Proposal for an Organ-Specific Chronic Inflammation–Remodeling–Carcinoma Sequence

Abstract
1. Introduction
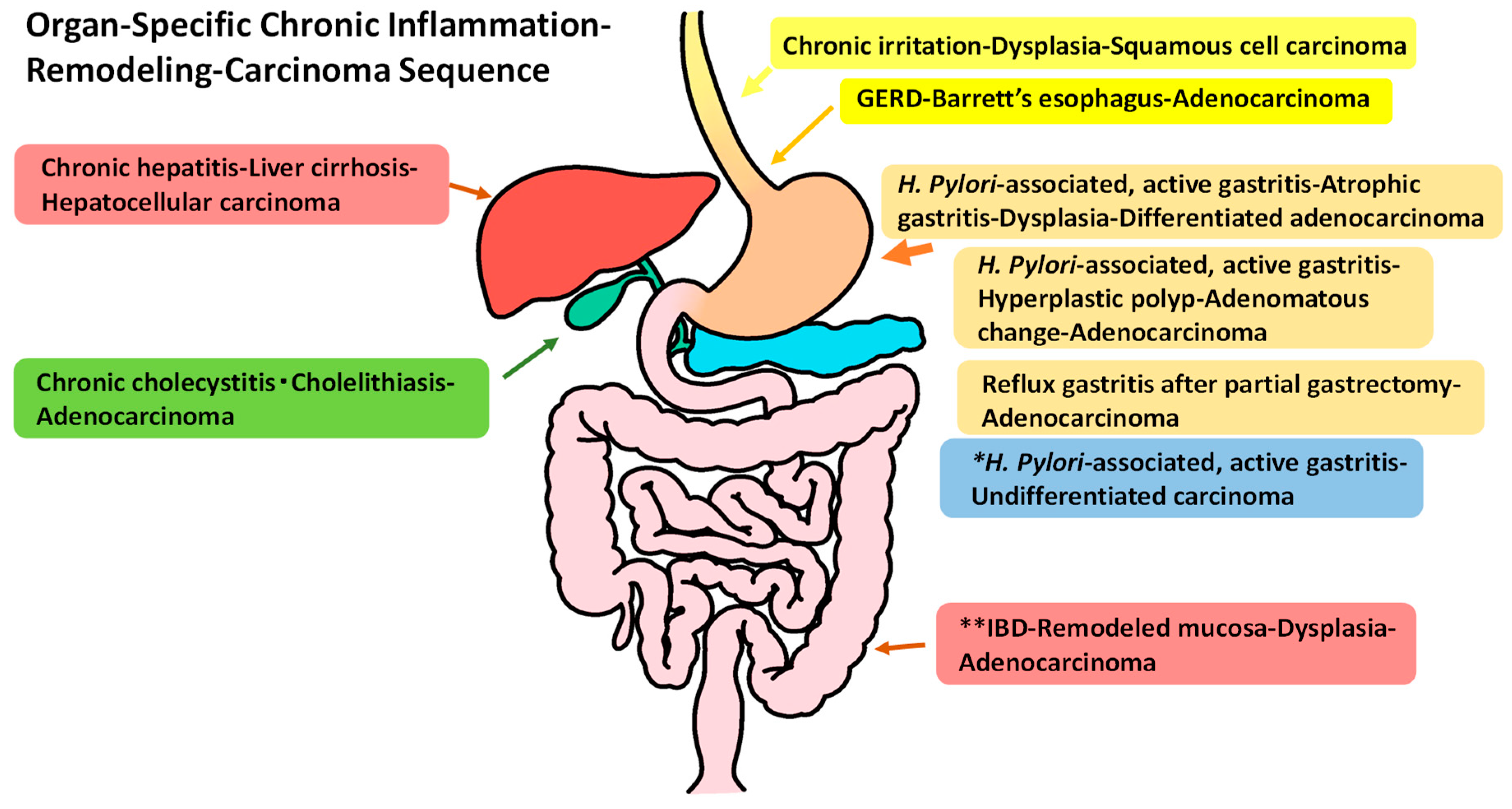
2. Organ-Specific Chronic Inflammation–Remodeling–Carcinoma Sequence
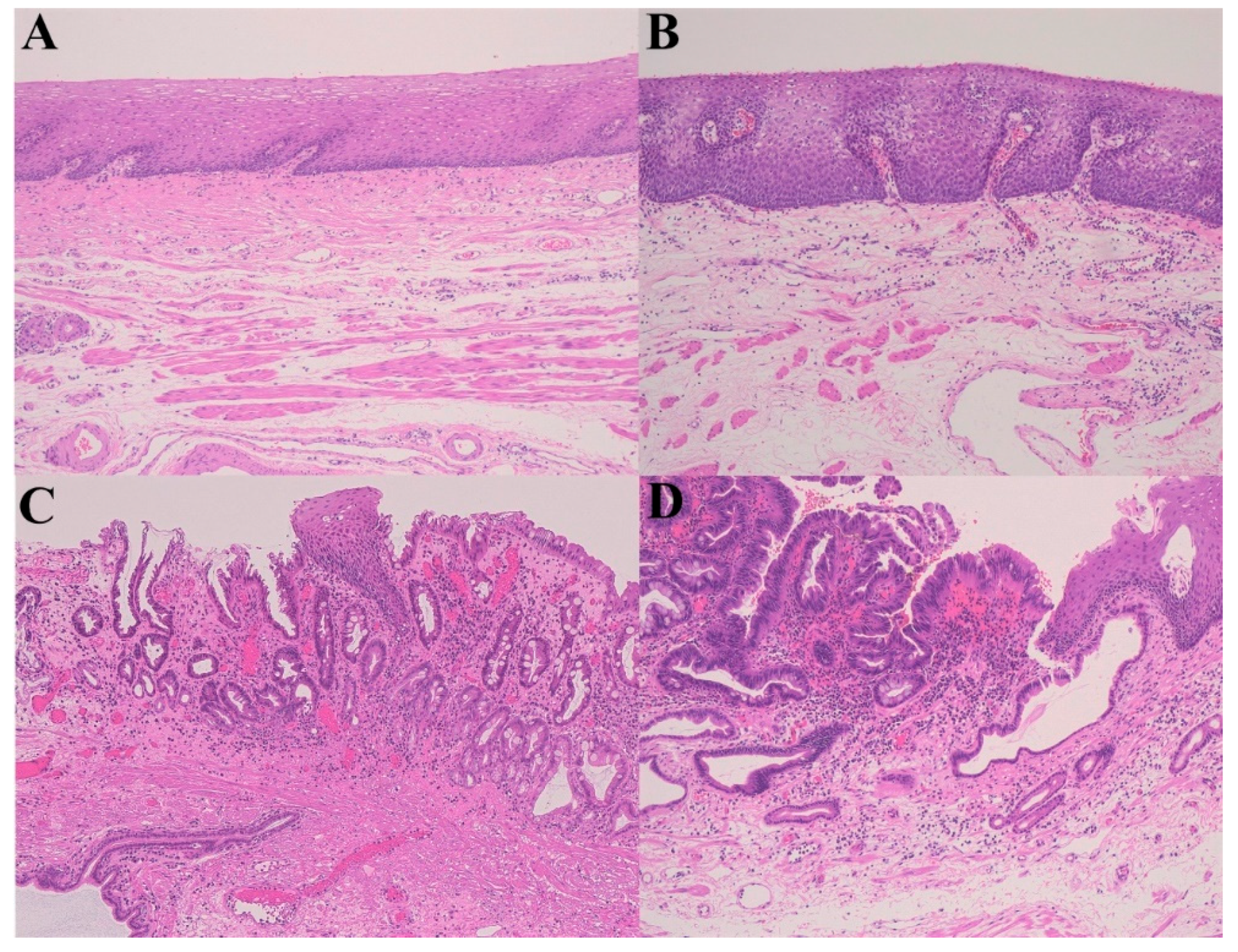
2.1. GERD–Barrett’s Esophagus (Mucosal Remodeling)–Adenocarcinoma Sequence)
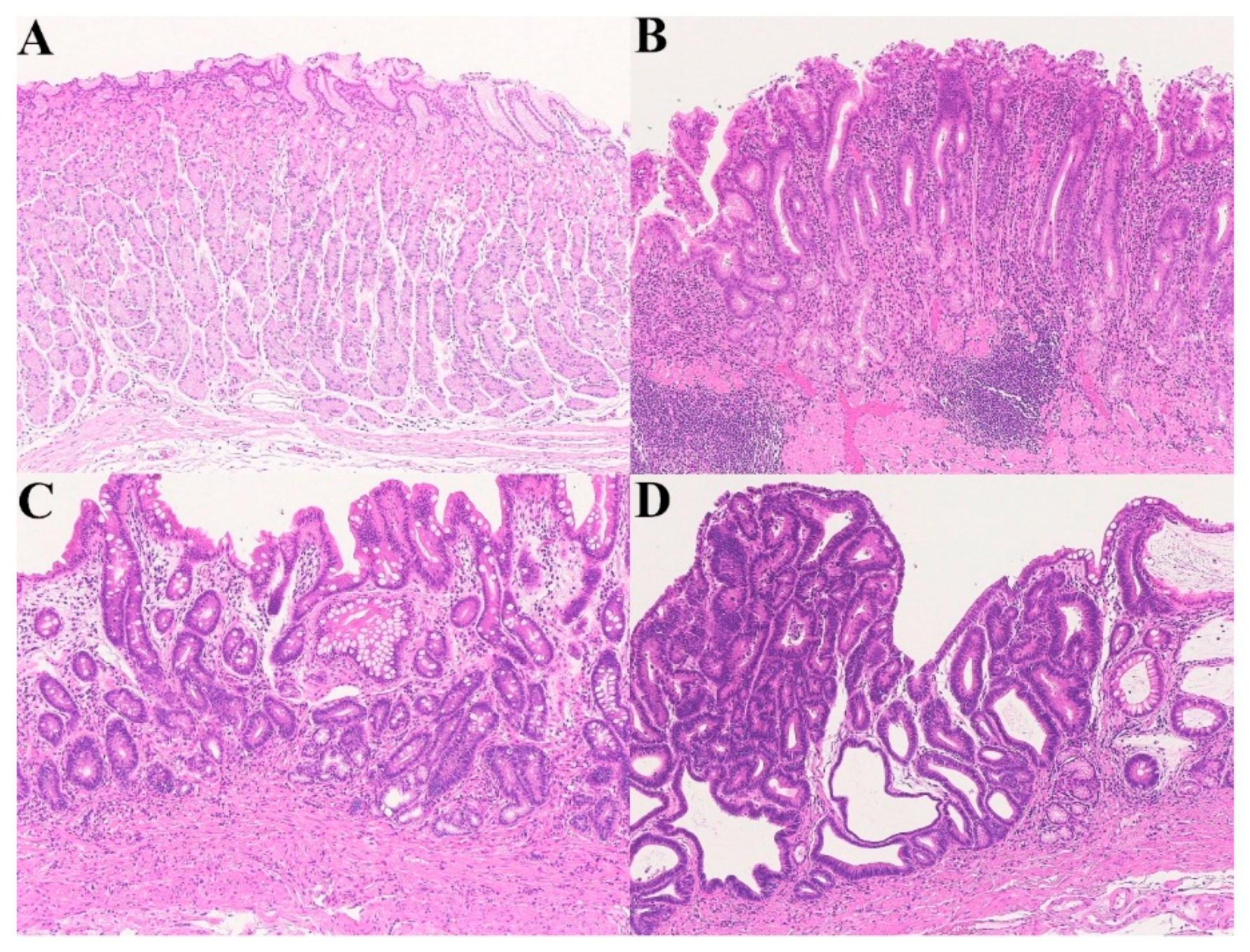
2.2. Chronic Active Gastritis–Atrophic Gastritis (Mucosal Remodeling)–Carcinoma Sequence
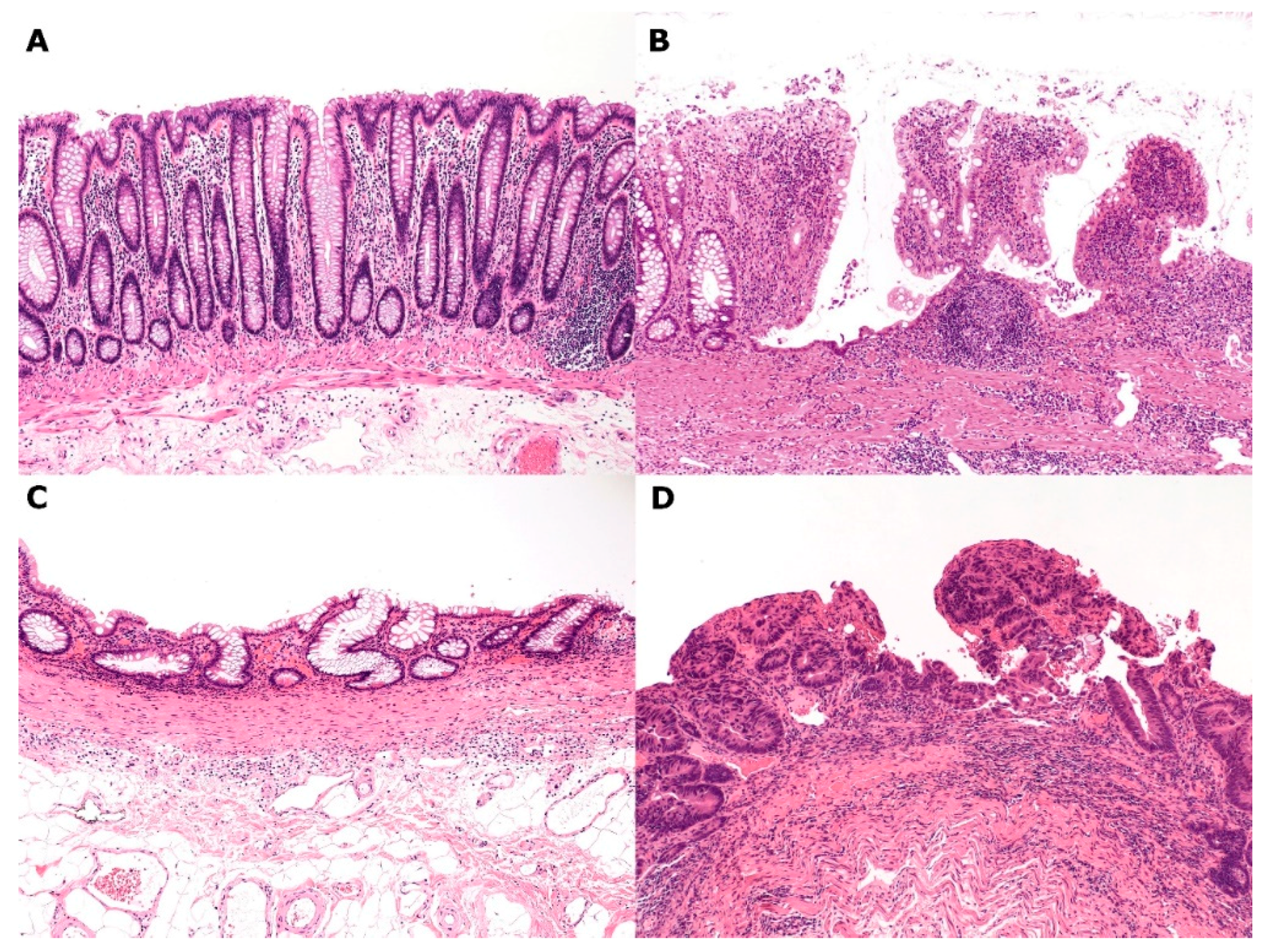
2.3. IBD (Ulcerative Colitis and Crohn’s Disease of the Colitis Type)–Regenerative Mucosa (Mucosal Remodeling)–Dysplasia–Carcinoma Sequence
3. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Okayasu, I. Development of ulcerative colitis and its associated colorectal neoplasia as a model of the organ-specific chronic inflammation-carcinoma sequence. Pathol. Int. 2012, 62, 368–380. [Google Scholar] [CrossRef] [PubMed]
- Okayasu, I.; Yoshida, T.; Mikami, T.; Mitsuhashi, J.; Ichinoe, M.; Yanagisawa, N.; Tokunaga, W.; Hana, K.; Ishibashi, Y. Mucosal remodeling and alteration of stromal microenvironment in ulcerative colitis as related to colorectal tumorigenesis. In Ulcerative Colitis-Epidemiology, Pathogenesis and Complications; O’Connor, M.B., Ed.; Intech Open Access Publisher: Rijeka, Croatia, 2011; pp. 241–256. ISBN 978-953-307-880-9. [Google Scholar]
- Yanagisawa, N.; Mikami, T.; Koike, M.; Okayasu, I. Enhanced cell kinetics, p53 accumulation and high p21WAF1 expression in chronic cholecystitis: Comparison with background mucosa of gallbladder carcinomas. Histopathology 2000, 36, 54–61. [Google Scholar] [CrossRef] [PubMed]
- Yanagisawa, N.; Mikami, T.; Yamashita, K.; Okayasu, I. Microsatellite instability in chronic cholecystitis is indicative of an early stage in gallbladder carcinogenesis. Am. J. Clin. Pathol. 2003, 120, 413–417. [Google Scholar] [CrossRef] [PubMed]
- Salimian, K.J.; Waters, K.M.; Eze, O.; Pezhouh, M.K.; Tarabishy, Y.; Shin, E.J.; Canto, M.I.; Voltaggio, L.; Montgomery, E.A. Definition of Barrett’s esophagus in the United States: Support for retention of a requirement for goblet cells. Am. J. Surg. Pathol. 2018, 42, 264–268. [Google Scholar] [CrossRef] [PubMed]
- Mukaisho, K.; Kanai, S.; Kushima, R.; Nakayama, T.; Hattori, T.; Sugihara, H. Barrett’s carcinogenesis. Pathol. Int. 2019, 69, 319–330. [Google Scholar]
- Bremner, C.G.; Lynch, V.P.; Ellis, F.H. Barrett’s oesophagus: Congenital or acquired? An experimental study of oesophageal mucosal regeneration in the dog. Surgery 1970, 68, 209–216. [Google Scholar] [PubMed]
- Tatsuta, T.; Mukaisho, K.; Sugihara, H.; Miwa, K.; Tani, K.; Hattori, T. Expression of Cdx2 in early GRCL of Barrett’s esophagus induced in rats by duodenal reflux. Dig. Dis. Sci. 2005, 50, 425–431. [Google Scholar] [CrossRef]
- Quante, M.; Bhagat, G.; Abrams, J.A.; Marache, F.; Good, P.; Lee, M.D.; Lee, Y.; Friedman, R.; Asfaha, S.; Dubeykovskaya, Z.; et al. Bile acid and inflammation activate gastric cardia stem cells in a mouse model of Barrett-like metaplasia. Cancer Cell 2012, 21, 36–51. [Google Scholar] [CrossRef]
- Kanai, S.; Mukaisho, K.; Yoshida, S.; Taniura, N.; Sugihara, H. Host factors influence Barrett’s carcinogenesis: Findings from a mouse gastroduodenal reflux model. Esophagus 2019, 16, 264–271. [Google Scholar] [CrossRef]
- Solaymani-Dodaran, M.; Logan, R.F.; West, J.; Card, T.; Coupland, C. Risk of oesophageal cancer in Barrett’s oesophagus and gastro-oesophageal reflux. Gut 2004, 53, 1070–1074. [Google Scholar] [CrossRef]
- Anaparthy, R.; Gaddam, S.; Kanakadandi, V.; Alsop, B.R.; Gupta, N.; Higbee, A.D.; Wani, S.B.; Singh, M.; Rastogi, A.; Bansal, A.; et al. Association between length of Barrett’s esophagus and risk of high-grade dysplasia or adenocarcinoma in patients without dysplasia. Clin. Gastroenterol. Hepatol. 2013, 11, 1430–1436. [Google Scholar] [CrossRef] [PubMed]
- Solanky, D.; Kirshnamoorthi, R.; Crews, N.; Johnson, M.; Wang, K.; Walfsen, H.; Fleischer, D.; Ramirez, F.C.; Katzka, D.; Buttar, N.; et al. Barrett esophagus length, nodularity, and low-grade dysplasia are predictive of progression to esophageal adenocarcinoma. J. Clin. Gastroenterol. 2019, 53, 361–365. [Google Scholar] [CrossRef] [PubMed]
