
In Situ Surface-Enhanced Raman Spectroscopy Investigation of the Passive Films That Form on Alloy 600, Alloy 690, Unalloyed Cr and Ni, and Alloys of Ni-Cr and Ni-Cr-Fe in Pressurized Water Nuclear Reactor Primary Water

Abstract
1. Introduction
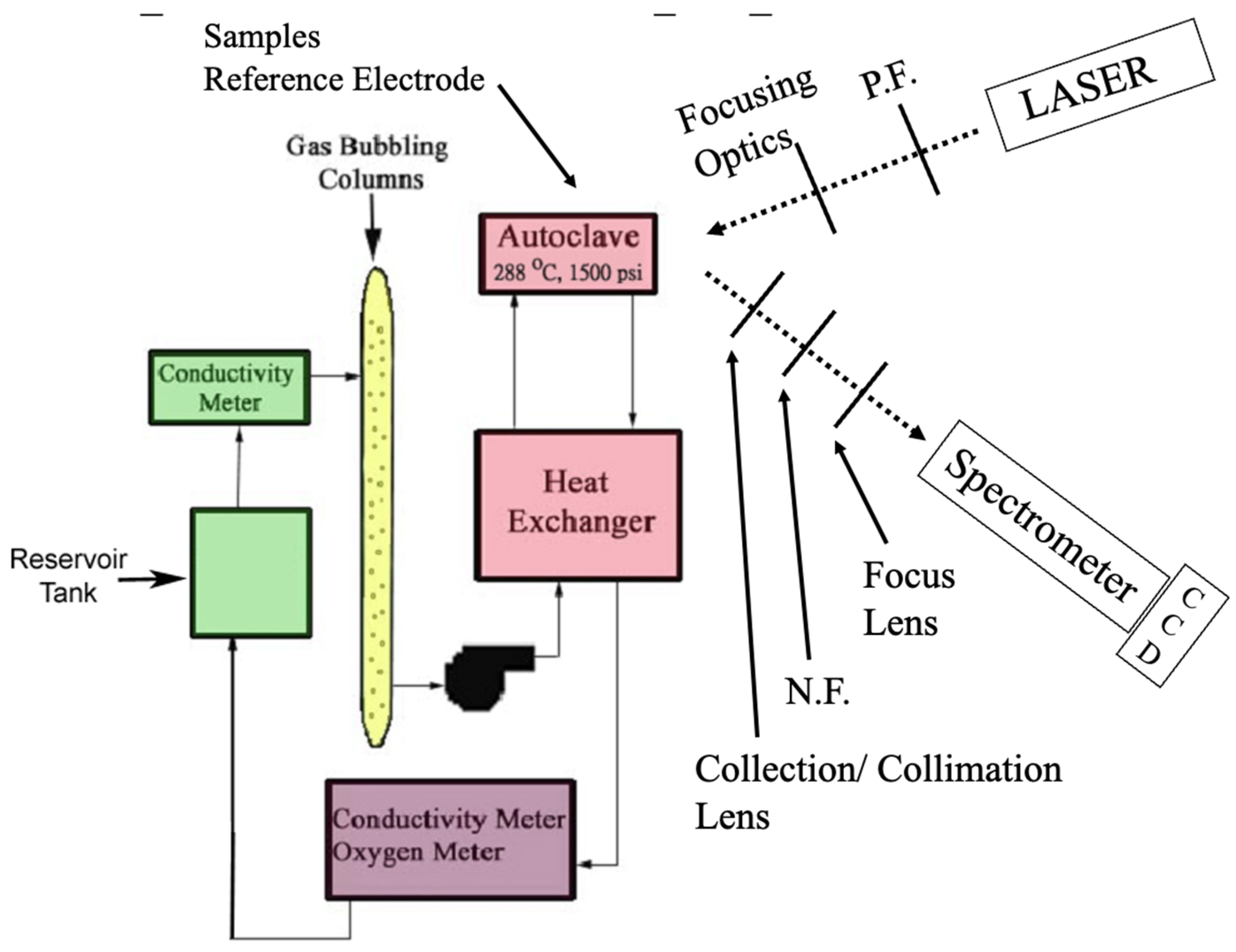
2. Experimental Procedure
2.1. Overview
2.2. Details of Experimental Procedure
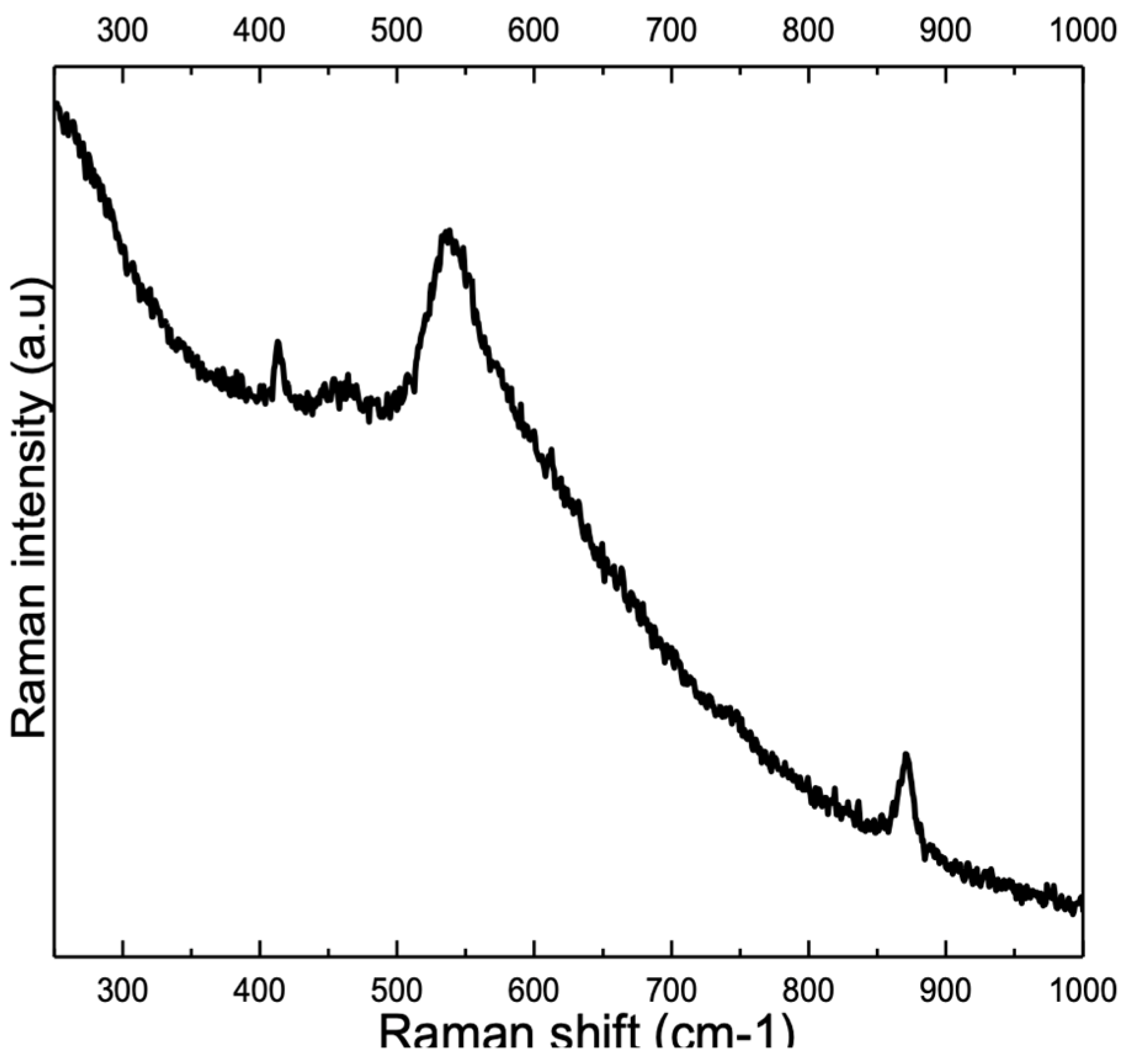
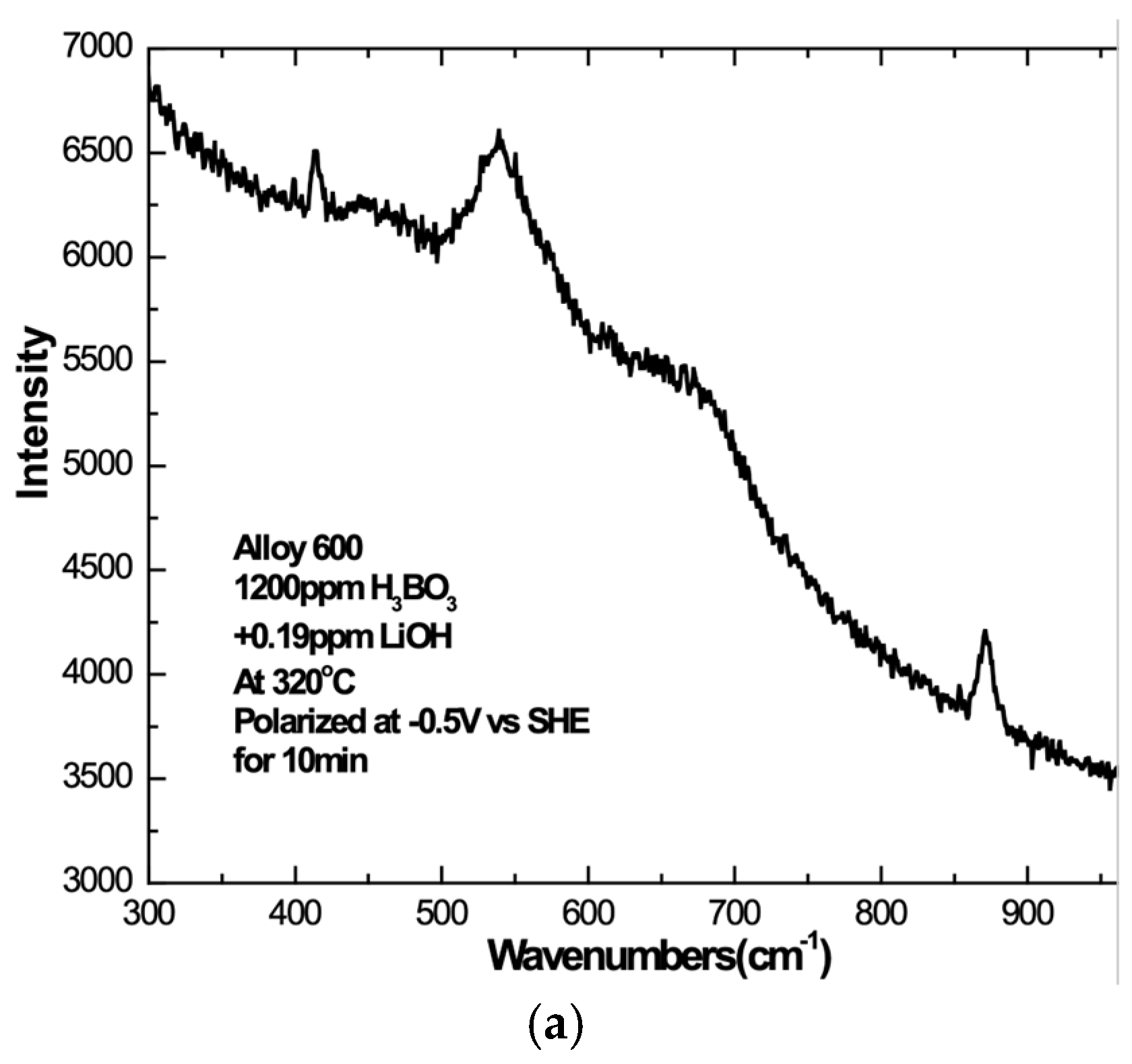
