Condensed Phase Guerbet Reactions of Ethanol/Isoamyl Alcohol Mixtures


Abstract
1. Introduction
2. Materials and Methods
2.1. Materials and Catalyst Preparation
2.2. Reactor Systems
2.2.1. Batch Reactor Experiments
2.2.2. Continuous Reactor Experiments
2.3. Analytical Methods
3. Results
3.1. Experimental Results
3.1.1. Batch Experiments
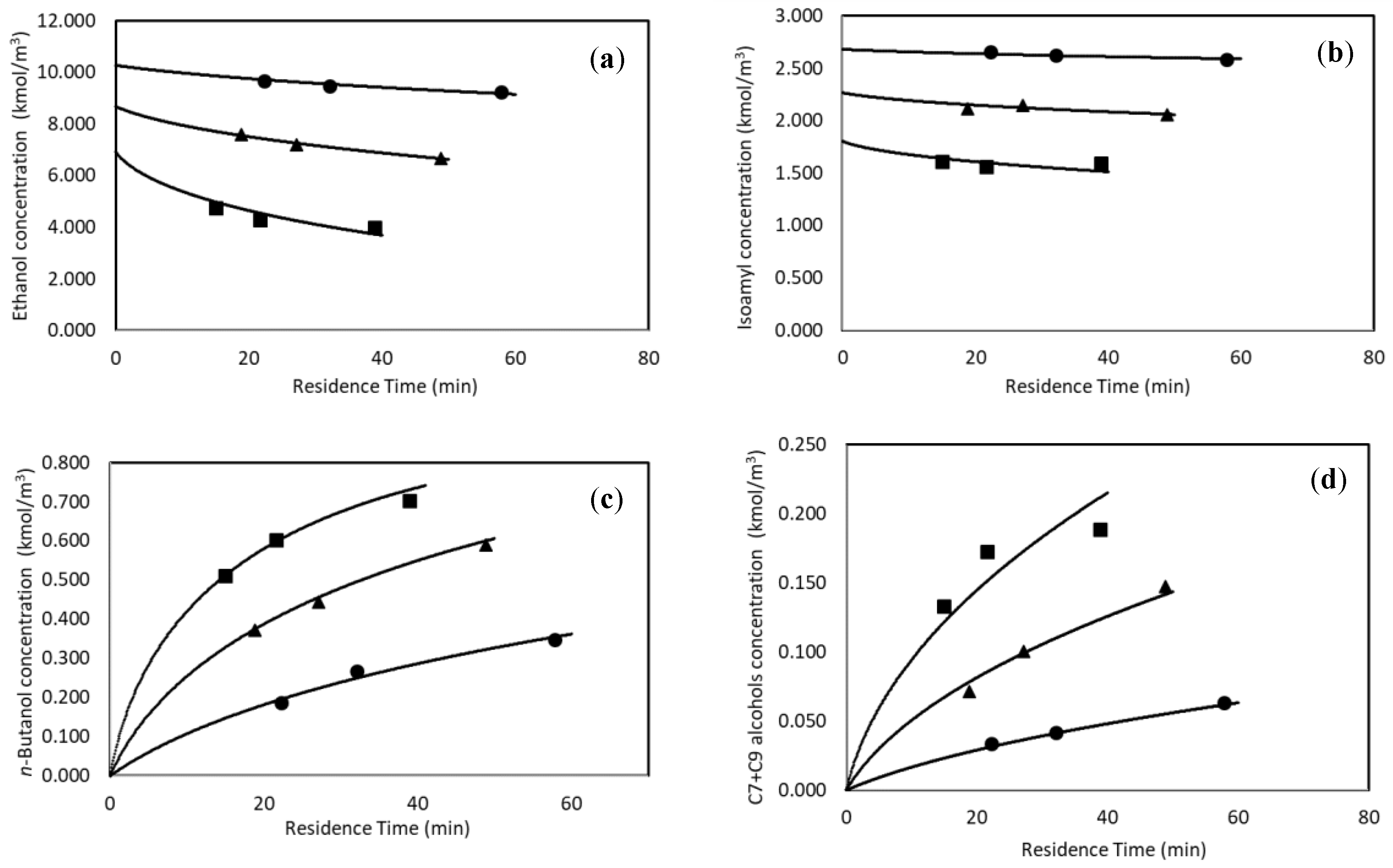
3.1.2. Continuous Experiments
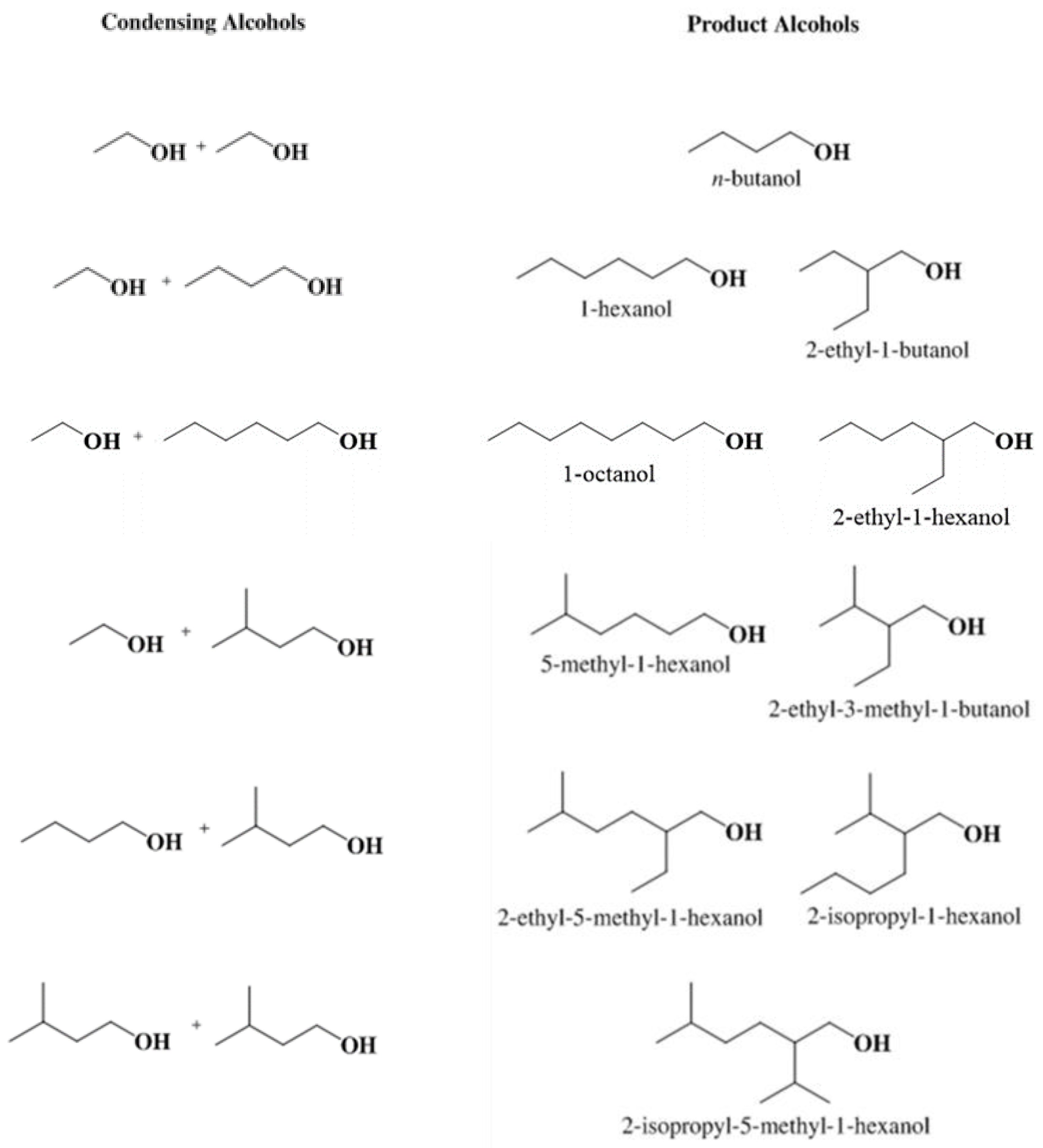
3.2. Kinetic Model Development
3.2.1. Continuous Reactor Modeling
3.2.2. Rate Constant Determination from Continuous Reactor Data
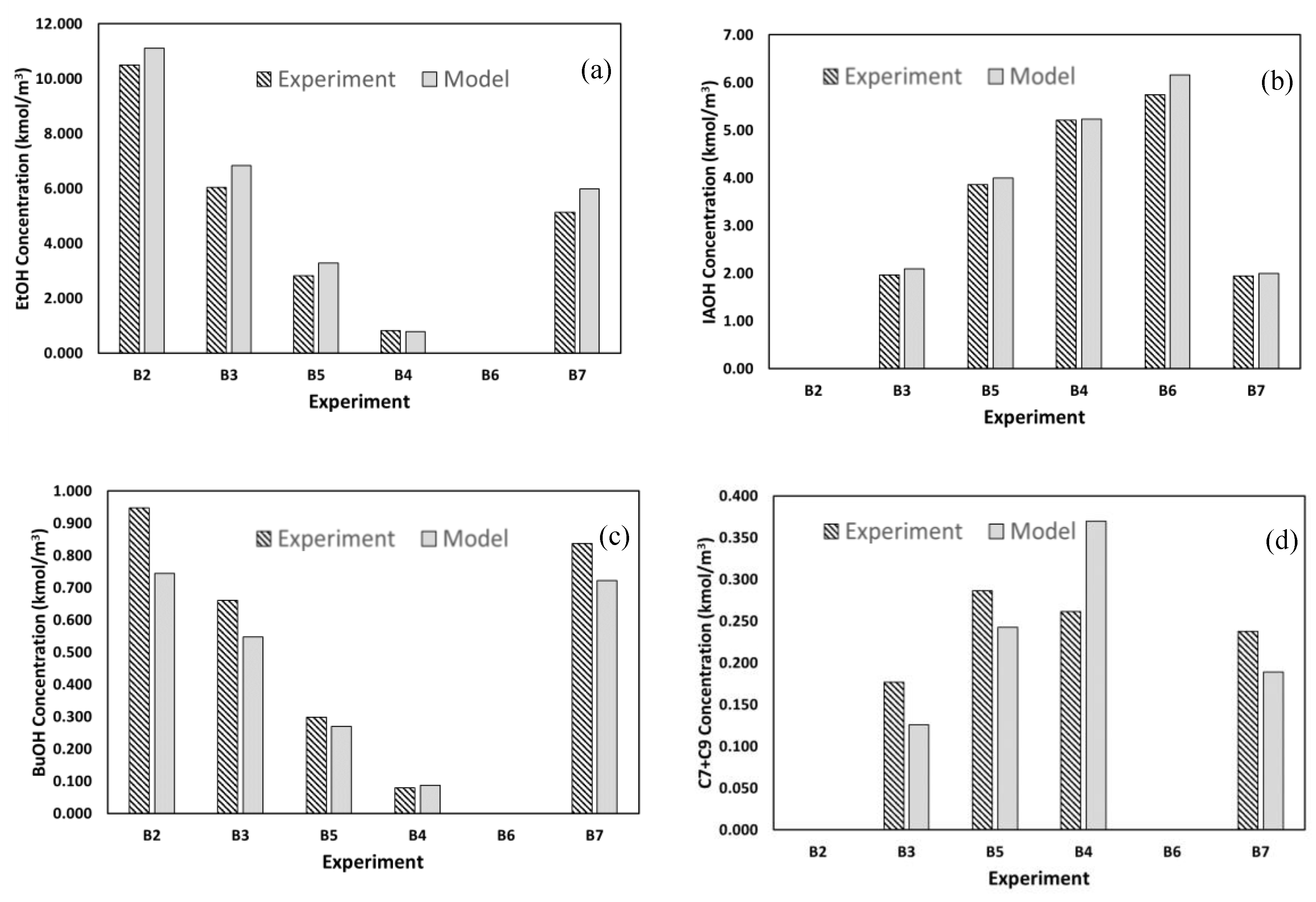
3.2.3. Comparison of Batch Experimental Data with Kinetic Model Simulation
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Al Abdallah, Q.; Nixon, B.T.; Fortwendel, J.R. The enzymatic conversion of major algal and cyanobacterial carbohydrates to bioethanol. Front. Energy Res. 2016, 4, 36. [Google Scholar] [CrossRef]
- Bergmann, J.C.; Trichez, D.; Sallet, L.P.; de Paula e Silva, F.C.; Almeida, J.R.M. Technological Advancements in 1G Ethanol Production and Recovery of By-Products Based on the Biorefinery Concept. In Advances in Sugarcane Biorefinery; Chandel, A.K., Luciano Silveira, M.H., Eds.; Elsevier: Amsterdam, The Netherlands, 2018; Chapter 4; pp. 73–95. [Google Scholar] [CrossRef]
- Hazelwood, L.A.; Daran, J.-M.; Van Maris, A.J.; Pronk, J.T.; Dickinson, J.R. The Ehrlich pathway for fusel alcohol production: A century of research on Saccharomyces cerevisiae metabolism. Appl. Environ. Microbiol. 2008, 74, 2259–2266. [Google Scholar] [CrossRef] [PubMed]
- Tesfaw, A.; Assefa, F. Current trends in bioethanol production by Saccharomyces cerevisiae: Substrate, inhibitor reduction, growth variables, coculture, and immobilization. Int. Sch. Res. Not. 2014, 2014, 532852. [Google Scholar] [CrossRef] [PubMed]
