
Ir-Catalyzed ortho-C-H Borylation of Aromatic C(sp2)-H Bonds of Carbocyclic Compounds Assisted by N-Bearing Directing Groups


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
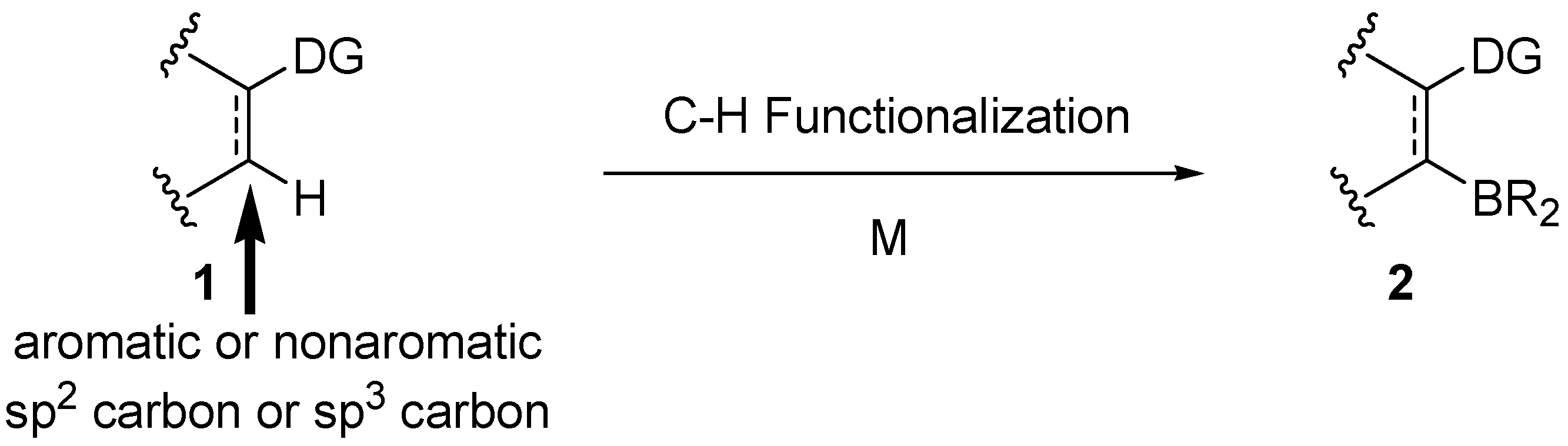
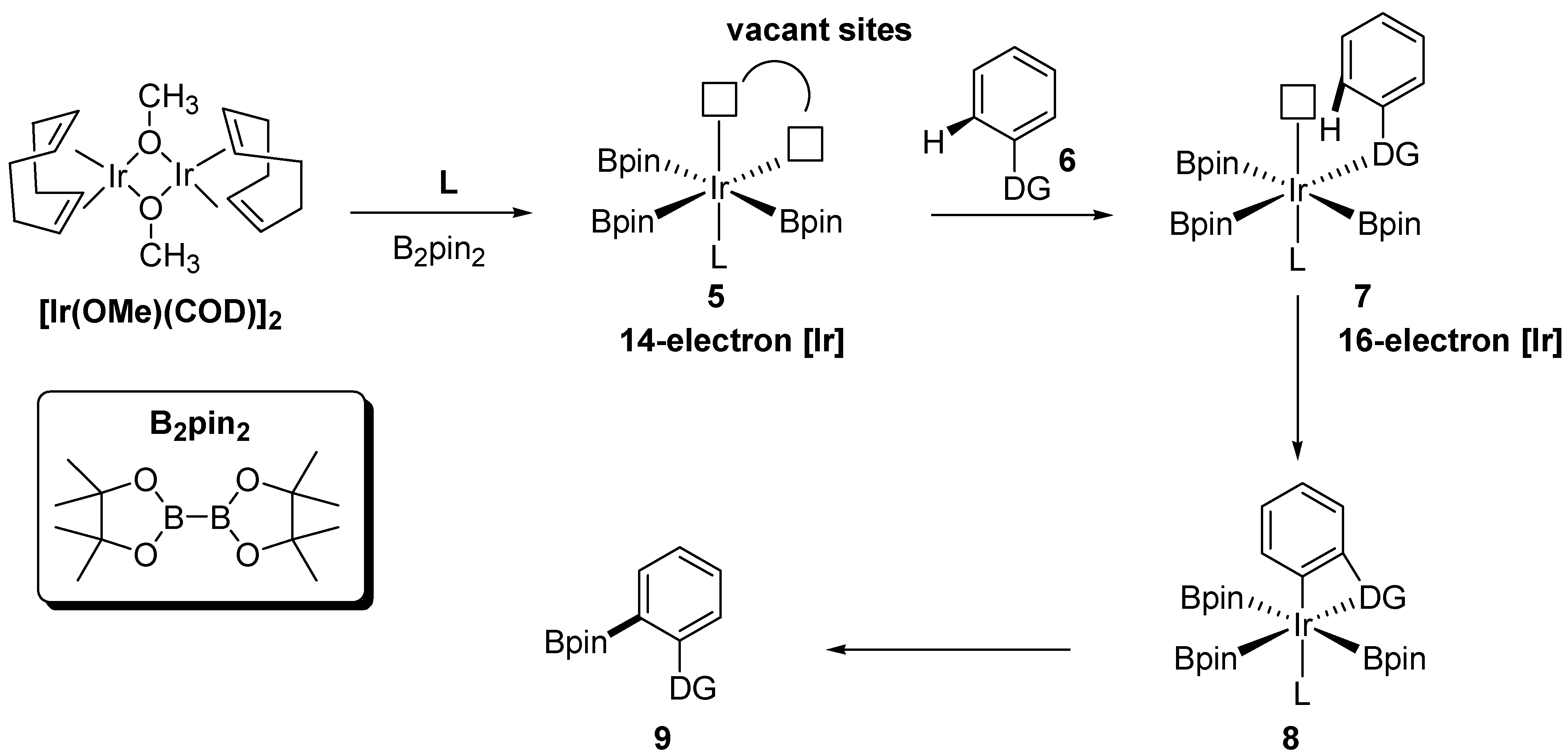
1. Introduction
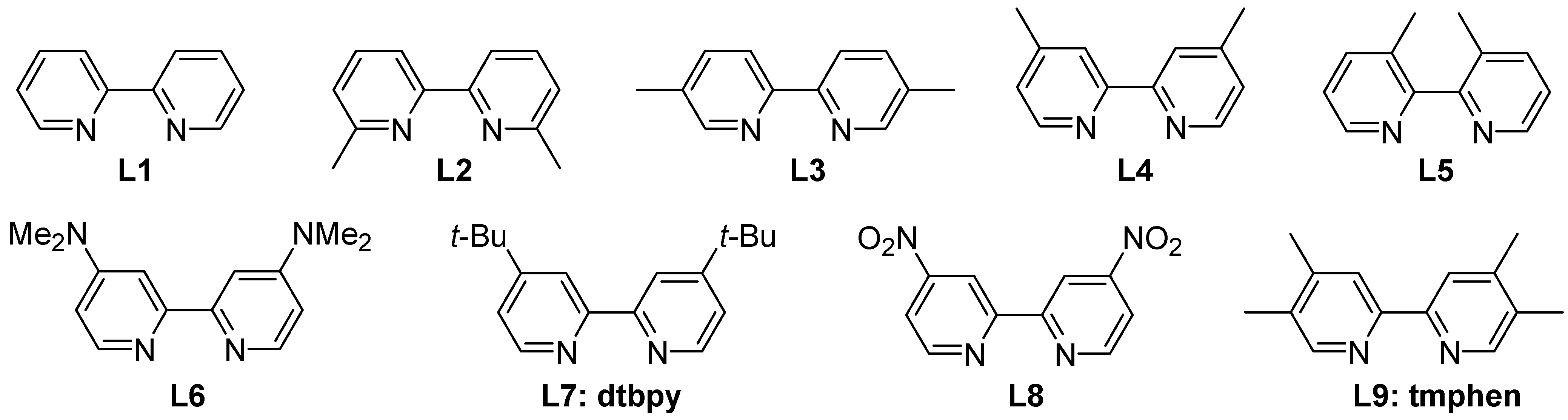
2. Discussion
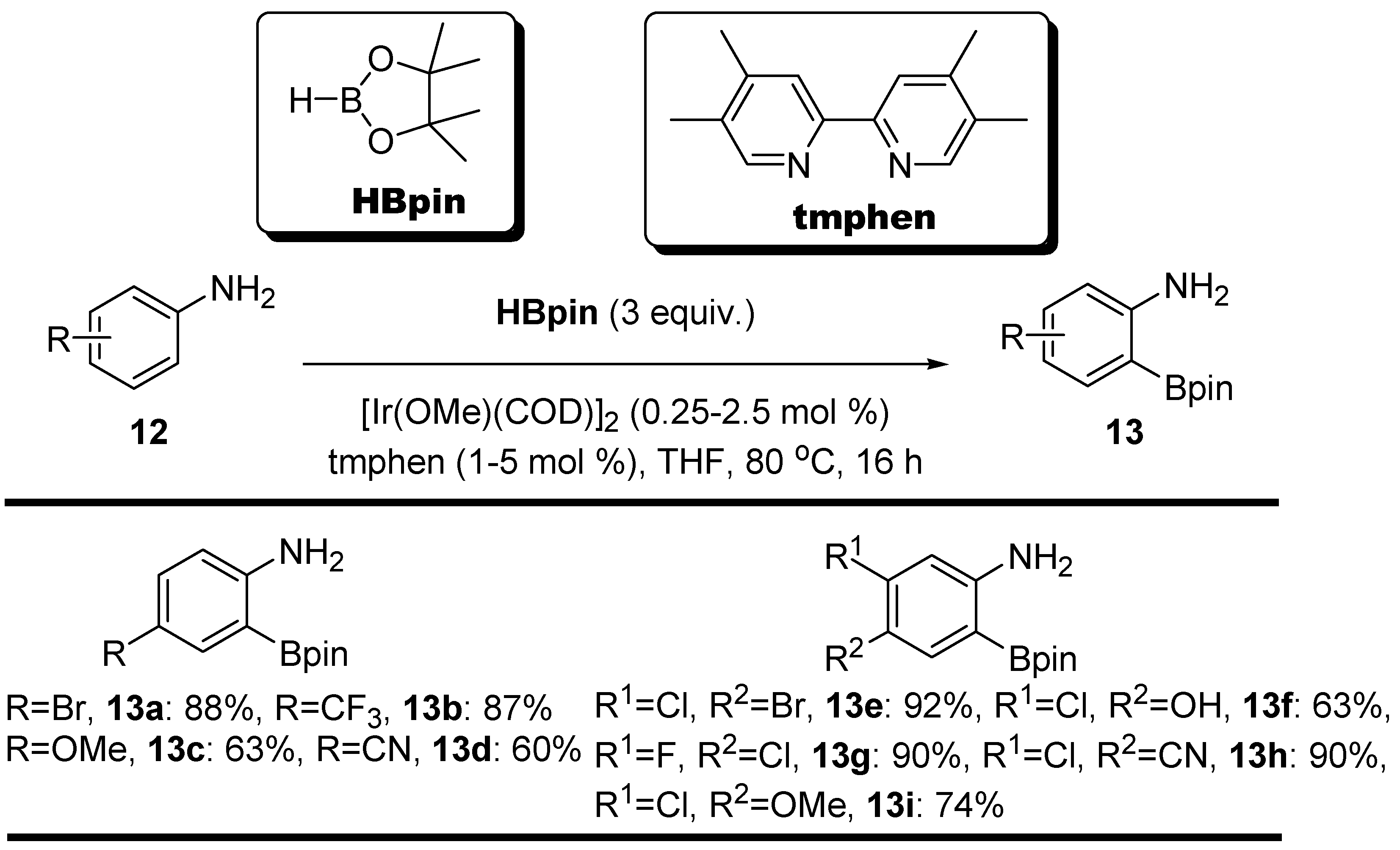
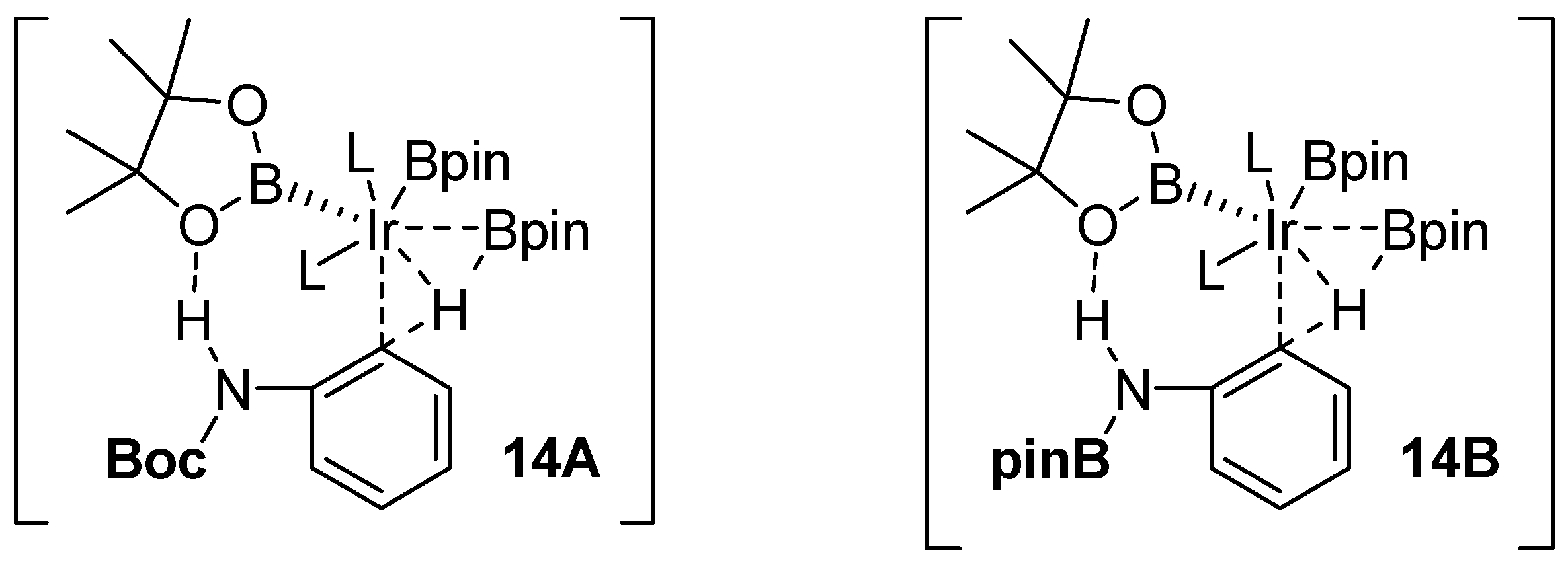
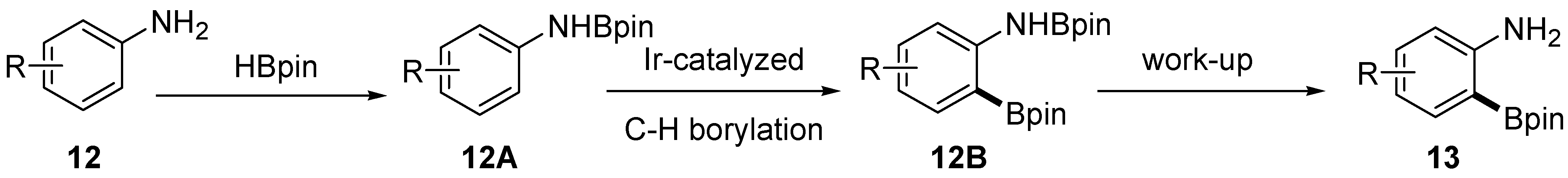
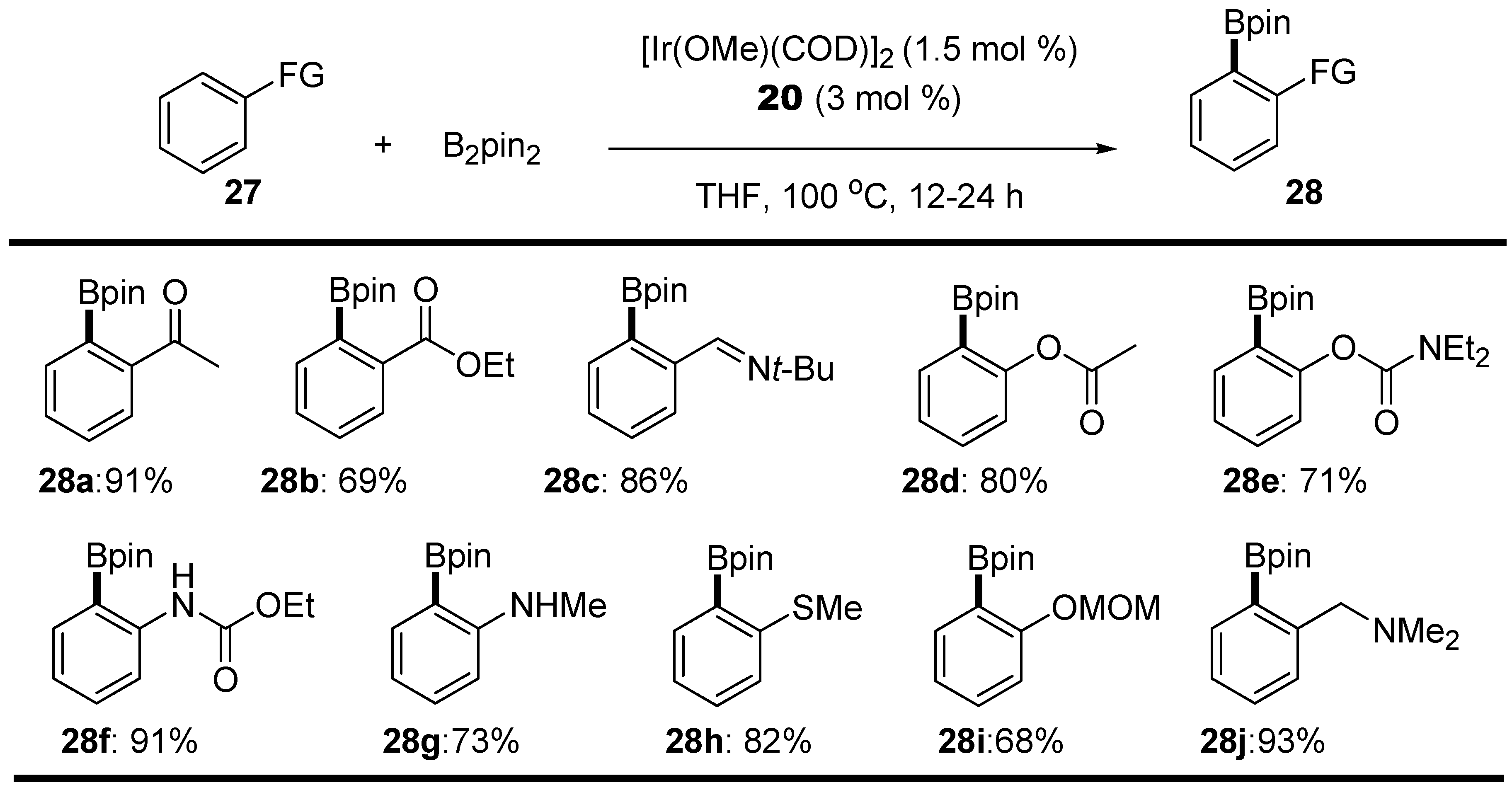
2.1. Anilines
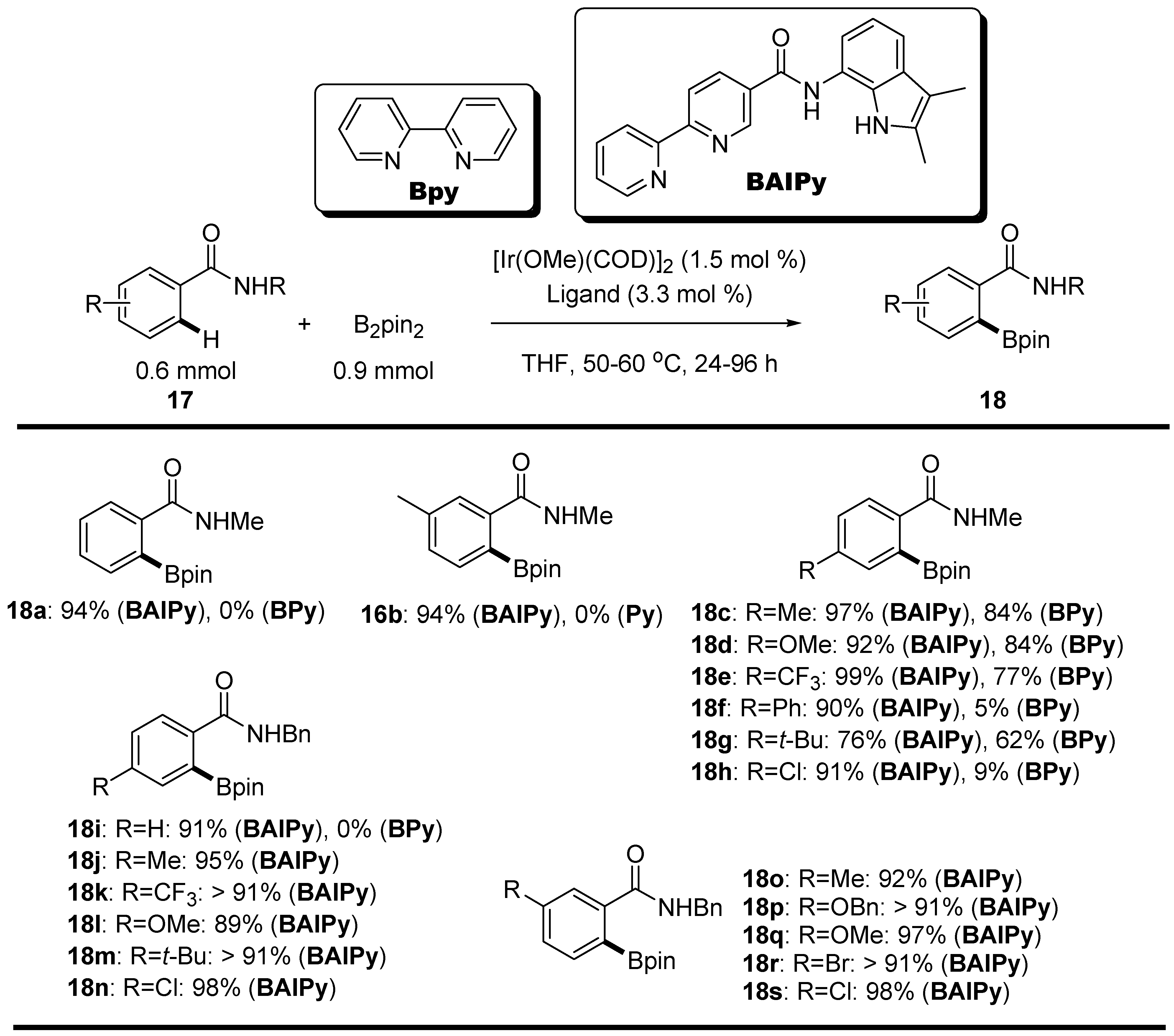
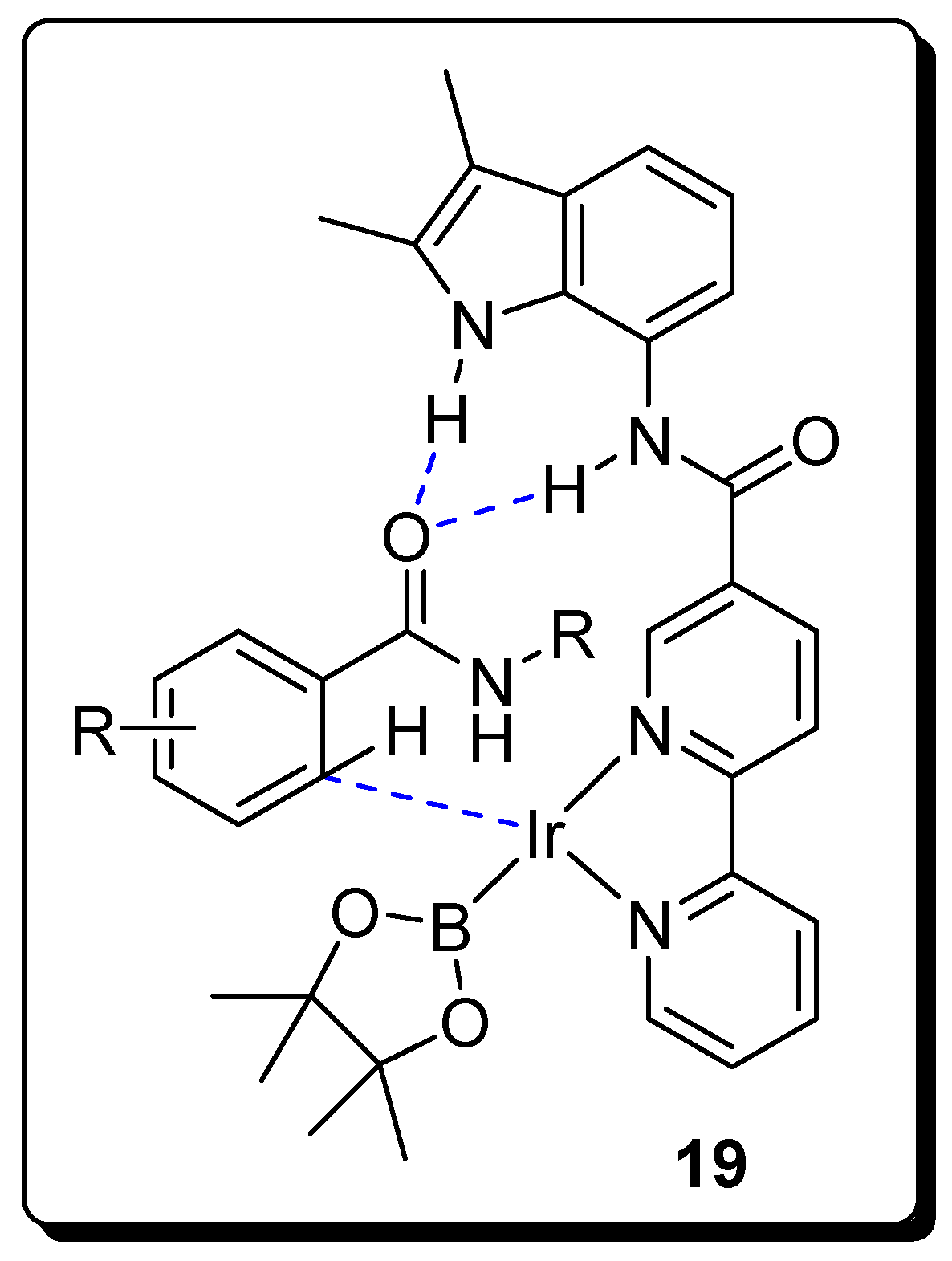
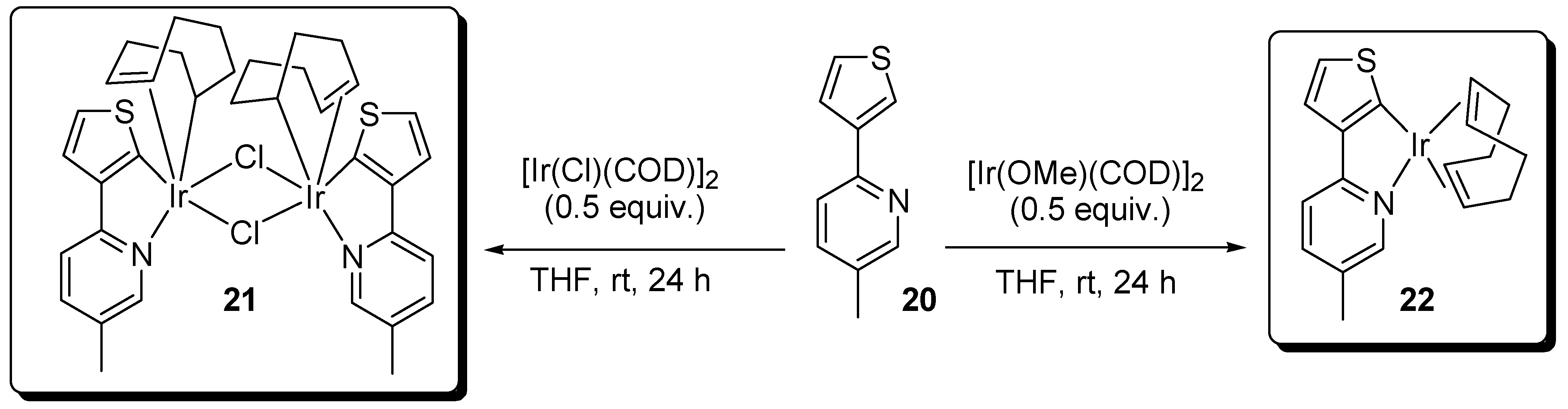
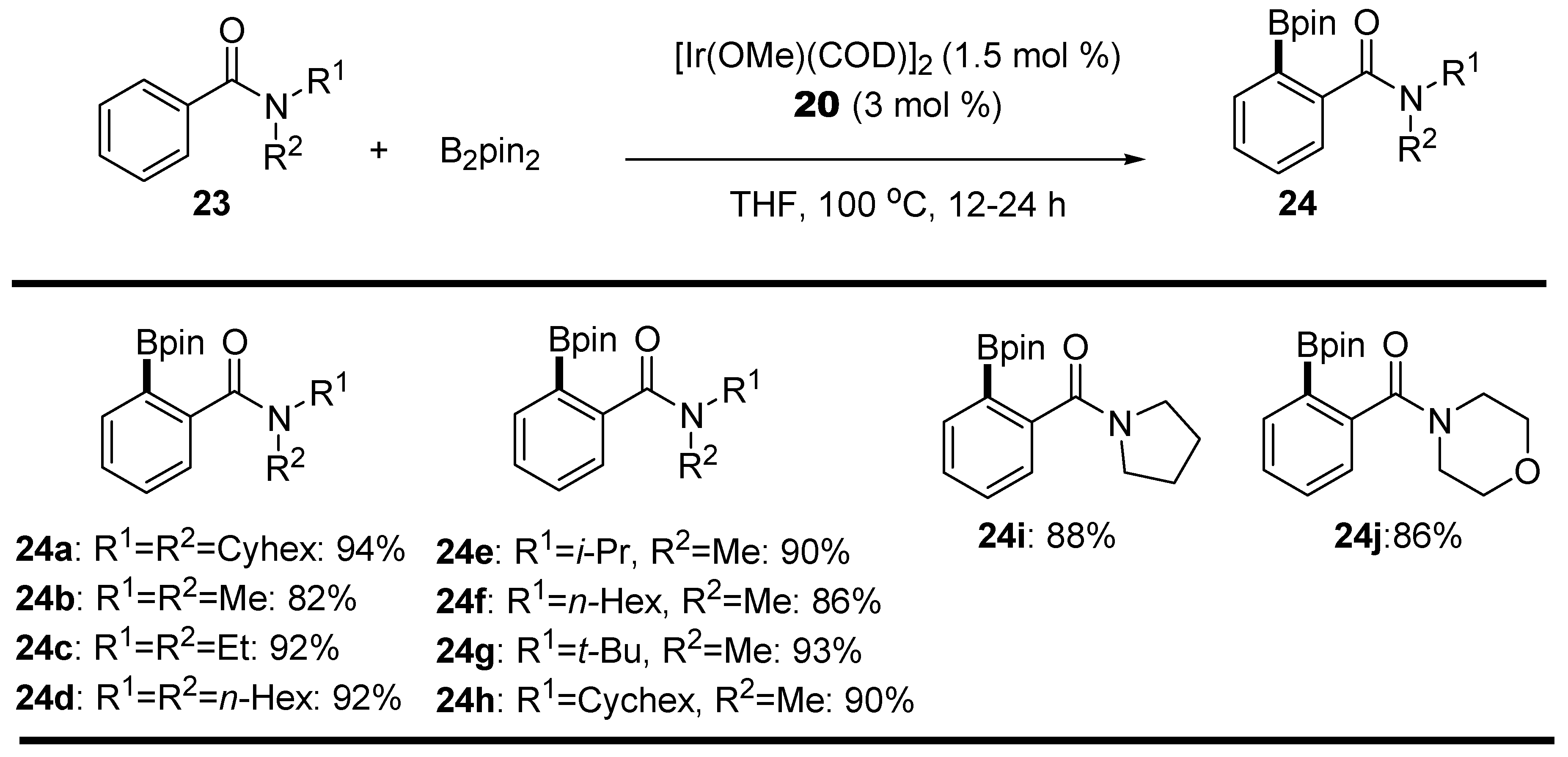
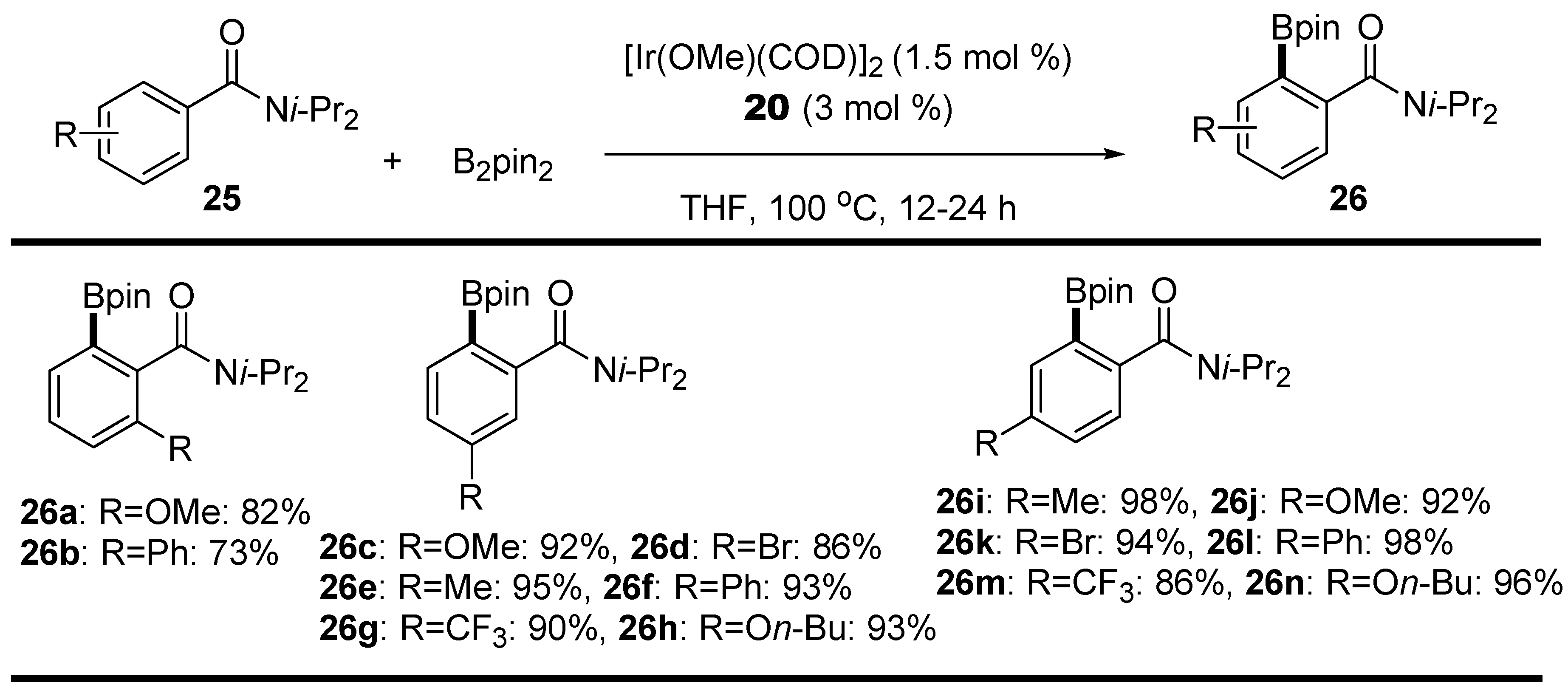
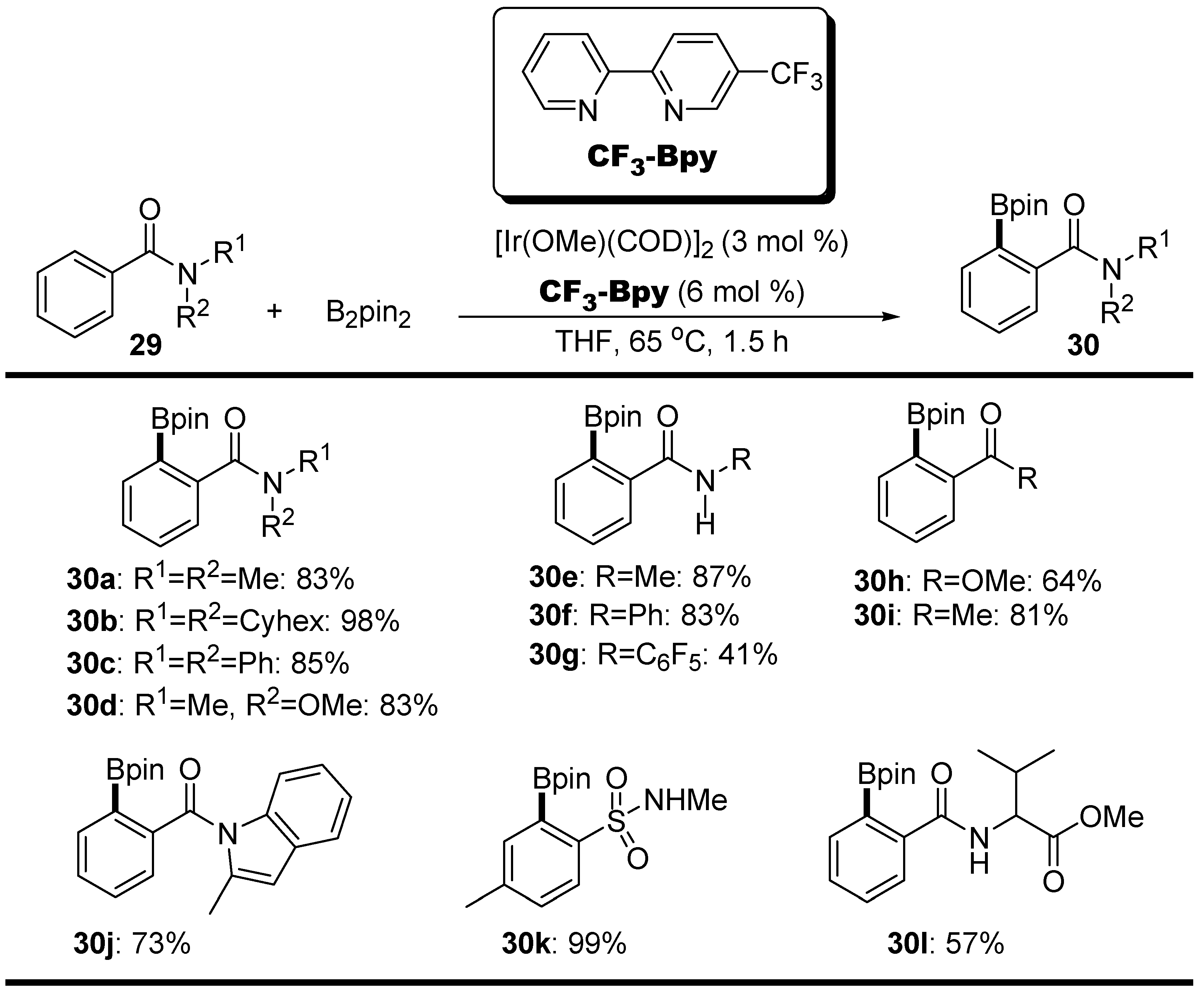
2.2. Amides
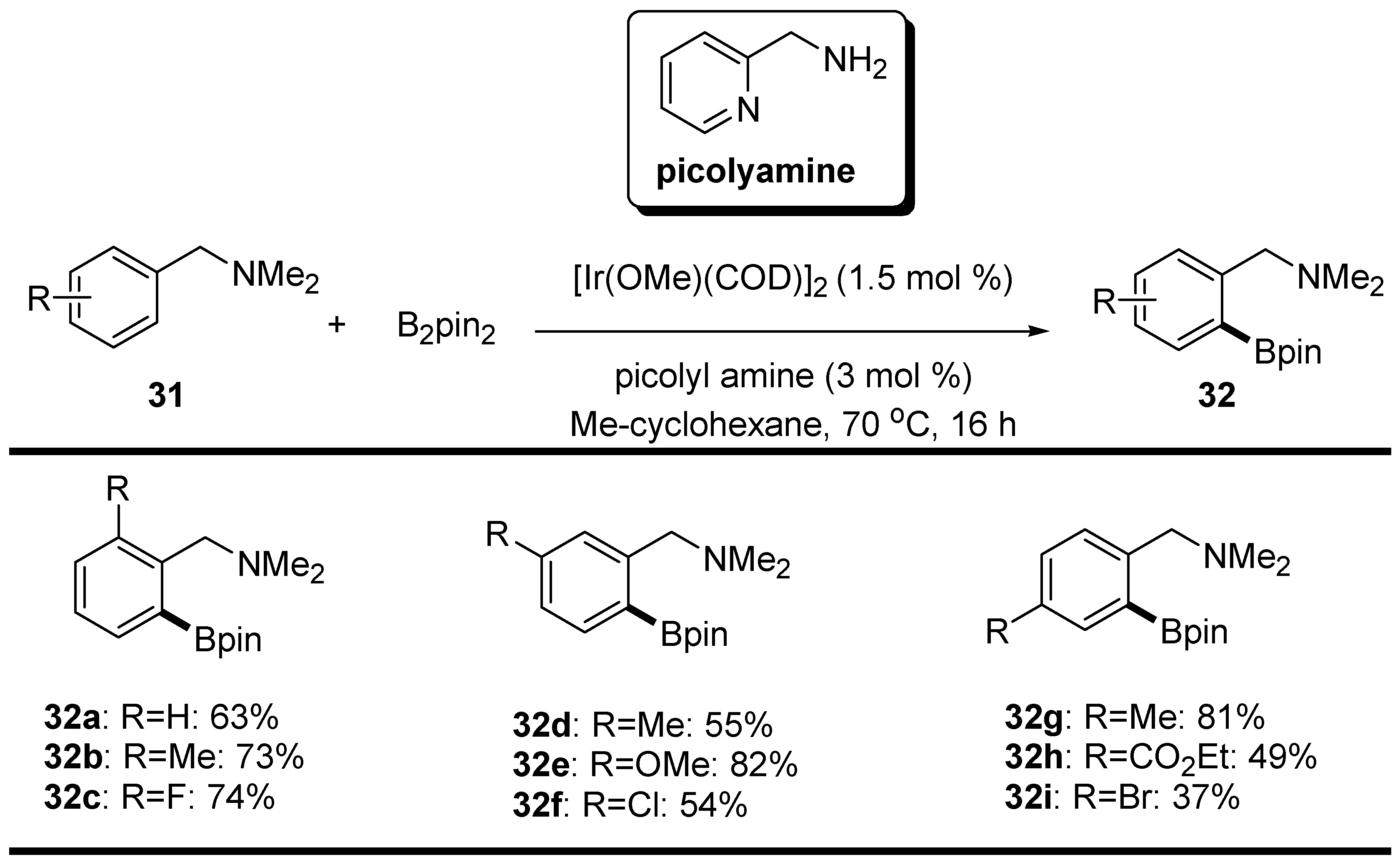
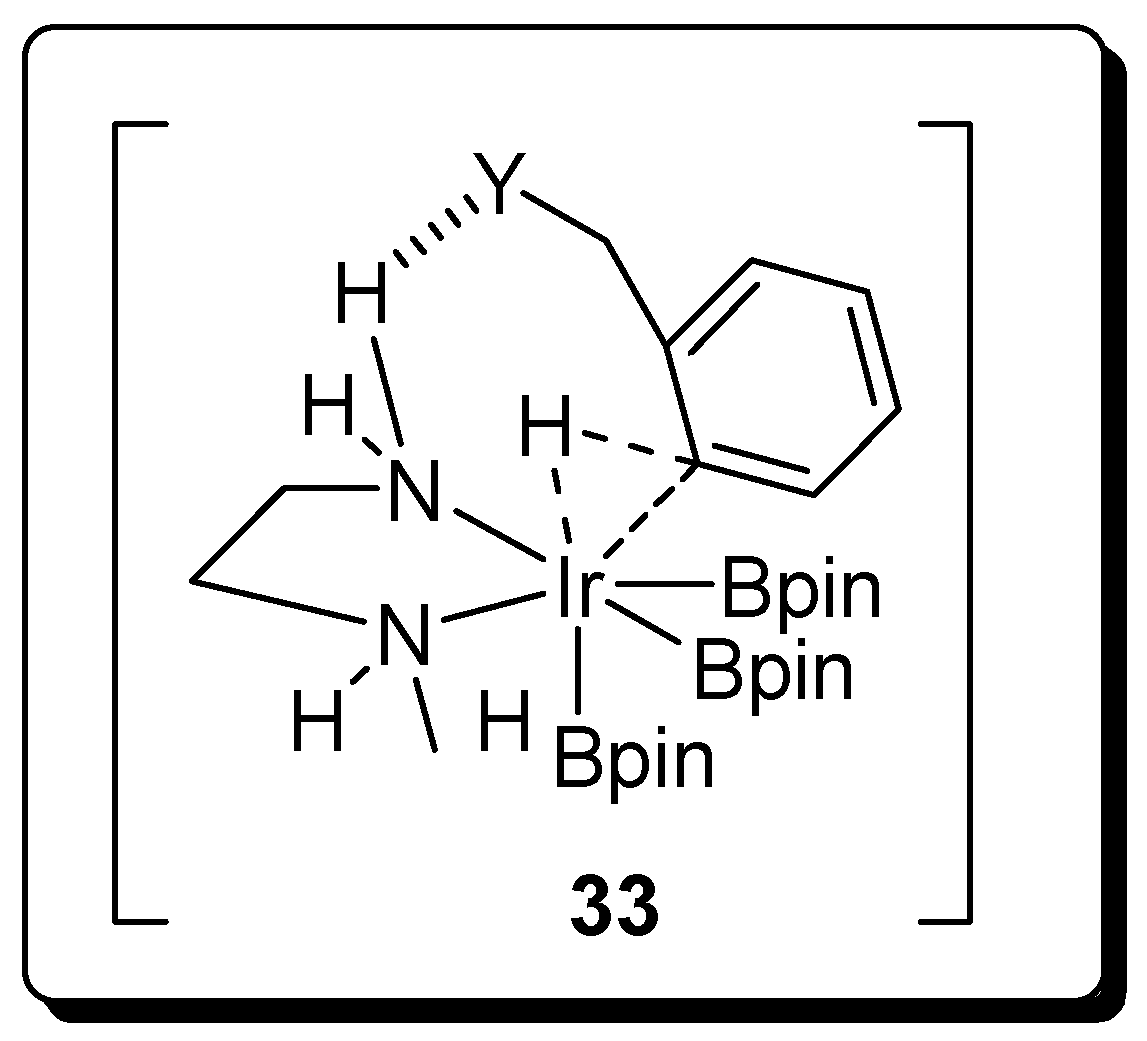
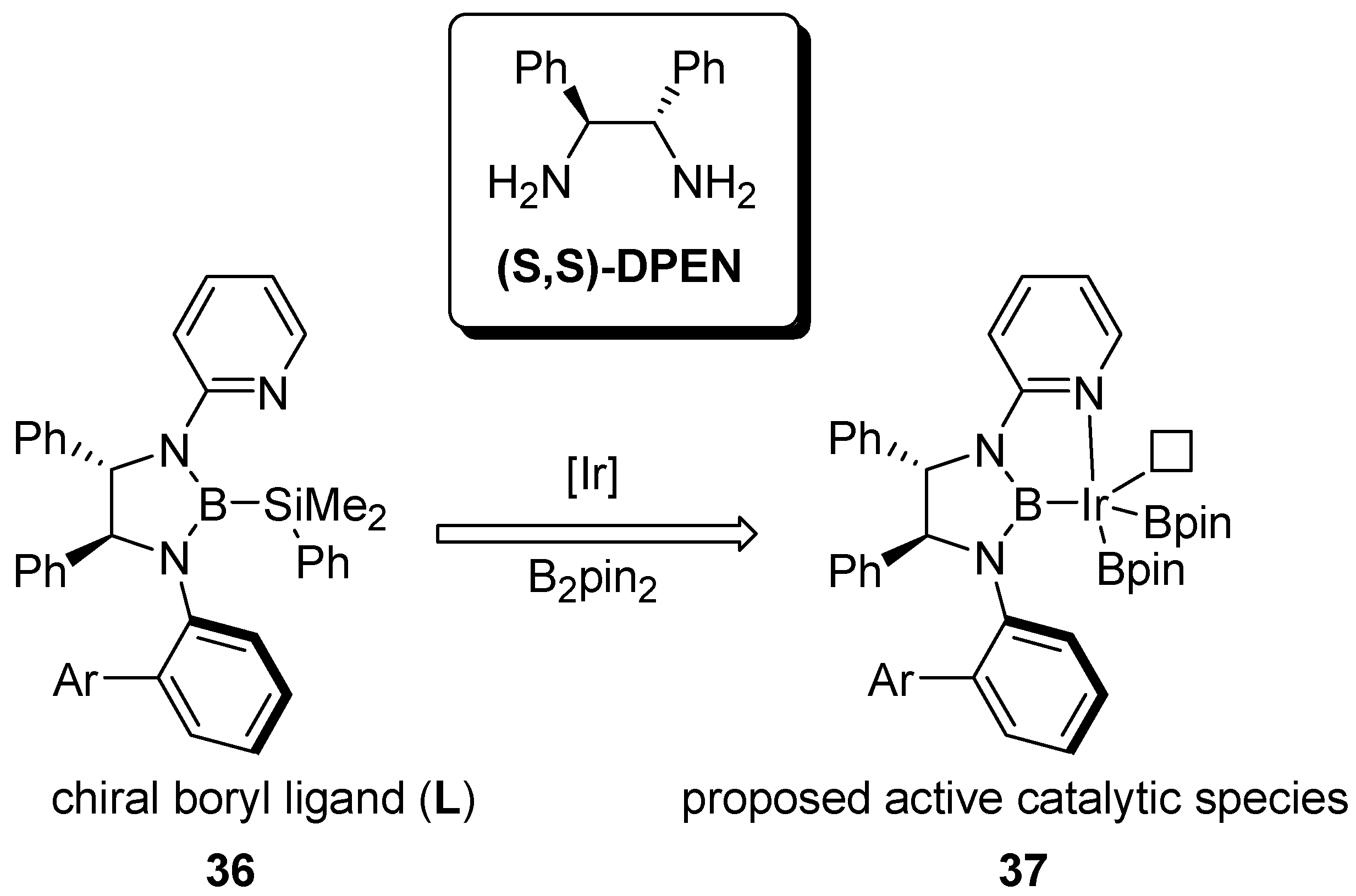
2.3. Benzyl Amines
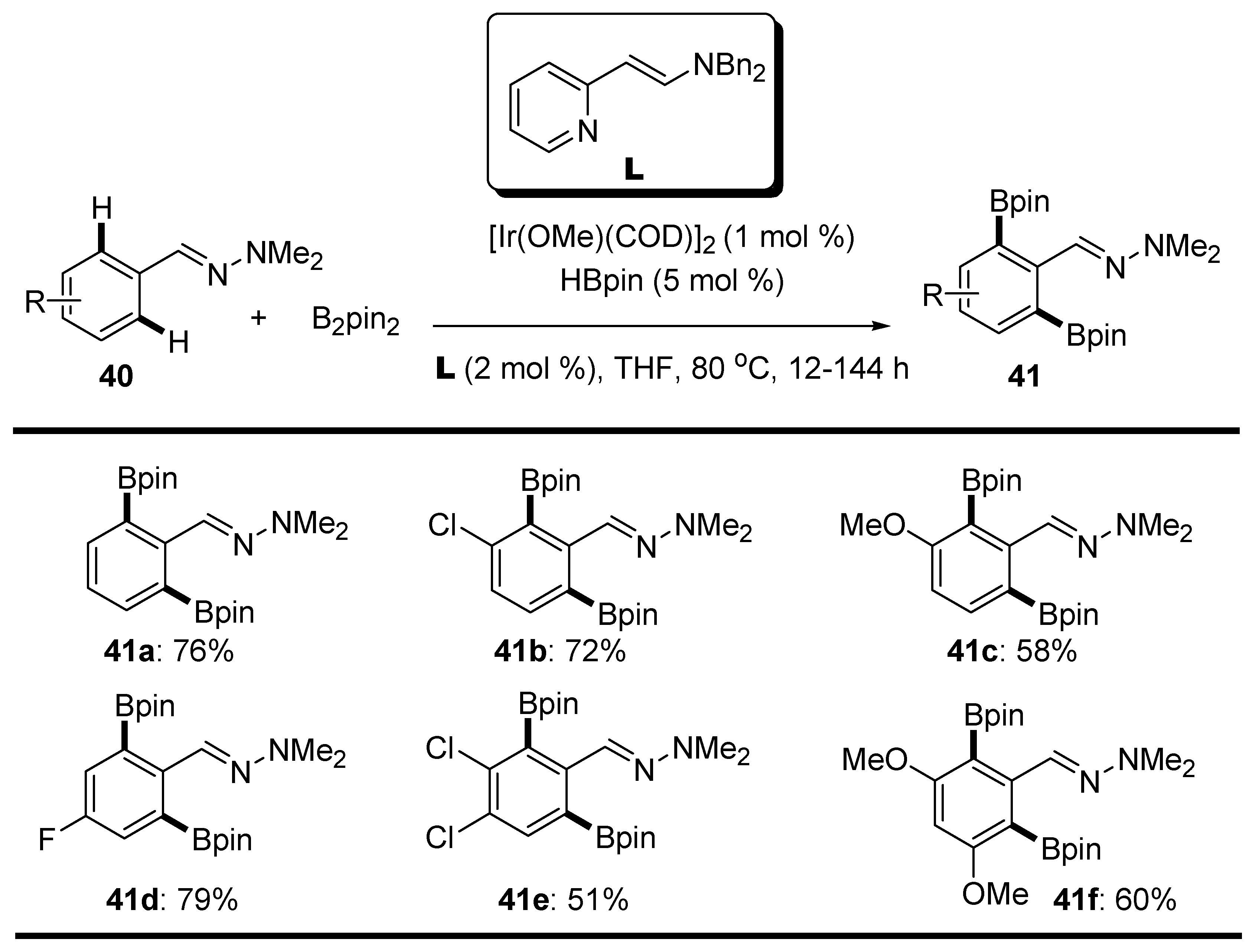
2.4. Hydrazones
2.5. Triazines
3. Conclusions
Funding
Conflicts of Interest
References
- Shilov, A.E.; Shul’pin, G.B. Activation of C–H Bonds by Metal Complexes. Chem. Rev. 1997, 97, 2879–2932. [Google Scholar] [CrossRef]
- Kakiuchi, F.; Chatani, N. Catalytic Methods for C–H Bond Functionalization: Application in Organic Synthesis. Adv. Synth. Catal. 2003, 345, 1077–1101. [Google Scholar] [CrossRef]
- Engle, K.M.; Mei, T.-S.; Wasa, M.; Yu, J.Q. Weak Coordination as a Powerful Means for Developing Broadly Useful C–H Functionalization Reactions. Acc. Chem. Res. 2012, 45, 788–802. [Google Scholar] [CrossRef]
- Rej, S.; Das, A.; Chatani, N. Strategic Evolution in Transition Metal-Catalyzed Directed C–H Bond Activation and Future Directions. Coord. Chem. Rev. 2021, 431, 213683–213719. [Google Scholar] [CrossRef]
- Al Mamari, H.H.; Štefane, B.; Žugelj, H.B. Metal-Catalyzed C–H Bond Functionalization of Phenol Derivatives. Tetrahedron 2020, 76, 130925–130936. [Google Scholar] [CrossRef]
- Docherty, J.H.; Lister, T.M.; Mcarthur, G.; Findlay, M.T.; Domingo-Legarda, P.; Jacob Kenyon, J.; Choudhary, S.; Igor Larrosa, I. Transition-Metal-Catalyzed C–H Bond Activation for the Formation of C–C Bonds in Complex Molecules. Chem. Rev. 2023, 123, 7692–7760. [Google Scholar] [CrossRef]
