Obesity and Related Type 2 Diabetes: A Failure of the Autonomic Nervous System Controlling Gastrointestinal Function?


Abstract
1. Introduction
Relationship between Obesity and Diabetes
2. Why Is the Spread of Obesity So Rapid?
2.1. Lifestyle Change
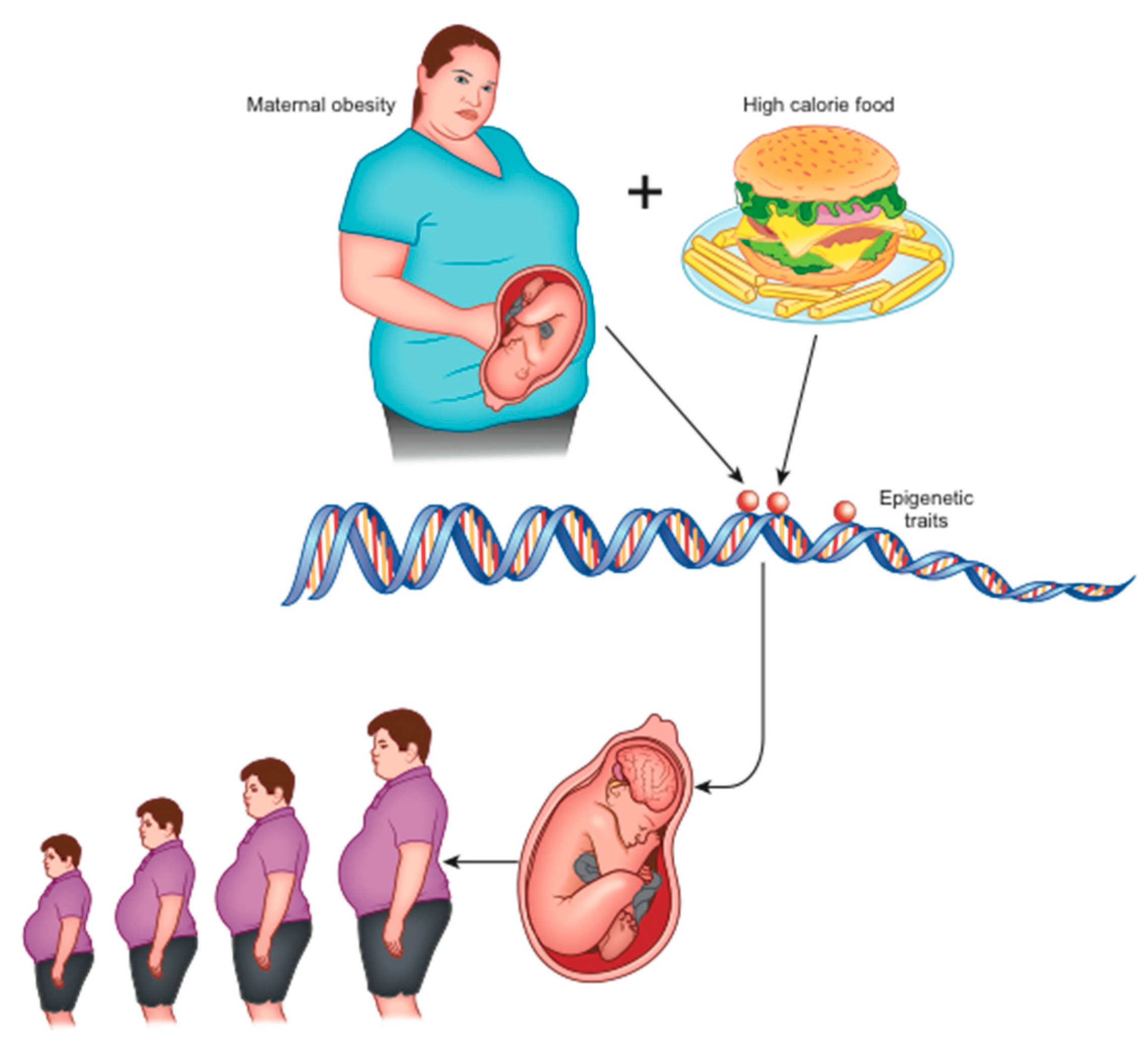
2.2. Role of Epigenetics: Development Programming
2.3. How Can Negative Environmental Conditions Influence The Development of Obesity?
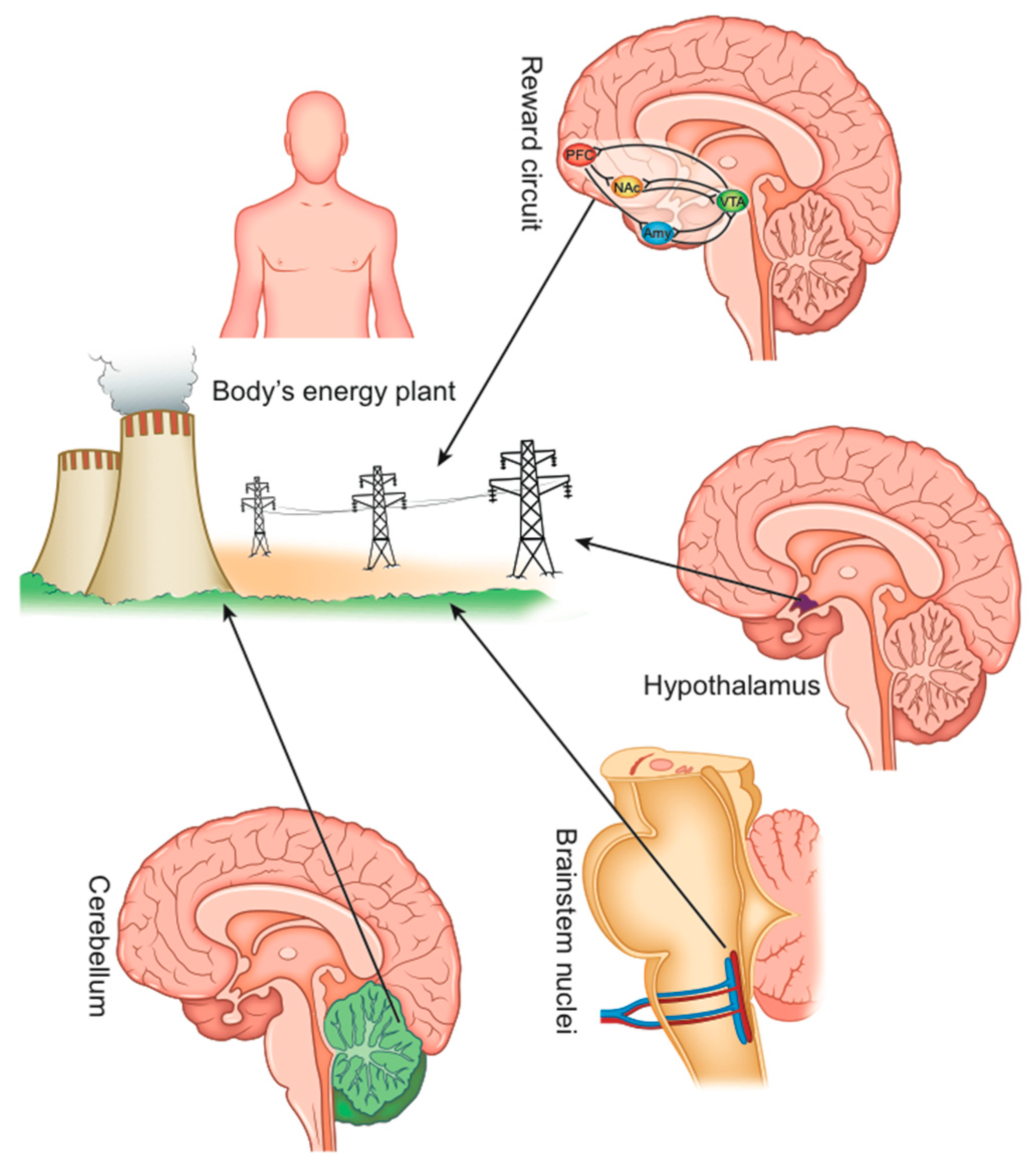
3. How Could the Dysfunction of Energy Homeostasis Control Be Responsible for Obesity?
3.1. What Is the Role of the Reward System Dysfunction? Can We Take into Account a Food Addiction?
3.2. Role of the Brainstem and the Gut–Brain Axis
3.3. Importance of Impulses from the Oropharynx
3.4. The Hypothalamus
3.5. The Cerebellum
3.6. Areas of the Cortex
3.7. Functional Brain Differences in Patients with Obesity as Evidenced by Neuroimaging
3.8. Prader–Willi Syndrome (PWS) as an Additional Source of Information
3.9. Another Rare Form of Childhood Obesity Related to Genetic Alterations Involving the CNS
4. Effectiveness of Metabolic Surgery: Which Could Be the Mechanisms?
4.1. Changes to Feeding
4.2. GLP-1: Modifications Following Bariatric Surgery in Light of Its Role in Obesity
4.3. Reactivation of the Vagus Afferent Neurocircuits Disrupted by HFD
4.4. Increased Insulin Response and Sensitivity
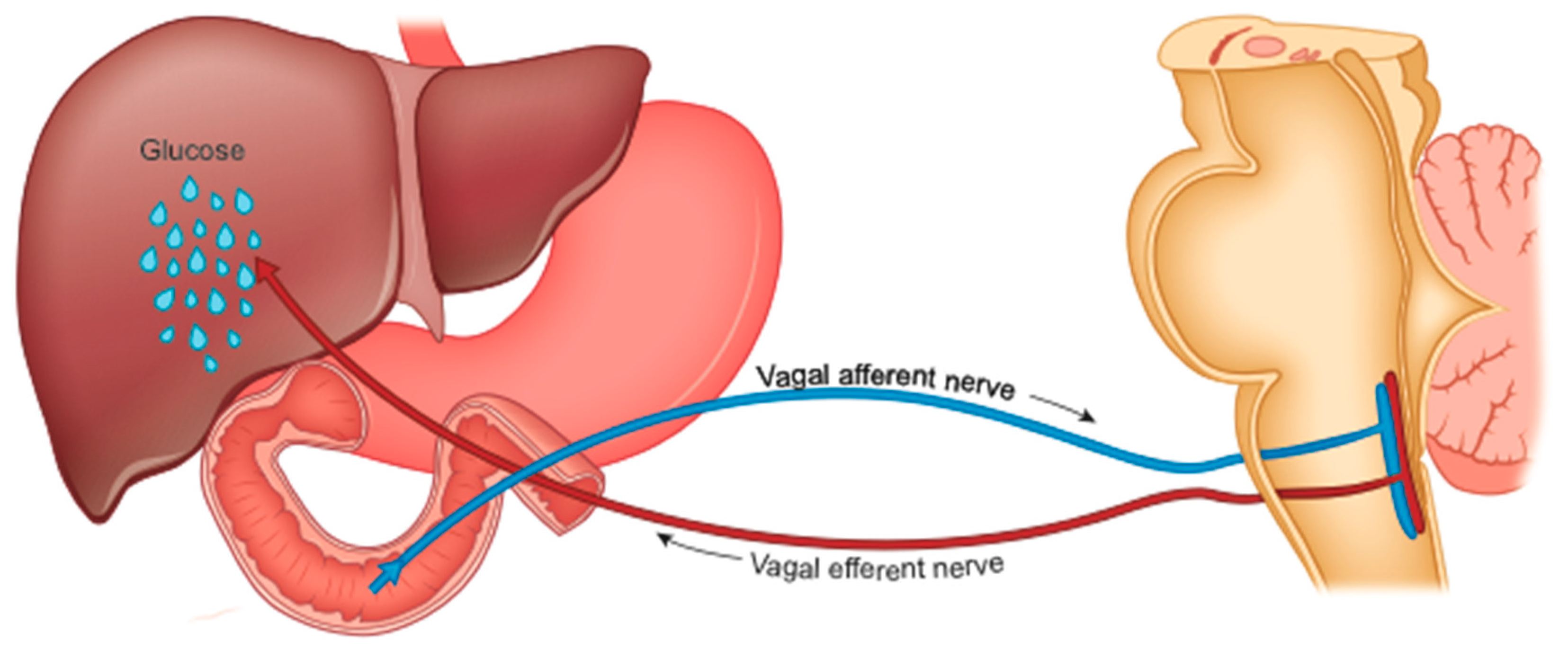
4.5. Reduced Hepatic Glucose Production (HGP)
4.5.1. HGP and Cholecystokinin
4.5.2. HGP and TOR Enzyme
4.5.3. HGP and Serotonin
4.5.4. Accelerated Gastric Emptying in Sleeve Gastrectomy (SG)
4.5.5. Conclusive Hypothesis on the Role of HGP
4.6. Modification of the Hypothalamic Set Point
4.7. Cerebellum
4.8. Alteration of Intestinal Bacteria
4.9. Possible Effect of Neck Radiation Therapy
5. What Could Be the Main Targets of Non-Conventional and Non-Surgical Therapeutic Approaches to Obesity?
5.1. The Synaptic Receptors
5.1.1. The N-Methyl-D-Aspartate Receptor (NMDA)
5.1.2. NMDA Receptor Co-Agonists
5.1.3. Gaba-Amino-Butyric Acid (GABA) Receptors
5.2. Other Potential Targets of Drug Treatment Beyond the Glutamatergic Signaling Circuits
5.2.1. Dopamine Receptors
5.2.2. 5-HT3 Agonists and Antagonists
5.2.3. Guanine Protein-Coupled Receptors (GPCR)
5.2.4. Inhibitor of Glycine Transporter 1 (Glyt1)
5.2.5. The melanocortin 4 Receptor (MC4R)
5.2.6. The Opioid System
5.3. Activation of Physical Activity through Stimulation of the Neuropeptide Orexin
5.4. The GIRK Channels
6. The Effects of Physical Instruments (Table 2)
6.1. Electrical Stimulation of The Vagus Nerve
6.1.1. Blocking of Vagal Activity

{kind=link}
{kind=link}
{kind=link}
| Main Sector | 1st Subgroup | 2nd Subgroup |
|---|---|---|
| Electrical stimulation of the vagus nerve | Blocking of vagal activity | Activation of the afferent vagus |
| Brain neuro-modulation | Deep brain stimulation (DBS) | Direct non-invasive stimulation (NIBS) with transcranial current |
| fMRI neurofeedback |
6.1.2. Activation of The Afferent Vagus
6.2. Brain Neuro-Modulation
6.2.1. Deep Brain Stimulation (DBS)
6.2.2. Direct Non-Invasive Stimulation (NIBS) with Transcranial Current
6.3. fMRI Neurofeedback
7. Treatment of Epigenetic Modifications
8. Conclusions
Funding
Conflicts of Interest
References
- Lim, S.S.; Vos, T.; Flaxman, A.D.; Danaei, G.; Shibuya, K.; Adair-Rohani, H.; Amann, M.; Anderson, H.R.; Andrews, K.G.; Aryee, M. A comparative risk assessment of burden of disease and injury attributable to 67 risk factors and risk factor clusters in 21 regions, 1990–2010: A systematic analysis for the global burden of disease study 2010. Lancet 2012, 380, 2224–2260. [Google Scholar] [CrossRef]
- Ward, Z.J.; Bleich, S.N.; Cradock, A.L.; Barrett, J.L.; Giles, C.M.; Flax, C.; Long, M.W.; Gortmaker, S.L. Projected U.S. State-Level Prevalence of Adult Obesity and Severe Obesity. N. Engl. J. Med. 2019, 381, 2440–2450. [Google Scholar] [CrossRef] [PubMed]
- Hossain, P.; Kawar, B.; El Nahas, M. Obesity and diabetes in the developing world—A growing challenge. N. Engl. J. Med. 2007, 356, 213–215. [Google Scholar] [CrossRef] [PubMed]
- Baldeweg, S.E.; Golay, A.; Natali, A.; Balkau, B.; Del Prato, S.; Coppack, S.W. On behalf of the European Group for the Study of Insulin Resistance (EGIR) Insulin resistance, lipid and fatty acid concentrations in 867 healthy Europeans. Eur. J. Clin. Investig. 2000, 30, 45–52. [Google Scholar] [CrossRef]
- Radaelli, M.G.; Martucci, F.; Perra, S.; Accornero, S.; Castoldi, G.; Lattuada, G.; Manzoni, G.; Perseghin, G. NAFLD/NASH in patients with type 2 diabetes and related treatment options. J. Endocrinol. Investig. 2018, 41, 509–521. [Google Scholar] [CrossRef] [PubMed]
- Dalton, M.; Finlayson, G. Psychobiological examination of liking and wanting for fat and sweet taste in trait binge eating females. Physiol. Behav. 2014, 136, 128–134. [Google Scholar] [CrossRef] [PubMed]
