.png)

Role of IL-33/ST2 Pathway in Inflammatory Bowel Disease: An Overview and Future Perspectives

 ,
,  , ,
, ,  , ,
, ,  ,
,  and
and {kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
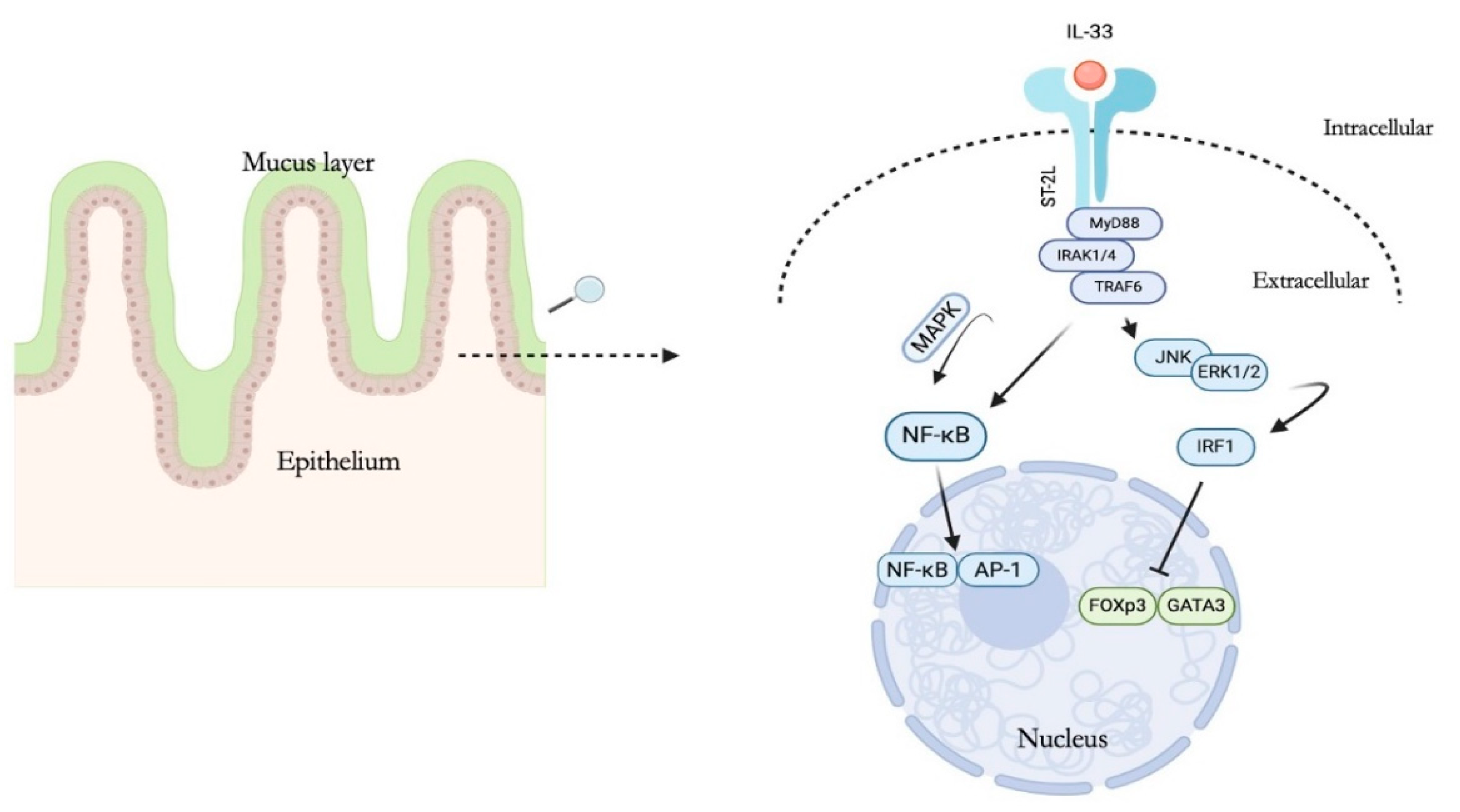
2. IL-33 Structure and Biological Function
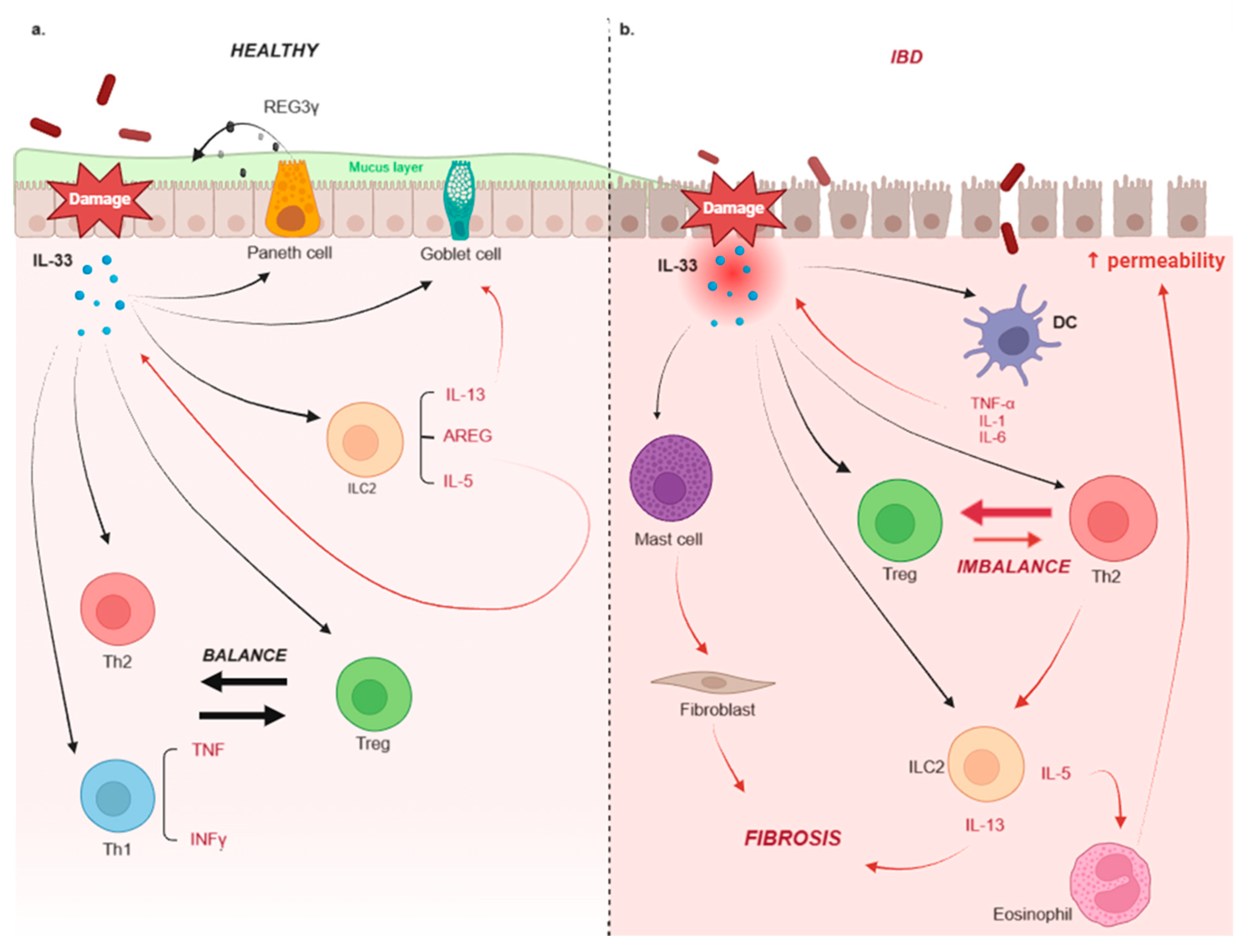
3. IL-33 and IBD
3.1. Ulcerative Colitis
3.2. Crohn’s Disease
4. IL-33 and IBD-Related CRC
5. Perspectives
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
References
- Borowitz, S.M. The epidemiology of inflammatory bowel disease: Clues to pathogenesis? Front. Pediatr. 2023, 10, 1103713. [Google Scholar] [CrossRef] [PubMed]
- Kofla-Dłubacz, A.; Pytrus, T.; Akutko, K.; Sputa-Grzegrzółka, P.; Piotrowska, A.; Dzięgiel, P. Etiology of IBD—Is It Still a Mystery? Int. J. Mol. Sci. 2022, 23, 12445. [Google Scholar] [CrossRef]
- Caviglia, G.P.; Garrone, A.; Bertolino, C.; Vanni, R.; Bretto, E.; Poshnjari, A.; Tribocco, E.; Frara, S.; Armandi, A.; Astegiano, M.; et al. Epidemiology of Inflammatory Bowel Diseases: A Population Study in a Healthcare District of North-West Italy. J. Clin. Med. 2023, 12, 641. [Google Scholar] [CrossRef] [PubMed]
- Ramos, G.P.; Papadakis, K.A. Mechanisms of Disease: Inflammatory Bowel Diseases. Mayo Clin. Proc. 2019, 94, 155–165. [Google Scholar] [CrossRef]
- Shouval, D.S.; Rufo, P.A. The Role of Environmental Factors in the Pathogenesis of Inflammatory Bowel Diseases: A Review. JAMA Pediatr. 2017, 171, 999–1005. [Google Scholar] [CrossRef] [PubMed]
- Nishida, A.; Inoue, R.; Inatomi, O.; Bamba, S.; Naito, Y.; Andoh, A. Gut Microbiota in the Pathogenesis of Inflammatory Bowel Disease. Clin. J. Gastroenterol. 2018, 11, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Chang, J.T. Pathophysiology of Inflammatory Bowel Diseases. N. Engl. J. Med. 2020, 383, 2652–2664. [Google Scholar] [CrossRef] [PubMed]
- Saez, A.; Herrero-Fernandez, B.; Gomez-Bris, R.; Sánchez-Martinez, H.; Gonzalez-Granado, J.M. Pathophysiology of Inflammatory Bowel Disease: Innate Immune System. Int. J. Mol. Sci. 2023, 24, 1526. [Google Scholar] [CrossRef] [PubMed]
- Liu, D.; Saikam, V.; Skrada, K.A.; Merlin, D.; Iyer, S.S. Inflammatory Bowel Disease Biomarkers. Med. Res. Rev. 2022, 42, 1856–1887. [Google Scholar] [CrossRef]
- Ungaro, R.; Mehandru, S.; Allen, P.B.; Peyrin-Biroulet, L.; Colombel, J.-F. Ulcerative Colitis. Lancet 2017, 389, 1756–1770. [Google Scholar] [CrossRef]
- Høivik, M.L.; Moum, B.; Solberg, I.C.; Henriksen, M.; Cvancarova, M.; Bernklev, T.; IBSEN Group. Work Disability in Inflammatory Bowel Disease Patients 10 Years after Disease Onset: Results from the IBSEN Study. Gut 2013, 62, 368–375. [Google Scholar] [CrossRef]
- Baumgart, D.C.; Sandborn, W.J. Crohn’s Disease. Lancet 2012, 380, 1590–1605. [Google Scholar] [CrossRef]
- Paudel, D.; Nair, D.V.T.; Joseph, G.; Castro, R.; Tiwari, A.K.; Singh, V. Gastrointestinal microbiota-directed nutritional and therapeutic interventions for inflammatory bowel disease: Opportunities and challenges. Gastroenterol. Rep. 2024, 12, goae033. [Google Scholar] [CrossRef]