- Dalton, T.; Faber, T.; Glorius, F. C–H Activation: Toward Sustainability and Applications. ACS Cent. Sci. 2021, 2, 245–261. [Google Scholar] [CrossRef]
- He, J.; Wasa, M.; Chan, K.S.L.; Shao, Q.; Yu, J.-Q. Palladium-Catalyzed Transformations of Alkyl C–H Bonds. Chem. Rev. 2017, 117, 8754–8786. [Google Scholar] [CrossRef]
- Chen, X.; Engle, K.M.; Wang, D.-H.; Yu, J.-Q. Palladium(II)-Catalyzed C–H Activation/C-C Cross-Coupling Reactions: Versatility and Practicality. Angew. Chem. Int. Ed. 2009, 48, 5094–5115. [Google Scholar] [CrossRef]
- Neufeldt, S.R.; Sanford, M.S. Controlling Site Selectivity in Palladium-Catalyzed C–H Bond Functionalization. Acc. Chem. Res. 2012, 45, 936–946. [Google Scholar] [CrossRef]
- Bay, K.L.; Yang, Y.-F.; Houk, K.N. Multiple Roles of Silver Salts in Palladium-Catalyzed C–H Activations. J. Organomet. Chem. 2018, 864, 19–25. [Google Scholar] [CrossRef]
- Colby, D.A.; Tsai, A.S.; Bergman, R.G.; Ellman, J.A. Rhodium Catalyzed Chelation-Assisted C–H Bond Functionalization Reactions. Acc. Chem. Res. 2012, 45, 814–825. [Google Scholar] [CrossRef]
- Rej, S.; Chatani, N. Rhodium-Catalyzed C(sp2)- or C(sp3)–H Bond Functionalization Assisted by Removable Directing Groups. Angew. Chem. Int. Ed. 2019, 58, 8304–8329. [Google Scholar] [CrossRef]
- Lewis, J.C.; Bergman, R.G.; Ellman, J.A. Direct Functionalization of Nitrogen Heterocycles via Rh-Catalyzed C–H Bond Activation. Acc. Chem. Res. 2008, 41, 1013–1025. [Google Scholar] [CrossRef]
- Vasquez-Cespedes, S.; Wang, X.; Glorius, F. Plausible Rh(V) Intermediates in Catalytic C–H Activation Reactions. ACS Catal. 2018, 8, 242–257. [Google Scholar] [CrossRef]
- Song, G.; Li, X. Substrate Activation Strategies in Rhodium(III)-Catalyzed Selective Functionalization of Arenes. Acc. Chem. Res. 2015, 48, 1007–1020. [Google Scholar] [CrossRef]
- Singh, K.S. Recent Advances in C–H Bond Functionalization with Ruthenium-Based Catalysts. Catalysts 2019, 9, 173/1–173/51. [Google Scholar] [CrossRef]
- Dana, S.; Yadav, M.R.; Sahon, A.K. Ruthenium-Catalyzed C-N and C–O Bond-Forming Processes from C–H Bond Functionalization. Top. Organomet. Chem. 2016, 55, 189–215. [Google Scholar]
- Ruiz, S.; Villuendas, P.; Urriolabeitia, E.P. Ru-catalyzed C-H functionalizations as a tool for selective organic synthesis. Tetrahedron Lett. 2016, 57, 3413–3432. [Google Scholar] [CrossRef]