- Nicola, S.M. Reassessing wanting and liking in the study of mesolimbic influence on food intake. Am. J. Physiol. Integr. Comp. Physiol. 2016, 311, R811–R840. [Google Scholar] [CrossRef] [PubMed]
- Morin, J.-P.; Rodríguez-Durán, L.F.; Guzmán-Ramos, K.; Perez-Cruz, C.; Ferreira, G.; Diaz-Cintra, S.; Pacheco-López, G. Palatable Hyper-Caloric Foods Impact on Neuronal Plasticity. Front. Behav. Neurosci. 2017, 11, 19. [Google Scholar] [CrossRef]
- Behary, P.; Miras, A.D. Brain responses to food and weight loss. Exp. Physiol. 2014, 99, 1121–1127. [Google Scholar] [CrossRef]
- Reynolds, C.M.; Segovia, S.A.; Vickers, M.H. Experimental Models of Maternal Obesity and Neuroendocrine Programming of Metabolic Disorders in Offspring. Front. Endocrinol. 2017, 8, 245. [Google Scholar] [CrossRef]
- Daxinger, L.; Whitelaw, E. Understanding transgenerational epigenetic inheritance via the gametes in mammals. Nat. Rev. Genet. 2012, 13, 153–162. [Google Scholar] [CrossRef] [PubMed]
- Schwartz, M.W.; Seeley, R.J.; Zeltser, L.M.; Drewnowski, A.; Ravussin, E.; Redman, L.M.; Leibel, R.L. Obesity Pathogenesis: An Endocrine Society Scientific Statement. Endocr. Rev. 2017, 38, 267–296. [Google Scholar] [CrossRef] [PubMed]
- Franzago, M.; Fraticelli, F.; Stuppia, L.; Vitacolonna, E. Nutrigenetics, epigenetics and gestational diabetes: Consequences in mother and child. Epigenetics 2019, 14, 215–235. [Google Scholar] [CrossRef] [PubMed]
- Crespi, B. Why and How Imprinted Genes Drive Fetal Programming. Front. Endocrinol. 2020, 10, 940–948. [Google Scholar] [CrossRef] [PubMed]
- Tucci, V.; Isles, A.R.; Kelsey, G.; Ferguson-Smith, A.C.; Bartolomei, M.S.; Benvenisty, N.; Bourc’His, D.; Charalambous, M.; Dulac, C.; Feil, R.; et al. Genomic Imprinting and Physiological Processes in Mammals. Cell 2019, 176, 952–965. [Google Scholar] [CrossRef]
- Montalvo-Martínez, L.; Maldonado-Ruiz, R.; Cárdenas-Tueme, M.; Reséndez-Pérez, D.; Camacho, A. Maternal Overnutrition Programs Central Inflammation and Addiction-Like Behavior in Offspring. BioMed Res. Int. 2018, 2018, 1–11. [Google Scholar] [CrossRef]
- Levin, B.E. Interaction of perinatal and pre-pubertal factors with genetic predisposition in the development of neural pathways involved in the regulation of energy homeostasis. Brain Res. 2010, 1350, 10–17. [Google Scholar] [CrossRef]
- Bhagat, R.; Fortna, S.R.; Browning, K.N. Exposure to a high fat diet during the perinatal period alters vagal motoneurone excitability, even in the absence of obesity. J. Physiol. 2014, 593, 285–303. [Google Scholar] [CrossRef]
- Fleming, T.P.; Watkins, A.J.; Velazquez, M.A.; Mathers, J.C.; Prentice, A.M.; Stephenson, J.; Barker, M.; Saffery, R.; Yajnik, C.S.; Eckert, J.J.; et al. Origins of lifetime health around the time of conception: Causes and consequences. Lancet 2018, 391, 1842–1852. [Google Scholar] [CrossRef]
- Barker, D.J. The fetal and infant origins of adult disease. BMJ 1990, 301, 1111. [Google Scholar] [CrossRef]
- Vickers, M.H. Developmental Programming and Transgenerational Transmission of Obesity. Ann. Nutr. Metab. 2014, 64, 26–34. [Google Scholar] [CrossRef]
- Rando, O.J.; Simmons, R.A. I’m Eating for Two: Parental Dietary Effects on Offspring Metabolism. Cell 2015, 161, 93–105. [Google Scholar] [CrossRef]
- Clyburn, C.; Howe, C.A.; Arnold, A.C.; Lang, C.H.; Travagli, R.A.; Browning, K.N. Perinatal high-fat diet alters development of GABAA receptor subunits in dorsal motor nucleus of vagus. Am. J. Physiol. Liver Physiol. 2019, 317, G40–G50. [Google Scholar] [CrossRef] [PubMed]
- Rinaman, L.; Banihashemi, L.; Koehnle, T.J. Early life experience shapes the functional organization of stress-responsive visceral circuits. Physiol. Behav. 2011, 104, 632–640. [Google Scholar] [CrossRef]
- Walker, C.-D. Development, brain plasticity and reward: Early high-fat diet exposure confers vulnerability to obesity—View from the chair. Int. J. Obes. Suppl. 2012, 2, S3–S6. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Vucetic, Z.; Kimmel, J.; Totoki, K.; Hollenbeck, E.; Reyes, T.M. Maternal High-Fat Diet Alters Methylation and Gene Expression of Dopamine and Opioid-Related Genes. Endocrinology 2010, 151, 4756–4764. [Google Scholar] [CrossRef]
- Day, J.; Savani, S.; Krempley, B.D.; Nguyen, M.; Kitlinska, J.B. Influence of paternal preconception exposures on their offspring: Through epigenetics to phenotype. Am. J. Stem Cells 2016, 5, 11–18. [Google Scholar]
- Smith, J.K. Exercise, Obesity and CNS Control of Metabolic Homeostasis: A Review. Front. Physiol. 2018, 9, 574. [Google Scholar] [CrossRef]
- Sasaki, T. Neural and Molecular Mechanisms Involved in Controlling the Quality of Feeding Behavior: Diet Selection and Feeding Patterns. Nutrients 2017, 9, 1151. [Google Scholar] [CrossRef] [PubMed]
- Hopf, F.W. Do specific NMDA receptor subunits act as gateways for addictive behaviors? Genes Brain Behav. 2016, 16, 118–138. [Google Scholar] [CrossRef] [PubMed]
- Adams, R.C.; Sedgmond, J.; Maizey, L.; Chambers, C.D.; Lawrence, N.S. Food Addiction: Implications for the Diagnosis and Treatment of Overeating. Nutrients 2019, 11, 2086. [Google Scholar] [CrossRef]