- Caygill, C.P.J.; Watson, A.; Lao-Sirieix, P.L.; Fitzgerald, R.C. Barrett’s oesophagus and adenocarcinoma. World J. Surg. Oncol. 2004, 2, 12. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Rahman, F.B.; Kadowaki, Y.; Ishihara, S.; Tobita, H.; Imaoka, H.; Fukuhara, H.; Aziz, M.M.; Furuta, K.; Amano, Y.; Kinoshita, Y. Fibroblast-derived HB-EGF promotes Cdx2 expression in esophageal squamous cells. Lab. Invest. 2010, 90, 1033–1048. [Google Scholar] [CrossRef]
- Soslow, R.A.; Petersen, C.G.; Remotti, H.; Altorki, N. Acidic fibroblast growth factor is expressed sequentially in the progression from Barrett’s esophagus to esophageal adenocarcinoma. Dis. Esophagus 2001, 14, 23–27. [Google Scholar] [CrossRef] [PubMed]
- Fléjou, J.F. Barrett’s oesophagus: From metaplasia to dysplasia and cancer. Gut 2005, 54, i6–i12. [Google Scholar] [CrossRef]
- Hamelin, R.; Flejou, J.F.; Muzeau, F.; Potet, F.; Laurent-Puig, P.; Fekete, F.; Thomas, G. TP53 gene mutations and p53 protein immunoreactivity in malignant and premalignant Barrett’s oesophagus. Gastroenterology 1994, 17, 1012–1018. [Google Scholar] [CrossRef]
- Casson, A.G.; Manolopoulos, B.; Troster, M.; Kerkvliet, N.; O’Malley, F.; Inculet, R.; Finley, R.; Roth, J.A. Clinical implications of 53 gene mutation in the progression of Barrett’s epithelium to invasive oesophageal cancer. Am. J. Surg. 1994, 167, 52–57. [Google Scholar] [CrossRef]
- Bani-Hani, K.; Martin, I.G.; Hardie, L.J.; Mapstone, N.; Briggs, J.A.; Forman, D.; Wild, C.P. Prospective study of cyclin D1 overexpression in Barrett’s oesophagus: Association with increased risk of adenocarcinoma. J. Natl. Cancer Inst. 2000, 2, 1316–1321. [Google Scholar] [CrossRef]
- Huang, Y.; Boynton, R.F.; Blount, P.L.; Silverstein, R.J.; Yin, J.; Tong, Y.; McDaniel, T.K.; Newkirk, C.; Resau, J.H.; Sridhara, R.; et al. Loss of heterozygosity involves multiple tumor suppressor genes in human esophageal cancers. Cancer Res. 1992, 52, 6525–6530. [Google Scholar]
- Sarbia, M.; Bektas, N.; Muller, W.; Heep, H.; Borchard, F.; Gabbert, H.E. Expression of cyclin E in dysplasia, carcinoma, and nonmalignant lesions of Barrett’s oesophagus. Cancer 1999, 86, 2597–2601. [Google Scholar] [CrossRef]
- Yu, M.; Maden, S.K.; Stachler, M.; Kaz, A.M.; Ayers, J.; Guo, Y.; Carter, K.T.; Wilbanks, A.; Heinzerling, T.J.; O’Leary, R.M.; et al. Subtypes of Barrett’s oesophagus and oesophageal adenocarcinoma based on genome-wide methylation analysis. Gut 2018. [Google Scholar] [CrossRef] [PubMed]
- Shiraishi, H.; Mikami, T.; Yoshida, T.; Tanabe, S.; Kobayashi, N.; Watanabe, M.; Okayasu, I. Early genetic instability of both epithelial and stromal cells in esophageal squamous cell carcinomas, contrasted with Barrett’s adenocarcinomas. J. Gastroenterol. 2006, 41, 1186–1196. [Google Scholar] [CrossRef] [PubMed]
- Shiraishi, H.; Mikami, T.; Aida, J.; Nakamura, K.; Izumiyama-Shimomur, N.; Arai, T.; Watanabe, M.; Okayasu, I.; Takubo, K. Telomere shortening in Barrett’s mucosa and esophageal adenocarcinoma and its association with loss of heterozygosity. Scand. J. Gastoenterol. 2009, 44, 538–544. [Google Scholar] [CrossRef] [PubMed]
- Altaf, K.; Xiong, J.J.; la Iglesia, D.; Hicke, L.; Kaul, A. Meta-analysis of biomarkers predicting risk of malignant progression in Barrett’s oesophagus. Br. J. Surg. 2017, 104, 493–502. [Google Scholar] [CrossRef] [PubMed]
- Castro, C.; Peleteiro, B.; Lunet, N. Modifiable factors and esophageal cancer: A systematic review of published meta-analyses. J. Gastroentrol. 2018, 53, 37–51. [Google Scholar] [CrossRef]
- Yang, X.; Chen, X.; Zhuang, M.; Yuan, Z.; Nie, S.; Lu, M.; Jin, L.; Ye, W. Smoking and alcohol drinking in relation to the risk of esophageal squamous cell carcinoma: A population-based case-control study in China. Sci. Rep. 2017, 7, 17249. [Google Scholar] [CrossRef]
- Sawada, G.; Niida, A.; Uchi, R.; Hirata, H.; Shimamua, T.; Suzuki, Y.; Shiraishi, Y.; Chiba, K.; Imoto, S.; Takahashi, Y.; et al. Genomic landscape of esophageal squamous cell carcinoma in a Japanese population. Gastroenterology 2016, 150, 1171–1182. [Google Scholar] [CrossRef]
- Takubo, K.; Fujita, M.; Izumiyama, N.; Nakamura, K.; Ishikawa, N.; Poon, S.S.; Fujiwara, M.; Sawabe, M.; Matsuura, M.; Grabsch, H.; et al. Q-FISH analysis of telomere and chromosome instability in the oesophagus with and without squamous cell carcinoma in situ. J. Pathol. 2010, 221, 201–209. [Google Scholar] [CrossRef]