- Li, B.; Dixneuf, P.H. Ruthenium(II)-Catalyzed sp2 C–H Bond Functionalization by C–C Bond Formation. Top. Organomet. Chem. 2015, 48, 119–193. [Google Scholar]
- Ackermann, L.; Vicente, R. Ruthenium-Catalyzed Direct Arylations through C–H Bond Cleavages. Top. Curr. Chem. 2010, 292, 211–229. [Google Scholar]
- Gandeepan, P.; Müller, T.; Zell, D.; Warratz, S.; Ackermann, L. 3d Transition Metals for C–H Activation. Chem. Rev. 2019, 119, 2192–2452. [Google Scholar] [CrossRef]
- Khake, S.M.; Chatani, N. Chelation-Assisted Nickel-Catalyzed C–H Functionalizations. Trends Chem. 2019, 1, 524–539. [Google Scholar] [CrossRef]
- Khake, S.M.; Chatani, N. Nickel-Catalyzed C–H Functionalization Using A Non-directed Strategy. Chem 2020, 6, 1056–1081. [Google Scholar] [CrossRef]
- Liu, Y.-H.; Xia, Y.-N.; Shi, B.-F. Ni-Catalyzed Chelation-Assisted Direct Functionalization of Inert C-H Bonds. Chin. J. Chem. 2020, 38, 635–662. [Google Scholar] [CrossRef]
- Cano, R.; Mackey, K.; McGlacken, G.P. Recent Advances in Manganese-Catalyzed C-H Activation: Scope and Mechanism. Catal. Sci. Technol. 2018, 8, 1251–1266. [Google Scholar] [CrossRef]
- Liu, W.; Ackermann, L. Manganese-Catalyzed C–H Activation. ACS Catal. 2016, 6, 3743–3752. [Google Scholar] [CrossRef]
- Lanzi, M.; Cera, G. Iron-Catalyzed C-H Functionalizations under Triazole-Assistance. Molecules 2020, 25, 1806. [Google Scholar] [CrossRef] [PubMed]
- Shang, R.; Ilies, L.; Nakamura, E. Iron-Catalyzed C–H Bond Activation. Chem. Rev. 2017, 117, 9086–9139. [Google Scholar] [CrossRef] [PubMed]
- Prakash, S.; Kuppusamy, R.; Cheng, C.H. Cobalt-Catalyzed Annulation Reactions via C–H Bond Activation. ChemCatChem 2018, 10, 683–705. [Google Scholar] [CrossRef]
- Wang, S.; Chen, S.-Y.; Yu, X.-Q. C–H Functionalization by High-Valent Cp*Co(III) Catalysis. Chem. Commun. 2017, 53, 3165–3180. [Google Scholar] [CrossRef] [PubMed]
- Yoshino, T.; Matsunaga, S. High-Valent Cobalt-Catalyzed C–H Bond Functionalization. Adv. Organomet. Chem. 2019, 68, 197–247. [Google Scholar] [CrossRef]
- Baccalini, A.; Vergura, S.; Dolui, P.; Zanoni, G.; Maiti, D. Recent Advances in Cobalt-Catalyzed C-H Functionalizations. Org. Biomol. Chem. 2019, 17, 10119–10141. [Google Scholar] [CrossRef] [PubMed]