- Vebr, M.; Pomahačová, R.; Sýkora, J.; Schwarz, J. A Narrative Review of Cytokine Networks: Pathophysiological and Therapeutic Implications for Inflammatory Bowel Disease Pathogenesis. Biomedicines 2023, 11, 3229. [Google Scholar] [CrossRef]
- Gajendran, M.; Loganathan, P.; Catinella, A.P.; Hashash, J.G. A Comprehensive Review and Update on Crohn’s Disease. Dis. Mon. 2018, 64, 20–57. [Google Scholar] [CrossRef]
- Burisch, J.; Bergemalm, D.; Halfvarson, J.; Domislovic, V.; Krznaric, Z.; Goldis, A.; Dahlerup, J.F.; Oksanen, P.; Collin, P.; Epi-IBD Group. The use of 5-aminosalicylate for patients with Crohn’s disease in a prospective European inception cohort with 5 years follow-up—An Epi-IBD study. United Eur. Gastroenterol. J. 2020, 8, 949–960. [Google Scholar] [CrossRef]
- Rousseaux, C.; Lefebvre, B.; Dubuquoy, L.; Lefebvre, P.; Romano, O.; Auwerx, J.; Metzger, D.; Wahli, W.; Desvergne, B.; Naccari, G.C.; et al. Intestinal Antiinflammatory Effect of 5-Aminosalicylic Acid Is Dependent on Peroxisome Proliferator–Activated Receptor-γ. J. Exp. Med. 2005, 201, 1205–1215. [Google Scholar] [CrossRef]
- Desreumaux, P.; Ghosh, S. Review Article: Mode of Action and Delivery of 5-Aminosalicylic Acid—New Evidence. Aliment. Pharmacol. Ther. 2006, 24, 2–9. [Google Scholar] [CrossRef]
- Bertin, B.; Dubuquoy, L.; Colombel, J.-F.; Desreumaux, P. PPAR-Gamma in Ulcerative Colitis: A Novel Target for Intervention. Curr. Drug Targets 2013, 14, 1501–1507. [Google Scholar] [CrossRef]
- Ramamoorthy, S.; Cidlowski, J.A. Corticosteroids: Mechanisms of Action in Health and Disease. Rheum. Dis. Clin. N. Am. 2016, 42, 15–31. [Google Scholar] [CrossRef]
- Dubois-Camacho, K.; Ottum, P.A.; Franco-Muñoz, D.; De la Fuente, M.; Torres-Riquelme, A.; Díaz-Jiménez, D.; Olivares-Morales, M.; Astudillo, G.; Quera, R.; Hermoso, M.A. Glucocorticosteroid Therapy in Inflammatory Bowel Diseases: From Clinical Practice to Molecular Biology. World J. Gastroenterol. 2017, 23, 6628–6638. [Google Scholar] [CrossRef] [PubMed]
- Sokic-Milutinovic, A.; Milosavljevic, T. IBD: From conventional immunosuppression to biological therapy. Dig. Dis. 2023, 1–11. [Google Scholar] [CrossRef]
- Guo, Y.; Lu, N.; Bai, A. Clinical Use and Mechanisms of Infliximab Treatment on Inflammatory Bowel Disease: A Recent Update. Biomed. Res. Int. 2013, 2013, 581631. [Google Scholar] [CrossRef]
- Krishnan, S.; Ding, Y.; Saedi, N.; Choi, M.; Sridharan, G.V.; Sherr, D.H.; Yarmush, M.L.; Alaniz, R.C.; Jayaraman, A.; Lee, K. Gut Microbiota-Derived Tryptophan Metabolites Modulate Inflammatory Response in Hepatocytes and Macrophages. Cell Rep. 2018, 23, 1099–1111. [Google Scholar] [CrossRef]
- Zhang, J.; Zhu, S.; Ma, N.; Johnston, L.J.; Wu, C.; Ma, X. Metabolites of Microbiota Response to Tryptophan and Intestinal Mucosal Immunity: A Therapeutic Target to Control Intestinal Inflammation. Med. Res. Rev. 2021, 41, 1061–1088. [Google Scholar] [CrossRef]
- Ma, N.; Zhang, J.; Reiter, R.J.; Ma, X. Melatonin Mediates Mucosal Immune Cells, Microbial Metabolism, and Rhythm Crosstalk: A Therapeutic Target to Reduce Intestinal Inflammation. Med. Res. Rev. 2020, 40, 606–632. [Google Scholar] [CrossRef]
- Aggeletopoulou, I.; Tsounis, E.P.; Triantos, C. Molecular Mechanisms Underlying IL-33-Mediated Inflammation in Inflammatory Bowel Disease. Int. J. Mol. Sci. 2023, 24, 623. [Google Scholar] [CrossRef]