- Takubo, K.; Fujii, S. Oesophageal squamous dysplasia. In Digestive System Tumours, 5th ed.; WHO Classification of Tumours; IARC: Lyon, France, 2019; pp. 36–37. [Google Scholar]
- Hibi, K.; Mitomi, H.; Koizumi, W.; Tanabe, S.; Saigenji, K.; Okayasu, I. Enhanced cellular proliferation and p53 accumulation in gastric mucosa chronically infected with Helicobacter pylori. Am. J. Clin. Pathol. 1997, 108, 26–34. [Google Scholar] [CrossRef]
- Ichinoe, M.; Mikami, T.; Hara, A.; Tsuruta, T.; Okayasu, I. Background submucosal cysts in early gastric cancer cases have unique clinicopathologic features suggestive of postgastritis and significant smoking association. Am. J. Clin. Pathol. 2007, 128, 746–762. [Google Scholar] [CrossRef] [PubMed]
- Koshida, Y.; Koizumi, W.; Sasabe, M.; Kato, Y.; Okayasu, I. Association of Helicobacter pylori-dependent gastritis with gastric carcinomas in young Japanese patients: Histopathological comparison of diffuse and intestinal type cancer cases. Histopathology 2000, 37, 124–130. [Google Scholar] [CrossRef] [PubMed]
- Horiuchi, Y.; Fujisaki, J.; Yamamoto, N.; Ishizuka, N.; Omae, M.; Ishiyama, A.; Yoshio, T.; Hirasawa, T.; Yamamoto, Y.; Nagahama, M.; et al. Mixed poorly differentiated adenocarcinoma in undifferentiated-type early gastric cancer predicts endoscopic noncurative resection. Gastric Cancer 2018, 21, 689–695. [Google Scholar] [CrossRef] [PubMed]
- Kwak, D.S.; Min, Y.W.; Lee, J.H.; Kang, S.H.; Jang, S.H.; Lee, H.; Min, B.H.; Kim, J.J.; Kim, K.M.; Sohn, T.S.; et al. Outcomes of endoscopic submucosal dissection for early gastric cancer with undifferentiated-type histology: A clinical simulation using a non-selected surgical cohort. Gut Liver 2018, 12, 23–270. [Google Scholar] [CrossRef] [PubMed]
- Ansari, S.; Gantuya, B.; Tuan, V.P.; Yamaoka, Y. Diffuse gastric cancer: A summary of analogous contributing factors for its molecular pathogenicity. Int. J. Mol. Sci. 2018, 19, 2424. [Google Scholar] [CrossRef] [PubMed]
- Yoshiura, K.; Kanai, Y.; Ochiai, A.; Shimoyama, Y.; Sugimura, T.; Hirohashi, S. Silencing of the E-cadherin invasion-suppressor gene by CpG methylation in human carcinomas. Proc. Natl. Acad. Sci. USA 1995, 92, 7416–7419. [Google Scholar] [CrossRef]
- Fukudome, Y.; Yanagihara, K.; Takeichi, M.; Ito, F.; Shibamoto, S. Characterization of a mutant E-cadherin protein encoded by a mutant gene frequently seen in diffuse-type human gastric carcinoma. Int. J. Cancer 2000, 88, 579–583. [Google Scholar] [CrossRef]
- Fricke, E.; Keller, G.; Becker, I.; Rosivatz, E.; Schott, C.; Plaschke, S.; Rudelius, M.; Hemannstăndter, C.; Busch, R.; Höfler, H.; et al. Relationship between E-cadherin gene mutation and p53 gene mutation, p53 accumulation, Bcl-2 expression and Ki-67 staining in diffuse-type gastric carcinoma. Int. J. Cancer 2003, 104, 60–65. [Google Scholar] [CrossRef]
- Huang, F.Y.; Chan, A.O.; Rashid, A.; Wong, D.K.; Cho, C.H.; Yuen, MF. Helicobacter pylori induces promoter methylation of E-cadherin via interleukin-1 activation of nitric oxide production in gastric cancer cells. Cancer 2012, 118, 4969–4980. [Google Scholar] [CrossRef]
- Chan, A.O.-O.; Lam, S.-K.; Wong, B.C.-Y.; Wong, W.-M.; Yuen, M.-F.; Yeung, Y.-H.; Hui, W.-M.; Rashid, A.; Kwong, Y.-L.. Promotor methylation of E-cadherin gene in gastric mucosa associated with Helicobacter pylori infection and in gastric cancer. Gut 2003, 52, 502–506. [Google Scholar] [CrossRef]
- Murakami, K.; Mitomi, H.; Yamashita, K.; Tanabe, S.; Saigenji, K.; Okayasu, I. p53, but not c-Ki-ras, mutation and down-regulation of p21WAF1/CIP1 and cyclin D1 are associated with malignant transformation in gastric hyperplastic polyps. Am. J. Clin. Pathol. 2001, 115, 224–234. [Google Scholar] [CrossRef] [PubMed]
- Tanigawa, H.; Uesugi, H.; Mitomi, H.; Saigenji, K.; Okayasu, I. Possible association of active gastritis, featuring accelerated cell turnover and p53 overexpression, with cancer development at anastomoses after gastrojejunostomy. Comparison with gastroduodenostomy. Am. J. Clin. Pathol. 2000, 114, 354–363. [Google Scholar] [CrossRef] [PubMed]
- Fukayama, M.; Kunita, A.; Kaneda, A. Gastritis-infection-cancer sequence of Epstein-Barr virus associated gastric cancer. Adv. Exp. Med. Biol. 2018, 1045, 437–457. [Google Scholar] [PubMed]