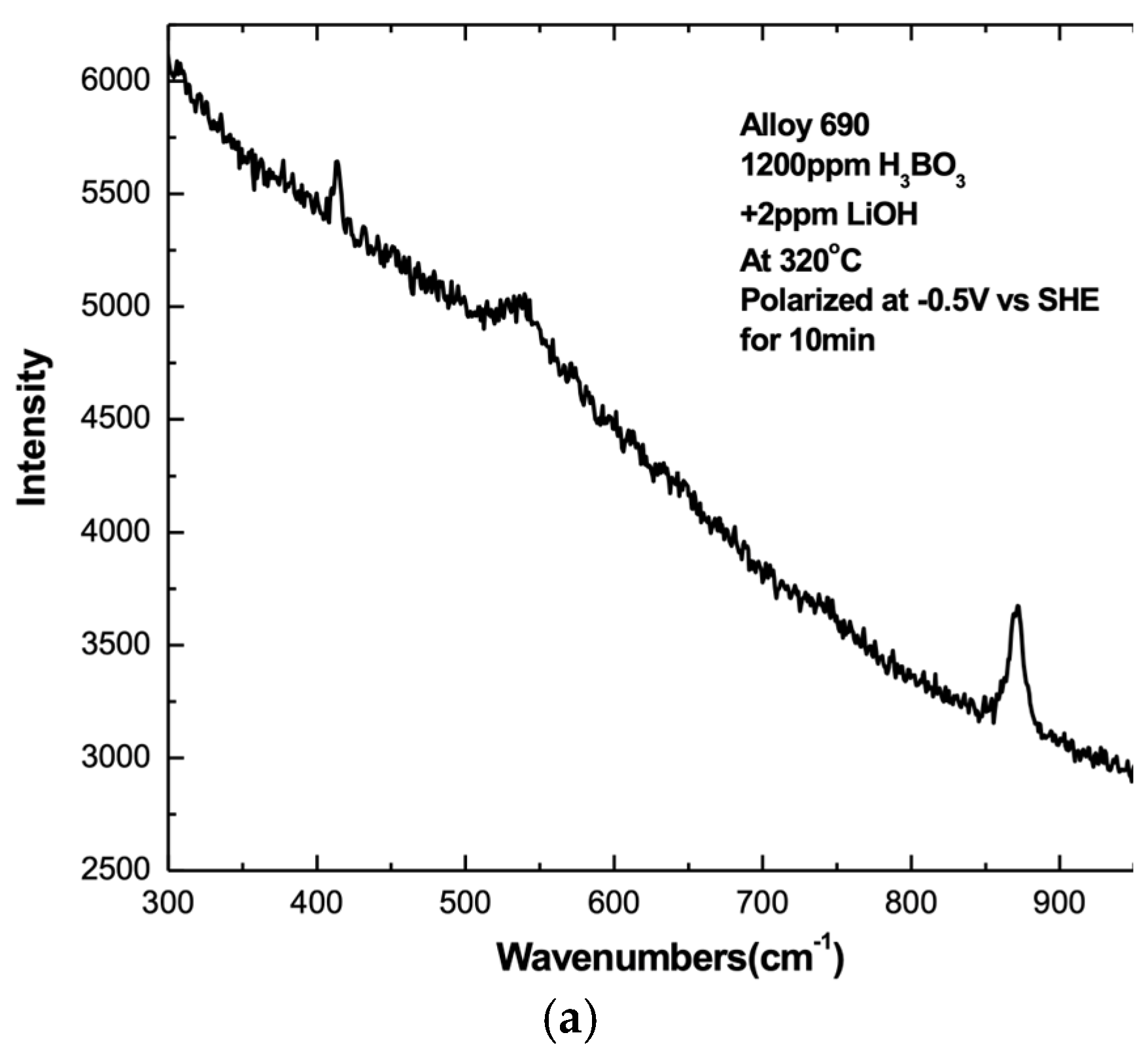
3. Results
3.1. Overview
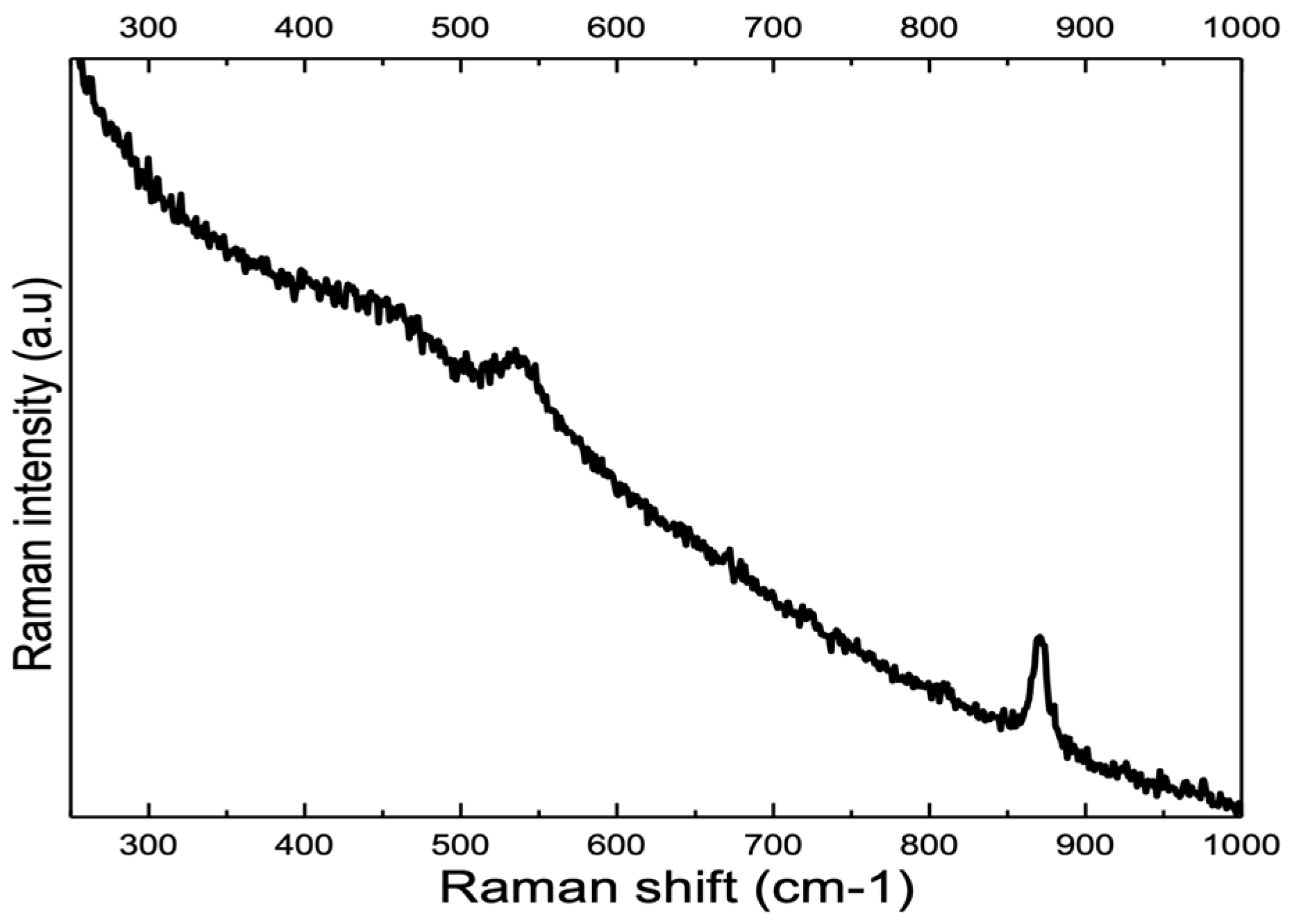
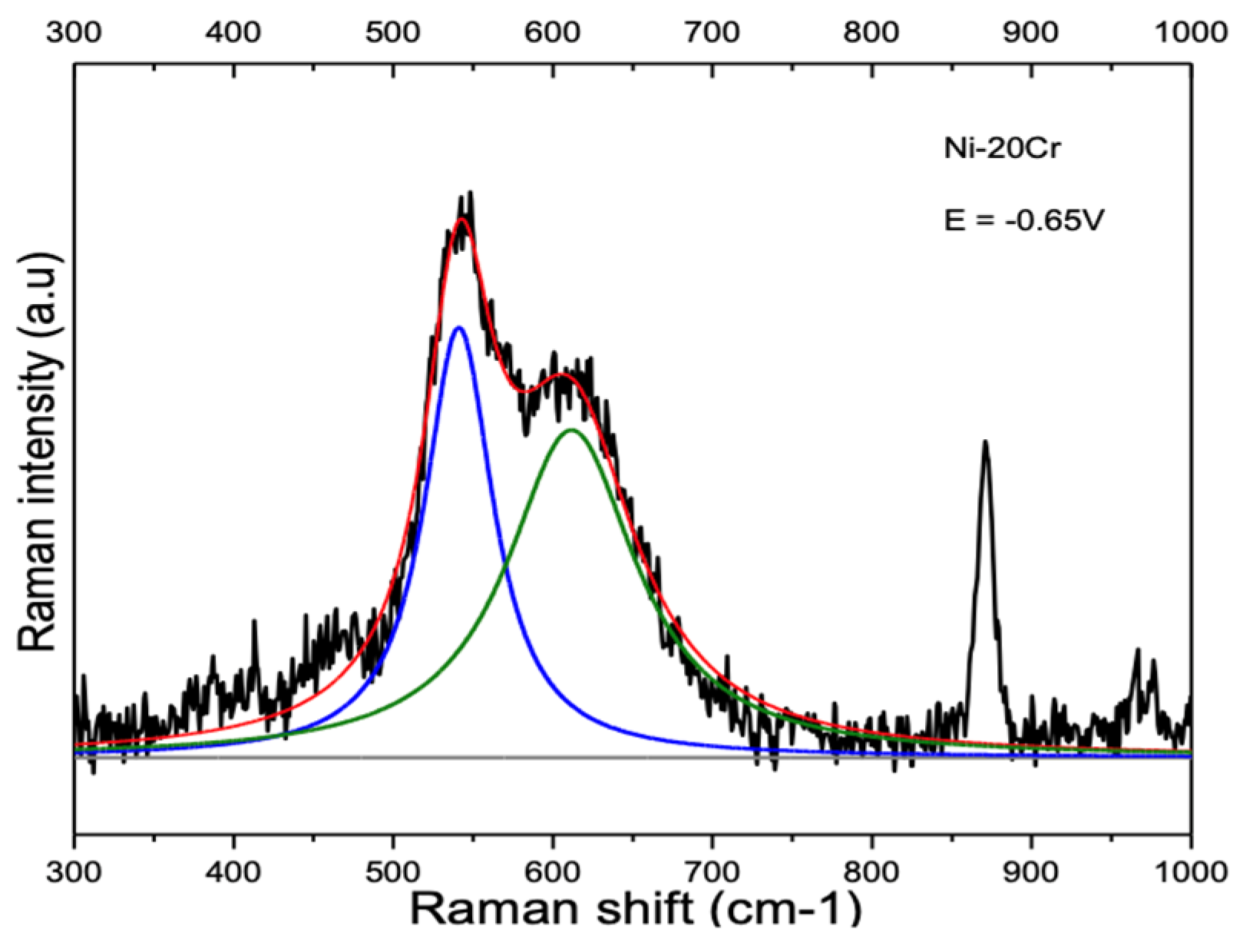
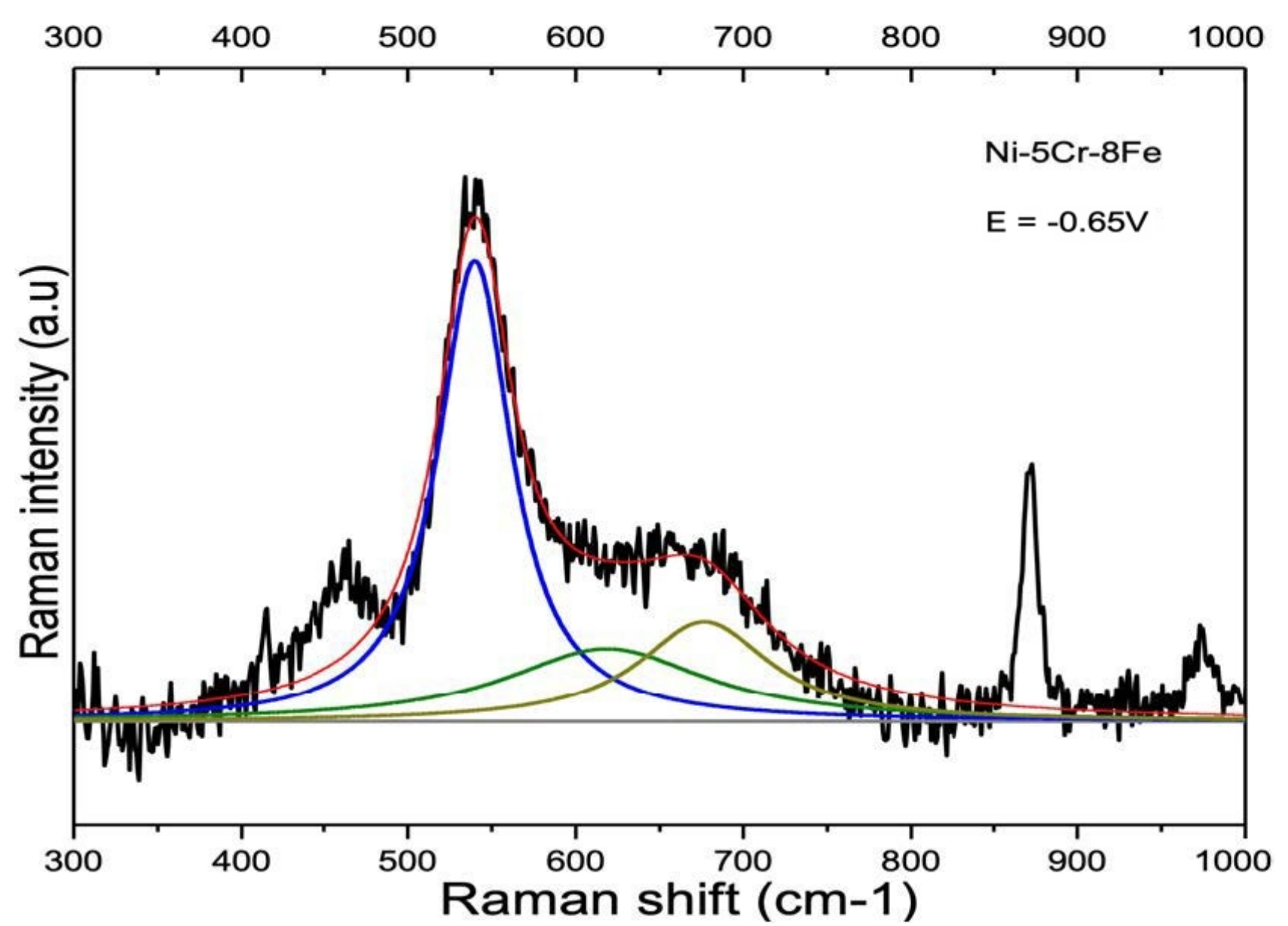
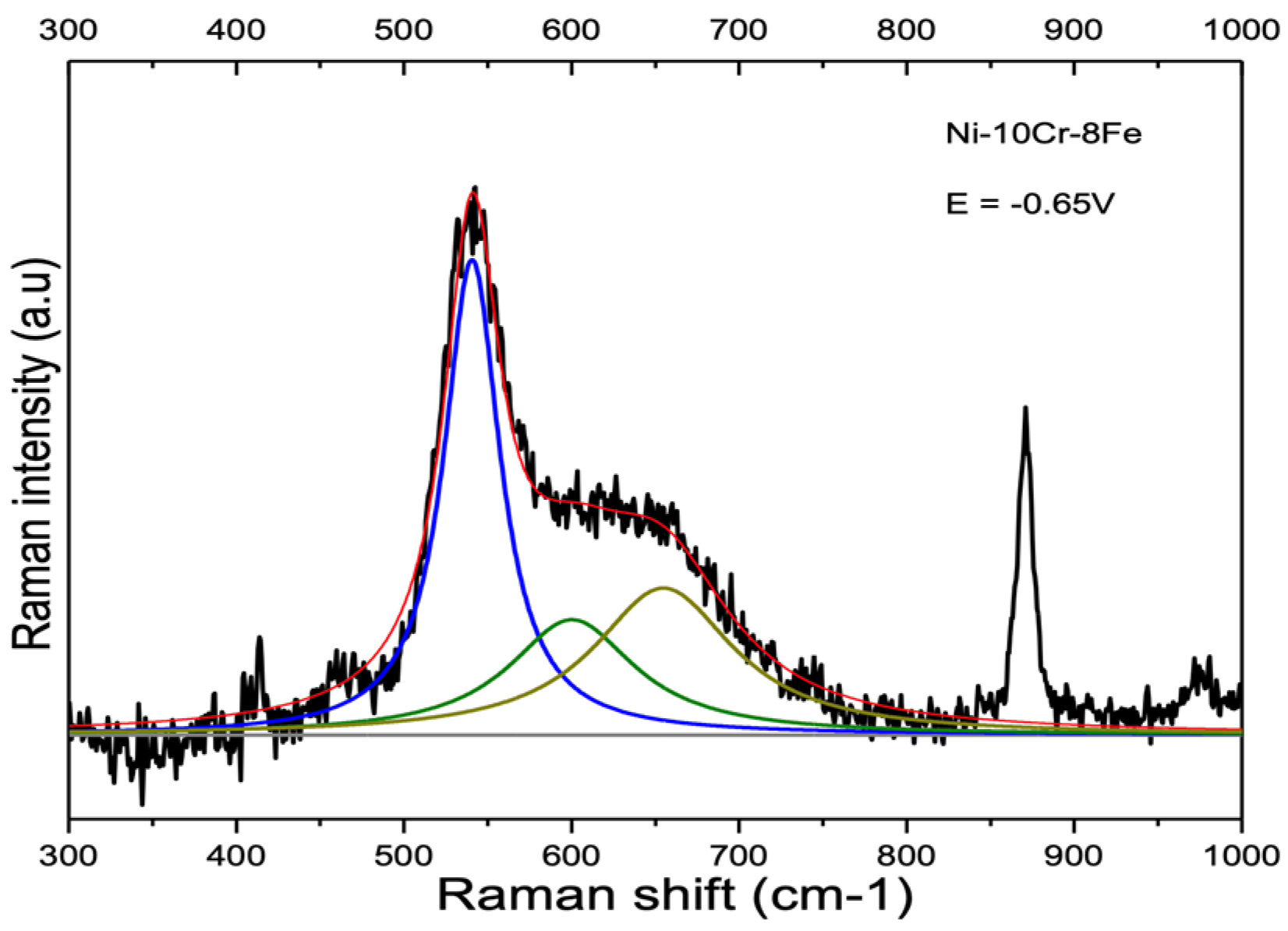
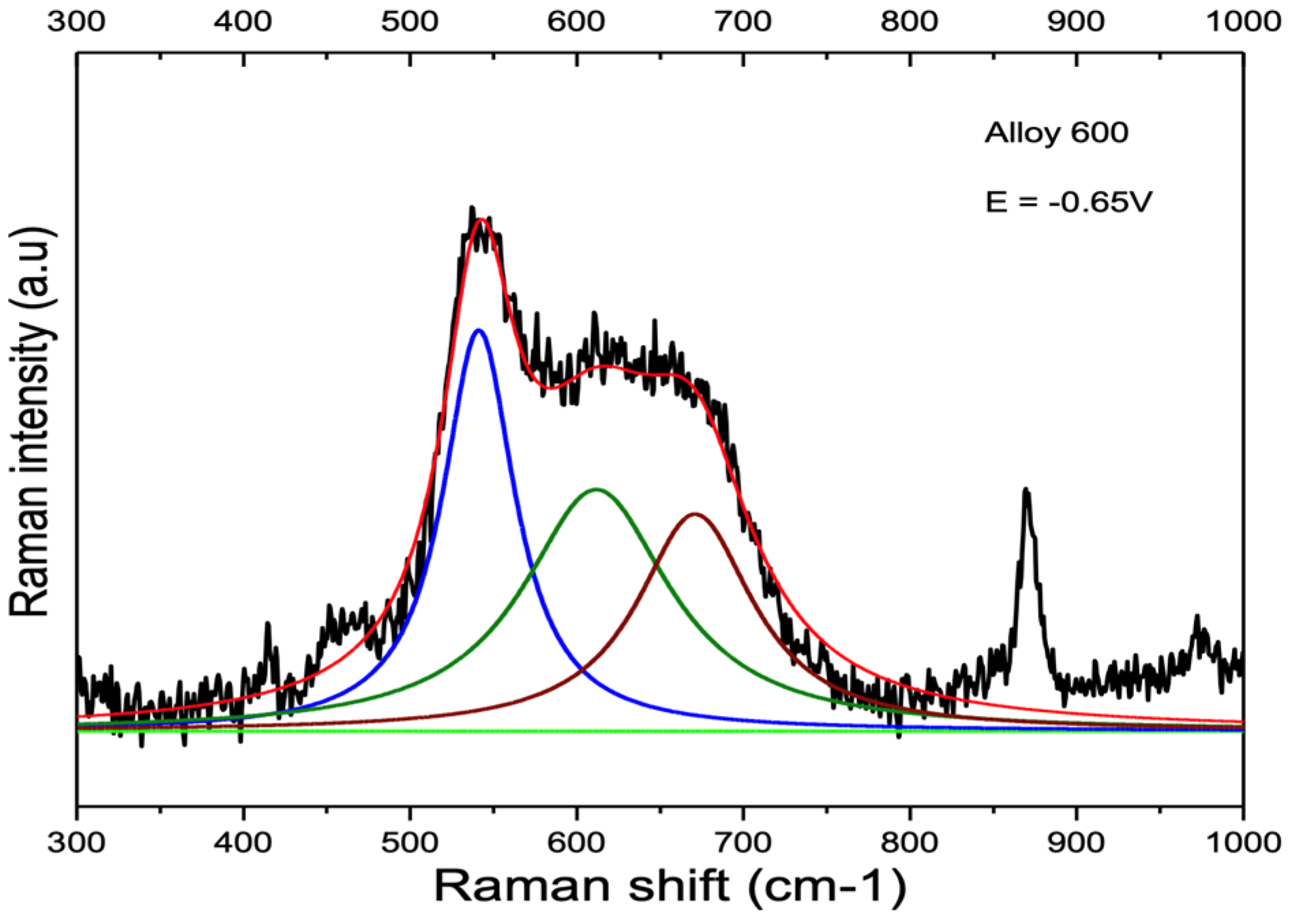
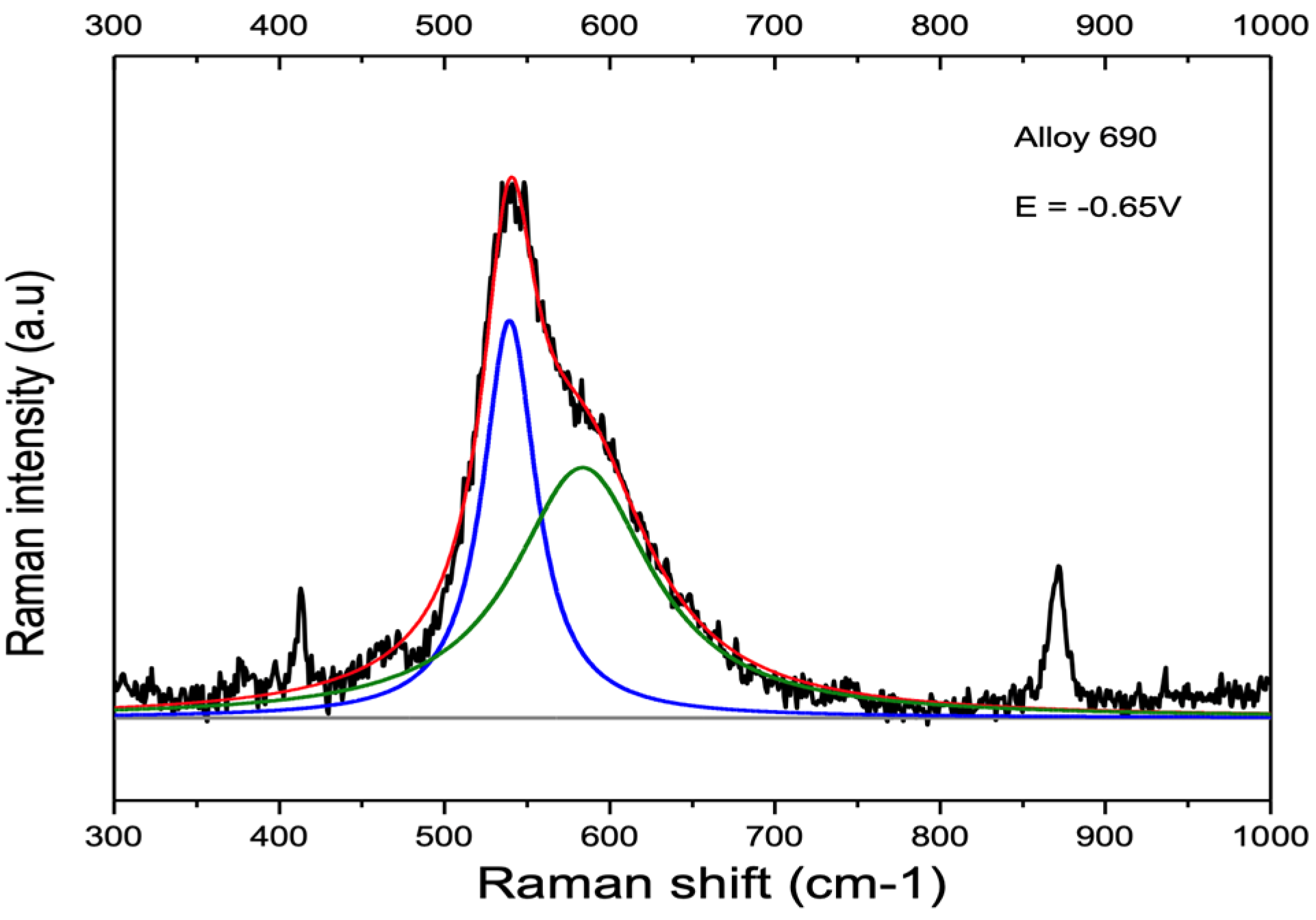
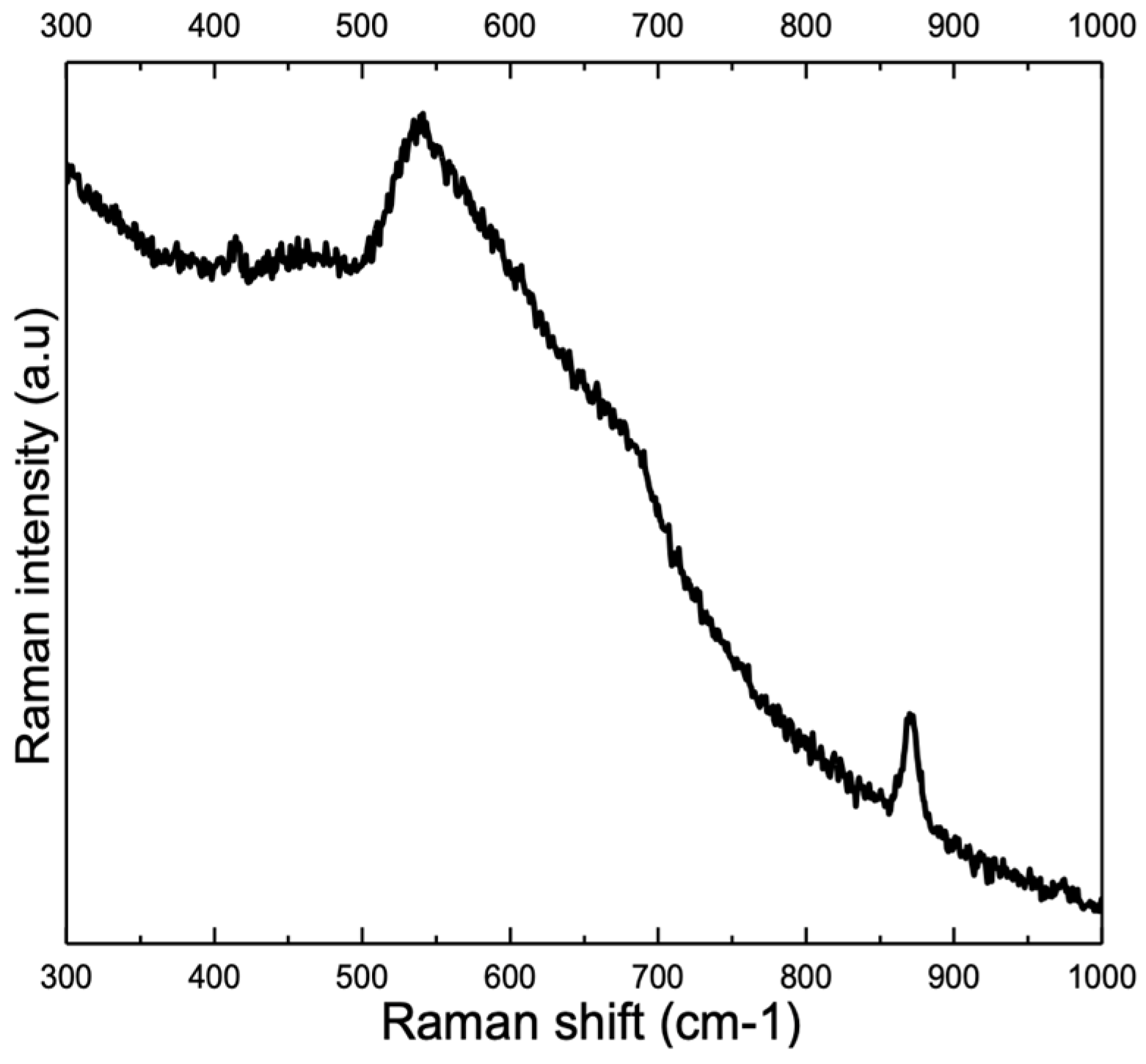
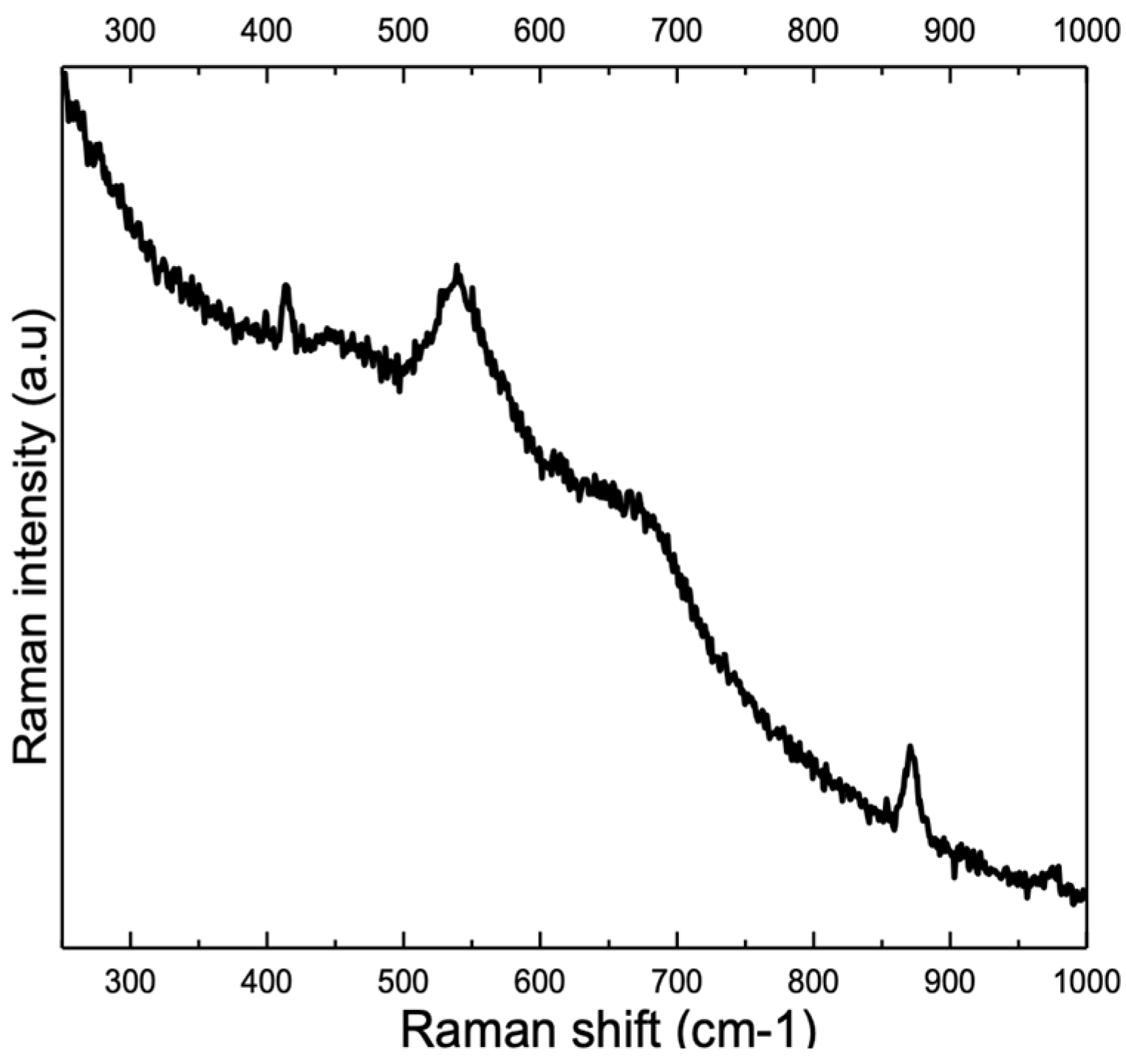