- Garlanda, C.; Dinarello, C.A.; Mantovani, A. The Interleukin-1 Family: Back to the Future. Immunity 2013, 39, 1003–1018. [Google Scholar] [CrossRef]
- Andoh, A.; Nishida, A. Pro- and Anti-Inflammatory Roles of Interleukin (IL)-33, IL-36, and IL-38 in Inflammatory Bowel Disease. J. Gastroenterol. 2023, 58, 69–78. [Google Scholar] [CrossRef]
- Chen, J.; He, Y.; Tu, L.; Duan, L. Dual immune functions of IL-33 in inflammatory bowel disease. Histol. Histopathol. 2020, 35, 137–146. [Google Scholar]
- Pastorelli, L.; Garg, R.R.; Hoang, S.B.; Spina, L.; Mattioli, B.; Scarpa, M.; Fiocchi, C.; Vecchi, M.; Pizarro, T.T. Epithelial-Derived IL-33 and Its Receptor ST2 Are Dysregulated in Ulcerative Colitis and in Experimental Th1/Th2 Driven Enteritis. Proc. Natl. Acad. Sci. USA 2010, 107, 8017–8022. [Google Scholar] [CrossRef] [PubMed]
- Liew, F.Y.; Girard, J.P.; Turnquist, H.R. Interleukin-33 in health and disease. Nat. Rev. Immunol. 2016, 16, 676–689. [Google Scholar] [CrossRef]
- Cayrol, C.; Girard, J.P. Interleukin-33 (IL-33): A nuclear cytokine from the IL-1 family. Immunol. Rev. 2018, 281, 154–168. [Google Scholar] [CrossRef] [PubMed]
- Murdaca, G.; Greco, M.; Tonacci, A. IL-33/IL-31 Axis in Immune-Mediated and Allergic Diseases. Int. J. Mol. Sci. 2019, 20, 5856. [Google Scholar] [CrossRef] [PubMed]
- Baekkevold, E.S.; Roussigne, M.; Yamanaka, T.; Johansen, F.E.; Jahnsen, F.L.; Amalric, F.; Brandtzaeg, P.; Erard, M.; Haraldsen, G.; Girard, J.P. Molecular characterization of NF-HEV, a nuclear factor preferentially expressed in human high endothelial venules. Am. J. Pathol. 2003, 163, 69–79. [Google Scholar] [CrossRef]
- Cayrol, C.; Girard, J.-P. Interleukin-33 (IL-33): A Critical Review of Its Biology and the Mechanisms Involved in Its Release as a Potent Extracellular Cytokine. Cytokine 2022, 156, 155891. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; He, C.; Xin, S.; Liu, X.; Zhang, S.; Qiao, B.; Shang, H.; Gao, L.; Xu, J. A Deep View of the Biological Property of Interleukin-33 and Its Dysfunction in the Gut. Int. J. Mol. Sci. 2023, 24, 13504. [Google Scholar] [CrossRef] [PubMed]
- Lefrancais, E.; Roga, S.; Gautier, V.; Gonzalez-de-Peredo, A.; Monsarrat, B.; Girard, J.P.; Cayrol, C. IL-33 is processed into mature bioactive forms by neutrophil elastase and cathepsin G. Proc. Natl. Acad. Sci. USA 2012, 109, 1673–1678. [Google Scholar] [CrossRef] [PubMed]
- Sedhom, M.A.K.; Pichery, M.; Murdoch, J.R.; Foligné, B.; Ortega, N.; Normand, S.; Mertz, K.; Sanmugalingam, D.; Brault, L.; Grandjean, T.; et al. Neutralisation of the Interleukin-33/ST2 Pathway Ameliorates Experimental Colitis through Enhancement of Mucosal Healing in Mice. Gut 2013, 62, 1714–1723. [Google Scholar] [CrossRef]
- Lopez-Casado, M.A.; Lorite, P.; Palomeque, T.; Torres, M.I. Potential role of the IL-33/ST2 axis in celiac disease. Cell. Mol. Immunol. 2017, 14, 285–292. [Google Scholar] [CrossRef]
- Cayrol, C.; Girard, J.P. The IL-1-like cytokine IL-33 is inactivated after maturation by caspase-1. Proc. Natl. Acad. Sci. USA 2009, 106, 9021–9026. [Google Scholar] [CrossRef]