- Boumba, V.A.; Ziavrou, K.S.; Vougiouklakis, T. Biochemical pathways generating post-mortem volatile compounds co-detected during forensic ethanol analyses. Forensic Sci. Int. 2008, 174, 133–151. [Google Scholar] [CrossRef]
- Tran, T.T.V.; Kongparakul, S.; Karnjanakom, S.; Reubroycharoen, P.; Guan, G.; Chanlek, N.; Samart, C. Selective production of green solvent (isoamyl acetate) from fusel oil using a sulfonic acid-functionalized KIT-6 catalyst. Mol. Catal. 2019, 110724. [Google Scholar] [CrossRef]
- Ehrlich, F. Über die Bedingungen der Fuselölbildung und über ihren Zusammenhang mit dem Eiweissaufbau der Hefe. Ber. Dtsch. Chem. Ges. 1907, 40, 1027–1047. [Google Scholar] [CrossRef]
- Jackson, R.S. 7—Fermentation. In Wine Science, 3rd ed.; Jackson, R.S., Ed.; Academic Press: San Diego, CA, USA, 2008; pp. 332–417. [Google Scholar] [CrossRef]
- Jackson, R.S. Qualitative Wine Assessment. In Wine Tasting, 3rd ed.; Jackson, R.S., Ed.; Academic Press: San Diego, CA, USA, 2017; Chapter 6; pp. 253–291. [Google Scholar] [CrossRef]
- Vilanova, M.; Pretorius, I.S.; Henschke, P.A. Influence of Diammonium Phosphate Addition to Fermentation on Wine Biologicals. In Processing and Impact on Active Components in Food; Preedy, V., Ed.; Academic Press: San Diego, CA, USA, 2015; Chapter 58; pp. 483–491. [Google Scholar] [CrossRef]
- Marullo, P.; Dubourdieu, D. Yeast selection for wine flavour modulation. In Managing Wine Quality: Oenology and Wine Quality; Elsevier: Amsterdam, The Netherlands, 2010; pp. 293–345. [Google Scholar] [CrossRef]
