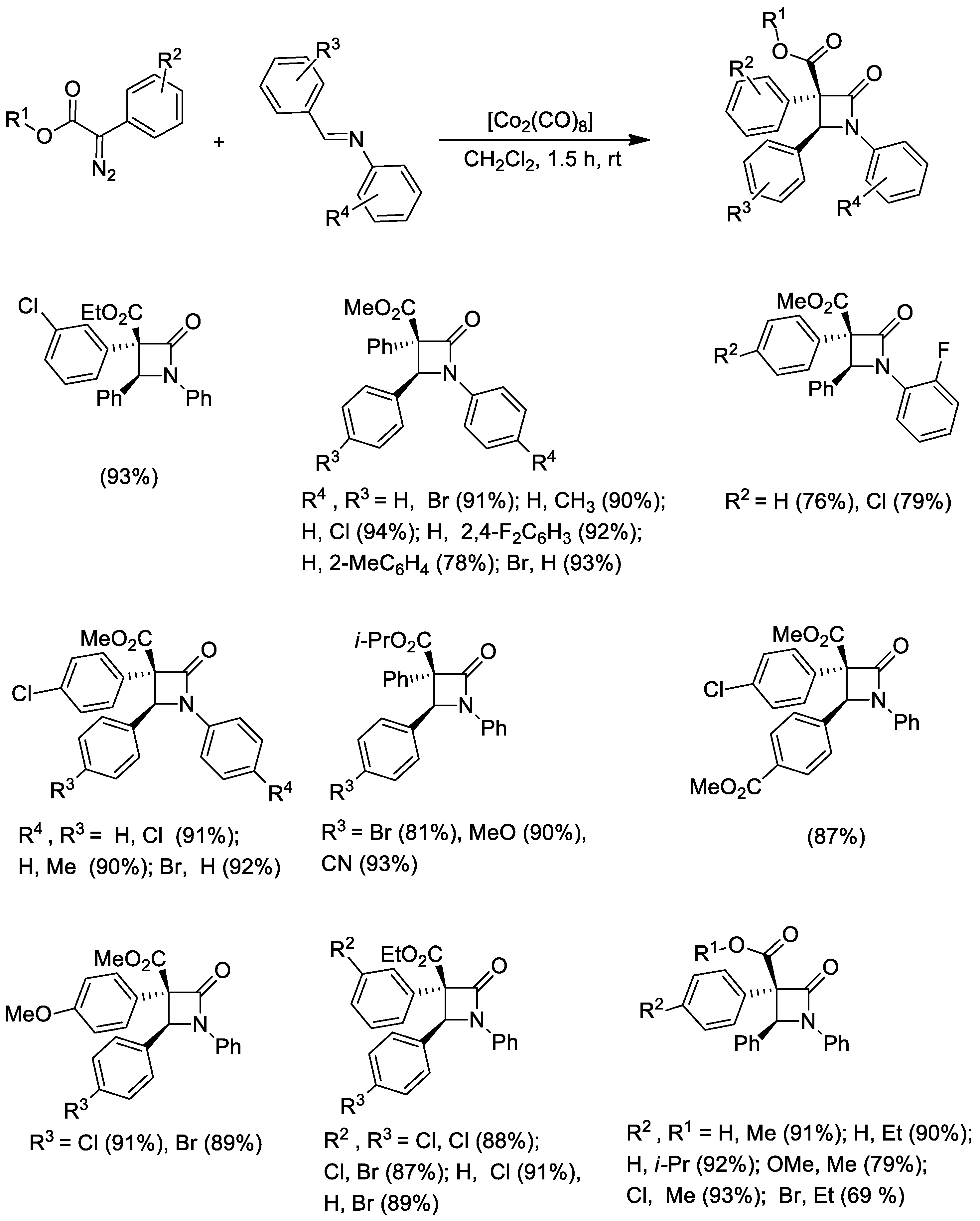
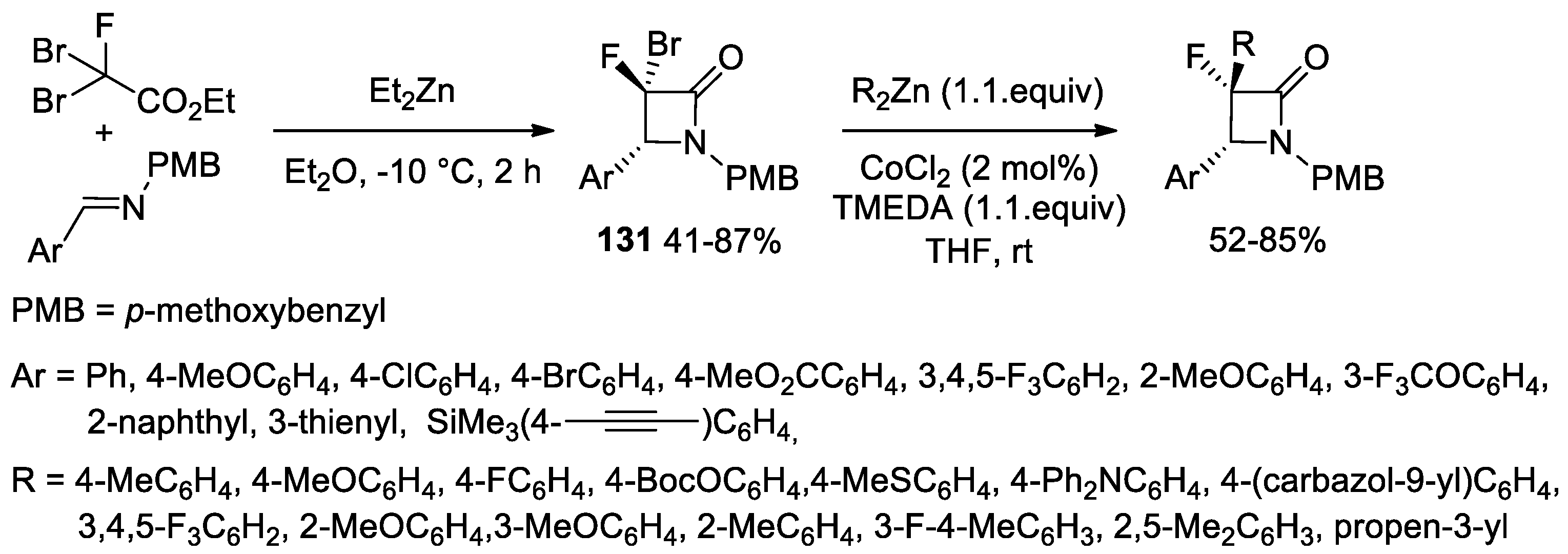
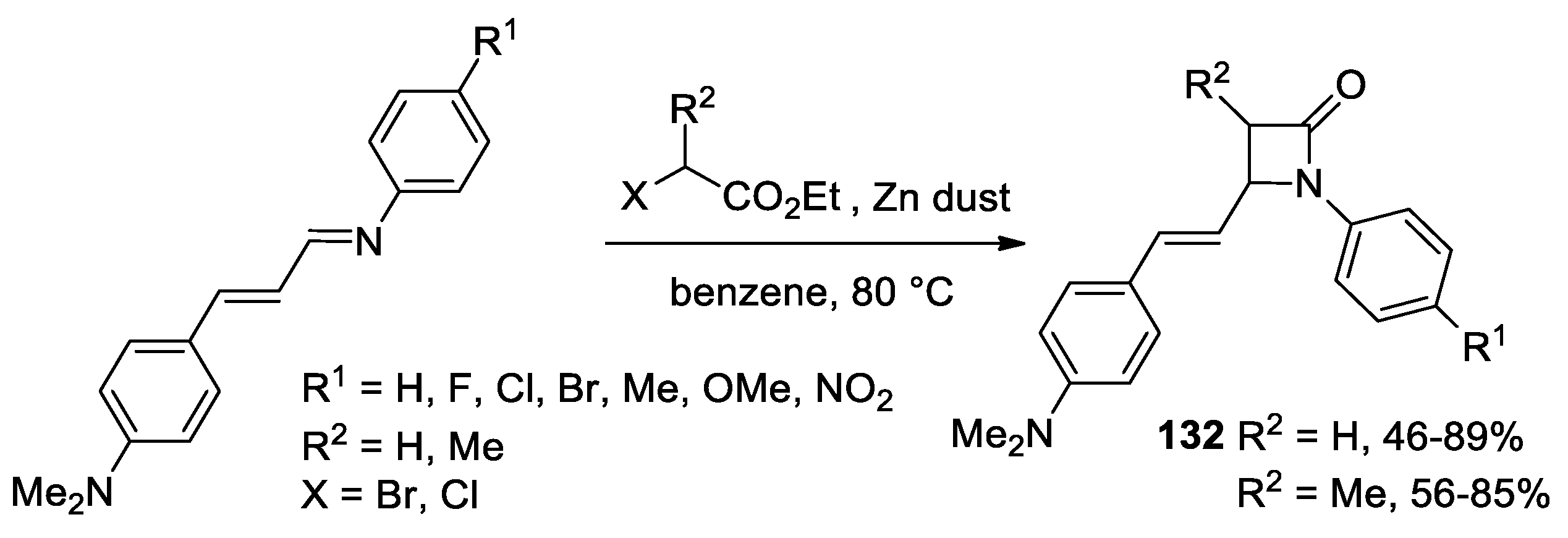
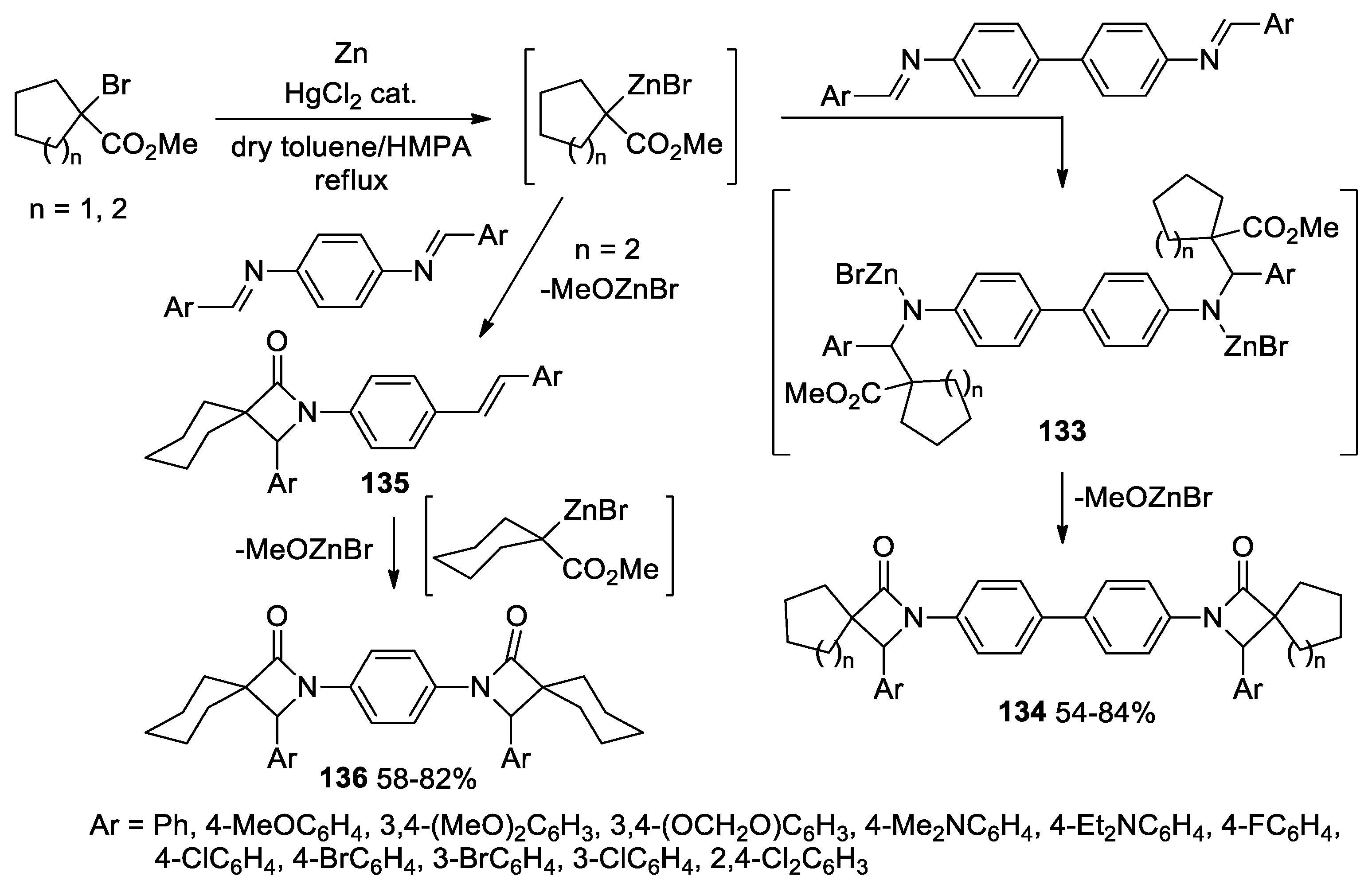