- Waddell, A.; Vallance, J.E.; Fox, S.; Rosen, M.J. IL-33 is produced by colon fibroblasts and differentially regulated in acute and chronic murine colitis. Sci. Rep. 2021, 11, 9575. [Google Scholar] [CrossRef] [PubMed]
- Cayrol, C. IL-33, an Alarmin of the IL-1 Family Involved in Allergic and Non Allergic Inflammation: Focus on the Mechanisms of Regulation of Its Activity. Cells 2021, 11, 107. [Google Scholar] [CrossRef] [PubMed]
- Yi, X.M.; Lian, H.; Li, S. Signaling and functions of interleukin-33 in immune regulation and diseases. Cell Insight. 2022, 1, 100042. [Google Scholar] [CrossRef] [PubMed]
- Seidelin, J.B.; Rogler, G.; Nielsen, O.H. A role for interleukin-33 in TH2-polarized intestinal inflammation? Mucosal Immunol. 2011, 4, 496–502. [Google Scholar] [CrossRef]
- Schmitz, J.; Owyang, A.; Oldham, E.; Song, Y.; Murphy, E.; McClanahan, T.K.; Zurawski, G.; Moshrefi, M.; Qin, J.; Li, X.; et al. IL-33, an interleukin-1-like cytokine that signals via the IL-1 receptor-related protein ST2 and induces T helper type 2-associated cytokines. Immunity 2005, 23, 479–490. [Google Scholar] [CrossRef]
- Fagundes, C.T.; Amaral, F.A.; Souza, A.L.S.; Vieira, A.T.; Xu, D.; Liew, F.Y.; Souza, D.G.; Teixeira, M.M. ST2, an IL-1R Family Member, Attenuates Inflammation and Lethality after Intestinal Ischemia and Reperfusion. J. Leukoc. Biol. 2007, 81, 492–499. [Google Scholar] [CrossRef]
- Palmer, G.; Lipsky, B.P.; Smithgall, M.D.; Meininger, D.; Siu, S.; Talabot-Ayer, D.; Gabay, C.; Smith, D.E. The IL-1 Receptor Accessory Protein (AcP) Is Required for IL-33 Signaling and Soluble AcP Enhances the Ability of Soluble ST2 to Inhibit IL-33. Cytokine 2008, 42, 358–364. [Google Scholar] [CrossRef]
- Griesenauer, B.; Paczesny, S. The ST2/IL-33 Axis in Immune Cells during Inflammatory Diseases. Front. Immunol. 2017, 8, 475. [Google Scholar] [CrossRef]
- Chen, Q.; Carroll, H.P.; Gadina, M. The Newest Interleukins: Recent Additions to the Ever-Growing Cytokine Family. Vitam. Horm. 2006, 74, 207–228. [Google Scholar]
- Bulek, K.; Swaidani, S.; Qin, J.; Lu, Y.; Gulen, M.F.; Herjan, T.; Min, B.; Kastelein, R.A.; Aronica, M.; Kosz-Vnenchak, M.; et al. The Essential Role of Single Ig IL-1 Receptor-Related Molecule/Toll IL-1R8 in Regulation of Th2 Immune Response. J. Immunol. 2009, 182, 2601–2609. [Google Scholar] [CrossRef] [PubMed]
- Bonanno, A.; Gangemi, S.; La Grutta, S.; Malizia, V.; Riccobono, L.; Colombo, P.; Cibella, F.; Profita, M. 25-Hydroxyvitamin D, IL-31, and IL-33 in Children with Allergic Disease of the Airways. Mediat. Inflamm. 2014, 2014, 520241. [Google Scholar] [CrossRef] [PubMed]
- Kaur, H.; Kaur, G.; Ali, S.A. IL-33’s role in the gut immune system: A comprehensive review of its crosstalk and regulation. Life Sci. 2023, 327, 121868. [Google Scholar] [CrossRef] [PubMed]
- Oboki, K.; Ohno, T.; Kajiwara, N.; Arae, K.; Morita, H.; Ishii, A.; Nambu, A.; Abe, T.; Kiyonari, H.; Matsumoto, K.; et al. IL-33 is a crucial amplifier of innate rather than acquired immunity. Proc. Natl. Acad. Sci. USA 2010, 107, 18581–18586. [Google Scholar] [CrossRef] [PubMed]
- Lamhamedi-Cherradi, S.E.; Zheng, S.J.; Maguschak, K.A.; Peschon, J.; Chen, Y.H. Defective thymocyte apoptosis and accelerated autoimmune diseases in TRAIL−/− mice. Nat. Immunol. 2003, 4, 255–260. [Google Scholar] [CrossRef]