- Moselage, M.; Lie, J.; Ackermann, L. Cobalt-Catalyzed C–H Activation. ACS Catal. 2016, 6, 498–525. [Google Scholar] [CrossRef]
- Sambiagio, C.; Schönbauer, D.; Blieck, R.; Dao-Huy, T.; Pototschnio, G.; Schaaf, P.; Wiesinger, T.; Zia, M.F.; Wencel-Delord, J.; Besset, T.; et al. A Comprehensive Overview of Directing Groups Applied in Metal-Catalyzed C–H Functionalization Chemistry. Chem. Soc. Rev. 2018, 47, 6603–6743. [Google Scholar] [CrossRef] [PubMed]
- Omae, I. Intramolecular five-membered ring compounds and their applications. Coord. Chem. Rev. 2004, 248, 995–1023. [Google Scholar] [CrossRef]
- Rouquet, G.; Chatani, N.N. Catalytic Functionalization of C(sp2)–H and C(sp3)–H Bonds by Using Bidentate Directing Groups. Angew. Chem. Int. Ed. 2013, 52, 11726–11743. [Google Scholar] [CrossRef] [PubMed]
- Hartwig, J.F. Borylation and Silylation of C–H Bonds: A Platform for Diverse C–H Bond Functionalizations. Acc. Chem. Res. 2012, 45, 864–873. [Google Scholar] [CrossRef] [PubMed]
- Mkhalid, I.A.I.; Barnard, J.H.; Marder, T.B.; Murphy, J.M.; Hartwig, J.F. C–H Activation for the Construction of C–B Bonds. Chem. Rev. 2010, 110, 890–931. [Google Scholar] [CrossRef]
- Neeve, E.C.; Geier, S.J.; Mkhalid, I.A.I.; Westcott, S.; Marder, T.B. Diboron(4) Compounds: From Structural Curiosity to Synthetic Workhorse. Chem. Rev. 2016, 110, 9091–9161. [Google Scholar] [CrossRef]
- Ishiyama, T.; Miyaura, N. Transition metal-catalyzed borylation of alkanes and arenes via C-H activation. J. Organometal. Chem. 2003, 680, 3–11. [Google Scholar] [CrossRef]
- Ishiyama, T.; Takagi, J.; Hartwig, J.F.; Miyaura, N. A Stoichiometric Aromatic C-H Borylation Catalyzed by Iridium(I)/2,2′-Bipyridine Complexes at Room Temperature. Angew. Chem. Int. Ed. 2002, 41, 3056–3058. [Google Scholar] [CrossRef]
- Kuroda, Y.; Nakao, Y. Catalyst-enabled Site-selectivity in the Iridium-catalyzed C–H Borylation of Arenes. Chem. Lett. 2019, 48, 1092–1100. [Google Scholar] [CrossRef]
- Li, Y.; Wu, X.-F. Direct C–H Bond Borylation of (Hetero)Arenes: Evolution from Noble Metal to Metal Free. Angew. Chem. Int. Ed. 2020, 59, 1770–1774. [Google Scholar] [CrossRef] [PubMed]
