Heterogeneously Catalyzed γ-Valerolactone Hydrogenation into 1,4-Pentanediol in Milder Reaction Conditions

 ,
, 
 ,
, 
Abstract
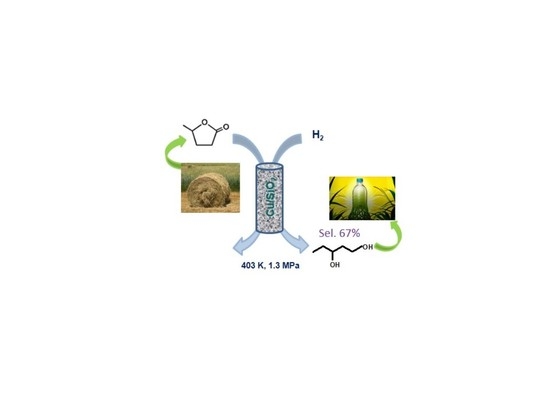
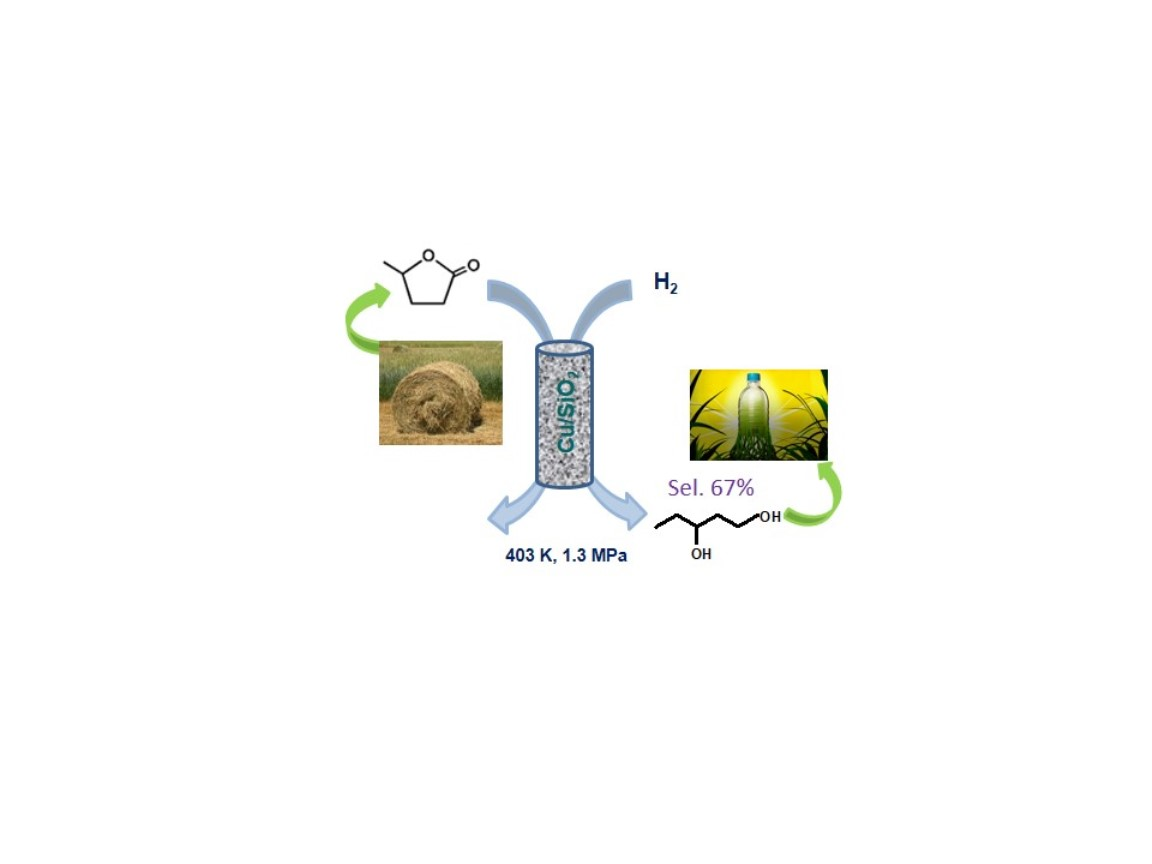
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Catalyst Preparation
2.3. Catalytic Experiments
2.4. Product Analysis
2.5. Catalysts Characterization