- Xiao, Y.; Huang, X.; Zhao, Y.; Chen, F.; Sun, M.; Yang, W.; Chen, L.; Yao, S.; Peniche, A.; Dann, S.M.; et al. Interleukin-33 Promotes REG3γ Expression in Intestinal Epithelial Cells and Regulates Gut Microbiota. Cell Mol. Gastroenterol. Hepatol. 2019, 8, 21–36. [Google Scholar] [CrossRef] [PubMed]
- Seo, D.H.; Che, X.; Kwak, M.S.; Kim, S.; Kim, J.H.; Ma, H.W.; Kim, D.H.; Kim, T.I.; Kim, W.H.; Kim, S.W.; et al. Interleukin-33 Regulates Intestinal Inflammation by Modulating Macrophages in Inflammatory Bowel Disease. Sci. Rep. 2017, 7, 851. [Google Scholar] [CrossRef] [PubMed]
- Kondo, Y.; Yoshimoto, T.; Yasuda, K.; Futatsugi-Yumikura, S.; Morimoto, M.; Hayashi, N.; Hoshino, T.; Fujimoto, J.; Nakanishi, K. Administration of IL-33 Induces Airway Hyperresponsiveness and Goblet Cell Hyperplasia in the Lungs in the Absence of Adaptive Immune System. Int. Immunol. 2008, 20, 791–800. [Google Scholar] [CrossRef] [PubMed]
- Schumacher, M.A.; Hsieh, J.J.; Liu, C.Y.; Appel, K.L.; Waddell, A.; Almohazey, D.; Katada, K.; Bernard, J.K.; Bucar, E.B.; Gadeock, S.; et al. Sprouty2 Limits Intestinal Tuft and Goblet Cell Numbers through GSK3β-Mediated Restriction of Epithelial IL-33. Nat. Commun. 2021, 12, 836. [Google Scholar] [CrossRef]
- Monticelli, L.A.; Osborne, L.C.; Noti, M.; Tran, S.V.; Zaiss, D.M.W.; Artis, D. IL-33 Promotes an Innate Immune Pathway of Intestinal Tissue Protection Dependent on Amphiregulin-EGFR Interactions. Proc. Natl. Acad. Sci. USA 2015, 112, 10762–10767. [Google Scholar] [CrossRef]
- Satoh-Takayama, N.; Kato, T.; Motomura, Y.; Kageyama, T.; Taguchi-Atarashi, N.; Kinoshita-Daitoku, R.; Kuroda, E.; Di Santo, J.P.; Mimuro, H.; Moro, K.; et al. Bacteria-Induced Group 2 Innate Lymphoid Cells in the Stomach Provide Immune Protection through Induction of IgA. Immunity 2020, 52, 635–649.e4. [Google Scholar] [CrossRef] [PubMed]
- Strober, W.; Fuss, I.J. Proinflammatory Cytokines in the Pathogenesis of Inflammatory Bowel Diseases. Gastroenterology 2011, 140, 1756–1767. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, T.; Minaga, K.; Kamata, K.; Sakurai, T.; Komeda, Y.; Nagai, T.; Kitani, A.; Tajima, M.; Fuss, I.J.; Kudo, M.; et al. RICK/RIP2 Is a NOD2-Independent Nodal Point of Gut Inflammation. Int. Immunol. 2019, 31, 669–683. [Google Scholar] [CrossRef] [PubMed]
- Kurimoto, M.; Watanabe, T.; Kamata, K.; Minaga, K.; Kudo, M. IL-33 as a Critical Cytokine for Inflammation and Fibrosis in Inflammatory Bowel Diseases and Pancreatitis. Front. Physiol. 2021, 12, 781012. [Google Scholar] [CrossRef] [PubMed]
- Sponheim, J.; Pollheimer, J.; Olsen, T.; Balogh, J.; Hammarström, C.; Loos, T.; Kasprzycka, M.; Sørensen, D.R.; Nilsen, H.R.; Küchler, A.M.; et al. Inflammatory Bowel Disease-Associated Interleukin-33 Is Preferentially Expressed in Ulceration-Associated Myofibroblasts. Am. J. Pathol. 2010, 177, 2804–2815. [Google Scholar] [CrossRef] [PubMed]
- Gundersen, M.D.; Goll, R.; Hol, J.; Olsen, T.; Rismo, R.; Sørbye, S.W.; Sundnes, O.; Haraldsen, G.; Florholmen, J. Loss of Interleukin 33 Expression in Colonic Crypts—A Potential Marker for Disease Remission in Ulcerative Colitis. Sci. Rep. 2016, 6, 35403. [Google Scholar] [CrossRef] [PubMed]