- Hall, D.G. Boronic Acids: Preparation and Applications in Organic Synthesis and Medicine; Wiley-VCH: Weinheim, Germany, 2005. [Google Scholar]
- Obligacion, J.V.; Semproni, S.P.; Chirik, P.J. Cobalt-Catalyzed C(sp2)-H Borylation. J. Am. Chem. Soc. 2014, 136, 4133–4136. [Google Scholar] [CrossRef] [PubMed]
- Palmer, W.N.; Obligacion, J.V.; Pappas, I.; Chirik, P.J. Cobalt-Catalyzed Benzylic Borylation: Enabling Polyborylation and Functionalization of Remote, Unactivated C(sp3)–H Bonds. J. Am. Chem. Soc. 2016, 138, 766–769. [Google Scholar] [CrossRef] [PubMed]
- Schaefer, B.A.; Margulieux, G.W.; Small, B.L.; Chirik, P.J.J. Evaluation of Cobalt Complexes Bearing Tridentate Pincer Ligands for Catalytic C–H Borylation. Organometallics 2015, 34, 1307–1320. [Google Scholar] [CrossRef]
- Furukawa, T.; Tobisu, M.; Chatani, N. Nickel-Catalyzed Borylation of Arenes and Indoles via C–H Bond Cleavage. Chem. Commun. 2015, 51, 6508–6511. [Google Scholar] [CrossRef] [PubMed]
- Dombray, T.; Werncke, C.G.; Jiang, S.; Grellier, M.; Vendier, L.; Bontemps, S.; Sortais, J.-B.; Sabo-Etienne, S.; Darcel, C. Iron-Catalyzed C-H Borylation of Arenes. J. Am. Chem. Soc. 2015, 137, 4062–4065. [Google Scholar] [CrossRef]
- Ros, A.; Fernández, R.; Lassaleta, J.M. Functional Group Directed C-H Borylation. Chem. Soc. Rev. 2014, 43, 3229–3243. [Google Scholar] [CrossRef]
- Bisht, R.; Halder, C.; Hassan, M.M.M.; Hoque, M.E.; Chaturvedi, J.; Chattopadhya, B. Metal-Catalyzed C-H Bond Activation and Borylation. Chem. Soc. Rev. 2022, 51, 5042–5100. [Google Scholar] [CrossRef] [PubMed]
- Itoh, H.; Kikuchi, T.; Ishiyama, T.; Miyaura, N. Iridium-catalyzed ortho-C–H Borylation of Aryl Ketones with Bis(pinacolato)diboron. Chem. Lett. 2011, 40, 1007–1008. [Google Scholar] [CrossRef]
- Xue, C.; Luo, Y.; Teng, H.; Ma, Y.; Nishiura, M.; Hou, Z. Ortho-Selective C–H Borylation of Aromatic Ethers with Pinacol-borane by Organo Rare-Earth Catalysts. ACS Catal. 2018, 8, 5017–5022. [Google Scholar] [CrossRef]
- Li, H.L.; Kuninobu, Y.; Kanai, M. Lewis Acid–Base Interaction-Controlled ortho-Selective C–H Borylation of Aryl Sulfides. Angew. Chem. Int. Ed. 2017, 56, 1495–1499. [Google Scholar] [CrossRef] [PubMed]