- Wang, G.-J.; Volkow, N.; Logan, J.; Pappas, N.R.; Wong, C.T.; Zhu, W.; Netusll, N.; Fowler, J.S. Brain dopamine and obesity. Lancet 2001, 357, 354–357. [Google Scholar] [CrossRef]
- Thanos, P.K.; Michaelides, M.; Subrize, M.; Miller, M.L.; Bellezza, R.; Cooney, R.N.; Leggio, L.; Wang, G.-J.; Rogers, A.M.; Volkow, N.D.; et al. Roux-en-Y Gastric Bypass Alters Brain Activity in Regions that Underlie Reward and Taste Perception. PLoS ONE 2015, 10, e0125570. [Google Scholar] [CrossRef]
- Daws, L.C.; Avison, M.J.; Robertson, S.D.; Niswender, K.D.; Galli, A.; Saunders, C. Insulin signaling and addiction. Neuropharmacology 2011, 61, 1123–1128. [Google Scholar] [CrossRef]
- Volkow, N.; Wang, G.-J.; Fowler, J.S.; Telang, F. Overlapping neuronal circuits in addiction and obesity: Evidence of systems pathology. Philos. Trans. R. Soc. B Biol. Sci. 2008, 363, 3191–3200. [Google Scholar] [CrossRef]
- Kalon, E.; Hong, J.; Tobin, C.; Schulte, T. Psychological and Neurobiological Correlates of Food Addiction. Int. Rev. Neurobiol. 2016, 129, 85–110. [Google Scholar] [CrossRef] [PubMed]
- Albaugh, V.L.; Flynn, C.R.; Tamboli, R.A.; Abumrad, N.N. Recent advances in metabolic and bariatric surgery. F1000Research 2016, 5, 978. [Google Scholar] [CrossRef]
- Jáuregui-Lobera, I.; Martínez-Quiñones, J.V. Neuromodulation in eating disorders and obesity: A promising way of treatment? Neuropsychiatr. Dis. Treat. 2018, 14, 2817–2835. [Google Scholar] [CrossRef] [PubMed]
- Stoeckel, L.E.; Kim, J.; Weller, R.E.; Cox, J.E.; Cook, E.W.; Horwitz, B. Effective connectivity of a reward network in obese women. Brain Res. Bull. 2009, 79, 388–395. [Google Scholar] [CrossRef]
- Sanak, M.; Filip, M. Cocaine-induced Changes in the Expression of NMDA Receptor Subunits. Curr. Neuropharmacol. 2019, 17, 1039–1055. [Google Scholar] [CrossRef]
- Stojek, M.M.; Fischer, S.; MacKillop, J. Stress, cues, and eating behavior. Using drug addiction paradigms to understand motivation for food. Appetite 2015, 92, 252–260. [Google Scholar] [CrossRef] [PubMed]
- Bove, C.; Travagli, R.A. The vagus nerve. In Encyclopedia of Gastroenterology, 2nd ed.; Academic Press: Oxford, UK, 2020; pp. 676–682. [Google Scholar]
- Liddle, R.A. Neuropods. Cell. Mol. Gastroenterol. Hepatol. 2019, 7, 739–747. [Google Scholar] [CrossRef] [PubMed]
- Raskov, H.; Burcharth, J.; Pommergaard, H.-C.; Rosenberg, J. Irritable bowel syndrome, the microbiota and the gut-brain axis. Gut Microbes 2016, 7, 365–383. [Google Scholar] [CrossRef] [PubMed]
- Bülbül, M.; Travagli, R.A. Novel transmitters in brain stem vagal neurocircuitry: New players on the pitch. Am. J. Physiol. Liver Physiol. 2018, 315, G20–G26. [Google Scholar] [CrossRef]
- Kentish, S.J.; Page, A.J. The role of gastrointestinal vagal afferent fibers in obesity. J. Physiol. 2015, 593, 775–786. [Google Scholar] [CrossRef] [PubMed]
- Blasi, C. The Role of the Vagal Nucleus Tractus Solitarius in the Therapeutic Effects of Obesity Surgery and Other Interventional Therapies on Type 2 Diabetes. Obes. Surg. 2016, 26, 3045–3057. [Google Scholar] [CrossRef]
- Schneeberger, M.; Gomis, R.; Claret, M. Hypothalamic and brainstem neuronal circuits controlling homeostatic energy balance. J. Endocrinol. 2014, 220, T25–T46. [Google Scholar] [CrossRef]
- Morton, G.J.; Cummings, D.E.; Baskin, D.G.; Barsh, G.S.; Schwartz, M.W. Central nervous system control of food intake and body weight. Nat. Cell Biol. 2006, 443, 289–295. [Google Scholar] [CrossRef]
- Van Der Klaauw, A.A. Neuropeptides in Obesity and Metabolic Disease. Clin. Chem. 2018, 64, 173–182. [Google Scholar] [CrossRef]
- Essner, R.A.; Smith, A.G.; Jamnik, A.A.; Ryba, A.R.; Trutner, Z.D.; Carter, M.E. AgRP Neurons Can Increase Food Intake during Conditions of Appetite Suppression and Inhibit Anorexigenic Parabrachial Neurons. J. Neurosci. 2017, 37, 8678–8687. [Google Scholar] [CrossRef]
- Perry, C.A.; Pravetoni, M.; Teske, J.A.; Aguado, C.; Erickson, D.J.; Medrano, J.F.; Luján, R.; Kotz, C.M.; Wickman, K. Predisposition to late-onset obesity in GIRK4 knockout mice. Proc. Natl. Acad. Sci. USA 2008, 105, 8148–8153. [Google Scholar] [CrossRef] [PubMed]
- Kloukina, V.; Herzer, S.; Karlsson, N.; Pérez, M.; Daraio, T.; Meister, B. G-protein-gated inwardly rectifying K+ channel 4 (GIRK4) immunoreactivity in chemically defined neurons of the hypothalamic arcuate nucleus that control body weight. J. Chem. Neuroanat. 2012, 44, 14–23. [Google Scholar] [CrossRef]
- Kotz, C.M.; Teske, J.A.; Billington, C.J. Neuroregulation of nonexercise activity thermogenesis and obesity resistance. Am. J. Physiol. Integr. Comp. Physiol. 2008, 294, R699–R710. [Google Scholar] [CrossRef]
- Bunney, P.; Zink, A.; Holm, A.; Billington, C.; Kotz, C. Orexin activation counteracts decreases in nonexercise activity thermogenesis (NEAT) caused by high-fat diet. Physiol. Behav. 2017, 176, 139–148. [Google Scholar] [CrossRef]