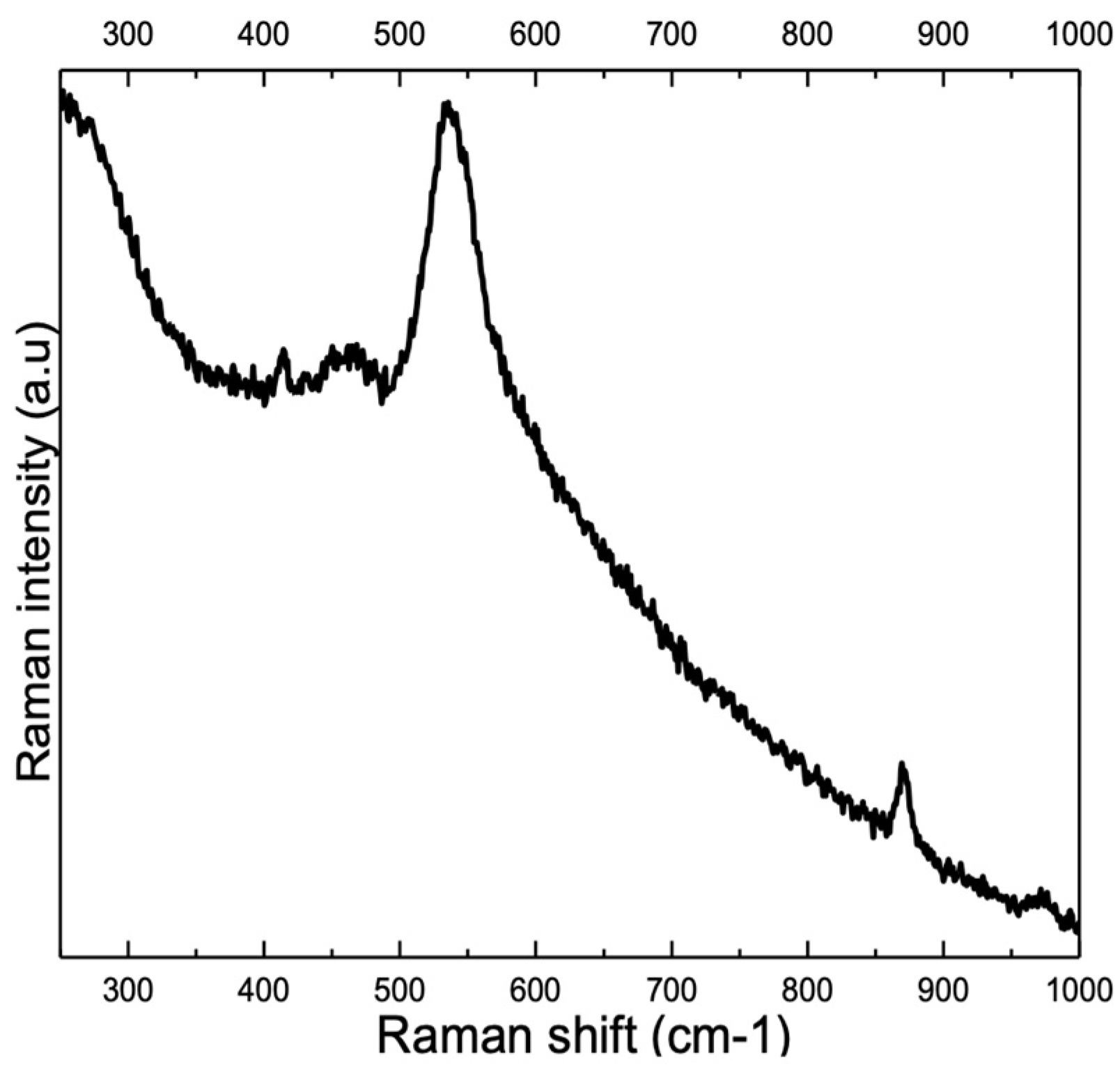
3.2. Influence of Alloy Composition
3.3. Influence of pH on SERS of Alloy 600 and Alloy 690
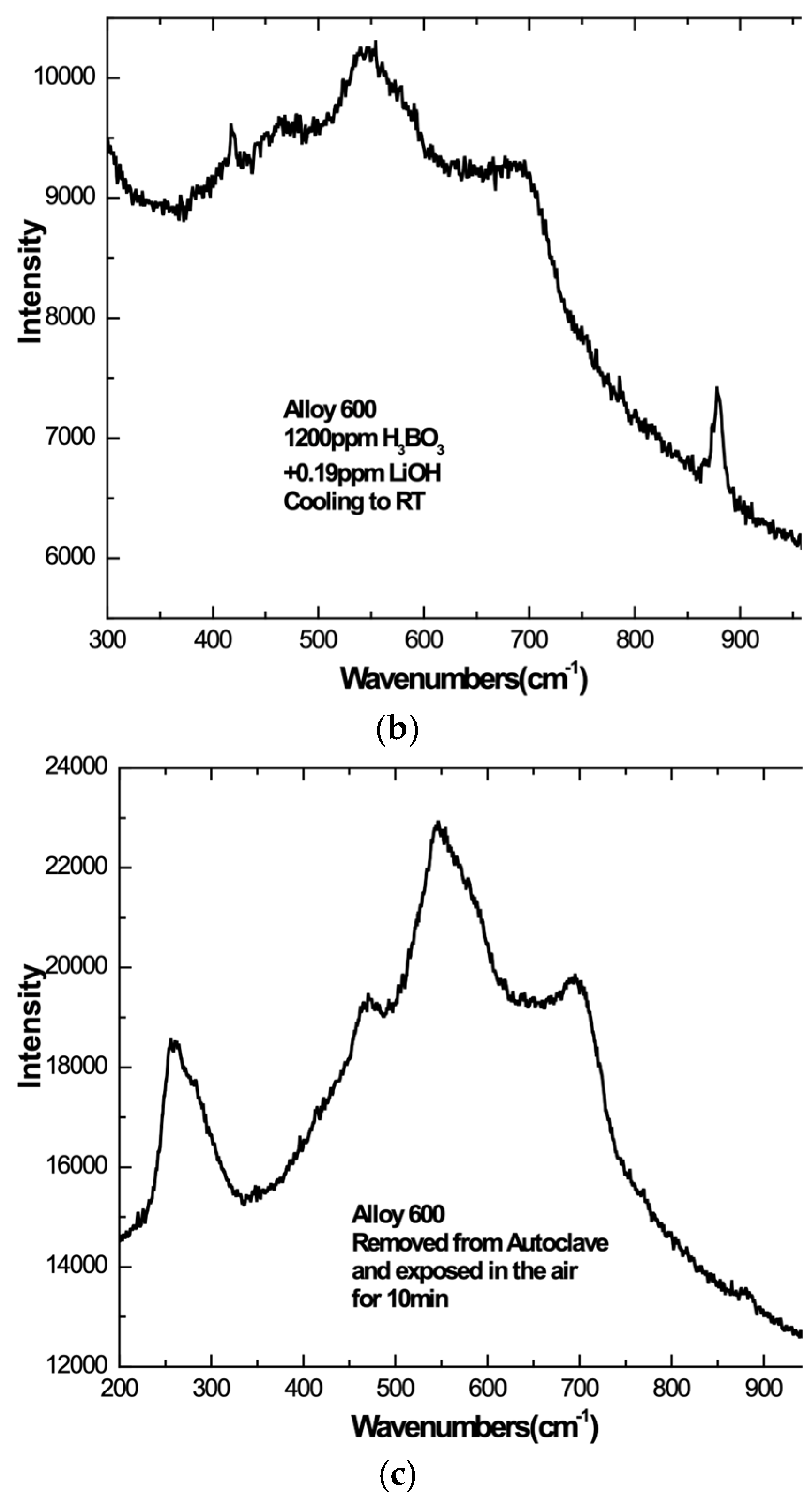
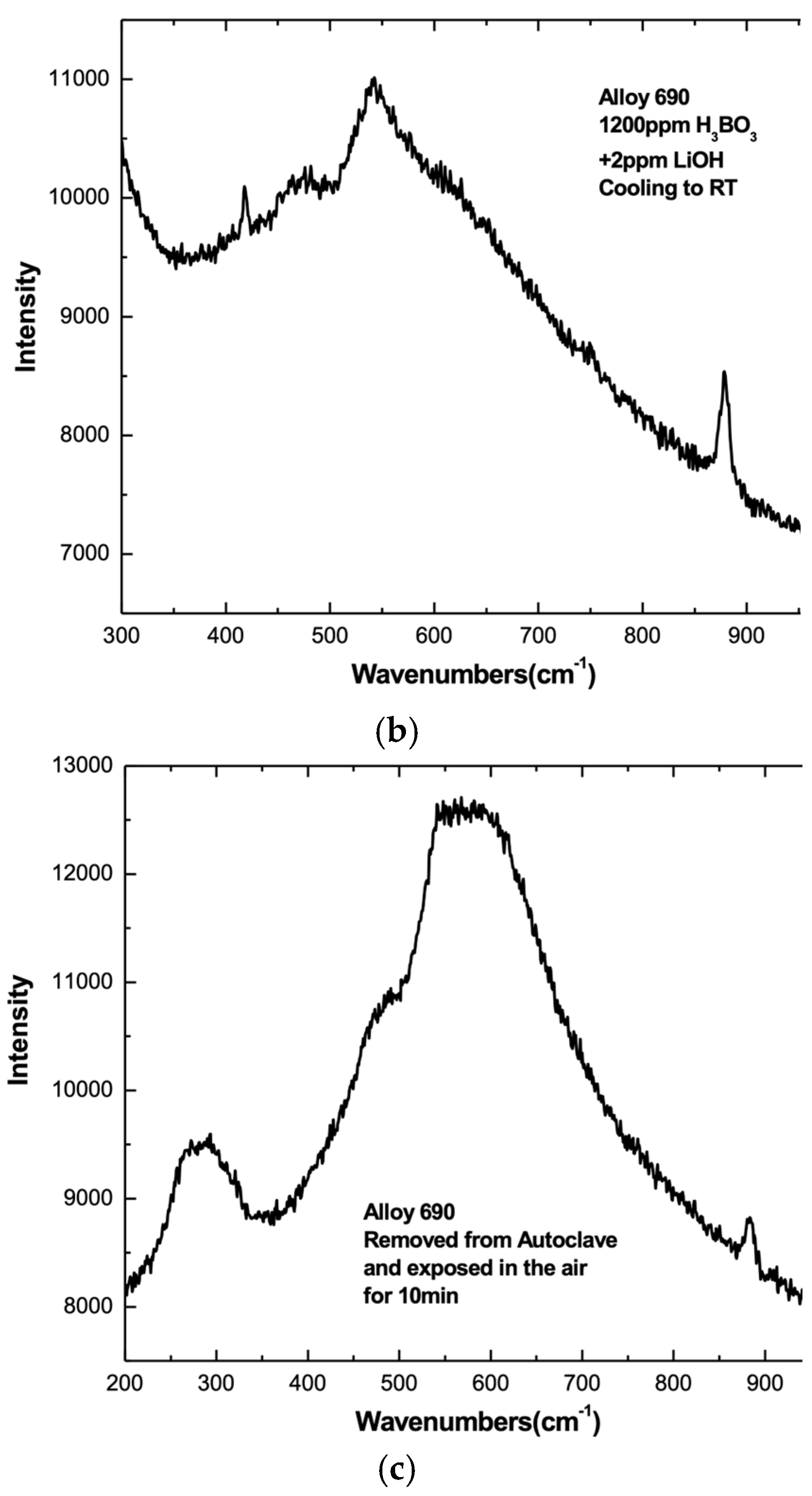
3.4. Cool-Down and Air Exposure
4. Discussion
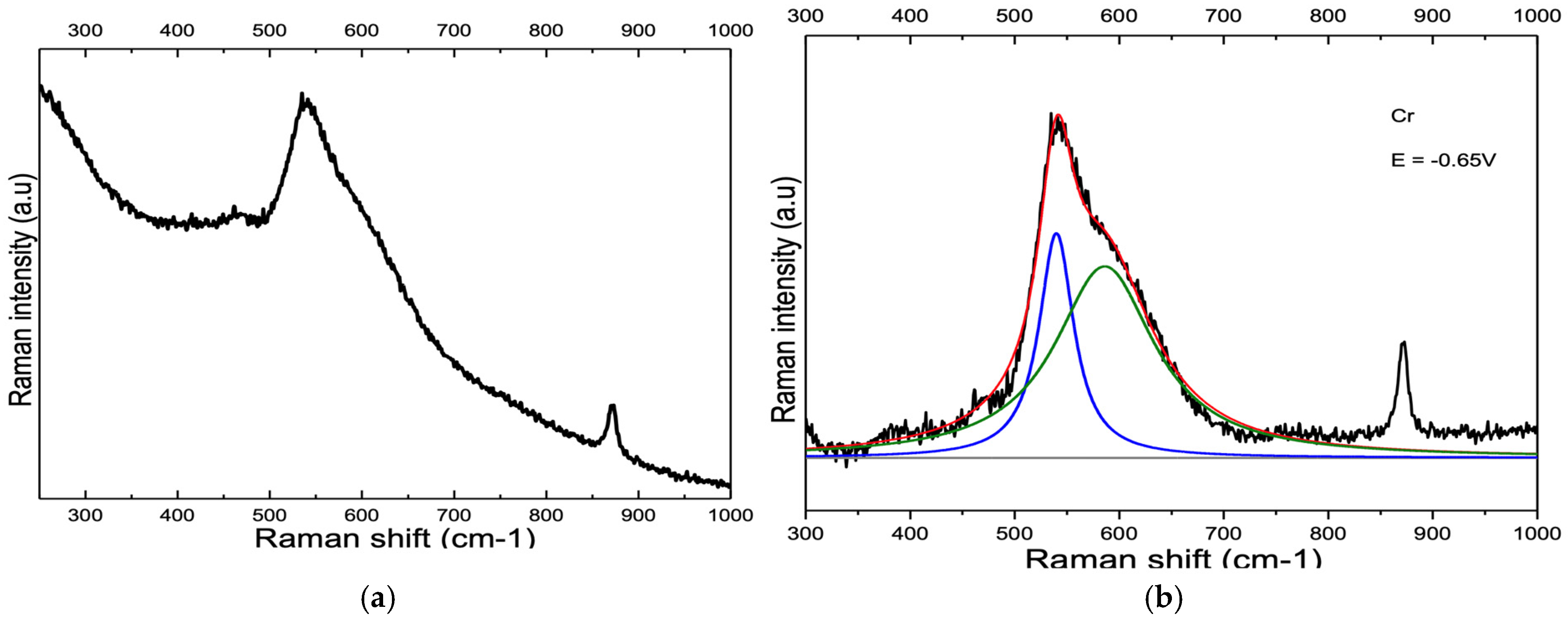
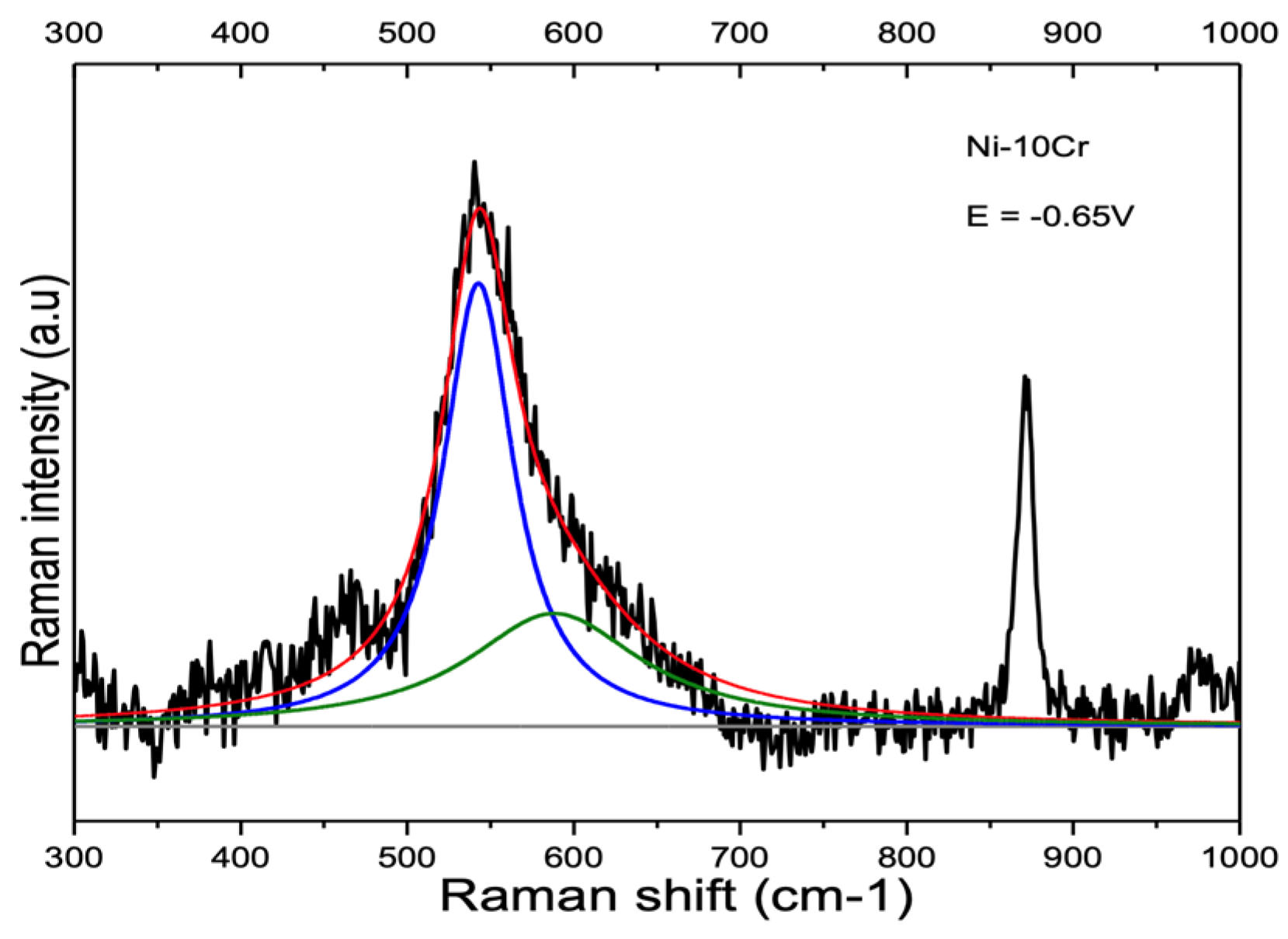
4.1. Peaks at 540 cm−1 and 580–610 cm−1
4.1.1. Nickel and Chromium
4.1.2. Ni-Cr and Ni-Cr-Fe Alloys