- Kobori, A.; Yagi, Y.; Imaeda, H.; Ban, H.; Bamba, S.; Tsujikawa, T.; Saito, Y.; Fujiyama, Y.; Andoh, A. Interleukin-33 Expression Is Specifically Enhanced in Inflamed Mucosa of Ulcerative Colitis. J. Gastroenterol. 2010, 45, 999–1007. [Google Scholar] [CrossRef] [PubMed]
- Pastorelli, L.; De Salvo, C.; Cominelli, M.A.; Vecchi, M.; Pizarro, T.T. Novel Cytokine Signaling Pathways in Inflammatory Bowel Disease: Insight into the Dichotomous Functions of IL-33 during Chronic Intestinal Inflammation. Ther. Adv. Gastroenterol. 2011, 4, 311–323. [Google Scholar] [CrossRef]
- Artis, D. Epithelial-Cell Recognition of Commensal Bacteria and Maintenance of Immune Homeostasis in the Gut. Nat. Rev. Immunol. 2008, 8, 411–420. [Google Scholar] [CrossRef]
- Malik, A.; Sharma, D.; Zhu, Q.; Karki, R.; Guy, C.S.; Vogel, P.; Kanneganti, T.-D. IL-33 Regulates the IgA-Microbiota Axis to Restrain IL-1α–Dependent Colitis and Tumorigenesis. J. Clin. Investig. 2016, 126, 4469–4481. [Google Scholar] [CrossRef]
- Tahaghoghi-Hajghorbani, S.; Ajami, A.; Ghorbanalipoor, S.; Hosseini-Khah, Z.; Taghiloo, S.; Khaje-Enayati, P.; Hosseini, V. Protective Effect of TSLP and IL-33 Cytokines in Ulcerative Colitis. Autoimmun. Highlights 2019, 10, 1. [Google Scholar] [CrossRef] [PubMed]
- Groβ, P.; Doser, K.; Falk, W.; Obermeier, F.; Hofmann, C. IL-33 Attenuates Development and Perpetuation of Chronic Intestinal Inflammation. Inflamm. Bowel Dis. 2012, 18, 1900–1909. [Google Scholar] [CrossRef]
- Ngo Thi Phuong, N.; Palmieri, V.; Adamczyk, A.; Klopfleisch, R.; Langhorst, J.; Hansen, W.; Westendorf, A.M.; Pastille, E. IL-33 Drives Expansion of Type 2 Innate Lymphoid Cells and Regulatory T Cells and Protects Mice From Severe, Acute Colitis. Front. Immunol. 2021, 12, 669787. [Google Scholar] [CrossRef]
- Tu, L.; Chen, J.; Xu, D.; Xie, Z.; Yu, B.; Tao, Y.; Shi, G.; Duan, L. IL-33-Induced Alternatively Activated Macrophage Attenuates the Development of TNBS-Induced Colitis. Oncotarget 2017, 8, 27704–27714. [Google Scholar] [CrossRef] [PubMed]
- Duan, L.; Chen, J.; Zhang, H.; Yang, H.; Zhu, P.; Xiong, A.; Xia, Q.; Zheng, F.; Tan, Z.; Gong, F.; et al. Interleukin-33 Ameliorates Experimental Colitis through Promoting Th2/Foxp3+ Regulatory T-Cell Responses in Mice. Mol. Med. 2012, 18, 753–761. [Google Scholar] [CrossRef]
- De Salvo, C.; Buela, K.-A.; Creyns, B.; Corridoni, D.; Rana, N.; Wargo, H.L.; Cominelli, C.L.; Delaney, P.G.; Rodriguez-Palacios, A.; Cominelli, F.; et al. NOD2 Drives Early IL-33-Dependent Expansion of Group 2 Innate Lymphoid Cells during Crohn’s Disease-like Ileitis. J. Clin. Investig. 2021, 131, e140624. [Google Scholar] [CrossRef]
- Baumann, C.; Bonilla, W.V.; Fröhlich, A.; Helmstetter, C.; Peine, M.; Hegazy, A.N.; Pinschewer, D.D.; Löhning, M. T-Bet- and STAT4-Dependent IL-33 Receptor Expression Directly Promotes Antiviral Th1 Cell Responses. Proc. Natl. Acad. Sci. USA 2015, 112, 4056–4061. [Google Scholar] [CrossRef]
- Bouma, G.; Strober, W. The Immunological and Genetic Basis of Inflammatory Bowel Disease. Nat. Rev. Immunol. 2003, 3, 521–533. [Google Scholar] [CrossRef] [PubMed]