- Koonthongkaew, J.; Toyokawa, Y.; Ohashi, M.; Large, C.R.L.; Dunham, M.J.; Takagi, H. Effect of the Ala234Asp replacement in mitochondrial branched-chain amino acid aminotransferase on the production of BCAAs and fusel alcohols in yeast. Appl. Microbiol. Biotechnol. 2020, 104, 7915–7925. [Google Scholar] [CrossRef]
- dos Santos, P.; Meireles, M.A.A.; Martínez, J. Production of isoamyl acetate by enzymatic reactions in batch and packed bed reactors with supercritical CO2. J. Supercrit. Fluids 2017, 127, 71–80. [Google Scholar] [CrossRef]
- Branduardi, P.; Porro, D. n-butanol: Challenges and solutions for shifting natural metabolic pathways into a viable microbial production. FEMS Microbiol. Lett. 2016, 363, 1–7. [Google Scholar] [CrossRef]
- Kang, A.; Lee, T.S. Secondary Metabolism for Isoprenoid-based Biofuels. In Biotechnology for Biofuel Production and Optimization; Eckert, C.A., Trinh, C.T., Eds.; Elsevier: Amsterdam, The Netherlands, 2016; Chapter 2; pp. 35–71. [Google Scholar] [CrossRef]
- Bessa, L.C.B.A.; Robustillo, M.D.; Silva, E.C.D.; Tadini, C.C.; Meirelles, A.J.D.A.; Pessôa Filho, P.D.A. Influence of Additives (Isoamyl Laurate or Isoamyl Nonanoate) in the Solid–Liquid Equilibrium of Fatty Acid Ethyl Esters. J. Chem. Eng. Data 2019, 64, 2062–2074. [Google Scholar] [CrossRef]
- Safieddin Ardebili, S.M.; Solmaz, H.; İpci, D.; Calam, A.; Mostafaei, M. A review on higher alcohol of fusel oil as a renewable fuel for internal combustion engines: Applications, challenges, and global potential. Fuel 2020, 279, 118516. [Google Scholar] [CrossRef]
- Ağbulut, Ü.; Sarıdemir, S.; Karagöz, M. Experimental investigation of fusel oil (isoamyl alcohol) and diesel blends in a CI engine. Fuel 2020, 267, 117042. [Google Scholar] [CrossRef]
- Weber, C.; Farwick, A.; Benisch, F.; Brat, D.; Dietz, H.; Subtil, T.; Boles, E. Trends and challenges in the microbial production of lignocellulosic bioalcohol fuels. Appl. Microbiol. Biotechnol. 2010, 87, 1303–1315. [Google Scholar] [CrossRef] [PubMed]