- Li, H.-L.; Kanai, M.; Kuninobu, Y. Iridium/Bipyridine- Catalyzed ortho-Selective C–H Borylation of Phenol and Aniline Derivatives. Org. Lett. 2017, 19, 5944–5947. [Google Scholar] [CrossRef]
- Chattopadhyay, B.; Dannatt, J.E.; Andujar-De Sanctis, I.L.; Gore, K.A.; Maleczka, R.E.; Singleton, D.A.; Smith, M.R. Ir-Catalyzed ortho-Borylation of Phenols Directed by Substrate–Ligand Electrostatic Interactions: A Combined Experimental/in Silico Strategy for Optimizing Weak Interactions. J. Am. Chem. Soc. 2017, 139, 7864–7871. [Google Scholar] [CrossRef] [PubMed]
- Yamazaki, K.; Kawamorita, S.; Ohmiya, H.; Sawamura, M. Directed Ortho Borylation of Phenol Derivatives Catalyzed by a Silica-Supported Iridium Complex. Org. Lett. 2010, 12, 3978–3981. [Google Scholar] [CrossRef] [PubMed]
- Sean, M.; Preshlock, S.M.; Ghaffari, B.; Peter, E.; Maligres, P.E.; Shane, W.; Krska, S.W.; Robert, E.; Maleczka, R.E., Jr.; Milton, R.; et al. High-Throughput Optimization of Ir-Catalyzed C–H Borylation: A Tutorial for Practical Applications. J. Am. Chem. Soc. 2013, 135, 7572–7582. [Google Scholar] [CrossRef] [PubMed]
- Raphael, J.; Oeschger, R.J.; Larsen, M.A.; Bismuto, A.; Hartwig, J.F. Origin of the Difference in Reactivity between Ir Catalysts for the Borylation of C–H Bonds. J. Am. Chem. Soc. 2019, 141, 16479–16485. [Google Scholar] [CrossRef]
- Boller, T.M.; Murphy, J.M.; Marko Hapke, M.; Tatsuo Ishiyama, T.; Miyaura, N.; Hartwig, J.F. Mechanism of the Mild Functionalization of Arenes by Diboron Reagents Catalyzed by Iridium Complexes. Intermediacy and Chemistry of Bipyridine-Ligated Iridium Trisboryl Complexes. J. Am. Chem. Soc. 2005, 127, 14263–14278. [Google Scholar] [CrossRef]
- Ishiyama, T.; Jun Takagi, J.; Kousaku Ishida, K.; Norio Miyaura, N.; Anastasi, N.R.; Hartwig, J.E. Mild Iridium-Catalyzed Borylation of Arenes. High Turnover Numbers, Room Temperature Reactions, and Isolation of a Potential Intermediate. J. Am. Chem. Soc. 2002, 124, 390–391. [Google Scholar] [CrossRef] [PubMed]
- Ochida, A.; Hara, K.; Ito, H.; Sawamura, M. Nonvolatile Me3P-like P-Donor Ligand: Synthesis and Properties of 4-Phenyl-1-phospha-4-silabicyclo[2.2.2]octane. Org. Lett. 2003, 5, 2671–2674. [Google Scholar] [CrossRef] [PubMed]