- Matsuo, T.; Ito, M.; Takata, S.; Tanaka, S.; Yoshihara, M.; Chayama, K. Low prevalence of Helicobacter pylori-negative gastric cancer among Japanese. Helicobacter 2011, 16, 415–419. [Google Scholar] [CrossRef] [PubMed]
- Kwak, H.W.; Choi, I.J.; Cho, S.J.; Lee, J.Y.; Kim, C.G.; Kook, M.C.; Ryu, K.W.; Kim, Y.W. Characteristics of gastric cancer according to Helicobacter pylori infection status. J. Gastroenterol. Hepatol. 2014, 29, 1671–1677. [Google Scholar] [CrossRef]
- Shinozaki-Ushiku, A.; Kunita, A.; Isogai, M.; Hibiya, T.; Ushiku, T.; Takada, K.; Fukayama, M. Profiling of virus-encoded microRNAs in Epstein-Barr virus-associated gastric carcinomas and their roles in gastric carcinogenesis. J. Viol. 2015, 89, 5581–5591. [Google Scholar] [CrossRef] [PubMed]
- Bayerdörffer, E.; Neubauer, A.; Rudolph, B.; Thiede, C.; Lehn, N.; Eidt, S.; Stolte, M.; MALT lymphoma study group. Regression of primary gastric lymphoma of mucosa-associated lymphoid tissue type after cure of Helicobacter pylori infection. Lancet 1995, 345, 1591–1594. [Google Scholar] [CrossRef]
- Parsonnet, J.; Fiedman, G.D.; Vandersteen, D.P.; Chang, Y.; Vogelman, J.H.; Orentreich, N.; Sibley, R.K. Helicobacter pylori infection and the risk of gastric carcinoma. N. Engl. J. Med. 1991, 325, 1127–1131. [Google Scholar] [CrossRef]
- Uemura, N.; Okamoto, S.; Yamamoto, S.; Matsumura, N.; Yamaguchi, S.; Yamakido, M.; Taniyama, K.; Sasaki, N.; Schlemper, R.J. Helicobacter pylori infection and the development of gastric cancer. N. Engl. J. Med. 2001, 345, 784–789. [Google Scholar] [CrossRef]
- Helicobacter and Cancer Collaborative Group. Gastric cancer and Helicobacter pylori: A combined analysis of 12 case control studies within prospective cohorts. Gut 2001, 49, 347–353. [Google Scholar] [CrossRef]
- Lee, Y.C.; Chiang, T.H.; Chou, C.K.; Tu, Y.K.; Liao, W.C.; Wu, M.S.; Graham, D.Y. Association between Helicobacter pylori eradication and gastric cancer incidence: A systemic review and meta-analysis. Gastroenterology 2016, 50, 1113–1124. [Google Scholar] [CrossRef] [PubMed]
- Inoue, M. Changing epidemiology of Helicobacter pylori in Japan. Gastric Cancer 2017, 20, S3–S7. [Google Scholar] [CrossRef] [PubMed]
- González, C.A.; Sanz-Anquela, J.M.; Companioni, O.; Bonet, C.; Berdasco, M.; López, C.; Mendoza, J.; Martin-Arranz, M.D.; Rey, E.; Poves, E.; et al. Incomplete type of intestinal metaplasia has the highest risk to progress to gastric cancer: Results of the Spanish follow-up multicenter study. J. Gastroenterol. Hepatol. 2016, 31, 953–958. [Google Scholar] [CrossRef] [PubMed]
- Li, D.; Bautista, M.C.; Jiang, S.F.; Daryani, P.; Brackett, M.; Armstrong, M.A.; Hung, Y.Y.; Postlethwaite, D.; Ladabaum, U. Risks and predictors of gastric adenocarcinoma in patients with gastric intestinal metaplasia and dysplasia: A population-based study. Am. J. Gastoenterol. 2016, 111, 1104–1113. [Google Scholar] [CrossRef] [PubMed]
- Lee, T.Y.; Wang, R.C.; Lee, Y.C.; Lin, J.T.; Ho, H.J.; Hsieh, M.C.; Wu, C.Y. The incidence of gastric adenocarcinoma among patients with gastric intestinal metaplasia: A long-term cohort study. Clin. Gastroenterol. 2016, 50, 532–537. [Google Scholar] [CrossRef] [PubMed]
- Salzman, N.H.; Hung, K.; Haribhai, D.; Chu, H.; Karisson-Sjöberg, J.; Amir, E.; Teggatz, P.; Barman, M.; Hayward, M.; Eastwood, D.; et al. Enteric defensins are essential regulators of intestinal microbial ecology. Nat. Immunol. 2010, 11, 76–83. [Google Scholar] [CrossRef] [PubMed]
- Gassler, N. Paneth cells in intestinal physiology and pathophysiology. World J. Gastrointest. Pathophysiol. 2017, 8, 150–160. [Google Scholar] [CrossRef]
- Chairatana, P.; Nolan, E.M. Defentins, lectins, mucins, and secretory immunoglobulin A: Microbe-binding biomolecules that contribute to mucosal immunity in the human gut. Crit. Rev. Biochem. Mol. Biol. 2017, 52, 45–56. [Google Scholar] [CrossRef]
- Camilo, V.; Garrido, M.; Valente, P.; Ricardo, S.; Amaral, A.L.; Barros, R.; Chaves, P.; Cameiro, F.; David, L.; Almeida, R. Differentiation reprogramming in gastric intestinal metaplasia and dysplasia: Role of SOX2 and CDX2. Histopathology 2015, 6, 243–350. [Google Scholar] [CrossRef]