- Mohsenzadeh, A.; Zamani, A.; Taherzadeh, M.J. Bioethylene Production from Ethanol: A Review and Techno-economical Evaluation. ChemBioEng Rev. 2017, 4, 75–91. [Google Scholar] [CrossRef]
- Xiang, N.; Xu, P.; Ran, N.; Ye, T. Production of acetic acid from ethanol over CuCr catalysts via dehydrogenation-(aldehyde–water shift) reaction. RSC Adv. 2017, 7, 38586–38593. [Google Scholar] [CrossRef]
- Nezam, I.; Peereboom, L.; Miller, D.J. Continuous condensed-phase ethanol conversion to higher alcohols: Experimental results and techno-economic analysis. J. Clean. Prod. 2019, 209, 1365–1375. [Google Scholar] [CrossRef]
- Gabriëls, D.; Hernández, W.Y.; Sels, B.; Van Der Voort, P.; Verberckmoes, A. Review of catalytic systems and thermodynamics for the Guerbet condensation reaction and challenges for biomass valorization. Catal. Sci. Technol. 2015, 5, 3876–3902. [Google Scholar] [CrossRef]
- Wu, X.; Fang, G.; Tong, Y.; Jiang, D.; Liang, Z.; Leng, W.; Liu, L.; Tu, P.; Wang, H.; Ni, J.; et al. Catalytic Upgrading of Ethanol to n-Butanol: Progress in Catalyst Development. ChemSusChem 2018, 11, 71–85. [Google Scholar] [CrossRef]
- Cheng, F.; Guo, H.; Cui, J.; Hou, B.; Xi, H.; Jia, L.; Li, D. Coupling of methanol and ethanol over CuMgAlOx catalysts: The roles of copper species and alkalinity. React. Kinet. Mech. Catal. 2019, 126, 119–136. [Google Scholar] [CrossRef]
- Benito, P.; Vaccari, A.; Antonetti, C.; Licursi, D.; Schiarioli, N.; Rodriguez-Castellón, E.; Raspolli Galletti, A.M. Tunable copper-hydrotalcite derived mixed oxides for sustainable ethanol condensation to n-butanol in liquid phase. J. Clean. Prod. 2019, 209, 1614–1623. [Google Scholar] [CrossRef]