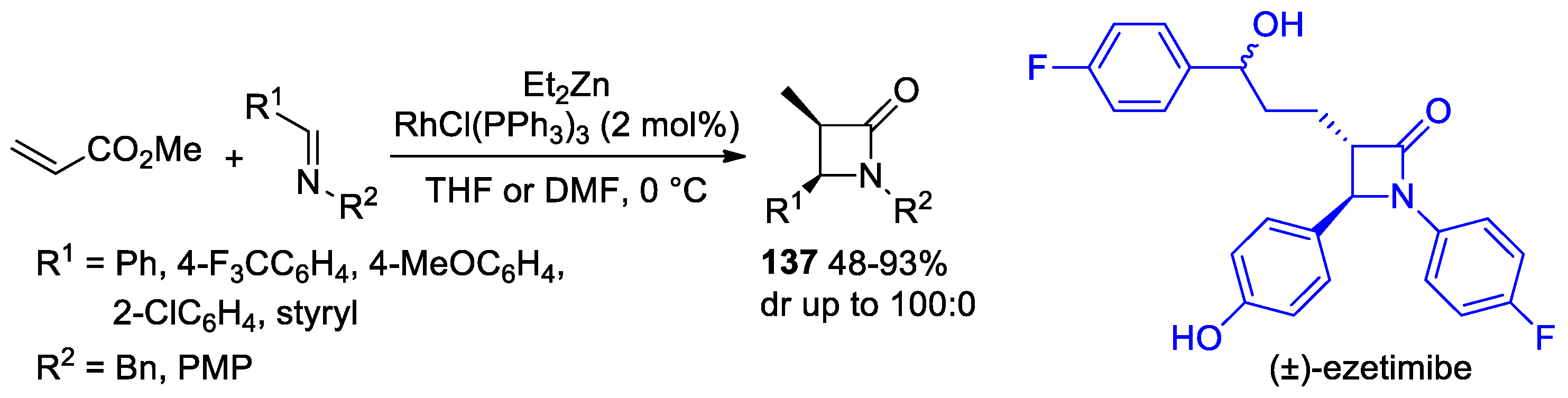




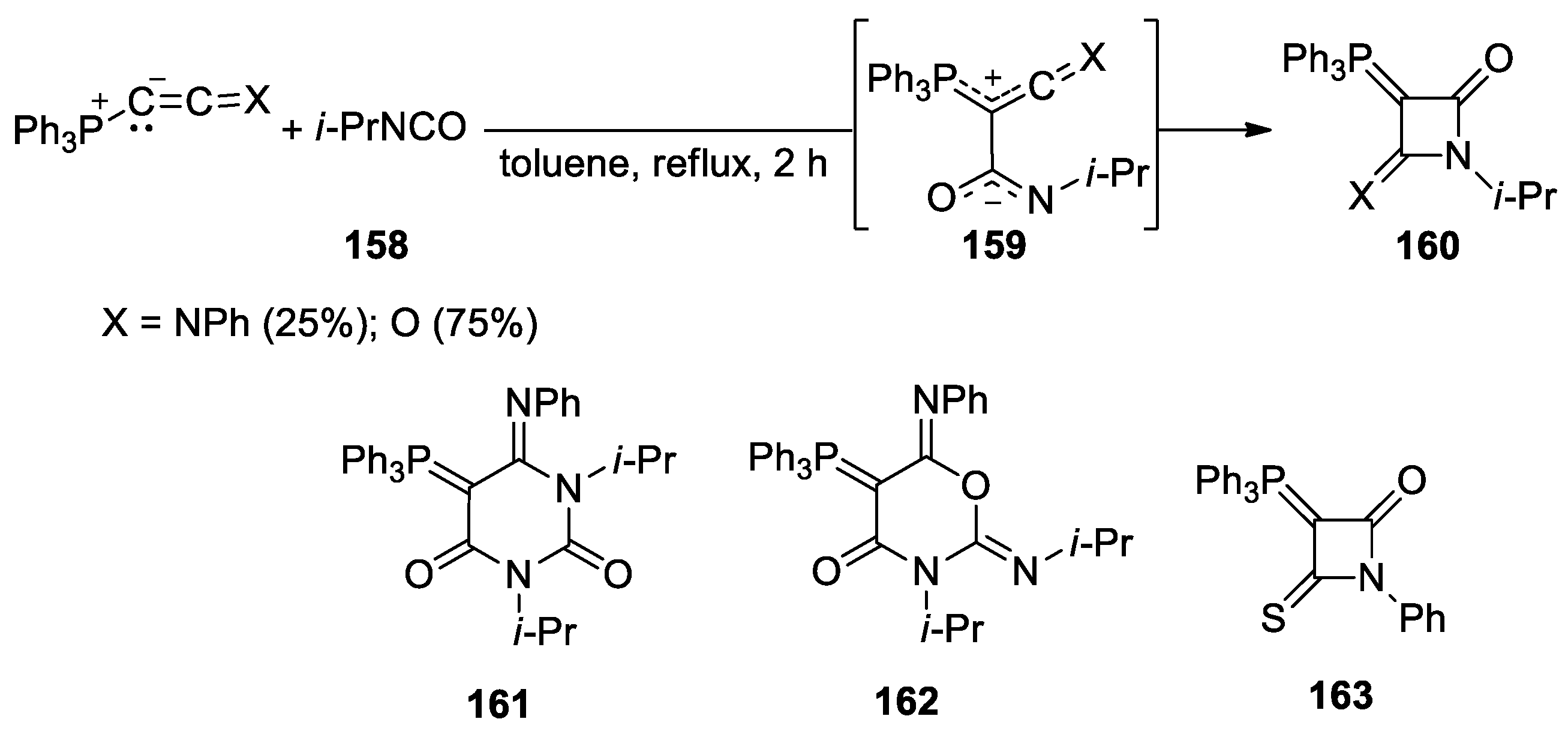
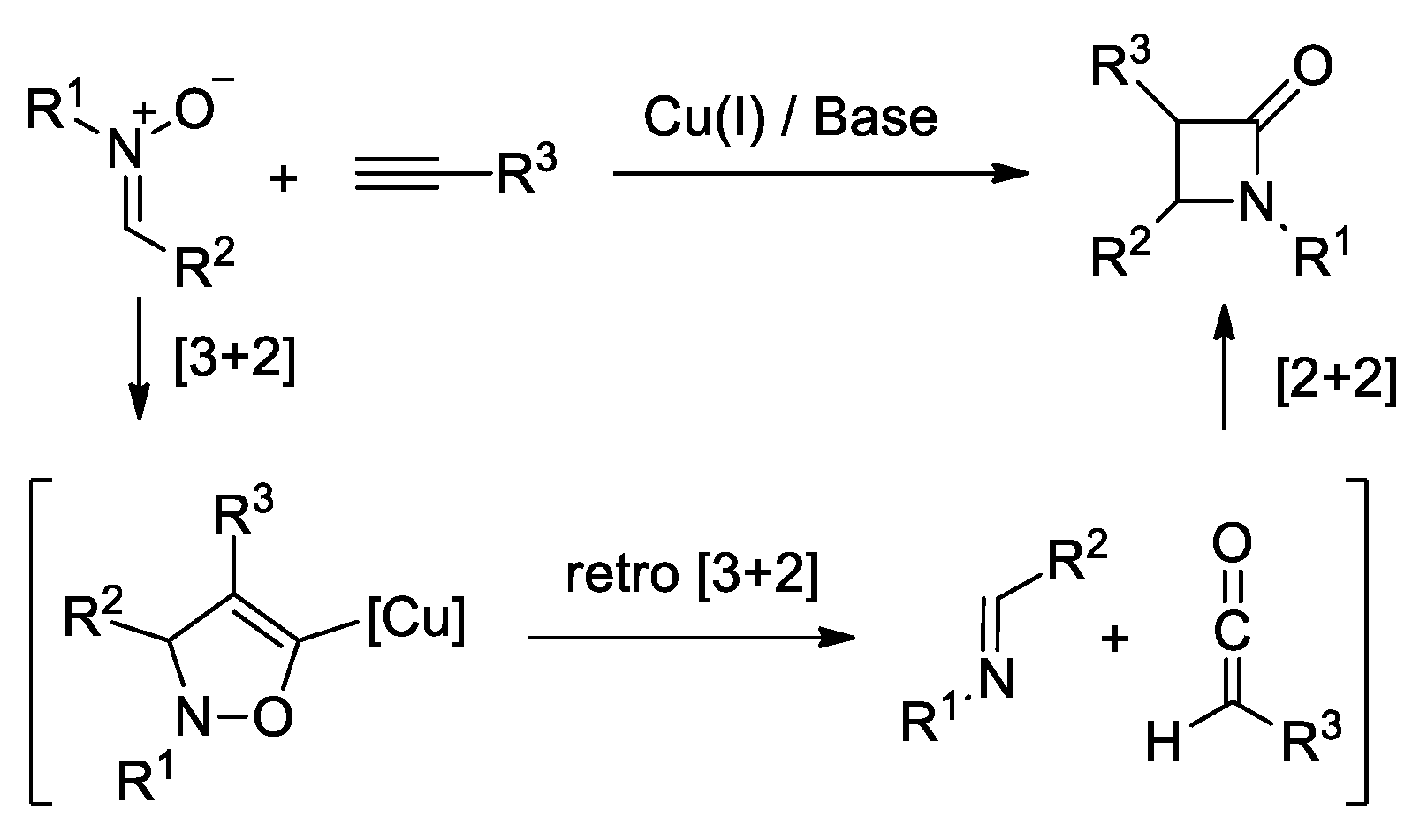