4.2. Peak at ≈680 cm−1
4.3. Effect of Temperature on SERS
4.4. In Situ vs. Ex Situ SERS
4.5. Miscellaneous
5. Summary and Conclusions
- The passive film that forms at 320 °C on unalloyed nickel is NiO and the film that forms on unalloyed chromium is a mixture of Cr2O3 and CrOOH. The relative amounts of Cr2O3 and CrOOH varied from sample to sample and ranged from instances in which Cr2O3 is clearly the dominant species to cases in which CrOOH is dominant and no Cr2O3 is detected.
- The most intense peaks of the SER spectra of NiO and of Cr2O3 are nearly identical and are located within 3.8 cm−1 of each other, which makes it very difficult to determine the relative amounts of NiO and Cr2O3 in the surface films of the Ni-Cr alloys, the Ni-Cr-Fe alloys, and Alloy 600 and Alloy 690.
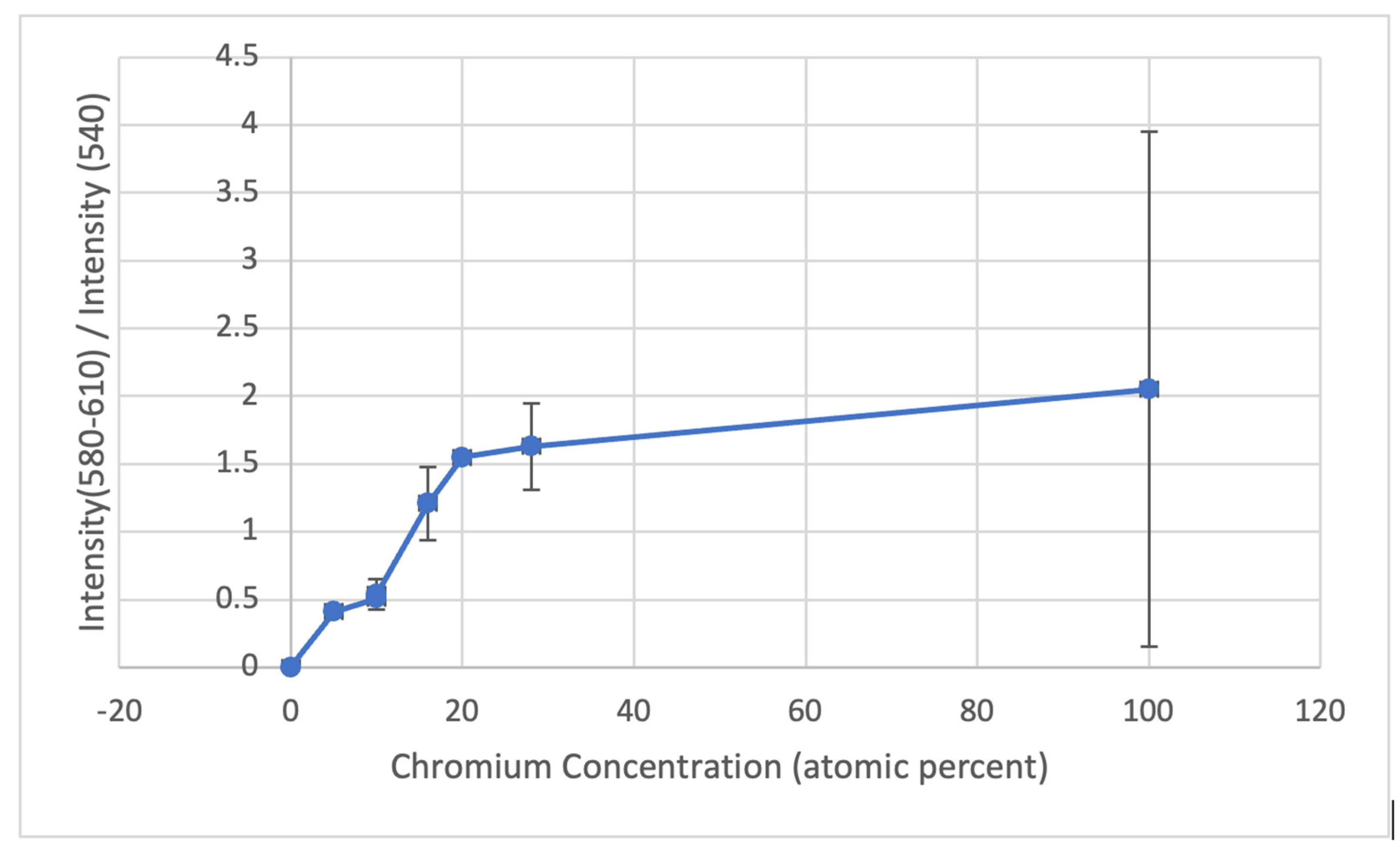