- Li, S.; Zhu, X.; An, H.; Zhao, X.; Wang, Y. Ethanol Guerbet Condensation to n-Butanol or C4-C8 Alcohols over Ni/TiO2 Catalyst. ChemistrySelect 2020, 5, 8669–8673. [Google Scholar] [CrossRef]
- Han, X.; Li, S.; Zhu, X.; An, H.; Zhao, X.; Wang, Y. Influence of noble metals on the catalytic performance of Ni/TiO2 for Ethanol Guerbet condensation. React. Kinet. Mech. Catal. 2020. [Google Scholar] [CrossRef]
- Matsu-ura, T.; Sakaguchi, S.; Obora, Y.; Ishii, Y. Guerbet Reaction of Primary Alcohols Leading to β-Alkylated Dimer Alcohols Catalyzed by Iridium Complexes. J. Org. Chem. 2006, 71, 8306–8308. [Google Scholar] [CrossRef] [PubMed]
- Busch, S.; Fleute-Schlachter, I.; Mack, S.; Mahnke, E.U.; Wick, A. Biocide Compositions Comprising Alkoxylation Products of Isoamyl Alcohol Derivatives. U.S. Patent Application No. 20130252982, 22 November 2016. [Google Scholar]
- Nezam, I.; Zak, J.; Miller, D.J. Condensed-Phase Ethanol Conversion to Higher Alcohols over Bimetallic Catalysts. Ind. Eng. Chem. Res. 2020, 59, 13906–13915. [Google Scholar] [CrossRef]
- Jordison, T.L.; Lira, C.T.; Miller, D.J. Condensed-Phase Ethanol Conversion to Higher Alcohols. Ind. Eng. Chem. Res. 2015, 54, 10991–11000. [Google Scholar] [CrossRef]
- Jordison, T.L.; Peereboom, L.; Miller, D.J. Impact of Water on Condensed Phase Ethanol Guerbet Reactions. Ind. Eng. Chem. Res. 2016, 55, 6579–6585. [Google Scholar] [CrossRef]
- Nezam, I.; Peereboom, L.; Miller, D.J. Enhanced Acrylate Production from 2-Acetoxypropanoic Acid Esters. Org. Proc. Res. Dev. 2017, 21, 715–719. [Google Scholar] [CrossRef]
- Vlasenko, N.V.; Kyriienko, P.I.; Valihura, K.V.; Kosmambetova, G.R.; Soloviev, S.O.; Strizhak, P.E. Yttria-Stabilized Zirconia as a High-Performance Catalyst for Ethanol to n-Butanol Guerbet Coupling. ACS Omega 2019, 21469–21476. [Google Scholar] [CrossRef]