- Bai, Y.Q.; Yamamoto, H.; Akiyama, Y.; Tanaka, H.; Takizawa, T.; Koike, M.; Yagi, K.O.; Saitoh, K.; Takeshita, K.; Iwai, T.; et al. Ectopic expression of homeodomain protein CDX2 in intestinal metaplasia and carcinoma of the stomach. Cancer Lett. 2002, 176, 47–55. [Google Scholar] [CrossRef]
- Yoda, T.; Takeshima, H.; Niwa, T.; Kim, J.G.; Ando, T.; Kushima, R.; Sugiyama, T.; Katai, H.; Noshiro, H.; Ushijima, T. Integrated analysis of cancer-related pathways affects by genetic and epigenetic alterations in gastric cancer. Gastric Cancer 2015, 18, 65–76. [Google Scholar] [CrossRef] [PubMed]
- Kiga, K.; Mimuro, H.; Suzuki, M.; Shinozaki-Ushiku, A.; Kobayashi, T.; Sanada, T.; Kim, M.; Ogawa, M.; Iwasaki, Y.W.; Kayo, H.; et al. Epigenetic silencing of miR-210 increases the proliferation of gastric epithelium during chronic Helicobacter pylori infection. Nat. Commun. 2014, 4497. [Google Scholar] [CrossRef] [PubMed]
- Muhammad, J.S.; Nanjo, S.; Ando, T.; Yamashita, S.; Maekita, T.; Ushijima, T.; Tabuchi, Y.; Sugiyama, T. Autophagy impairment by Helicobacter pylori-induced methylation silencing of MAP1LC3Av1 promotes gastric carcinogenesis. Int. J. Cancer 2017, 140, 2272–2283. [Google Scholar] [CrossRef] [PubMed]
- Wu, M.S.; Lee, C.W.; Shun, C.T.; Wang, H.P.; Lee, W.J.; Chang, M.C.; Sheu, J.C.; Lin, J.T. Distinct clinicopathologic and genetic profiles in sporadic gastric cancer with different mutator phenotypes. Genes Chromosomes Cancer 2000, 27, 403–411. [Google Scholar] [CrossRef]
- Zou, X.P.; Zhang, B.; Zhang, X.Q.; Chen, M.; Cao, J.; Liu, W.J. Promoter hypermethylation of multiple genes in early gastric adenocarcinoma and precancerous lesions. Hum. Pathol. 2009, 40, 1534–1542. [Google Scholar] [CrossRef] [PubMed]
- Mutoh, H.; Sakurai, S.; Satoh, K.; Osawa, H.; Tomiyama, T.; Kita, H.; Yoshida, T.; Tamada, K.; Yamamoto, H.; Isoda, N.; et al. Pericryptal fibroblast sheath in intestinal metaplasia and gastric carcinoma. Gut 2015, 54, 33–39. [Google Scholar] [CrossRef]
- Mutoh, H.; Sashikawa, M.; Hayakawa, H.; Sugano, H. Monocyte chemoattractant protein-1 is generated via TGF-beta by myofibroblasts in gastric intestinal metaplasia and carcinoma without H. pylori infection. Cancer Sci. 2010, 101, 1783–1789. [Google Scholar] [CrossRef] [PubMed]
- Kinoshita, H.; Hirata, Y.; Nakagawa, H.; Sakamoto, K.; Hayakawa, Y.; Takahashi, R.; Nakata, W.; Sakitani, K.; Serizawa, T.; Hikiba, Y.; et al. Interleukin-6 mediates epithelial-stromal interactions and promotes gastric tumorigenesis. PLoS ONE 2013, 8, e60914. [Google Scholar] [CrossRef]
- Lutgens, M.W.; van Oijen, M.G.; van der Heijden, G.J.; Vleggaar, F.P.; Sersema, P.D.; Oldenburg, B. Declining risk of colorectal cancer in inflammatory bowel disease: An updated meta-analysis of population-based cohort studies. Inflamm. Bowel Dis. 2013, 19, 789–799. [Google Scholar] [CrossRef]
- Beaugerie, L.; Svrcek, M.; Seksik, P.; Bouvier, A.M.; Simon, T.; Allez, M.; Brixi, H.; Gornet, J.M.; Altwegg, R.; Beau, P.; et al. Risk of colorectal high-grade dysplasia and cancer in a prospective observational cohort of patients with inflammatory bowel disease over 30 years. Gastroenterology 2013, 145, 166–175. [Google Scholar] [CrossRef]
- Beaugerie, L.; Itzkowitz, S.H. Cancers complicating inflammatory bowel disease. N. Engl. J. Med. 2015, 372, 1441–1452. [Google Scholar] [CrossRef] [PubMed]
- Arai, N.; Mitomi, H.; Ohtani, Y.; Igarashi, M.; Kakita, A.; Okayasu, I. Enhanced epithelial cell turnover associated with p53 accumulation and high p21WAF1/CIP1 expression in ulcerative colitis. Mod. Pathol. 1999, 12, 604–611. [Google Scholar] [PubMed]
- Mitsuhashi, J.; Mikami, T.; Saigenji, K.; Okayasu, I. Significant correlation of morphological remodeling in ulcerative colitis with disease duration and between elevated p53 and p21 expression in rectal mucosa and neoplastic development. Pathol. Int. 2005, 55, 113–121. [Google Scholar] [CrossRef]
- Yoshida, T.; Matsumoto, N.; Mikami, T.; Okayasu, I. Upregulation of p16INK4A and Bax in p53 wild/p53-overexpressing crypts in ulcerative colitis-associated tumours. Br. J. Cancer 2004, 91, 1081–1088. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Grivennikov, S.; Karin, E.; Terzic, J.; Mucida, D.; Yu, G.Y.; Vallabhapurapu, S.; Scheller, J.; Rose-John, S.; Cheroutre, H.; Eckmann, L.; et al. IL-6 and Stat3 are required for survival of intestinal epithelial cells and development of colitis-associated cancer. Cancer Cell 2009, 15, 103–113. [Google Scholar] [CrossRef] [PubMed]