- Li, B.; Zhuang, Q.-X.; Gao, H.-R.; Wang, J.-J.; Zhu, J.-N. Medial cerebellar nucleus projects to feeding-related neurons in the ventromedial hypothalamic nucleus in rats. Brain Struct. Funct. 2016, 222, 957–971. [Google Scholar] [CrossRef] [PubMed]
- Mahler, P.; Guastavino, J.-M.; Jacquart, G.; Strazielle, C. An unexpected role of the cerebellum: Involvement in nutritional organization. Physiol. Behav. 1993, 54, 1063–1067. [Google Scholar] [CrossRef]
- Van Der Laan, L.N.; De Ridder, D.T.D.; Viergever, M.A.; Smeets, P.A.M. The first taste is always with the eyes: A meta-analysis on the neural correlates of processing visual food cues. NeuroImage 2011, 55, 296–303. [Google Scholar] [CrossRef]
- Gautier, J.F.; Chen, K.; Salbe, A.D.; Bandy, D.; Pratley, R.E.; Heiman, M.; Ravussin, E.; Reiman, E.M.; Tataranni, P.A. Differential brain responses to satiation in obese and lean men. Diabetes 2000, 49, 838–846. [Google Scholar] [CrossRef]
- Mueller, K.; Sacher, J.; Arelin, K.; Holiga, Š.; Kratzsch, J.; Villringer, A.; Schroeter, M.L. Overweight and obesity are associated with neuronal injury in the human cerebellum and hippocampus in young adults: A combined MRI, serum marker and gene expression study. Transl. Psychiatry 2012, 2, e200. [Google Scholar] [CrossRef]
- Heinitz, S.; Reinhardt, M.; Piaggi, P.; Weise, C.M.; Diaz, E.; Stinson, E.J.; Venti, C.; Votruba, S.B.; Wassermann, E.M.; Alonso-Alonso, M.; et al. Neuromodulation directed at the prefrontal cortex of subjects with obesity reduces snack food intake and hunger in a randomized trial. Am. J. Clin. Nutr. 2017, 106, 1347–1357. [Google Scholar] [CrossRef] [PubMed]
- Le, D.S.N.T.; Pannacciulli, N.; Chen, K.; Del Parigi, A.; Salbe, A.D.; Reiman, E.M.; Krakoff, J. Less activation of the left dorsolateral prefrontal cortex in response to a meal: A feature of obesity. Am. J. Clin. Nutr. 2006, 84, 725–731. [Google Scholar] [CrossRef] [PubMed]
- Carnell, S.; Gibson, C.; Benson, L.; Ochner, C.N.; Geliebter, A. Neuroimaging and obesity: Current knowledge and future directions. Obes. Rev. 2011, 13, 43–56. [Google Scholar] [CrossRef] [PubMed]
- Elliott, J.A.; Reynolds, J.V.; Le Roux, C.W.; Docherty, N.G. Physiology, pathophysiology and therapeutic implications of enteroendocrine control of food intake. Expert Rev. Endocrinol. Metab. 2016, 11, 475–499. [Google Scholar] [CrossRef] [PubMed]
- Clark, L. Disordered gambling: The evolving concept of behavioral addiction. Ann. N. Y. Acad. Sci. 2014, 1327, 46–61. [Google Scholar] [CrossRef]
- Pak, K.; Kim, S.-J.; Kim, I.J. Obesity and Brain Positron Emission Tomography. Nucl. Med. Mol. Imaging 2017, 52, 16–23. [Google Scholar] [CrossRef]
- Zhang, Y.E.; Wang, J.; Zhang, G.; Zhu, Q.; Cai, W.; Tian, J.; Miller, J.L.; Wen, X.; Ding, M.; Gold, M.S.; et al. The neurobiological drive for overeating implicated in Prader–Willi syndrome. Brain Res. 2015, 1620, 72–80. [Google Scholar] [CrossRef]
- Angulo, M.A.; Butler, M.G.; Cataletto, M.E. Prader-Willi syndrome: A review of clinical, genetic, and endocrine findings. J. Endocrinol. Investig. 2015, 38, 1249–1263. [Google Scholar] [CrossRef]
- Salminen, I.I.; Crespi, B.J.; Mokkonen, M. Baby food and bedtime: Evidence for opposite phenotypes from different genetic and epigenetic alterations in Prader-Willi and Angelman syndromes. SAGE Open Med. 2019, 7. [Google Scholar] [CrossRef]
- Miller, J.L.; James, G.A.; Goldstone, A.P.; Couch, J.A.; He, G.; Driscoll, D.J.; Liu, Y. Enhanced activation of reward mediating prefrontal regions in response to food stimuli in Prader-Willi syndrome. J. Neurol. Neurosurg. Psychiatry 2007, 78, 615–619. [Google Scholar] [CrossRef]
- Ogura, K.; Fujii, T.; Abe, N.; Hosokai, Y.; Shinohara, M.; Fukuda, H.; Mori, E. Regional cerebral blood flow and abnormal eating behavior in Prader–Willi syndrome. Brain Dev. 2013, 35, 427–434. [Google Scholar] [CrossRef]
- Grüters-Kieslich, A.; Reyes, M.; Sharma, A.; Demirci, C.; DeClue, T.J.; Lankes, E.; Tiosano, D.; Schnabel, D.; Jüppner, H. Early-Onset Obesity: Unrecognized First Evidence for GNAS Mutations and Methylation Changes. J. Clin. Endocrinol. Metab. 2017, 102, 2670–2677. [Google Scholar] [CrossRef] [PubMed]
- Cornejo-Pareja, I.; Clemente-Postigo, M.; Tinahones, F.J. Metabolic and Endocrine Consequences of Bariatric Surgery. Front. Endocrinol. 2019, 10, 626. [Google Scholar] [CrossRef] [PubMed]
- Peters, J.H.; Gallaher, Z.R.; Ryu, V.; Czaja, K. Withdrawal and restoration of central vagal afferents within the dorsal vagal complex following subdiaphragmatic vagotomy. J. Comp. Neurol. 2013, 521, 3584–3599. [Google Scholar] [CrossRef] [PubMed]
- Raghow, R. Gut-brain crosstalk regulates craving for fatty food. World J. Diabetes 2017, 8, 484–488. [Google Scholar] [CrossRef]
- Kittrell, H.; Graber, W.; Mariani, E.; Czaja, K.; Hajnal, A.; DiLorenzo, P. Taste and odor preferences following Roux-en-Y surgery in humans. PLoS ONE 2018, 13, e0199508. [Google Scholar] [CrossRef]