- Ochida, A.; Ito, S.; Miyahara, T.; Ito, H.; Sawamura, M. Electronically Tunable Compact Trialkylphosphines: SMAPs-bridged Bicyclic Phosphines. Chem. Lett. 2006, 35, 294–295. [Google Scholar] [CrossRef]
- Kawamorita, S.; Ohmiya, H.; Hara, K.; Fukuoka, A.; Sawamura, M. Directed Ortho Borylation of Functionalized Arenes Catalyzed by a Silica-Supported Compact Phosphine–Iridium System. J. Am. Chem. Soc. 2009, 131, 5058–5059. [Google Scholar] [CrossRef] [PubMed]
- Kawamorita, S.; Ohmiya, H.; Sawamura, M. Ester-Directed Regioselective Borylation of Heteroarenes Catalyzed by a Silica-Supported Iridium Complex. J. Org. Chem. 2010, 75, 3855–3858. [Google Scholar] [CrossRef] [PubMed]
- Kawamorita, S.; Murakami, R.; Iwai, T.; Sawamura, M. Synthesis of Primary and Secondary Alkylboronates through Site-Selective C(sp3)–H Activation with Silica-Supported Monophosphine–Ir Catalysts. J. Am. Chem. Soc. 2013, 135, 2947–2950. [Google Scholar] [CrossRef] [PubMed]
- Konishi, S.; Kawamorita, S.; Iwai, T.; Steel, P.G.; Marder, T.B.; Sawamura, M. Site-Selective C–H Borylation of Quinolines at the C8 Position Catalyzed by a Silica-Supported Phosphane–Iridium System. Chem. Asian. J. 2014, 9, 434–438. [Google Scholar] [CrossRef]
- Roosen, P.C.; Kallepalli, V.A.; Chattopadhyay, B.; Singleton, D.A.; Maleczka, R.E., Jr.; Smith, M.R., III. Outer-Sphere Direction in Iridium C–H Borylation. J. Am. Chem. Soc. 2012, 134, 11350–11353. [Google Scholar] [CrossRef] [PubMed]
- Preshlock, S.M.; Plattner, D.L.; Maligres, P.E.; Krska, S.W.; Maleczka, R.E., Jr.; Smith, M.R., III. A Traceless Directing Group for C-H Borylation. Angew. Chem. Int. Ed. 2013, 52, 12915–12919. [Google Scholar] [CrossRef]
- Bai, S.-T.; Bheeter, C.B.; Reek, J.N.H. Hydrogen Bond Directed ortho-Selective C-H Borylation of Secondary Aromatic Amides. Angew. Chem. Int. Ed. 2019, 58, 13039–13043. [Google Scholar] [CrossRef]
- Hoque, M.M.; Hassan, M.M.M.; Chattopadhyay, B. Remarkably Efficient Iridium Catalysts for Directed C(sp2)-H and C(sp3)-H Borylation of Diverse Classes of Substrates. J. Am. Chem. Soc. 2021, 143, 5022–5037. [Google Scholar] [CrossRef] [PubMed]
- Marcos-Atanes, D.; Vidal, C.; Navo, C.D.; Peccati, F.; Jiménez-Osés, G.; Mascareñas, J.L. Iridium-Catalyzed ortho-Selective Borylation of Aromatic Amides Enabled by 5-Trifluoromethylated Bipyridine Ligands. Angew. Chem. Int. Ed. 2023, 62, e202214510. [Google Scholar] [CrossRef]