- Hirai, F.; Andoh, A.; Ueno, F.; Watanabe, K.; Ohmiya, N.; Nakase, H.; Kato, S.; Esaki, M.; Endo, Y.; Yamamoto, H.; et al. Efficacy of Endoscopic Balloon Dilation for Small Bowel Strictures in Patients With Crohn’s Disease: A Nationwide, Multi-Centre, Open-Label, Prospective Cohort Study. J. Crohn’s Colitis 2018, 12, 394–401. [Google Scholar] [CrossRef]
- Nakayama, T.; Hirahara, K.; Onodera, A.; Endo, Y.; Hosokawa, H.; Shinoda, K.; Tumes, D.J.; Okamoto, Y. Th2 Cells in Health and Disease. Annu. Rev. Immunol. 2017, 35, 53–84. [Google Scholar] [CrossRef]
- Eberl, G.; Colonna, M.; Di Santo, J.P.; McKenzie, A.N.J. Innate Lymphoid Cells. Innate Lymphoid Cells: A New Paradigm in Immunology. Science 2015, 348, aaa6566. [Google Scholar] [CrossRef]
- Bailey, J.R.; Bland, P.W.; Tarlton, J.F.; Peters, I.; Moorghen, M.; Sylvester, P.A.; Probert, C.S.J.; Whiting, C.V. IL-13 Promotes Collagen Accumulation in Crohn’s Disease Fibrosis by down-Regulation of Fibroblast MMP Synthesis: A Role for Innate Lymphoid Cells? PLoS ONE 2012, 7, e52332. [Google Scholar] [CrossRef] [PubMed]
- Nishida, A.; Andoh, A.; Imaeda, H.; Inatomi, O.; Shiomi, H.; Fujiyama, Y. Expression of Interleukin 1-like Cytokine Interleukin 33 and Its Receptor Complex (ST2L and IL1RAcP) in Human Pancreatic Myofibroblasts. Gut 2010, 59, 531–541. [Google Scholar] [CrossRef] [PubMed]
- Pisani, L.F.; Teani, I.; Vecchi, M.; Pastorelli, L. Interleukin-33: Friend or Foe in Gastrointestinal Tract Cancers? Cells 2023, 12, 1481. [Google Scholar] [CrossRef] [PubMed]
- Eissmann, M.F.; Dijkstra, C.; Wouters, M.A.; Baloyan, D.; Mouradov, D.; Nguyen, P.M.; Davalos-Salas, M.; Putoczki, T.L.; Sieber, O.M.; Mariadason, J.M.; et al. Interleukin 33 Signaling Restrains Sporadic Colon Cancer in an Interferon-γ-Dependent Manner. Cancer Immunol. Res. 2018, 6, 409–421. [Google Scholar] [CrossRef]
- Pastille, E.; Wasmer, M.H.; Adamczyk, A.; Vu, V.P.; Mager, L.F.; Phuong, N.N.T.; Palmieri, V.; Simillion, C.; Hansen, W.; Kasper, S.; et al. The IL-33/ST2 pathway shapes the regulatory T cell phenotype to promote intestinal cancer. Mucosal Immunol. 2019, 12, 990–1003. [Google Scholar] [CrossRef]
- Akimoto, M.; Takenaga, K. Role of the IL-33/ST2L axis in colorectal cancer progression. Cell Immunol. 2019, 343, 103740. [Google Scholar] [CrossRef]
- Liu, X.; Zhu, L.; Lu, X.; Bian, H.; Wu, X.; Yang, W.; Qin, Q. IL-33/ST2 pathway contributes to metastasis of human colorectal cancer. Biochem. Biophys. Res. Commun. 2014, 453, 486–492. [Google Scholar] [CrossRef]
- Van der Jeught, K.; Sun, Y.; Fang, Y.; Zhou, Z.; Jiang, H.; Yu, T.; Yang, J.; Kamocka, M.M.; So, K.M.; Li, Y.; et al. ST2 as Checkpoint Target for Colorectal Cancer Immunotherapy. JCI Insight 2020, 5, e136073. [Google Scholar] [CrossRef]
- Ercolano, G.; Gomez-Cadena, A.; Dumauthioz, N.; Vanoni, G.; Kreutzfeldt, M.; Wyss, T.; Michalik, L.; Loyon, R.; Ianaro, A.; Ho, P.-C.; et al. PPARɣ Drives IL-33-Dependent ILC2 pro-Tumoral Functions. Nat. Commun. 2021, 12, 2538. [Google Scholar] [CrossRef]
- Li, Y.; Shi, J.; Qi, S.; Zhang, J.; Peng, D.; Chen, Z.; Wang, G.; Wang, Z.; Wang, L. IL-33 Facilitates Proliferation of Colorectal Cancer Dependent on COX2/PGE2. J. Exp. Clin. Cancer Res. 2018, 37, 196. [Google Scholar] [CrossRef] [PubMed]