2.6. Copper Surface Determination
3. Results and Discussion
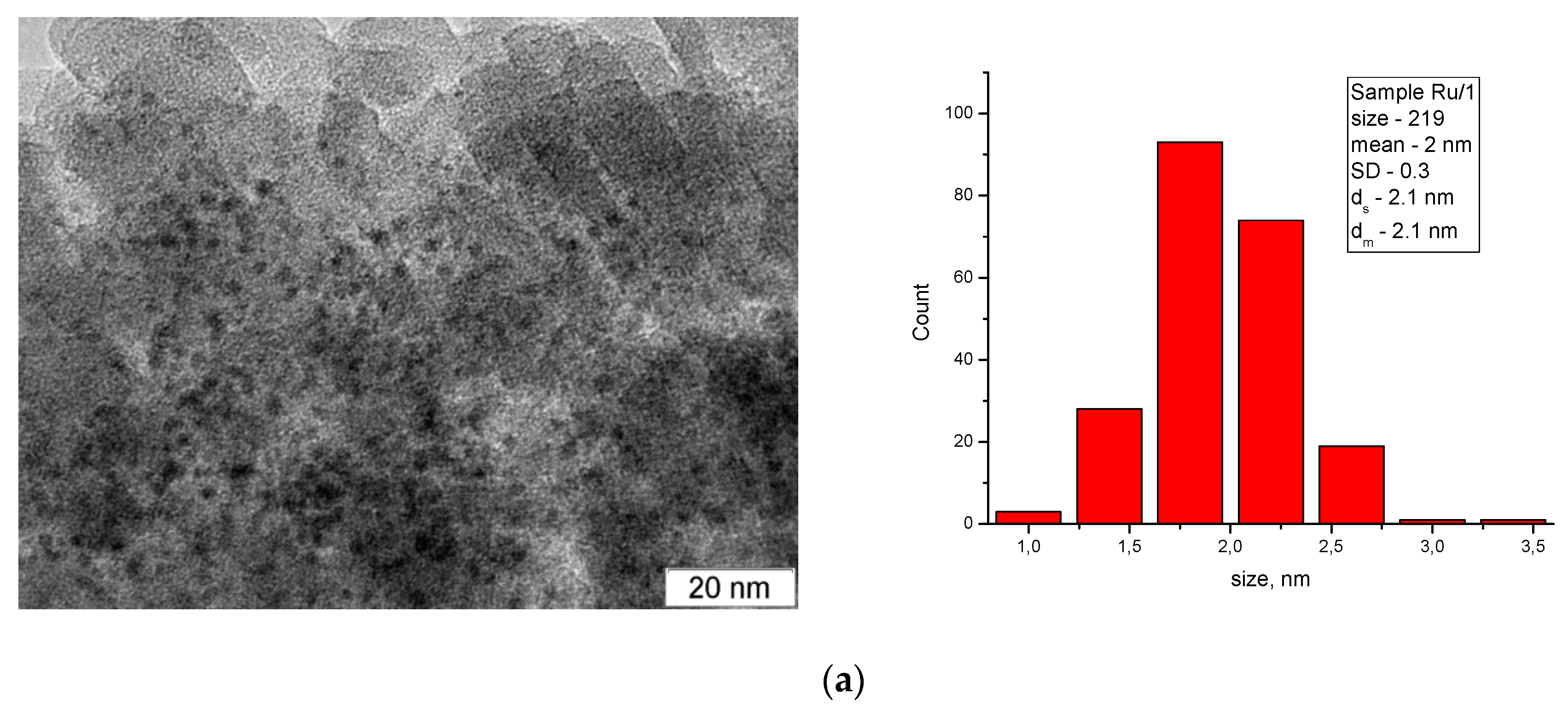
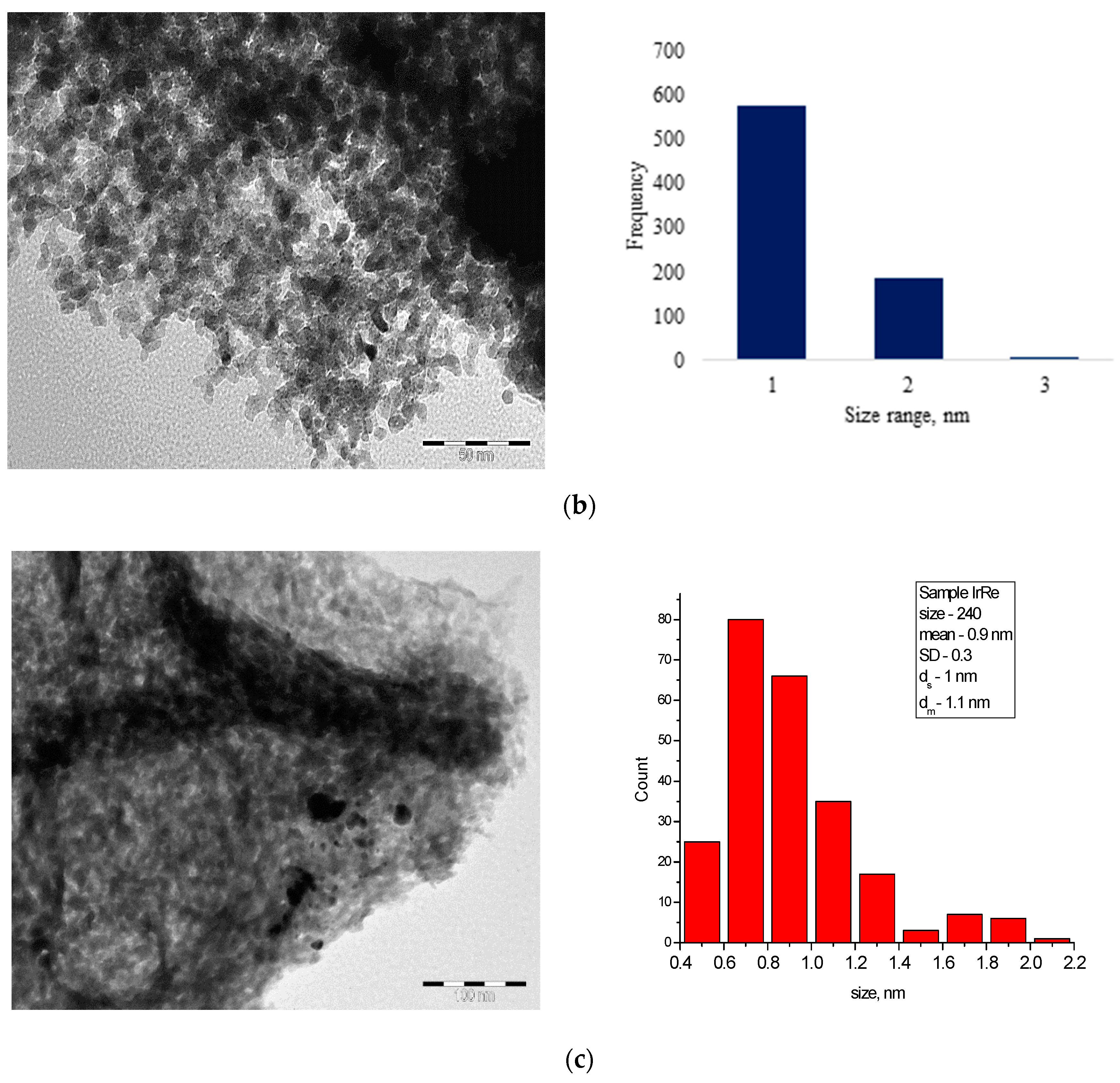
3.1. Characterization of Catalysts
3.2. Preliminary Catalyst Screening
3.3. Continuous Hydrogenation
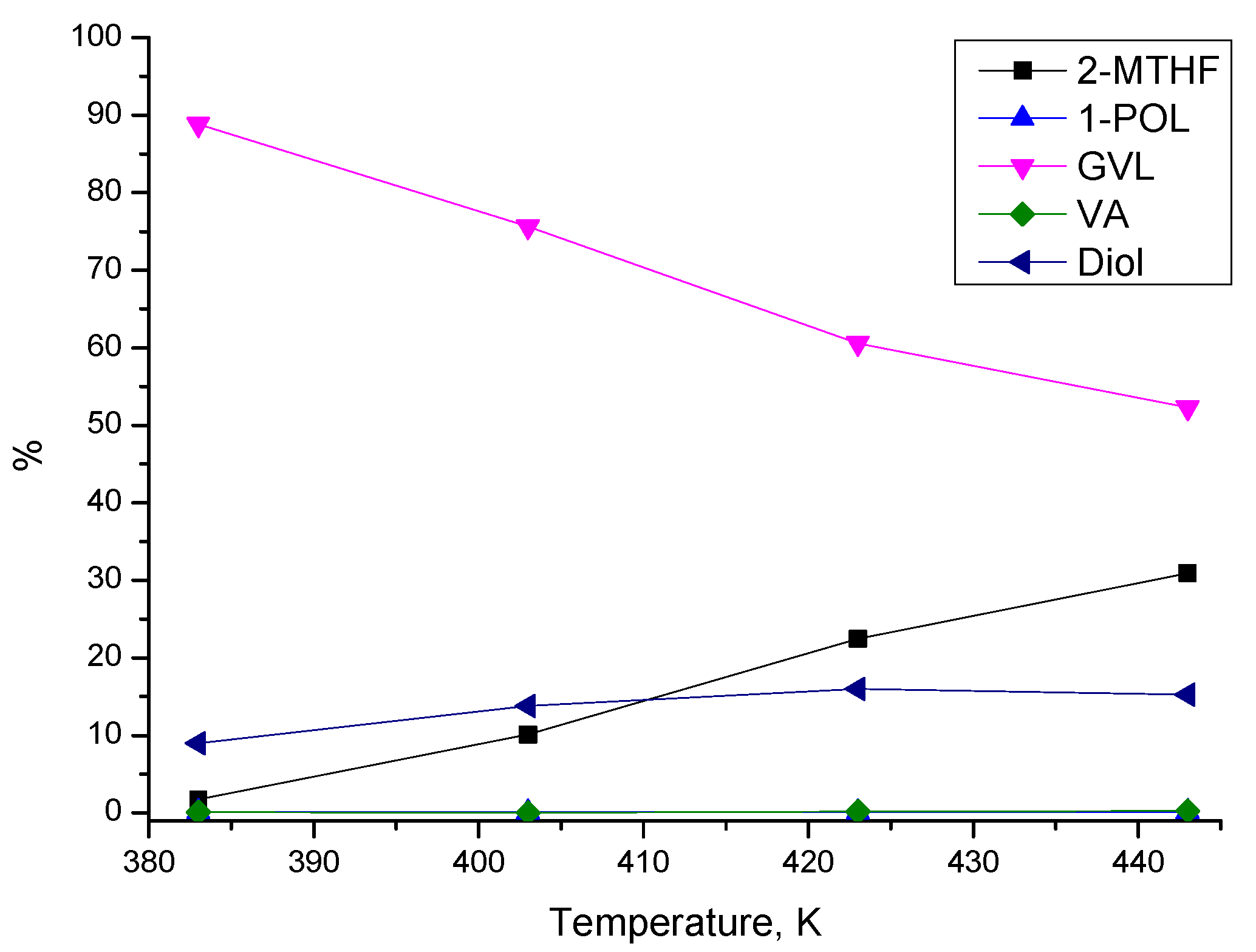
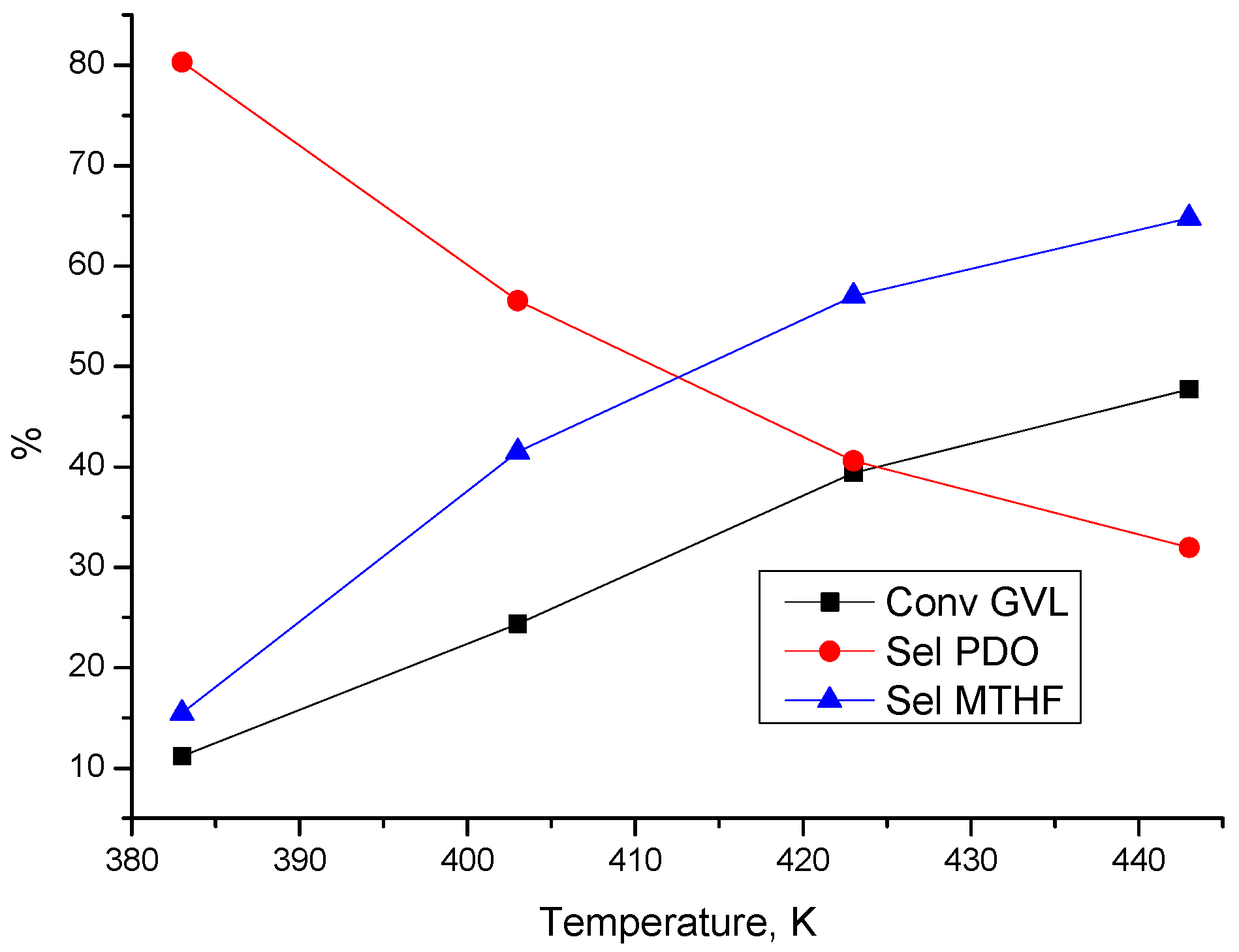
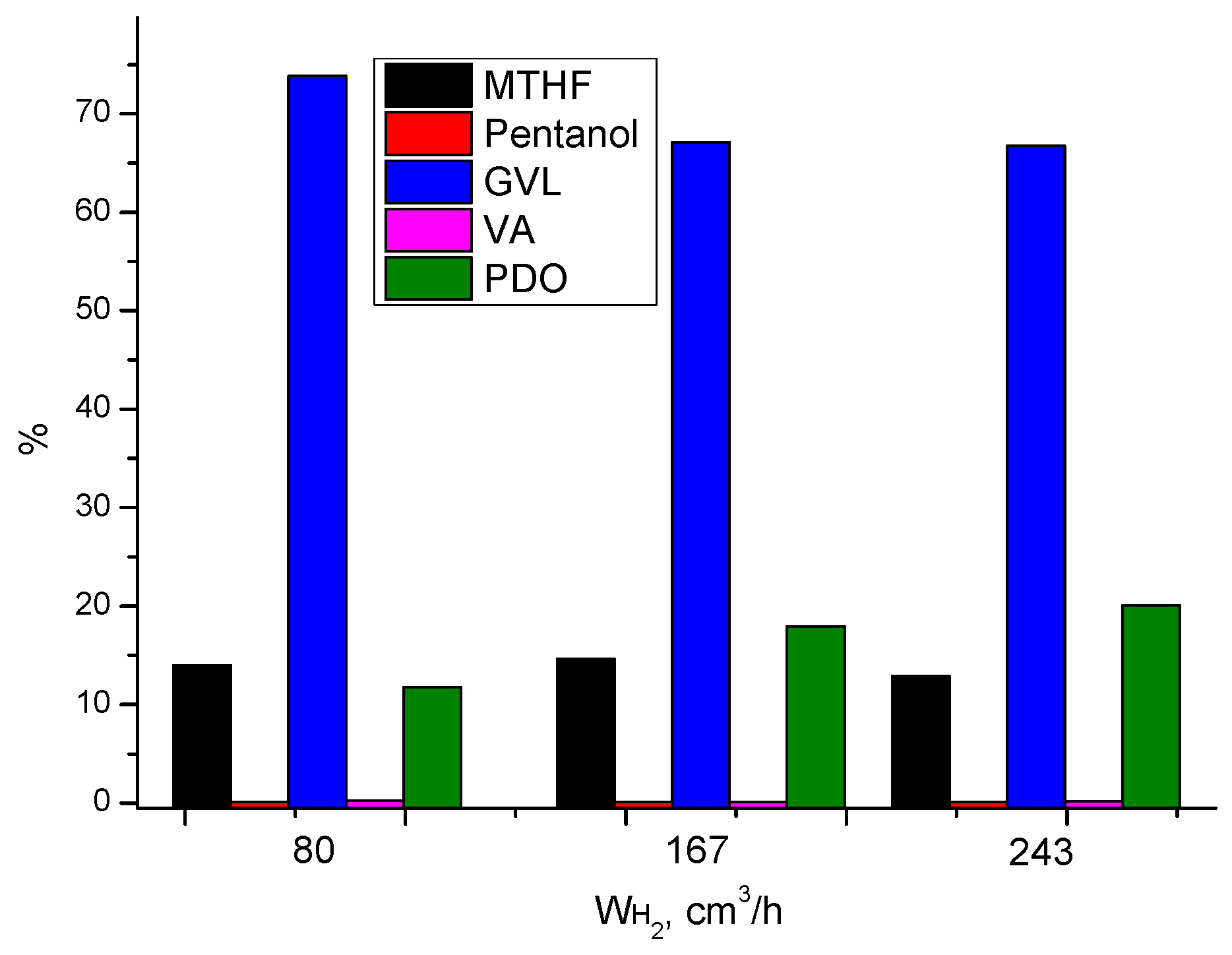
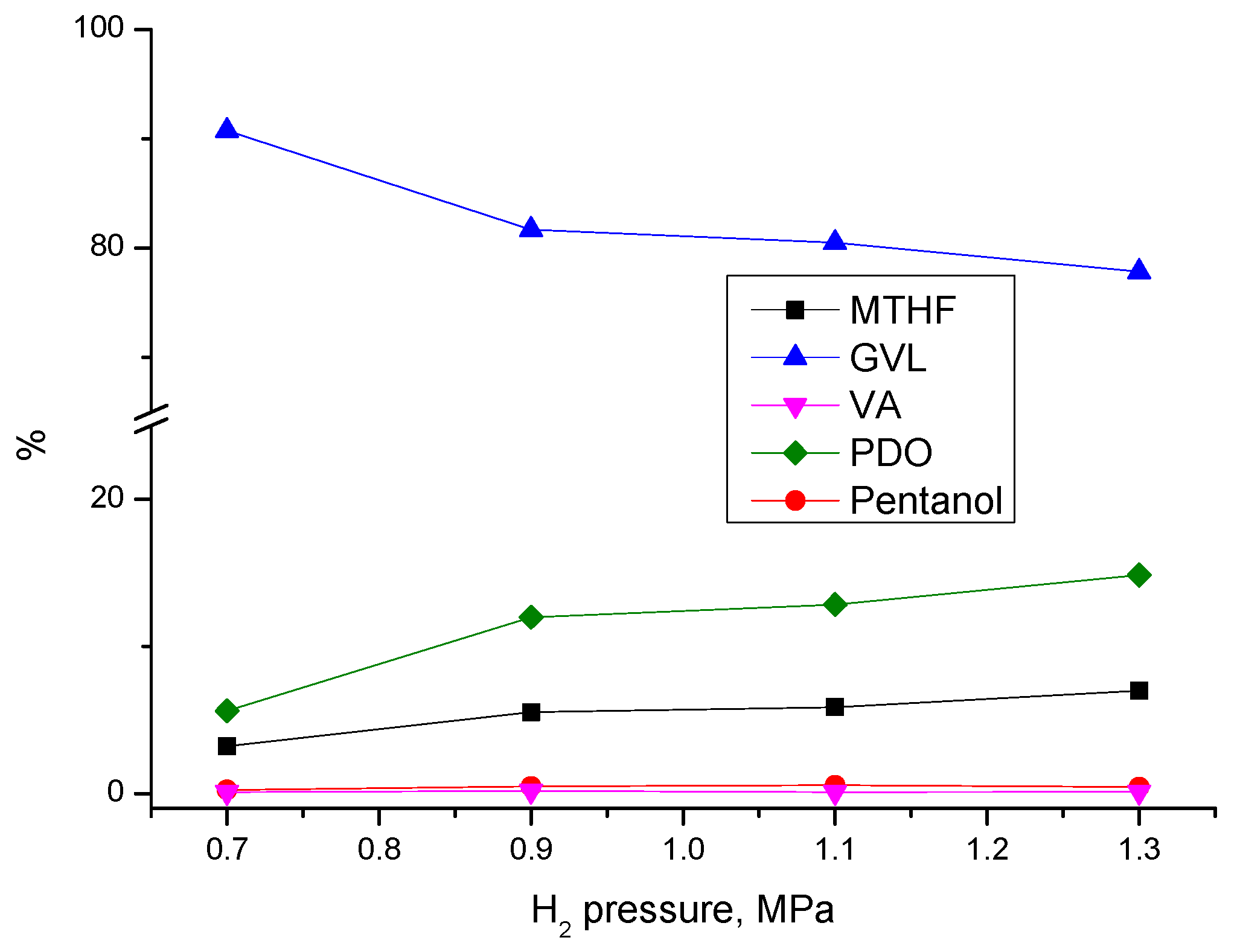