- Xi, X.-Y.; Sun, Z.-H.; Cao, H.-T.; Pei, Y.-T.; ten Brink, G.H.; Deuss, P.J.; Barta, K.; Heeres, H.J. Catalyst Performance Studies on the Guerbet Reaction in a Continuous Flow Reactor Using Mono-and Bi-Metallic Cu-Ni Porous Metal Oxides. Catalysts 2020, 10, 996. [Google Scholar] [CrossRef]
- Neumann, C.N.; Rozeveld, S.J.; Yu, M.; Rieth, A.J.; Dincă, M. Metal–Organic Framework-Derived Guerbet Catalyst Effectively Differentiates between Ethanol and Butanol. J. Am. Chem. Soc. 2019, 141, 17477–17481. [Google Scholar] [CrossRef]
- Larina, O.V.; Valihura, K.V.; Kyriienko, P.I.; Vlasenko, N.V.; Balakin, D.Y.; Khalakhan, I.; Čendak, T.; Soloviev, S.O.; Orlyk, S.M. Successive vapour phase Guerbet condensation of ethanol and 1-butanol over Mg-Al oxide catalysts in a flow reactor. Appl. Catal. A Gen. 2019, 588, 117265. [Google Scholar] [CrossRef]
- Zakumbaeva, G.D.; Omashev, K.G.; Khan, C. Influence of water on the heat of hydrogen adsorption on nickel. React. Kinet. Catal. Lett. 1977, 6, 363–369. [Google Scholar] [CrossRef]
- Renouprez, A.J.; Fouilloux, P.; Candy, J.P.; Tomkinson, J. Chemisorption of water on nickel surfaces. Surf. Sci. 1979, 83, 285–295. [Google Scholar] [CrossRef]
- Rahman, M.M.; Davidson, S.D.; Sun, J.; Wang, Y. Effect of Water on Ethanol Conversion over ZnO. Top. Catal. 2016, 59, 37–45. [Google Scholar] [CrossRef]
- Cabello González, G.M.; Concepción, P.; Villanueva Perales, A.L.; Martínez, A.; Campoy, M.; Vidal-Barrero, F. Ethanol conversion into 1,3-butadiene over a mixed Hf-Zn catalyst: Effect of reaction conditions and water content in ethanol. Fuel Process. Technol. 2019, 193, 263–272. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Exp. a | Initial Molar Ratio EtOH/IAOH | Conversion (%) | Selectivity to Higher Alcohols (%) | Selectivity to Liquid Byproducts (%) | Selectivity to Gas Byproducts (%) | Overall Carbon Recovery (%) | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| EtOH | IAOH | EtOH Products | Cross Products | IAOH Prod. | ||||||||||
| w.r.t. EtOH | w.r.t. IAOH | |||||||||||||
| C4 | C6 | C8 | C7 | C9 | C7 | C9 | C10 | |||||||
| B2 | 100/0 | 21.1 | - | 67.5 | 22.0 | 5.0 | - | - | - | - | - | 3.4 | 2.1 | 105.3 |
| B3 | 79/21 | 30.1 | 13.7 | 50.8 | 15.5 | 3.3 | 6.0 | 0.0 | 50.1 | 6.8 | 0.0 | 26.9 | 2.2 | 96.7 |
| B5 | 50/50 | 35.2 | 11.4 | 39.0 | 9.6 | 3.0 | 16.8 | 0.3 | 52.1 | 5.6 | 0.9 | 33.8 | 2.6 | 100.8 |
| B4 | 20/80 | 43.3 | 9.7 | 25.5 | 3.2 | 8.6 | 39.5 | 2.9 | 43.8 | 2.9 | 3.2 | 39.2 | 3.5 | 102.9 |
| B6 | 0/100 | - | 11.1 | - | - | - | - | - | - | - | 4.8 | 86.9 | 8.3 | 97.3 |
| B7 | 79/21 | 40.5 | 14.6 | 47.9 | 15.9 | 3.6 | 5.9 | 0.1 | 62.9 | 8.7 | 0.6 | 25.7 | 1.2 | 101.0 |
| Conditions | Conversion (%) | Selectivity to Higher Alcohols (%) | Selectivity to Liquid Byproducts (%) | Selectivity to Gas Byproducts (%) | Overall Carbon Recovery (%) | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| EtOH Products | EtOH + IAOH Cross Products | IAOH Products | |||||||||||||
| w.r.t. EtOH | w.r.t. IAOH | ||||||||||||||
| T (°C) | WHSV a | Τ b | EtOH | IAOH | C4 | C6 | C8 | C7 | C9 | C7 | C9 | C10 | |||
| 210 | 0.8 | 0.96 | 10.2 | 3.7 | 65.7 | 11.6 | 1.4 | 5.1 | 0.9 | 52.8 | 5.2 | 0.0 | 12.2 | 6.0 | 104.2 |
| 1.4 | 0.54 | 8.0 | 2.3 | 64.9 | 8.7 | 0.7 | 4.4 | 0.6 | 62.7 | 4.6 | 0.0 | 11.9 | 8.5 | 104.7 | |
| 2.1 | 0.37 | 5.7 | 1.5 | 62.7 | 6.4 | 0.2 | 3.8 | 0.5 | 59.0 | 3.8 | 0.0 | 10.2 | 10.1 | 105.1 | |
| 230 | 0.8 | 0.82 | 23.6 | 9.6 | 57.6 | 14.3 | 2.6 | 6.2 | 1.4 | 60.8 | 6.8 | 0.3 | 12.4 | 8.2 | 96.8 |