- Bollrath, J.; Phesse, T.J.; von Burstin, V.A.; Putoczki, T.; Bennecke, M.; Bateman, T.; Nebelsiek, T.; Lungren-May, T.; Canli, Ö.; Schwitalla, S.; et al. gp-130-mediated Stat3 activation in enterocytes regulates cell survival and cell-cell cycle progression during colitis-associated tumorigenesis. Cancer Cell 2009, 15, 91–102. [Google Scholar] [CrossRef] [PubMed]
- Schmitz, H.; Fromm, M.; Bode, H.; Scholz, P.; Riecken, E.O.; Schulzke, J.D. Tumor necrosis factor-α induces Cl− and K+ secretion in human distal colon driven by prostaglandin E2. Am. J. Physiol. 1996, 271, G669–G674. [Google Scholar] [CrossRef]
- Bode, H.; Schmitz, H.; Fromm, M.; Scholz, P.; Riecken, E.O.; Schulzke, J.D. IL-1β and TNF-α, but not INF-α, IFN-γ, IL-6 or IL-8, are secretory mediators in human distal colon. Cytokine 1998, 10, 457–465. [Google Scholar] [CrossRef]
- Otani, T.; Yamaguchi, K.; Scherl, E.; Du, B.; Tai, H.H.; Greifer, M.; Petrovic, L.; Daikoku, T.; Dey, S.K.; Subbaramaiah, K.; et al. Levels of NAD(+)-dependent 15-hydroxyprostaglandin dehydrogenase are reduced in inflammatory bowel disease: Evidence for involvement of TNF-α. Am. J. Physiol. Gastrointest. Liver Physiol. 2006, 290, G361–G368. [Google Scholar] [CrossRef]
- Li, Y.; Soendergaad, C.; Bergenheim, F.H.; Aronoff, D.M.; Milne, G.; Riis, L.B.; Seidelin, J.B.; Jensen, K.B.; Nielsen, O.H. COX-2-PGE2 signaling impairs intestinal epithelial regeneration and associates with TNF inhibitor responsiveness in ulcerative colitis. EBioMedicine 2018, 36, 497–507. [Google Scholar] [CrossRef]
- Sasaki, S.; Hirata, I.; Maemura, K.; Hamamoto, N.; Murano, M.; Toshina, K.; Katsu, K. Prostaglandin E2 inhibits lesion formation in dextran sodium sulphate-induced colitis in rats and reduces the levels of mucosal inflammatory cytokines. Scand. J. Immunol. 2000, 51, 23–28. [Google Scholar] [CrossRef] [PubMed]
- Kawamata, S.; Matsuzaki, K.; Murata, M.; Seki, T.; Matsuoka, K.; Iwao, Y.; Hibi, T.; Okazaki, K. Oncogenic Smad3 signaling induced by chronic inflammation is an early event in ulcerative colitis-associated carcinogenesis. Inflamm. Bowel Dis. 2011, 17, 683–695. [Google Scholar] [CrossRef] [PubMed]
- Brentnall, T.A.; Cripin, D.A.; Rabinovitch, P.S.; Haggitt, R.C.; Rubin, C.E.; Sevens, A.C.; Burmer, G.C. Mutations in the p53 gene: An early marker of neoplastic progression in ulcerative colitis. Gastroenterology 1994, 107, 369–378. [Google Scholar] [CrossRef]
- Yoshida, T.; Mikami, T.; Mitomi, H.; Okayasu, I. Diverse p53 alterations in ulcerative colitis-associated low-grade dysplasia: Full-length gene sequencing in microdissected single crypts. J. Pathol. 2003, 199, 166–175. [Google Scholar] [CrossRef] [PubMed]
- Seril, D.N.; Liao, J.; Yang, G.Y.; Yang, C.S. Oxidative stress and ulcerative colitis-associated carcinogenesis: Studies in human and animal models. Carcinogenesis 2003, 24, 353–362. [Google Scholar] [CrossRef]
- Nishikawa, M.; Oshitani, N.; Matsumoto, T.; Nishigami, T.; Arakawa, T.; Inoue, M. Accumulation of mitochondrial DNA mutation with colorectal carcinogenesis in ulcerative colitis. Br. J. Cancer 2005, 93, 331–337. [Google Scholar] [CrossRef]
- Okayasu, I.; Hana, K.; Yoshida, T.; Mikami, T.; Kanno, J.; Fujiwara, M. Significant increase of colonic mutated crypts in ulcerative colitis correlatively with duration of illness. Cancer Res. 2002, 62, 2236–2238. [Google Scholar]
- Yoshida, T.; Haga, S.; Numata, Y.; Yamashita, K.; Mikami, T.; Ogawa, T.; Ohkusa, T.; Okayasu, I. Disruption of the p53-p53R2 DNrepair system in ulcerative colitis contributes to colon tumorigenesis. Int. J. Cancer 2006, 118, 1395–1403. [Google Scholar] [CrossRef]
- Risques, R.A.; Lai, L.A.; Brentnall, T.A.; Li, L.; Feng, Z.; Gallaher, J.; Mandelson, M.T.; Potter, J.D.; Bronner, M.P.; Ravinovitch, P.S. Ulcerative colitis is a disease of accelerated colon aging: Evidence from telomere attrition and DNA damage. Gastroenterolgy 2008, 135, 410–418. [Google Scholar] [CrossRef]
- Risques, R.A.; Lai, L.A.; Himmetoglu, C.; Ebasee, A.; Li, L.; Feng, Z.; Bronner, M.P.; Al-kahham, B.; Kowdley, K.V.; Lindor, K.D.; et al. Ulcerative colitis-associated colorectal cancer arises in a field of short telomeres, senescence, and inflammation. Cancer Res. 2011, 71, 1669–1679. [Google Scholar] [CrossRef]