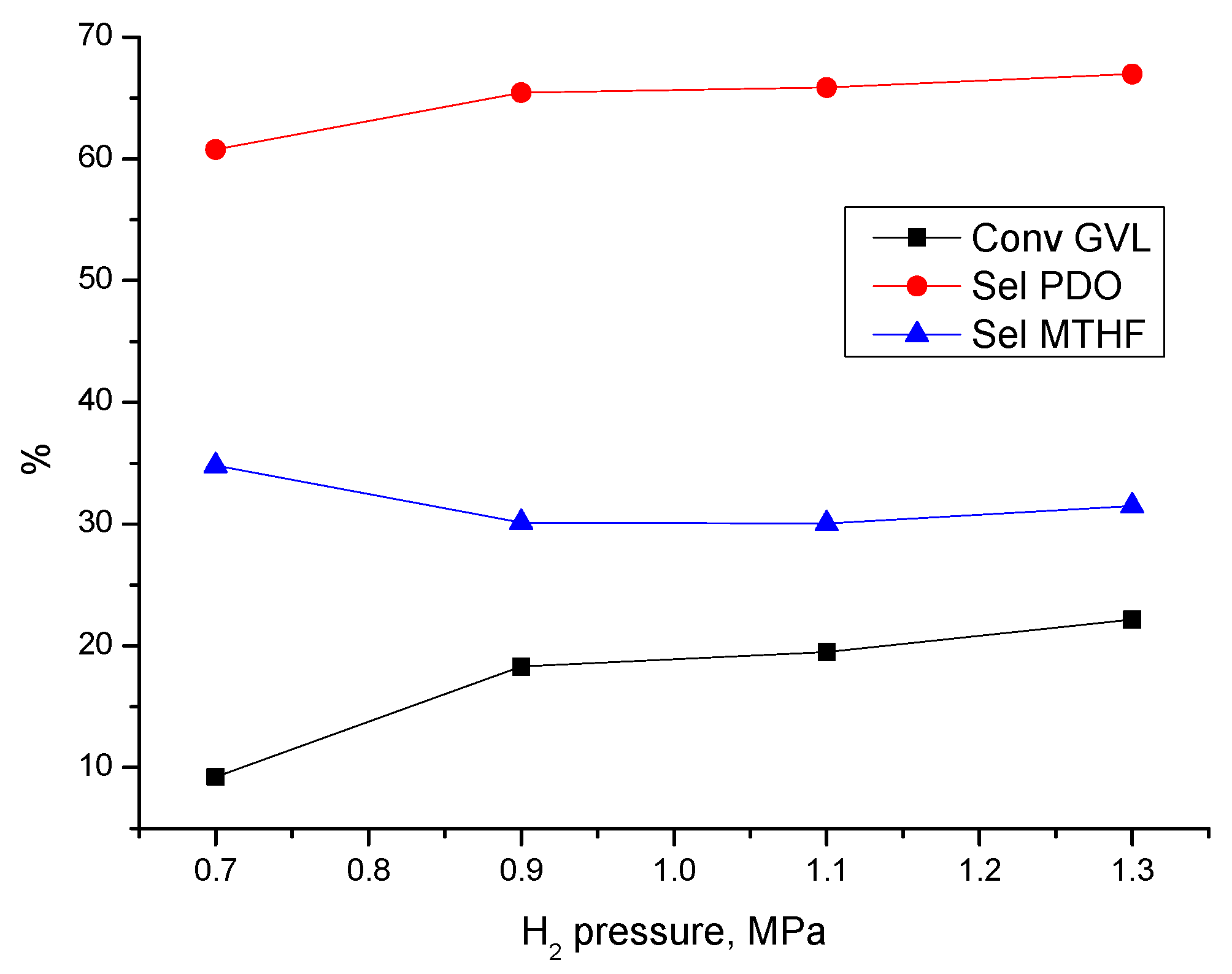
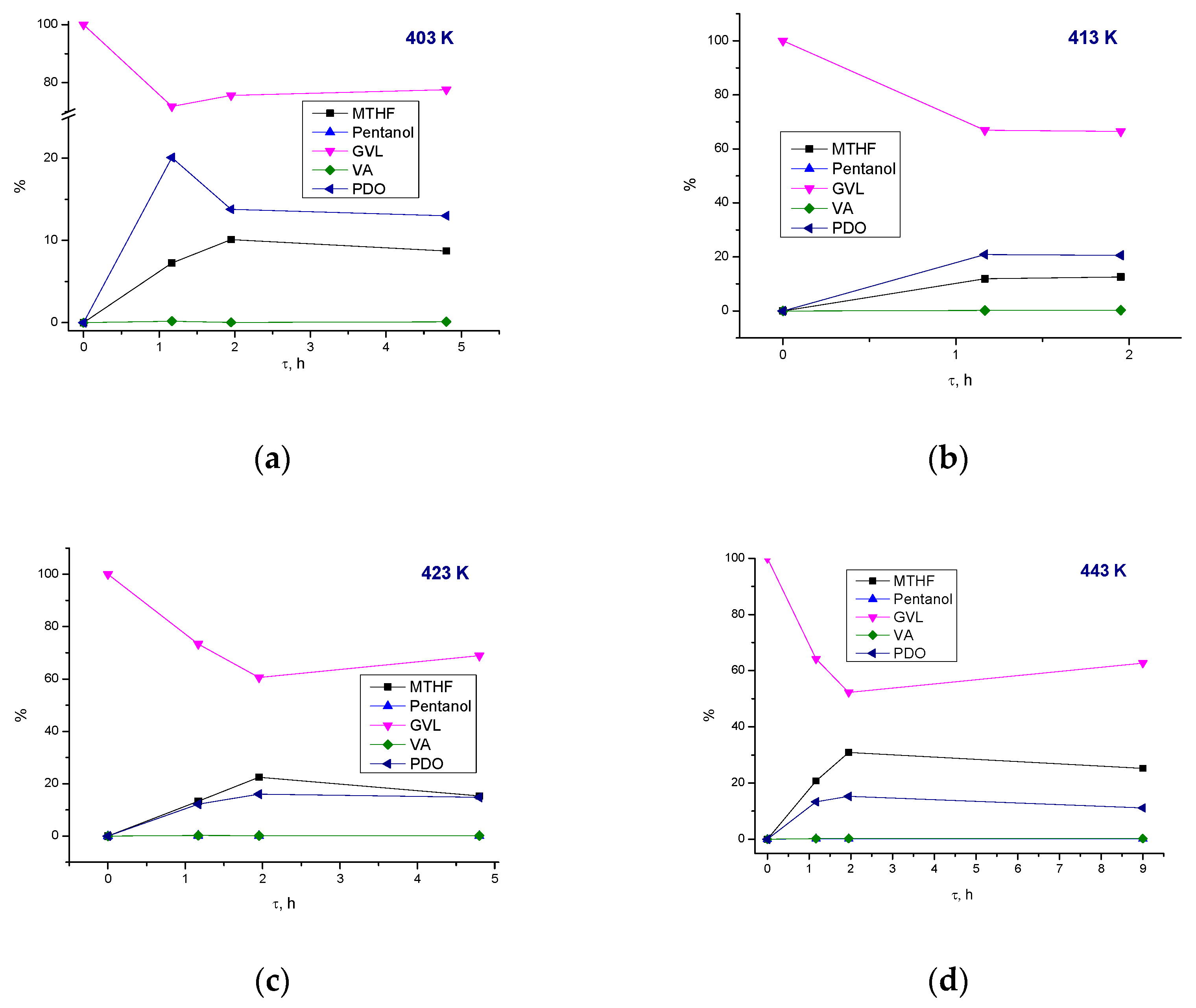
3.3.1. Effect of Process Parameters
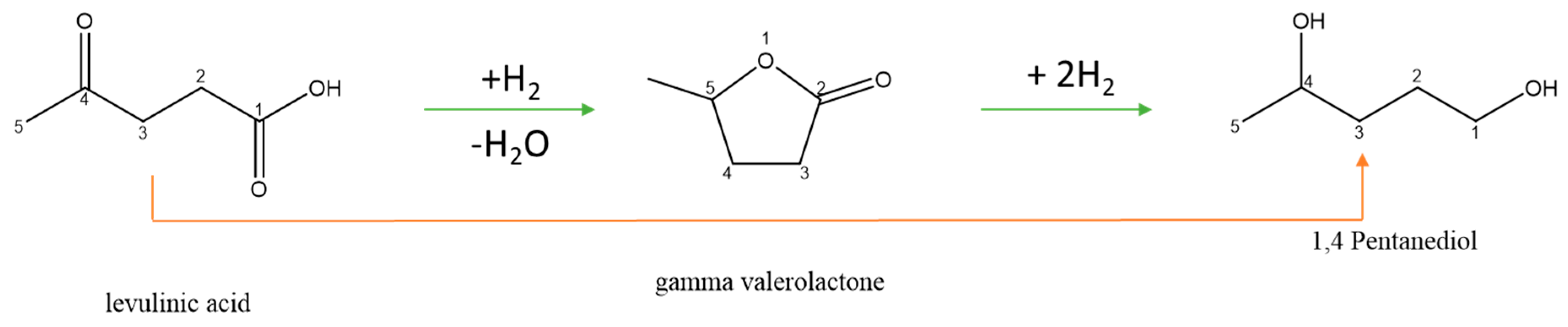
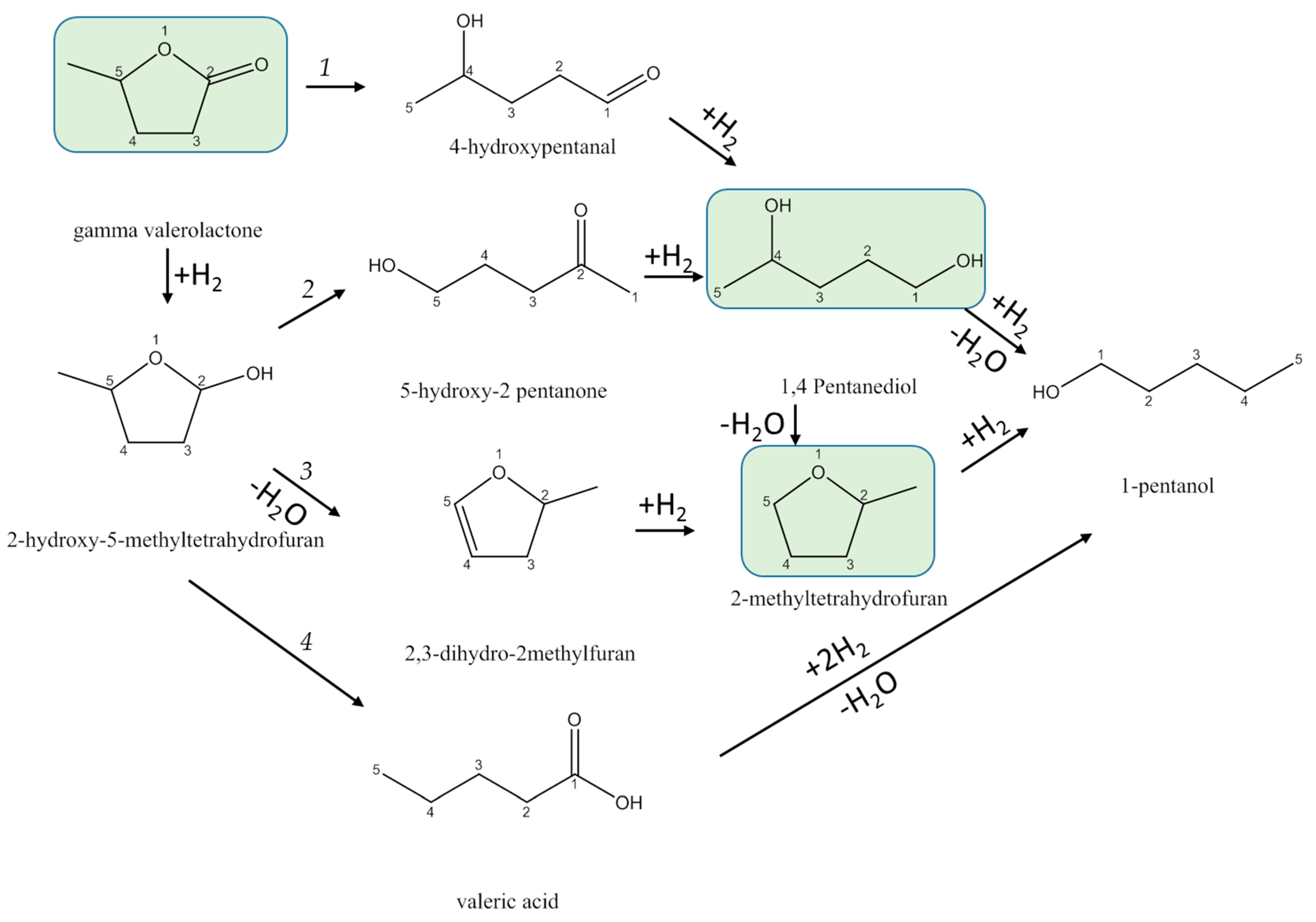
3.3.2. Reaction Pathways
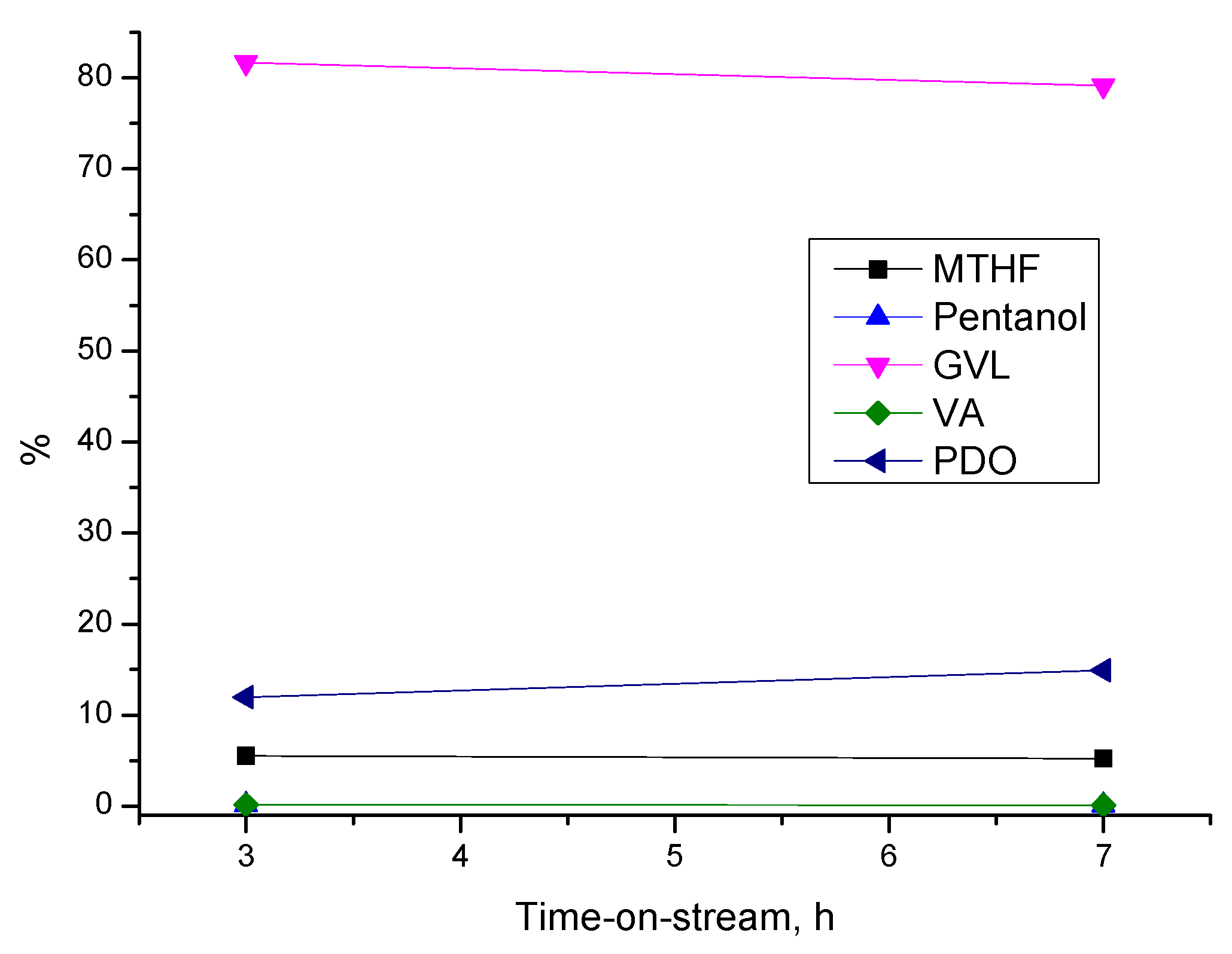
3.3.3. Stability of Cu/SiO2 Catalyst
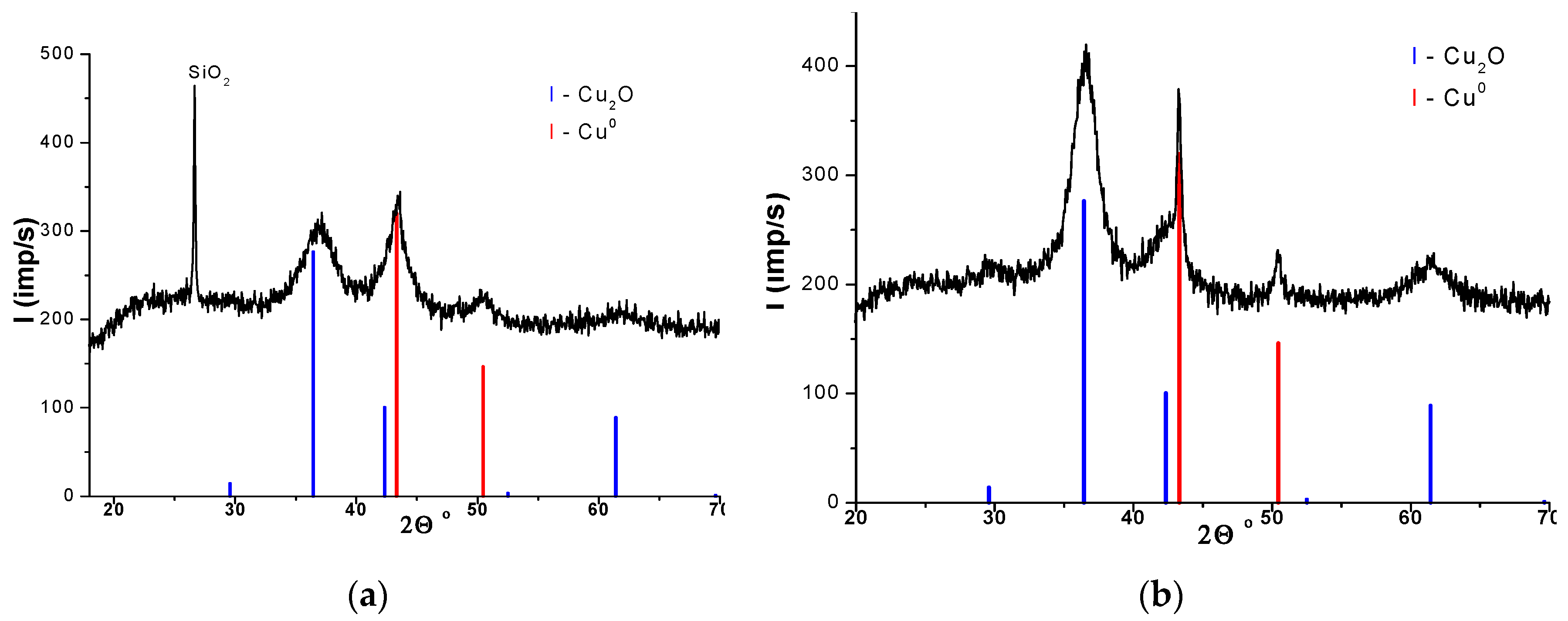