- Ochner, C.N.; Stice, E.; Hutchins, E.; Afifi, L.; Geliebter, A.; Hirsch, J.; Teixeira, J. Relation between changes in neural responsivity and reductions in desire to eat high-calorie foods following gastric bypass surgery. Neuroscience 2012, 209, 128–135. [Google Scholar] [CrossRef]
- Eickhoff, H. Central Modulation of Energy Homeostasis and Cognitive Performance After Bariatric Surgery. Perinat. Program. Neurodev. 2017, 19, 213–236. [Google Scholar] [CrossRef]
- Nauck, M.A.; Meier, J.J. Incretin hormones: Their role in health and disease. Diabetes Obes. Metab. 2018, 20, 5–21. [Google Scholar] [CrossRef]
- Brown, E.; Cuthbertson, D.J.; Wilding, J.P. Newer GLP-1 receptor agonists and obesity-diabetes. Peptides 2018, 100, 61–67. [Google Scholar] [CrossRef]
- Pi-Sunyer, X.; Astrup, A.; Fujioka, K.; Greenway, F.; Halpern, A.; Krempf, M.; Lau, D.C.; Le Roux, C.W.; Violante Ortiz, R.; Jensen, C.B.; et al. SCALE Obesity and prediabetes NN8022-1839 study group. A randomized, controlled trial of 3.0 mg of liraglutide in weight management. N. Engl. J. Med. 2015, 373, 11–22. [Google Scholar] [CrossRef]
- O’Neil, P.M.; Birkenfeld, A.L.; McGowan, B.; Mosenzon, O.; Pedersen, S.D.; Wharton, S.; Carson, C.G.; Jepsen, C.H.; Kabisch, M.; Wilding, J.P.H. Efficacy and safety of semaglutide compared with liraglutide and placebo for weight loss in patients with obesity: A randomised, double-blind, placebo and active controlled, dose-ranging, phase 2 trial. Lancet 2018, 392, 637–649. [Google Scholar] [CrossRef]
- Hutch, C.R.; Sandoval, D.A. The Role of GLP-1 in the Metabolic Success of Bariatric Surgery. Endocrinology 2017, 158, 4139–4151. [Google Scholar] [CrossRef] [PubMed]
- Ye, J.; Hao, Z.; Mumphrey, M.B.; Townsend, R.L.; Patterson, L.M.; Stylopoulos, N.; Münzberg, H.; Morrison, C.D.; Drucker, D.J.; Berthoud, H.-R. GLP-1 receptor signaling is not required for reduced body weight after RYGB in rodents. Am. J. Physiol. Integr. Comp. Physiol. 2014, 306, R352–R362. [Google Scholar] [CrossRef] [PubMed]
- El Khoury, L.; Chouillard, E.; Chahine, E.; Saikaly, E.; Debs, T.; Kassir, R. Metabolic Surgery and Diabesity: A Systematic Review. Obes. Surg. 2018, 28, 2069–2077. [Google Scholar] [CrossRef]
- Browning, K.N.; Fortna, S.R.; Hajnal, A. Roux-en-Y gastric bypass reverses the effects of diet-induced obesity to inhibit the responsiveness of central vagal motoneurones. J. Physiol. 2013, 591, 2357–2372. [Google Scholar] [CrossRef]
- Huang, K.P.; Goodson, M.L.; Vang, W.; Li, H.; Page, A.J.; Raybould, H.E. Leptin signaling in vagal afferent neurons supports the absorption and storage of nutrients from high-fat diet [published online ahead of print, 2020 Sep 11]. Int. J. Obes. 2020, 10, 103. [Google Scholar]
- Kim, J.; Kirkland, R.; Lee, S.; Cawthon, C.; Rzepka, K.; Minaya, D.; De Lartigue, G.; Czaja, K.; De La Serre, C. Gut microbiota composition modulates inflammation and structure of the vagal afferent pathway. Physiol. Behav. 2020, 225, 113082. [Google Scholar] [CrossRef]
- Pérez-Pevida, B.; Escalada, J.; Miras, A.D.; Frühbeck, G. Mechanisms Underlying Type 2 Diabetes Remission After Metabolic Surgery. Front. Endocrinol. 2019, 10, 641. [Google Scholar] [CrossRef]
- Meyers, E.E.; Kronemberger, A.; Lira, V.; Rahmouni, K.; Stauss, H.M. Contrasting effects of afferent and efferent vagal nerve stimulation on insulin secretion and blood glucose regulation. Physiol. Rep. 2016, 4, e12718. [Google Scholar] [CrossRef]
- Laessle, C.; Nenova, G.; Marjanovic, G.; Seifert, G.; Kousoulas, L.; Jaenigen, B.; Fichtner-Feigl, S.; Fink, J.M. Duodenal Exclusion but Not Sleeve Gastrectomy Preserves Insulin Secretion, Making It the More Effective Metabolic Procedure. Obes. Surg. 2017, 28, 1408–1416. [Google Scholar] [CrossRef]
- Rubino, F. Is Type 2 Diabetes an Operable Intestinal Disease: A provocative yet reasonable hypothesis. Diabetes Care 2008, 31. [Google Scholar] [CrossRef] [PubMed]
- Gero, D.; Steinert, R.E.; Hosa, H.; Cummings, D.E.; Bueter, M. Appetite, Glycemia, and Entero-Insular Hormone Responses Differ Between Oral, Gastric-Remnant, and Duodenal Administration of a Mixed-Meal Test After Roux-en-Y Gastric Bypass. Diabetes Care 2018, 41, 1295–1298. [Google Scholar] [CrossRef]
- Duca, F.A.; Bauer, P.V.; Hamr, S.C.; Lam, T.K. Glucoregulatory Relevance of Small Intestinal Nutrient Sensing in Physiology, Bariatric Surgery, and Pharmacology. Cell Metab. 2015, 22, 367–380. [Google Scholar] [CrossRef] [PubMed]
- Scarlett, J.M.; Schwartz, M.W. Gut-brain mechanisms controlling glucose homeostasis. F1000Prime Rep. 2015, 7, 12. [Google Scholar] [CrossRef]
- Uno, K. Roles of the inter-organ neuronal network in the development of metabolic syndrome. Diabetol. Int. 2016, 7, 205–211. [Google Scholar] [CrossRef]
- Lin, Y.; Liang, Z.; He, L.; Yang, M.; Liu, D.; Gu, H.F.; Liu, H.; Zhu, Z.; Zheng, H.; Li, L.; et al. Gut ghrelin regulates hepatic glucose production and insulin signaling via a gut-brain-liver pathway. Cell Commun. Signal. 2019, 17, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Perry, R.J.; Peng, L.; Cline, G.W.; Wang, Y.; Rabin-Court, A.; Song, J.D.; Zhang, D.; Zhang, X.-M.; Nozaki, Y.; Dufour, S.; et al. Mechanisms by which a Very-Low-Calorie Diet Reverses Hyperglycemia in a Rat Model of Type 2 Diabetes. Cell Metab. 2018, 27, 210–217.e3. [Google Scholar] [CrossRef]