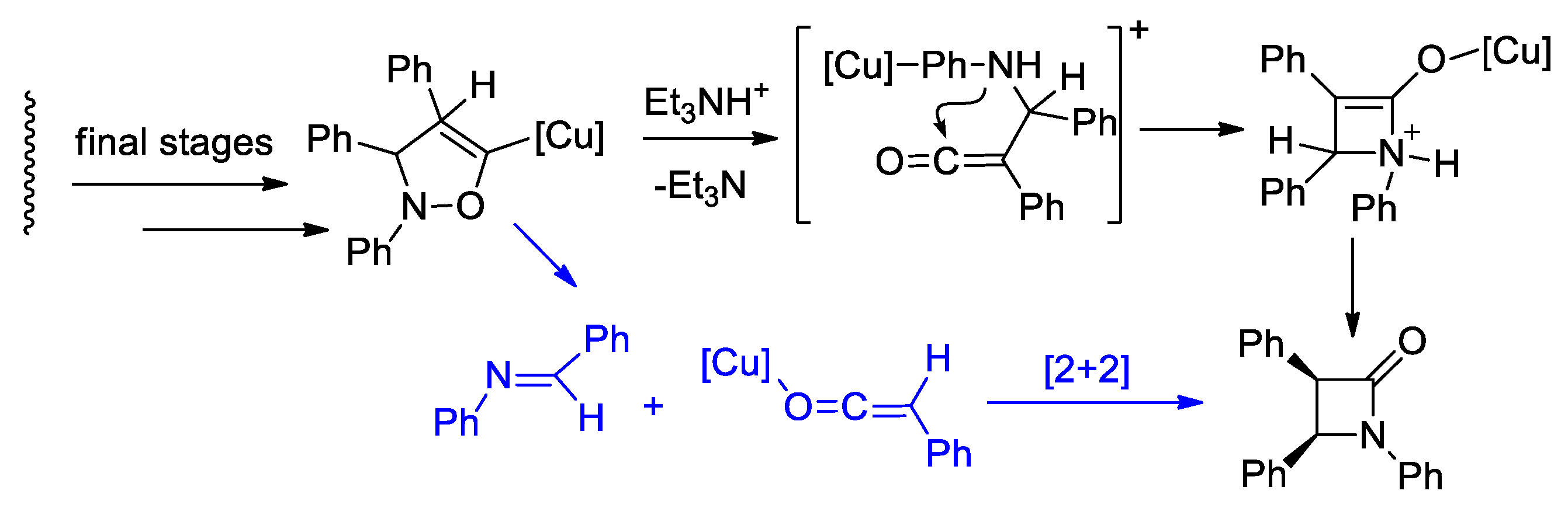
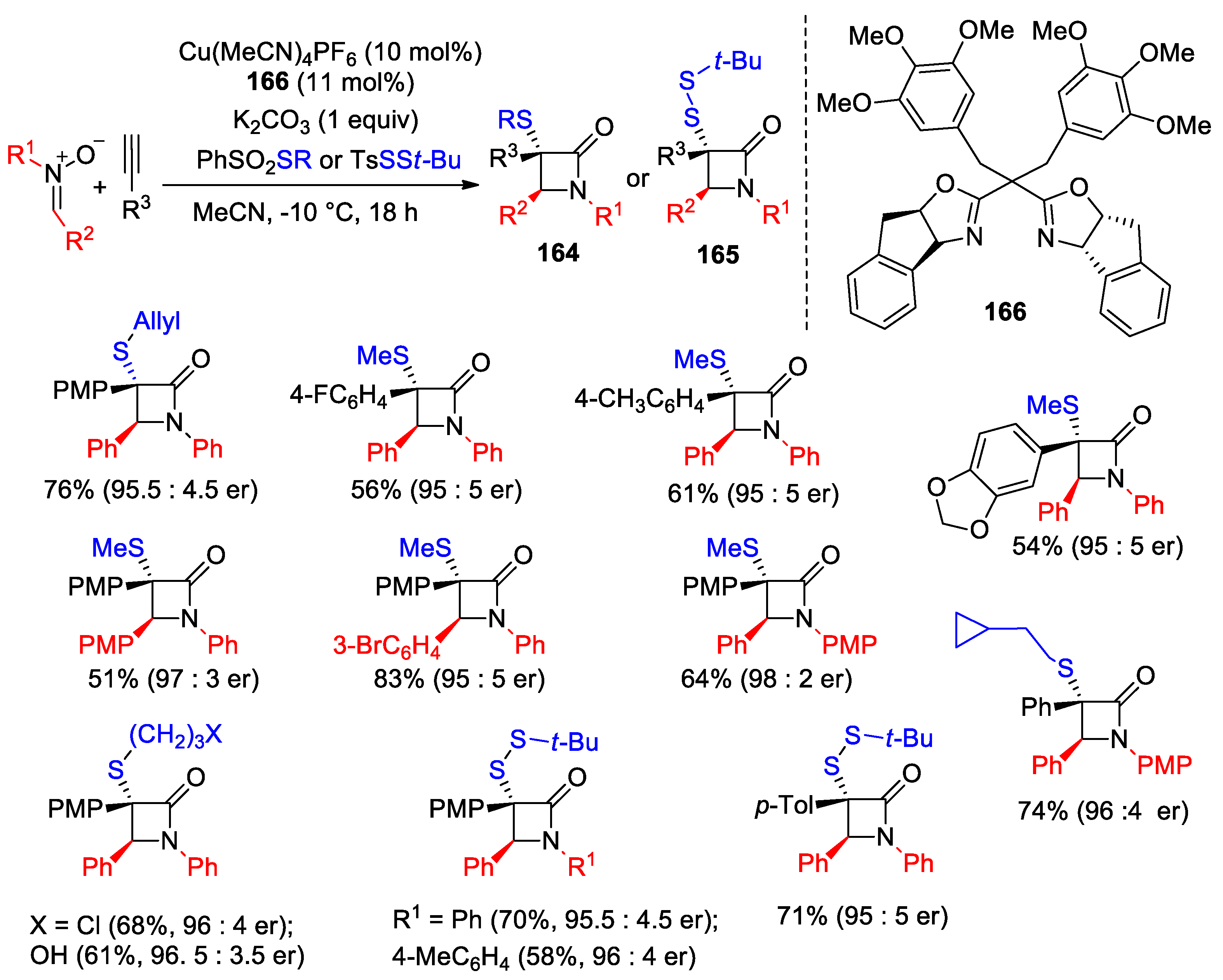


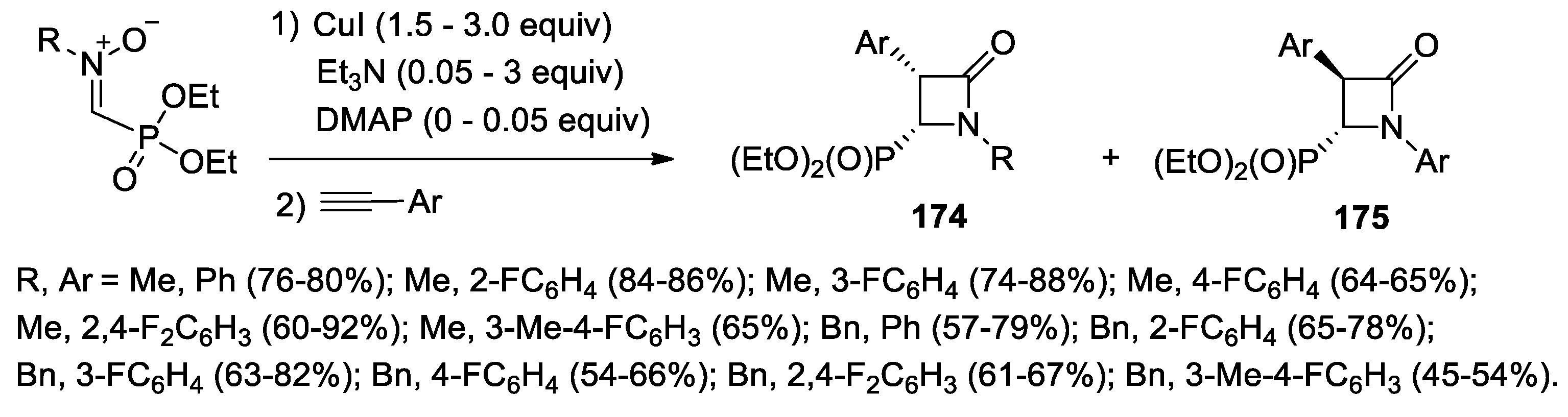

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cordero, F.M.; Giomi, D.; Machetti, F. Synthesis of 2-Azetidinones via Cycloaddition Approaches: An Update. Reactions 2024, 5, 492-566. https://doi.org/10.3390/reactions5030026
Cordero FM, Giomi D, Machetti F. Synthesis of 2-Azetidinones via Cycloaddition Approaches: An Update. Reactions. 2024; 5(3):492-566. https://doi.org/10.3390/reactions5030026
Chicago/Turabian StyleCordero, Franca Maria, Donatella Giomi, and Fabrizio Machetti. 2024. "Synthesis of 2-Azetidinones via Cycloaddition Approaches: An Update" Reactions 5, no. 3: 492-566. https://doi.org/10.3390/reactions5030026
APA StyleCordero, F. M., Giomi, D., & Machetti, F. (2024). Synthesis of 2-Azetidinones via Cycloaddition Approaches: An Update. Reactions, 5(3), 492-566. https://doi.org/10.3390/reactions5030026