3.4. XRD of Cu/SiO2 before and after the Reaction
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Mäki-Arvela, P.; Holmbom, B.; Salmi, T.; Murzin, D.Y. Recent progress in synthesis of fine and specialty chemicals from wood and other biomass by heterogeneous catalytic processes. Catal. Rev. Sci. Eng. 2007, 49, 197–340. [Google Scholar] [CrossRef]
- Bozell, J.J.; Moens, L.; Elliott, D.C.; Wang, Y.; Neuenscwander, G.G.; Fitz-Patrick, S.W.; Bilski, R.J.; Jarnefeld, J.L. Production of levulinic acid and use as a platform chemical for derived products. Resour. Conserv. Recycl. 2000, 28, 227–239. [Google Scholar] [CrossRef]
- De Souza, R.O.M.A.; Miranda, L.S.M.; Luque, R. Bio (chemo) technological strategies for biomass conversion into bioethanol and key carboxylic acids. Green Chem. 2014, 16, 2386–2405. [Google Scholar] [CrossRef]
- Werpy, T.; Petersen, G. Top Value Added Chemicals from Biomass: Volume I—Results of Screening for Potential Candidates from Sugars and Synthesis Gas (No. DOE/GO-102004-1992); US Department of Energy: Golden, CO, USA, 2004. Available online: https://www.nrel.gov/docs/fy04osti/35523.pdf (accessed on 15 October 2020).