- Abbasi, J. Unveiling the “Magic” of Diabetes Remission After Weight-Loss Surgery. JAMA 2017, 317, 571. [Google Scholar] [CrossRef]
- Rasmussen, B.A.; Breen, D.M.; Luo, P.; Cheung, G.W.; Yang, C.S.; Sun, B.; Kokorovic, A.; Rong, W.; Lam, T.K. Duodenal Activation of cAMP-Dependent Protein Kinase Induces Vagal Afferent Firing and Lowers Glucose Production in Rats. Gastroenterology 2012, 142, 834–843.e3. [Google Scholar] [CrossRef]
- Cheung, G.W.; Kokorovic, A.; Lam, C.K.; Chari, M.; Lam, T.K. Intestinal Cholecystokinin Controls Glucose Production through a Neuronal Network. Cell Metab. 2009, 10, 99–109. [Google Scholar] [CrossRef]
- Waise, T.M.Z.; Rasti, M.; Duca, F.A.; Zhang, S.-Y.; Bauer, P.V.; Rhodes, C.J.; Lam, T.K.T. Inhibition of upper small intestinal mTOR lowers plasma glucose levels by inhibiting glucose production. Nat. Commun. 2019, 10, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Kezic, A.; Popovic, L.; Lalic, N.M. mTOR Inhibitor Therapy and Metabolic Consequences: Where Do We Stand? Oxidative Med. Cell. Longev. 2018, 2018, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Lumsden, A.L.; Martin, A.M.; Sun, E.W.; Schober, G.; Isaacs, N.J.; Pezos, N.; Wattchow, D.A.; De Fontgalland, D.; Rabbitt, P.; Hollington, P.; et al. Sugar Responses of Human Enterochromaffin Cells Depend on Gut Region, Sex, and Body Mass. Nutrients 2019, 11, 234. [Google Scholar] [CrossRef]
- Benaiges, D.; Más-Lorenzo, A.; Goday, A.; Ramon, J.M.; Benaiges, D.; Pedro-Botet, J.; Roux, J.A.F.-L. Laparoscopic sleeve gastrectomy: More than a restrictive bariatric surgery procedure? World J. Gastroenterol. 2015, 21, 11804–11814. [Google Scholar] [CrossRef] [PubMed]
- Chambers, A.P.; Smith, E.P.; Begg, D.P.; Grayson, B.E.; Sisley, S.; Greer, T.; Sorrell, J.; Lemmen, L.; LaSance, K.; Woods, S.C.; et al. Regulation of gastric emptying rate and its role in nutrient-induced GLP-1 secretion in rats after vertical sleeve gastrectomy. Am. J. Physiol. Metab. 2014, 306, E424–E432. [Google Scholar] [CrossRef]
- Anderson, B.; Switzer, N.J.; Almamar, A.; Shi, X.; Birch, D.W.; Karmali, S. The Impact of Laparoscopic Sleeve Gastrectomy on Plasma Ghrelin Levels: A Systematic Review. Obes. Surg. 2013, 23, 1476–1480. [Google Scholar] [CrossRef]
- Chieffi, S.; Carotenuto, M.; Monda, V.; Valenzano, A.; Villano, I.; Precenzano, F.; Tafuri, D.; Salerno, M.; Filippi, N.; Nuccio, F.; et al. Orexin System: The Key for a Healthy Life. Front. Physiol. 2017, 8, 357. [Google Scholar] [CrossRef]
- Aron-Wisnewsky, J.; Prifti, E.; Belda, E.; Ichou, F.; Kayser, B.D.; Dao, M.C.; Verger, E.O.; Hedjazil, L.; Bouillot, J.-L.; Chevallier, J.-M.; et al. Major microbiota dysbiosis in severe obesity: Fate after bariatric surgery. Gut 2019, 68, 70–82. [Google Scholar] [CrossRef]
- Green, M.; Arora, K.; Prakash, S. Microbial Medicine: Prebiotic and Probiotic Functional Foods to Target Obesity and Metabolic Syndrome. Int. J. Mol. Sci. 2020, 21, 2890. [Google Scholar] [CrossRef]
- Vaughn, A.C.; Cooper, E.M.; DiLorenzo, P.M.; O’Loughlin, L.J.; Konkel, M.E.; Peters, J.H.; Hajnal, A.; Sen, T.; Lee, S.H.; De La Serre, C.B.; et al. Energy‑dense diet triggers changes in gut microbiota, reorganization of gut‑brain vagal communication and increases body fat accumulation. Acta Neurobiol. Exp. 2017, 77, 18–30. [Google Scholar] [CrossRef]
- Maqsood, R.; Stone, T.W. The Gut-Brain Axis, BDNF, NMDA and CNS Disorders. Neurochem. Res. 2016, 41, 2819–2835. [Google Scholar] [CrossRef]
- Goldman, J.M.; Wheeler, M.F. Remission of Diabetes After Irradiation of Head and Neck. Diabetes Care 1987, 10, 137–138. [Google Scholar] [CrossRef] [PubMed]
- Raheja, B.S.; Motwani, B.T.; Mehta, A.R.; Kumar, V.D. Remission of NIDDM After Irradiation of Metastatic Cervical Lymph Nodes. Diabetes Care 1986, 9, 101–103. [Google Scholar] [CrossRef] [PubMed]
- Rex, D.; Duckworth, W.C. Remission of Overt Diabetes Mellitus After Removal of an Oral Epidermoid Carcinoma. Am. J. Med. Sci. 1984, 287, 43–45. [Google Scholar] [CrossRef]
- Rubino, F.; Nathan, D.M.; Eckel, R.H. Metabolic surgery in the treatment algorithm for type 2 diabetes: A joint statement by international diabetes organizations. Obes. Surg. 2017, 27, 2–21. [Google Scholar] [CrossRef] [PubMed]
- Üner, A.; Gonçalves, G.H.; Li, W.; Porceban, M.; Caron, N.; Schönke, M.; Delpire, E.; Sakimura, K.; Bjorbaek, C. The role of GluN2A and GluN2B NMDA receptor subunits in AgRP and POMC neurons on body weight and glucose homeostasis. Mol. Metab. 2015, 4, 678–691. [Google Scholar] [CrossRef] [PubMed]
- Wu, Q.; Zheng, R.; Srisai, D.; McKnight, G.S.; Palmiter, R.D. NR2B subunit of the NMDA glutamate receptor regulates appetite in the parabrachial nucleus. Proc. Natl. Acad. Sci. USA 2013, 110, 14765–14770. [Google Scholar] [CrossRef] [PubMed]