- Araki, K.; Mikami, T.; Yoshida, T.; Kikuchi, M.; Sato, Y.; Oh-ishi, M.; Kodera, Y.; Maeda, T.; Okayasu, I. High expression of HSP47 in ulcerative colitis-associated carcinomas: Proteomic approach. Br. J. Cancer 2009, 101, 492–497. [Google Scholar] [CrossRef] [PubMed]
- Okayasu, I.; Mikami, T.; Yoshida, T.; Hana, K.; Yokozawa, M.; Sada, M.; Fujiwara, M.; Kawada, N. Cytoglobin expression of rectal subepithelial myofibroblasts: Significant alterations of cytoglobin + stromal cells in long-standing ulcerative colitis. Histol. Histopathol. 2011, 26, 679–688. [Google Scholar] [PubMed]
- Okayasu, I.; Yoshida, T.; Mikami, T.; Hana, K.; Yokozawa, M.; Araki, K.; Mitsuhashi, J.; Kikuchi, M.; Adachi, E.; Sada, N. Mucosal remodeling in long-standing ulcerative colitis with colorectal neoplasia: Significant alterations of NCAM+ or SMA+ subepithelial myofibroblasts and interstitial cells. Pathol. Int. 2009, 59, 701–711. [Google Scholar] [CrossRef] [PubMed]
- Okayasu, I.; Hana, K.; Nemoto, N.; Yoshida, T.; Saegusa, M.; Yokota-Nakatsuma, A.; Song, S.-Y.; Iwata, M. Vitamin A inhibits development of dextran sulfate sodium-induced colitis and colon cancer in a mouse model. BioMed. Res. Int. 2016, 2016, 4874809. [Google Scholar] [CrossRef] [PubMed]
- Okayasu, I.; Fujiwara, M.; Yoshida, T. The role of vitamin A-storing cells (stellate cells) in inflammation and tumorigenesis. In Vitamin A; Zepka, L.Q., Ed.; INTEC Open Access Publisher: London, UK, 2019. [Google Scholar] [CrossRef]
- Samuel, S.; Walsh, R.; Webb, J.; Robins, A.; Potten, C.; Mahida, Y.R. Characterization of putative stem cells in isolate human colonic crypt epithelial cells and their interaction with myofibroblasts. Am. J. Physiol. Cell Physiol. 2009, 296, C296–C305. [Google Scholar] [CrossRef] [PubMed]
- Yeung, T.M.; Chia, L.A.; Kosinski, C.M.; Kuo, C.J. Regulation of self-renewal and differentiation by the intestinal stem cell niche. Cell. Mol. Life Sci. 2011, 68, 2513–2523. [Google Scholar] [CrossRef]
- Kosinski, C.; Li, V.S.; Chan, A.S.; Zhang, J.; Ho, C.; Tsui, W.Y.; Chan, T.L.; Mifflin, R.C.; Powell, D.W.; Yuen, S.T. Gene expression patterns of human colon tops and basal and BMP antagonist as intestinal stem cell niche factors. Proc. Natl. Acad. Sci. USA 2007, 104, 15418–15423. [Google Scholar] [CrossRef]
- Shaker, A.; Swietlicki, E.A.; Wang, L.; Jiang, S.; Onal, B.; Bala, S.; DeSchryver, K.; Newberry, R.; Levin, M.S.; Rubin, D.C. Epimorphin deletion protects mice from inflammation-induced colon carcinogenesis and alters stem cell niche myofibroblast secretion. J. Clin. Investig. 2010, 120, 2081–2093. [Google Scholar] [CrossRef][Green Version]
- Matsumoto, N.; Yoshida, T.; Okayasu, I. High epithelial and stromal genetic instability of chromosome 17 in ulcerative colitis-associated carcinogenesis. Cancer Res. 2003, 63, 6153–6161. [Google Scholar]
- Yagishita, H.; Yoshida, T.; Ishiguro, K.; Numata, Y.; Okayasu, I. Epithelial and stromal genetic instability linked to tumor suppressor genes in ulcerative colitis-associated tumorigenesis. Scand. J. Gastroenterol. 2008, 43, 559–566. [Google Scholar] [CrossRef]
- Mikami, T.; Yoshida, T.; Numata, Y.; Kikuchi, M.; Araki, K.; Nakada, N.; Okayasu, I. Invasive behavior of ulcerative colitis-associated carcinoma is related to reduced expression of CD44 extracellular domain: Comparison with sporadic colon carcinoma. Diagn. Pathol. 2011, 6, 30. [Google Scholar] [CrossRef] [PubMed]
- Mikami, T.; Yoshida, T.; Numata, Y.; Shiraishi, H.; Araki, K.; Guiot, M.C.; Jass, J.R.; Okayasu, I. Low frequency of promoter methylation of O6-methylguanine DNA methyltransferase (MGMT) and hMLH1 in ulcerative colitis-associated tumors: Comparison with sporadic colonic tumors. Am. J. Clin. Pathol. 2007, 127, 366–373. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Mikami, T.; Mitomi, H.; Hara, A.; Yanagisawa, N.; Yoshida, T.; Tsuruta, O.; Okayasu, I. Decreased expression of CD44, alpha-catenin, and deleted colon carcinoma and altered expression of beta-catenin in ulcerative colitis-associated dysplasia and carcinoma, as compared with sporadic colon neoplasms. Cancer 2000, 89, 733–740. [Google Scholar] [CrossRef]