- Simakova, I.L.; Murzin, D.Y. Transformation of bio-derived acids into fuel-like alkanes via ketonic decarboxylation and hydrodeoxygenation: Design of multifunctional catalyst, kinetic and mechanistic aspects. J. Energy Chem. 2016, 25, 208–224. [Google Scholar] [CrossRef]
- Han, J. Process design and techno-economic evaluation for catalytic production of cellulosic γ-valerolactone using lignin derived propyl guaiacol. J. Ind. Eng. Chem. 2017, 52, 218–223. [Google Scholar] [CrossRef]
- Hong, M.; Chen, E.Y.-X. Chemically recyclable polymers: A circular economy approach to sustainability. Green Chem. 2017, 19, 3692–3706. [Google Scholar] [CrossRef]
- Westhues, S.; Idel, J.; Klankermayer, J. Molecular catalyst systems as key enablers for tailored polyesters and polycarbonate recycling concepts. Sci. Adv. 2018, 4, eaat9669. [Google Scholar] [CrossRef]
- Du, X.-L.; Bi, Q.-Y.; Liu, Y.-M.; Cao, Y.; He, H.-Y.; Fan, K.-N. Tunable copper-catalyzed chemoselective hydrogenolysis of biomass-derived γ-valerolactone into 1,4-pentanediol or 2-methyltetrahydrofuran. Green Chem. 2012, 14, 935–939. [Google Scholar] [CrossRef]
- Ahn, Y.-C.; Han, J. Catalytic production of 1,4-pentanediol from corn stover. Bioresour. Technol. 2017, 245, 442–448. [Google Scholar] [CrossRef]
- Geilen, F.M.A.; Engendahl, B.; Harwardt, A.; Marquardt, W.; Klankermayer, J.; Leitner, W. Selective and flexible transformation of biomass-derived platform chemicals by a multifunctional catalytic system. Angew. Chem. 2010, 122, 5642–5646. [Google Scholar] [CrossRef]
- Delidovich, I.; Leonhard, K.; Palkovits, R. Cellulose and hemicellulose valorisation: An integrated challenge of catalysis and reaction engineering. Energy Environ. Sci. 2014, 7, 2803–2830. [Google Scholar] [CrossRef]
- Vom Stein, T.; Meuresch, M.; Limper, D.; Schmitz, M.; Hoelscher, M.; Coetzee, J.; Cole-Hamilton, D.J.; Klankermayer, J.; Leitner, W. Highly versatile catalytic hydrogenation of carboxylic and carbonic acid derivatives using a Ru-triphos complex: Molecular control over selectivity and substrate scope. J. Am. Chem. Soc. 2011, 36, 13217–13225. [Google Scholar]
- Li, W.; Xie, J.-H.; Yuan, M.-L.; Zhou, Q.-L. Ruthenium complexes of tetradentate bipyridine ligands: Highly efficient catalysts for the hydrogenation of carboxylic esters and lactones. Green Chem. 2014, 16, 4081–4085. [Google Scholar] [CrossRef]
- Krstanje, T.J.; van der Vlugt, J.I.; Elsevier, C.J.; de Bruin, B. Hydrogenation of carboxylic acids with a homogeneous cobalt catalyst. Science 2015, 350, 298–302. [Google Scholar] [CrossRef]
- Srimani, D.; Mukherjee, A.; Goldberg, A.F.G.; Leitus, G.; Diskin-Posner, Y.; Shimon, L.J.W.; David, Y.B.; Milstein, D. Cobalt-catalyzed hydrogenation of esters to alcohols: Unexpected reactivity trend indicates ester enolate intermediacy. Angew. Chem. Int. Ed. 2015, 54, 12357–12360. [Google Scholar] [CrossRef]
- Elangovan, S.; Wendt, B.; Topf, C.; Bachmann, S.; Scalone, M.; Spannenberg, A.; Jiao, H.; Baumann, W.; Junge, K.; Beller, M. Improved second generation iron pincer complexes for effective ester hydrogenation. Adv. Synth. Catal. 2016, 358, 820–825. [Google Scholar] [CrossRef]
- Al-Shaal, M.G.; Dzierbinski, A.; Palkovits, R. Solvent-free γ-valerolactone hydrogenation to 2-methyltetrahydrofuran catalysed by Ru/C: A reaction network analysis. Green Chem. 2014, 16, 1358–1364. [Google Scholar] [CrossRef]
- Mizugaki, T.; Nagatsu, Y.; Togo, K.; Maeno, Z.; Mitsudome, T.; Jitsukawa, K.; Kaneda, K. Selective hydrogenation of levulinic acid to 1,4-pentanediol in water using a hydroxyapatite-supported Pt–Mo bimetallic catalyst. Green Chem. 2015, 17, 5136–5139. [Google Scholar] [CrossRef]
- Li, M.; Li, G.; Li, N.; Wang, A.; Dong, W.; Wang, X.; Cong, Y. Aqueous phase hydrogenation of levulinic acid to 1,4-pentanediol. Chem. Commun. 2014, 50, 1414–1416. [Google Scholar] [CrossRef]
- Sun, D.; Saito, T.; Yamada, Y.; Chen, X.; Sato, S. Hydrogenation of γ-valerolactone to 1,4-pentanediol in a continuous flow reactor. Appl. Catal. A Gen. 2017, 542, 289–295. [Google Scholar] [CrossRef]
- Xue-jiao, Z.; Chuang, L.; Xin, D.; Dong-dong, Y.; Chang-hai, L. Preparation of Cu/MgO catalysts for γ-valerolactone hydrogenation to 1,4-pentanediol by MOCVD. J. Fuel Chem. Technol. 2017, 45, 537–546. [Google Scholar]
- Xu, Q.; Li, X.; Pan, T.; Yu, C.; Deng, J.; Guo, Q.; Fu, Y. Supported copper catalysts for highly efficient hydrogenation of biomass-derived levulinic acid and γ-valerolactone. Green Chem. 2016, 18, 1287 1294. [Google Scholar] [CrossRef]
- Wu, J.; Gao, G.; Li, Y.; Sun, P.; Wang, J.; Li, F. Highly chemoselective hydrogenation of lactone to diol over efficient copper-based bifunctional nanocatalysts. Appl. Catal. B Environ. 2019, 245, 251–261. [Google Scholar] [CrossRef]
- Simakova, I.L.; Demidova, Y.S.; Simonov, M.N.; Niphadkar, P.S.; Bokade, V.V.; Devi, N.; Dhepe, P.L.; Murzin, D.Y. Carbon supported size-controlled ru catalysts for selective levulinic acid hydrogenation into γ-valerolactone. J. Sib. Fed. Univ. Chem. 2020, 13, 5–16. [Google Scholar] [CrossRef]
- Simakova, I.L.; Demidova, Y.S.; Simonov, M.N.; Niphadkar, P.S.; Bokade, V.V.; Devi, N.; Dhepe, P.L.; Murzin, D.Y. Mesoporous carbon and microporous zeolite supported Ru catalysts for selective levulinic acid hydrogenation into γ-valerolactone. Catal. Sustain. Energy 2019, 6, 38–49. [Google Scholar] [CrossRef]
- Yurieva, T.M.; Kustova, G.N.; Minyukova, T.P.; Poels, E.K.; Bliek, A.; Demeshkina, M.P.; Plyasova, L.M.; Kriger, T.A.; Zaikovskii, V.I. Non-hydrothermal synthesis of copper-, zinc and copper-zinc hydrosilicates. Mater. Res. Innov. 2001, 5, 3–11. [Google Scholar] [CrossRef]