| 1.4 | 0.45 | 17.2 | 5.5 | 59.1 | 12.2 | 1.8 | 5.9 | 1.1 | 73.6 | 7.2 | 0.2 | 11.6 | 8.9 | 101.0 | |
| 2.1 | 0.31 | 12.8 | 7.0 | 66.5 | 12.3 | 1.7 | 5.6 | 1.1 | 41.1 | 3.9 | 0.0 | 10.3 | 10.2 | 103.7 | |
| 250 | 0.8 | 0.65 | 42.6 | 12.1 | 47.6 | 13.7 | 2.8 | 5.4 | 1.4 | 76.6 | 9.6 | 0.5 | 9.6 | 14.8 | 93.7 |
| 1.4 | 0.36 | 38.2 | 13.9 | 45.5 | 14.2 | 3.2 | 5.5 | 1.5 | 60.6 | 8.3 | 0.4 | 14.9 | 14.8 | 97.3 | |
| 2.1 | 0.25 | 31.2 | 10.9 | 47.4 | 13.0 | 2.6 | 5.2 | 1.3 | 59.5 | 7.6 | 0.2 | 13.6 | 17.5 | 101.2 | |
| unit | i | 1 | 2 | 3 | 4 | 5 | 6 | KW | |
|---|---|---|---|---|---|---|---|---|---|
| Ea,i | (kJ/mol) | 127.7 | 140.2 | 176.0 | 82.53 | 167.2 | 128.0 | −7.2 * | |
| ln(ko,i) | (m6/kg cat/kmol/h) | 22.48 | 25.06 | 27.36 | 13.01 | 31.42 | 21.22 | −0.67 + | |
| ki (230 °C) | (m6/kg cat/kmol/h) | 3.18 × 10−4 | 2.10 × 10−4 | 4.00 × 10−7 | 1.20 × 10−3 | 1.91 × 10−4 | 8.45 × 10−5 | 2.9 + |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nezam, I.; Peereboom, L.; Miller, D.J. Condensed Phase Guerbet Reactions of Ethanol/Isoamyl Alcohol Mixtures. Reactions 2020, 1, 102-114. https://doi.org/10.3390/reactions1020009
Nezam I, Peereboom L, Miller DJ. Condensed Phase Guerbet Reactions of Ethanol/Isoamyl Alcohol Mixtures. Reactions. 2020; 1(2):102-114. https://doi.org/10.3390/reactions1020009
Chicago/Turabian StyleNezam, Iman, Lars Peereboom, and Dennis J. Miller. 2020. "Condensed Phase Guerbet Reactions of Ethanol/Isoamyl Alcohol Mixtures" Reactions 1, no. 2: 102-114. https://doi.org/10.3390/reactions1020009
APA StyleNezam, I., Peereboom, L., & Miller, D. J. (2020). Condensed Phase Guerbet Reactions of Ethanol/Isoamyl Alcohol Mixtures. Reactions, 1(2), 102-114. https://doi.org/10.3390/reactions1020009