- Mikami, T.; Yoshida, T.; Shiraishi, H.; Tokuyama, W.; Motoori, T.; Okayasu, I. Bottom-up cell proliferation with cyclin A and p27Kip1 expression in ulcerative colitis-associated dysplasia. Pathol. Int. 2006, 56, 10–16. [Google Scholar] [CrossRef] [PubMed]
- Mikami, T.; Yoshida, T.; Akino, F.; Motoori, T.; Yajima, M.; Okayasu, I. Apoptosis regulation differs between ulcerative colitis-associated and sporadic colonic tumors. Association with surviving and bcl-2. Am. J. Clin. Pathol. 2003, 119, 723–730. [Google Scholar] [CrossRef] [PubMed]
- Choi, P.M.; Zelig, M.P. Similarity of colorectal cancer in Crohn’s disease and ulcerative colitis: Implications for carcinogenesis and prevention. Gut 1994, 35, 950–954. [Google Scholar] [CrossRef] [PubMed]
- Canavan, C.; Abrams, K.R.; Mayberry, J. Meta-analysis: Colorectal and small bowel cancer risk in patients with Crohn’s disease. Aliment. Pharmacol. Ther. 2006, 23, 1097–1104. [Google Scholar] [CrossRef]
- Scaringi, S.; Di Martino, C.; Zambonin, D.; Fazi, M.; Canonico, G.; Leo, F.; Ficari, F.; Tonelli, F. Colorectal cancer and Crohn’s colitis: Clinical implications from 313 surgical patients. World J. Surg. 2013, 37, 902–910. [Google Scholar] [CrossRef]
- Eluri, S.; Shaheen, N.J. Barrett’s esophagus: Diagnosis and management. Gastrointest. Endosc. 2017, 85, 889–903. [Google Scholar] [CrossRef]
- Peters, Y.; Al-Kaabi, A.; Shaheen, N.J.; Chak, A.; Blum, A.; Souza, R.F.; Di Pietro, M.; Lyer, P.G.; Pech, O.; Fitzgerald, R.C.; et al. Barrett’s oesophagus. Nat. Rev. Dis. Primers 2019, 5, 35. [Google Scholar] [CrossRef]
- Tonkic, A.; Tonkic, M.; Lehours, P.; Mégraud, F. Epidemiology and diagnosis of Helicobacter pylori infection. Helicobacter 2012, 17, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Hamashima, C. Current issue and future perspectives of gastric cancer screening. World J. Gastroenterol. 2014, 20, 13767–13774. [Google Scholar] [CrossRef] [PubMed]
- Arai, Y.; Arihiro, S.; Matsuura, T.; Kato, T.; Matsuoka, M.; Saruta, M.; Mitsunaga, M.; Matsuura, M.; Fujiwara, M.; Okayasu, I.; et al. Prostaglandin E-major urinary metabolite as a reliable surrogate marker for mucosal inflammation in ulcerative colitis. Inflamm. Bowel Dis. 2014, 20, 1208–1216. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Chronic Inflammation | Gastritis and Remodeling | Cancer | Notes |
|---|---|---|---|
| H. pylori-associated gastritis | Atrophy, intestinal metaplasia Fibrosis | Differentiated type (Lauren’s intestinal-type) Adenocarcinoma | Relatively elderly patients |
| H. pylori-associated gastritis | Active gastritis without mucosal atrophy No mucosal remodeling | Undifferentiated type (Lauren’s diffuse-type) Adenocarcinoma [34,35,36,37,38,39,40,41,42] | Relatively young patients |
| H. pylori-associated gastritis | Hyperplastic polyp-adenomatous change (Dysplasia) | Adenocarcinoma [43] | p53 mutation (41%) |
| Reflux gastritis after partial gastrectomy | Hyperplastic gastritis DNA damage, foveolar cell hyperplasia, EB virus infection | Adenocarcinoma in remnant stomach [44] | High risk after gastro-jejunostomy, enterogastric reflux |
| EB virus-associated gastritis | Atrophic gastritis, lympho-epithelioma-like histology | Adenocarcinoma [45,46,47,48] in a proximal location | Predominance among males |
| H. pylori-associated gastritis | Chronic severe gastritis | * MALT type lymphoma [49] |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Okayasu, I.; Ichinoe, M.; Yoshida, T. Proposal for an Organ-Specific Chronic Inflammation–Remodeling–Carcinoma Sequence. Gastrointest. Disord. 2019, 1, 341-357. https://doi.org/10.3390/gidisord1030028
Okayasu I, Ichinoe M, Yoshida T. Proposal for an Organ-Specific Chronic Inflammation–Remodeling–Carcinoma Sequence. Gastrointestinal Disorders. 2019; 1(3):341-357. https://doi.org/10.3390/gidisord1030028
Chicago/Turabian StyleOkayasu, Isao, Masaaki Ichinoe, and Tsutomu Yoshida. 2019. "Proposal for an Organ-Specific Chronic Inflammation–Remodeling–Carcinoma Sequence" Gastrointestinal Disorders 1, no. 3: 341-357. https://doi.org/10.3390/gidisord1030028
APA StyleOkayasu, I., Ichinoe, M., & Yoshida, T. (2019). Proposal for an Organ-Specific Chronic Inflammation–Remodeling–Carcinoma Sequence. Gastrointestinal Disorders, 1(3), 341-357. https://doi.org/10.3390/gidisord1030028