- Minyukova, T.P.; Khassin, A.A.; Yurieva, T.M. Controlling the catalytic properties of copper-containing oxide catalysts. Kinet. Catal. 2018, 59, 112–122. [Google Scholar] [CrossRef]
- Simonov, M.N.; Zaikin, P.A.; Simakova, I.L. Highly selective catalytic propylene glycol synthesis from alkyl lactate over copper on silica: Performance and mechanism. Appl. Catal. B Environ. 2012, 119, 340–347. [Google Scholar] [CrossRef]
- Bart, J.C.J.; Sneeden, R.P.A. Copper-zinc oxide-alumina methanol catalysts revisited. Catal. Today 1987, 2, 1–124. [Google Scholar] [CrossRef]
- Turek, T.; Trimm, D.L.; Cant, N.W. The catalytic hydrogenolysis of esters to alcohols. Catal. Rev. Sci. Eng. 1994, 36, 645–683. [Google Scholar] [CrossRef]
- Rhodes, C.; Hutchings, G.J.; Ward, A.M. Water-gas shift reaction: Finding the mechanistic boundary. Catal. Today 1995, 23, 43–58. [Google Scholar] [CrossRef]
- Shao, Y.; Sun, K.; Li, Q.; Liu, Q.; Zhang, S.; Liu, Q.; Hu, G.; Hu, X. Copper-based catalysts with tunable acidic and basic sites for the selective conversion of levulinic acid/ester to γ-valerolactone or 1,4-pentanediol. Green Chem. 2019, 21, 4499–4511. [Google Scholar] [CrossRef]
- Nieminen, V.; Karhu, H.; Kumar, N.; Heinma, I.; Ek, P.; Samoson, A.; Salmi, T.; Murzin, D.Y. Physico-chemical and catalytic properties of Zr- and Cu-Zr ion-exchanged H-MCM-41. Phys. Chem. Chem. Phys. 2004, 6, 4062–4069. [Google Scholar] [CrossRef]
- Evans, J.W.; Wainwright, M.S.; Bridgewater, A.J.; Young, D.J. On the determination of copper surface area by reaction with nitrous oxide. Appl. Catal. A Gen. 1983, 7, 75–83. [Google Scholar] [CrossRef]
- Simakova, O.A.; Simonov, P.A.; Romanenko, A.V.; Simakova, I.L. Preparation of Pd/C catalysts via deposition of palladium hydroxide onto Sibunit carbon and their application to partial hydrogenation of rapeseed oil. React. Kinet. Catal. Lett. 2008, 95, 3–12. [Google Scholar] [CrossRef]
- Zaytseva, Y.A.; Panchenko, V.N.; Simonov, M.N.; Shutilov, A.A.; Zenkovets, G.A.; Renz, M.; Simakova, I.L.; Parmon, V.N. Effect of gas atmosphere on catalytic behaviour of zirconia, ceria and ceria-zirconia catalysts in valeric acid ketonization. Top. Catal. 2013, 56, 846–855. [Google Scholar] [CrossRef]
- Mäki-Arvela, P.; Martin, G.; Simakova, I.; Tokarev, A.; Wärnå, J.; Hemming, J.; Holmbom, B.; Salmi, T.; Murzin, D.Y. Kinetics, catalyst deactivation and modeling in the hydrogenation of β-sitosterol to β-sitostanol over microporous and mesoporous carbon supported Pd catalysts. Chem. Eng. J. 2009, 154, 45–51. [Google Scholar]
- Panchenko, V.N.; Paukshtis, E.A.; Murzin, D.Y.; Simakova, I.L. Solid base assisted n-pentanol coupling over VIII group metals: Elucidation of the Guerbet reaction mechanism by DRIFTS. Ind. Eng. Chem. Res. 2017, 56, 13310–13321. [Google Scholar] [CrossRef]
- Simonov, M.N.; Simakova, I.L.; Minyukova, T.P.; Khassin, A.A. Hydrogenation of lactic acid on reduced copper-containing catalysts. Russ. Chem. Bull. 2009, 58, 1114–1118. [Google Scholar] [CrossRef]
- Yurieva, T.M.; Minyukova, T.P.; Kustova, G.N.; Plyasova, L.M.; Kriger, T.A.; Demeshkina, M.P.; Zaikovskii, V.I.; Malakhov, V.V.; Dovlitova, L.S. Copper ions distribution in synthetic copper-zinc hydrosilicate. Mater. Res. Innov. 2001, 5, 74–80. [Google Scholar] [CrossRef]
- Huang, X.; Cant, N.W.; Wainwright, M.S.; Ma, L. The dehydrogenation of methanol to methyl Formate—Part I: Kinetic studies using copper-based catalysts. Chem. Eng. Process. 2005, 44, 393–402. [Google Scholar]
- Sun, D.; Takahashi, Y.; Yamada, Y.; Sato, S. Efficient formation of angelica lactones in a vapor-phase conversion of levulinic acid. Appl. Catal. A Gen. 2016, 526, 62–69. [Google Scholar] [CrossRef]
- Akiyama, M.; Sato, S.; Takahashi, R.; Inui, K.; Yokota, M. Dehydration–hydrogenation of glycerol into 1,2-propanediol at ambient hydrogen pressure. Appl. Catal. A Gen. 2009, 371, 60–66. [Google Scholar] [CrossRef]













{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Catalyst | CSD, nm | Phase Content, wt.% | Molar Ratio of Phases | Unit Cell Parameter a = b = c, Å, Determined/Standard | |||
|---|---|---|---|---|---|---|---|
| Cu0 | Cu2O | Cu0 | Cu2O | Cu0/Cu2O | Cu0 | Cu2O | |
| Cu/SiO2 fresh | 6.5 | 2.5 | 30 | 70 | ~1/1 | 3.613(2)/3.615 | 4.225(3)/4.269 |
| Cu/SiO2 spent | 31.0 | 4.0 | 7 | 93 | ~1/6 | 3.615(1)/3.615 | 4.266(2)/4.269 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Simakova, I.; Demidova, Y.; Simonov, M.; Prikhod’ko, S.; Niphadkar, P.; Bokade, V.; Dhepe, P.; Murzin, D.Y. Heterogeneously Catalyzed γ-Valerolactone Hydrogenation into 1,4-Pentanediol in Milder Reaction Conditions. Reactions 2020, 1, 54-71. https://doi.org/10.3390/reactions1020006
Simakova I, Demidova Y, Simonov M, Prikhod’ko S, Niphadkar P, Bokade V, Dhepe P, Murzin DY. Heterogeneously Catalyzed γ-Valerolactone Hydrogenation into 1,4-Pentanediol in Milder Reaction Conditions. Reactions. 2020; 1(2):54-71. https://doi.org/10.3390/reactions1020006
Chicago/Turabian StyleSimakova, Irina, Yulia Demidova, Mikhail Simonov, Sergey Prikhod’ko, Prashant Niphadkar, Vijay Bokade, Paresh Dhepe, and Dmitry Yu. Murzin. 2020. "Heterogeneously Catalyzed γ-Valerolactone Hydrogenation into 1,4-Pentanediol in Milder Reaction Conditions" Reactions 1, no. 2: 54-71. https://doi.org/10.3390/reactions1020006
APA StyleSimakova, I., Demidova, Y., Simonov, M., Prikhod’ko, S., Niphadkar, P., Bokade, V., Dhepe, P., & Murzin, D. Y. (2020). Heterogeneously Catalyzed γ-Valerolactone Hydrogenation into 1,4-Pentanediol in Milder Reaction Conditions. Reactions, 1(2), 54-71. https://doi.org/10.3390/reactions1020006