- Minaya, D.M.; Larson, R.W.; Podlasz, P.; Czaja, K. Glutamate-dependent regulation of food intake is altered with age through changes in NMDA receptor phenotypes on vagal afferent neurons. Physiol. Behav. 2018, 189, 26–31. [Google Scholar] [CrossRef]
- Debacker, J.; Hawken, E.R.; Normandeau, C.P.; Jones, A.A.; Di Prospero, C.; Mechefske, E.; Gregory, J.G.; Hayton, S.J.; Dumont, É.C. GluN2B-Containing NMDA Receptors Blockade Rescues Bidirectional Synaptic Plasticity in the Bed Nucleus of the Stria Terminalis of Cocaine Self-Administering Rats. Neuropsychopharmacology 2014, 40, 394–405. [Google Scholar] [CrossRef]
- Goldsmith, P.J. NMDAR PAMs: Multiple Chemotypes for Multiple Binding Sites. Curr. Top. Med. Chem. 2019, 19, 2239–2253. [Google Scholar] [CrossRef]
- Hackos, D.H.; Hanson, J.E. Diverse modes of NMDA receptor positive allosteric modulation: Mechanisms and consequences. Neuropharmacology 2017, 112, 34–45. [Google Scholar] [CrossRef] [PubMed]
- Nickols, H.H.; Conn, P.J. Development of allosteric modulators of GPCRs for treatment of CNS disorders. Neurobiol. Dis. 2014, 61, 55–71. [Google Scholar] [CrossRef] [PubMed]
- Agabio, R.; Colombo, G. GABAB receptor as therapeutic target for drug addiction: From baclofen to positive allosteric modulators. Psychiatr. Pol. 2015, 49, 215–223. [Google Scholar] [CrossRef] [PubMed]
- Martel, J.C.; McArthur, S.G. Dopamine Receptor Subtypes, Physiology and Pharmacology: New Ligands and Concepts in Schizophrenia. Front. Pharmacol. 2020, 11, 1003. [Google Scholar] [CrossRef] [PubMed]
- Botticelli, L.; Di Bonaventura, E.M.; Del Bello, F.; Giorgioni, G.; Piergentili, A.; Eromano, A.; Quaglia, W.; Cifani, C.; Di Bonaventura, M.V.E. Underlying Susceptibility to Eating Disorders and Drug Abuse: Genetic and Pharmacological Aspects of Dopamine D4 Receptors. Nutrients 2020, 12, 2288. [Google Scholar] [CrossRef] [PubMed]
- Astrup, A.; Madsbad, S.; Breum, L.; Jensen, T.J.; Kroustrup, J.P.; Larsen, T. Effect of tesofensine on bodyweight loss, body composition, and quality of life in obese patients: A randomised, double-blind, placebo-controlled trial. Lancet 2008, 372, 1906–1913. [Google Scholar] [CrossRef]
- Urban, D.J.; Roth, B.L. DREADDs (Designer Receptors Exclusively Activated by Designer Drugs): Chemogenetic Tools with Therapeutic Utility. Annu. Rev. Pharmacol. Toxicol. 2015, 55, 399–417. [Google Scholar] [CrossRef] [PubMed]
- Bohula, E.A.; Scirica, B.M.; Inzucchi, S.E.; McGuire, D.K.; Keech, A.C.; Smith, S.R.; Kanevsky, E.; Murphy, A.S.; Leiter, A.L.; Dwyer, J.P.; et al. Effect of lorcaserin on prevention and remission of type 2 diabetes in overweight and obese patients (CAMELLIA-TIMI 61): A randomised, placebo-controlled trial. Lancet 2018, 392, 2269–2279. [Google Scholar] [CrossRef]
- Faris, P.L.; Kim, S.W.; Meller, W.H.; Goodale, R.L.; Oakman, A.S.; Hofbauer, R.D.; Marshall, A.M.; Daughters, R.S.; Banerjee-Stevens, D.; Eckert, E.D.; et al. Effect of decreasing afferent vagal activity with ondansetron on symptoms of bulimia nervosa: A randomised, double-blind trial. Lancet 2000, 355, 792–797. [Google Scholar] [CrossRef]
- Zhu, H.; Roth, B.L. Silencing Synapses with DREADDs. Neuron 2014, 82, 723–725. [Google Scholar] [CrossRef]
- Zink, A.N.; Bunney, E.P.; Holm, A.A.; Billington, C.J.; Kotz, C.M. Neuromodulation of orexin neurons reduces diet-induced adiposity. Int. J. Obes. 2017, 42, 737–745. [Google Scholar] [CrossRef]
- Ferguson, S.M.; Neumaier, J.F. Grateful DREADDs: Engineered Receptors Reveal How Neural Circuits Regulate Behavior. Neuropsychopharmacology 2011, 37, 296–297. [Google Scholar] [CrossRef] [PubMed]
- Yue, J.T.Y.; Abraham, M.A.; Bauer, P.V.; Lapierre, M.P.; Wang, P.; Duca, F.A.; Filippi, B.M.; Chan, O.; Lam, T.K. Inhibition of glycine transporter-1 in the dorsal vagal complex improves metabolic homeostasis in diabetes and obesity. Nat. Commun. 2016, 7, 13501. [Google Scholar] [CrossRef] [PubMed]
- Campos, C.A.; Wright, J.S.; Czaja, K.; Ritter, R.C. CCK-Induced Reduction of Food Intake and Hindbrain MAPK Signaling Are Mediated by NMDA Receptor Activation. Endocrinology 2012, 153, 2633–2646. [Google Scholar] [CrossRef] [PubMed]
- Wikberg, J.E.S.; Mutulis, F. Targeting melanocortin receptors: An approach to treat weight disorders and sexual dysfunction. Nat. Rev. Drug Discov. 2008, 7, 307–323. [Google Scholar] [CrossRef]
- Johannessen, H.; Revesz, D.; Kodama, Y.; Cassie, N.; Skibicka, K.P.; Barrett, P.; Dickson, S.; Holst, J.; Rehfeld, J.; Van Der Plasse, G.; et al. Vagal Blocking for Obesity Control: A Possible Mechanism-Of-Action. Obes. Surg. 2016, 27, 177–185. [Google Scholar] [CrossRef]
- Prologo, J.D. Percutaneous CT-Guided Cryovagotomy. Tech. Vasc. Interv. Radiol. 2020, 23, 100660. [Google Scholar] [CrossRef] [PubMed]
- Hays, S.A.; Rennaker, R.L.; Kilgard, M.P. Targeting Plasticity with Vagus Nerve Stimulation to Treat Neurological Disease. Chang. Brains Appl. Brain Plast. Adv. Recover Hum. Abil. 2013, 207, 275–299. [Google Scholar] [CrossRef]