- Saliba, J.; Coutaud, B.; Makhani, K.; Epstein Roth, N.; Jackson, J.; Park, J.Y.; Gagnon, N.; Costa, P.; Jeyakumar, T.; Bury, M.; et al. Loss of NFE2L3 Protects against Inflammation-Induced Colorectal Cancer through Modulation of the Tumor Microenvironment. Oncogene 2022, 41, 1563–1575. [Google Scholar] [CrossRef]
- Zhang, Y.; Davis, C.; Shah, S.; Hughes, D.; Ryan, J.C.; Altomare, D.; Peña, M.M.O. IL-33 Promotes Growth and Liver Metastasis of Colorectal Cancer in Mice by Remodeling the Tumor Microenvironment and Inducing Angiogenesis. Mol. Carcinog. 2017, 56, 272–287. [Google Scholar] [CrossRef] [PubMed]
- Cui, G.; Yuan, A.; Pang, Z.; Zheng, W.; Li, Z.; Goll, R. Contribution of IL-33 to the Pathogenesis of Colorectal Cancer. Front. Oncol. 2018, 8, 561. [Google Scholar] [CrossRef]
- O’Donnell, C.; Mahmoud, A.; Keane, J.; Murphy, C.; White, D.; Carey, S.; O’Riordain, M.; Bennett, M.W.; Brint, E.; Houston, A. An Antitumorigenic Role for the IL-33 Receptor, ST2L, in Colon Cancer. Br. J. Cancer 2016, 114, 37–43. [Google Scholar] [CrossRef] [PubMed]
- Oliveira, E.C.S.; Quaglio, A.E.V.; Magro, D.O.; Di Stasi, L.C.; Sassaki, L.Y. Intestinal Microbiota and miRNA in IBD: A Narrative Review about Discoveries and Perspectives for the Future. Int. J. Mol. Sci. 2023, 24, 7176. [Google Scholar] [CrossRef] [PubMed]
- Suri, K.; Bubier, J.A.; Wiles, M.V.; Shultz, L.D.; Amiji, M.M.; Hosur, V. Role of microRNA in inflammatory bowel disease: Clinical evidence and the development of preclinical animal models. Cells 2021, 10, 2204. [Google Scholar] [CrossRef] [PubMed]
- Masi, L.; Capobianco, I.; Magrì, C.; Marafini, I.; Petito, V.; Scaldaferri, F. MicroRNAs as Innovative Biomarkers for Inflammatory Bowel Disease and Prediction of Colorectal Cancer. Int. J. Mol. Sci. 2022, 23, 7991. [Google Scholar] [CrossRef]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Giordano, W.; Ricciardi, G.; Casciaro, M.; Fiorentino, V.; Pizzimenti, C.; Viola, A.; Martini, M.; Tuccari, G.; Ieni, A. Role of IL-33/ST2 Pathway in Inflammatory Bowel Disease: An Overview and Future Perspectives. Gastrointest. Disord. 2024, 6, 446-460. https://doi.org/10.3390/gidisord6020030
Giordano W, Ricciardi G, Casciaro M, Fiorentino V, Pizzimenti C, Viola A, Martini M, Tuccari G, Ieni A. Role of IL-33/ST2 Pathway in Inflammatory Bowel Disease: An Overview and Future Perspectives. Gastrointestinal Disorders. 2024; 6(2):446-460. https://doi.org/10.3390/gidisord6020030
Chicago/Turabian StyleGiordano, Walter, Gabriele Ricciardi, Marco Casciaro, Vincenzo Fiorentino, Cristina Pizzimenti, Anna Viola, Maurizio Martini, Giovanni Tuccari, and Antonio Ieni. 2024. "Role of IL-33/ST2 Pathway in Inflammatory Bowel Disease: An Overview and Future Perspectives" Gastrointestinal Disorders 6, no. 2: 446-460. https://doi.org/10.3390/gidisord6020030
APA StyleGiordano, W., Ricciardi, G., Casciaro, M., Fiorentino, V., Pizzimenti, C., Viola, A., Martini, M., Tuccari, G., & Ieni, A. (2024). Role of IL-33/ST2 Pathway in Inflammatory Bowel Disease: An Overview and Future Perspectives. Gastrointestinal Disorders, 6(2), 446-460. https://doi.org/10.3390/gidisord6020030