- Roering, A.J.; Hale, L.V.A.; Squier, P.A.; Ringgold, M.A.; Wiederspan, E.R.; Clark, T.B. Iridium-Catalyzed, Substrate-Directed C-H Borylation Reactions of Benzylic Amines. Org. Lett. 2012, 14, 3558–3561. [Google Scholar] [CrossRef] [PubMed]
- Zou, X.; Zhao, H.; Li, Y.; Gao, Q.; Ke, Z.; Senmiao Xu, S. Chiral Bidentate Boryl Ligand Enabled Iridium-Catalyzed Asymmetric C(sp2)–H Borylation of Diarylmethylamines. J. Am. Chem. Soc. 2019, 141, 5334–5342. [Google Scholar] [CrossRef] [PubMed]
- Ros, A.; López-Rodríguez, R.; Estepa, B.; Álvarez, E.; Fernández, R.; Lassaletta, J.M. Hydrazone as the Directing Group for Ir-Catalyzed Arene Diborylations and Sequential Functionalizations. J. Am. Chem. Soc. 2012, 134, 4573–4576. [Google Scholar] [CrossRef]
- Mao, S.; Yuan, B.; Wang, X.; Zhao, Y.; Wang, L.; Yang, X.-Y.; Chen, Y.-M.; Zhang, S.-Q.; Li, P. Triazene as the Directing Group Achieving Highly Ortho-Selective Diborylation and Sequential Functionalization. Org. Lett. 2022, 24, 3594–3598. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions, and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions, or products referred to in the content. |





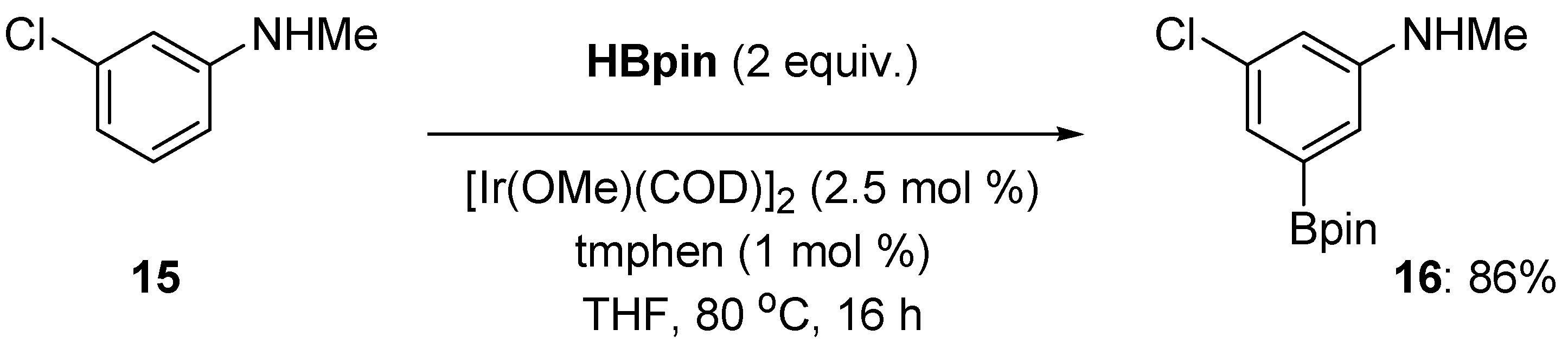
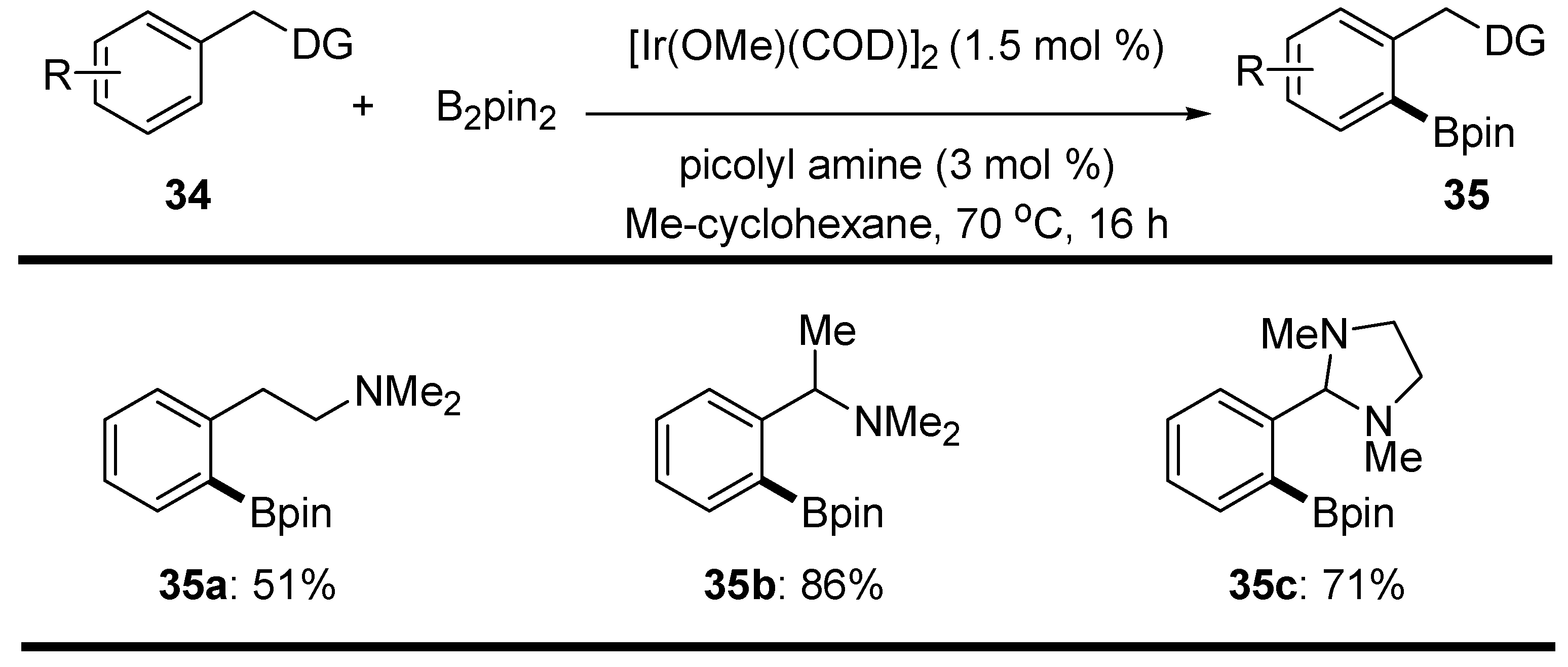


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Al Mamari, H.H. Ir-Catalyzed ortho-C-H Borylation of Aromatic C(sp2)-H Bonds of Carbocyclic Compounds Assisted by N-Bearing Directing Groups. Reactions 2024, 5, 318-337. https://doi.org/10.3390/reactions5020016
Al Mamari HH. Ir-Catalyzed ortho-C-H Borylation of Aromatic C(sp2)-H Bonds of Carbocyclic Compounds Assisted by N-Bearing Directing Groups. Reactions. 2024; 5(2):318-337. https://doi.org/10.3390/reactions5020016
Chicago/Turabian StyleAl Mamari, Hamad H. 2024. "Ir-Catalyzed ortho-C-H Borylation of Aromatic C(sp2)-H Bonds of Carbocyclic Compounds Assisted by N-Bearing Directing Groups" Reactions 5, no. 2: 318-337. https://doi.org/10.3390/reactions5020016
APA StyleAl Mamari, H. H. (2024). Ir-Catalyzed ortho-C-H Borylation of Aromatic C(sp2)-H Bonds of Carbocyclic Compounds Assisted by N-Bearing Directing Groups. Reactions, 5(2), 318-337. https://doi.org/10.3390/reactions5020016