- Childs, J.E.; Alvarez-Dieppa, A.C.; McIntyre, C.K.; Kroener, S. Vagus Nerve Stimulation as a Tool to Induce Plasticity in Pathways Relevant for Extinction Learning. J. Vis. Exp. 2015, 102, e53032. [Google Scholar] [CrossRef]
- Han, W.; Tellez, L.A.; Perkins, M.H.; Perez, I.O.; Qu, T.; Ferreira, J.; Ferreira, T.L.; Quinn, D.; Liu, Z.W.; Gao, X.B.; et al. A neural circuit for gut-induced reward. Cell 2018, 175, 665–678. [Google Scholar] [CrossRef]
- Dalton, B.L.; Campbell, I.C.; Schmidt, U.H. Neuromodulation and neurofeedback treatments in eating disorders and obesity. Curr. Opin. Psychiatry 2017, 30, 458–473. [Google Scholar] [CrossRef] [PubMed]
- Whiting, A.C.; Oh, M.Y.; Whiting, D.M. Deep brain stimulation for appetite disorders: A review. Neurosurg. Focus 2018, 45, E9. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Wei, N.-L.; Wang, Y.; Wang, X.; Zhang, J.-G.; Zhang, K. Deep brain stimulation of the nucleus accumbens shell induces anti-obesity effects in obese rats with alteration of dopamine neurotransmission. Neurosci. Lett. 2015, 589, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Franco, R.R.; Fonoff, E.T.; Alvarenga, P.G.; Lopes, A.C.; Miguel, E.C.; Teixeira, M.J.; Damiani, D.; Hamani, C. DBS for Obesity. Brain Sci. 2016, 6, 21. [Google Scholar] [CrossRef]
- Harat, M.; Rudaś, M.; Zieliński, P.; Birska, J.; Sokal, P. Nucleus accumbens stimulation in pathological obesity. Neurol. Neurochir. Pol. 2016, 50, 207–210. [Google Scholar] [CrossRef]
- Gorelick, D.A.; Zangen, A.; George, M.S. Transcranial magnetic stimulation in the treatment of substance addiction. Ann. N. Y. Acad. Sci. 2014, 1327, 79–93. [Google Scholar] [CrossRef]
- Ljubisavljevic, M.; Maxood, K.; Bjekic, J.; Oommen, J.; Nagelkerke, N. Long-Term Effects of Repeated Prefrontal Cortex Transcranial Direct Current Stimulation (tDCS) on Food Craving in Normal and Overweight Young Adults. Brain Stimul. 2016, 9, 826–833. [Google Scholar] [CrossRef]
- Lee, D.J.; Elias, G.J.; Lozano, A.M. Neuromodulation for the treatment of eating disorders and obesity. Ther. Adv. Psychopharmacol. 2017, 8, 73–92. [Google Scholar] [CrossRef]
- Hall, P.A.; Vincent, C.M.; Burhan, A.M. Non-invasive brain stimulation for food cravings, consumption, and disorders of eating: A review of methods, findings and controversies. Appetite 2018, 124, 78–88. [Google Scholar] [CrossRef]
- López-Camarillo, C.; Gallardo-Rincón, D.; Álvarez-Sánchez, M.E.; Marchat, L.A. Pharmaco-epigenomics: On the Road of Translation Medicine. Adv. Exp. Med. Biol. 2019, 1168, 31–42. [Google Scholar] [CrossRef]
- Chandhok, N.S.; Prebet, T. Insights into novel emerging epigenetic drugs in myeloid malignancies. Ther. Adv. Hematol. 2019, 10. [Google Scholar] [CrossRef] [PubMed]
- Stephenson, J.; Heslehurst, N.; Hall, J.; Schoenaker, D.A.; Hutchinson, J.; Cade, J.E.; Poston, L.; Barrett, G.; Crozier, S.R.; Barker, M.; et al. Before the beginning: Nutrition and lifestyle in the preconception period and its importance for future health. Lancet 2018, 391, 1830–1841. [Google Scholar] [CrossRef]



| Main Sector | 1st Subgroup | 2nd Subgroup |
|---|---|---|
| Post-synaptic glutamate and GABA receptors | NMDA receptors | Subunits GluN2A and GluN2B |
| NMDA receptor co-agonists | Inhibition of glycine transporters or block of the glycine site | |
| Memantine: blocks excessive activation of the NMDA receptor by glutamate | ||
| Gamma-amino-butyric acid (GABA) receptors | GABAB’s positive allosteric modulators (PAM ADX71441) | |
| 5-HT3 agonists and antagonists | 5-HT2C agonist lorcaserin | Type 3 serotonin receptor antagonist Ondansetron |
| Guanine protein-coupled receptors (GPCR) | Activation of designer drug receptors (DREADD) | Muscarinic M3 DREADD receptor (hM4Di) |
| Glycine transporter 1 (GlyT1) | Inhibitor of GlyT1 | |
| Melanocortin 4 receptor (MC4R) | MC3/4R agonists | |
| Opioid system | Opioid antagonist, Naltrexone μ receptor agonists | |
| Neuropeptide orexin | DREADD-dependent activation of orexin neurons | |
| GIRK4-containing channels |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Blasi, C. Obesity and Related Type 2 Diabetes: A Failure of the Autonomic Nervous System Controlling Gastrointestinal Function? Gastrointest. Disord. 2020, 2, 423-447. https://doi.org/10.3390/gidisord2040039
Blasi C. Obesity and Related Type 2 Diabetes: A Failure of the Autonomic Nervous System Controlling Gastrointestinal Function? Gastrointestinal Disorders. 2020; 2(4):423-447. https://doi.org/10.3390/gidisord2040039
Chicago/Turabian StyleBlasi, Claudio. 2020. "Obesity and Related Type 2 Diabetes: A Failure of the Autonomic Nervous System Controlling Gastrointestinal Function?" Gastrointestinal Disorders 2, no. 4: 423-447. https://doi.org/10.3390/gidisord2040039
APA StyleBlasi, C. (2020). Obesity and Related Type 2 Diabetes: A Failure of the Autonomic Nervous System Controlling Gastrointestinal Function? Gastrointestinal Disorders, 2(4), 423-447. https://doi.org/10.3390/gidisord2040039