- The films that form on Ni-Cr binary alloys and Ni-Cr-Fe ternary alloys consist of NiO and a mixture of Cr2O3 and CrOOH. The amounts of CrOOH relative to the combined amounts of Cr2O3 and NiO are very reproducible and monotonically increase as the alloy’s chromium concentration increases. When combined with earlier TEM investigations, the SERS results suggest a monotonic decrease in the amount of NiO in the surface film as the alloy’s chromium concentration increases.
- With the exception of Alloy 690, all alloys with 8% Fe exhibited OLs with spinel FeCr2O4—M(Fe,Cr)2O4 along with NiO. The absence of spinel in the OL of the surface film of Alloy 690 indicates the greater protectiveness of 690’s IL of Cr2O3 + CrOOH compared to Alloy 600’s IL of Cr2O3 + CrOOH. The greater protectiveness is associated with Alloy 690’s IL’s greater resistance to the transport of iron cations.
- There is a greater amount of spinel in the OL of Alloy 600 formed at pH 6.2 than at pH 7.2. No spinel is formed in the passive film of Alloy 690 at pH 7.2 and pH 6.2.
- The SERS background has a significant effect on the location, size, and shape of the Raman peaks of the surface films. In addition, The SERS background impairs analyses of surface films by swamping the intensities of secondary peaks in the SER spectra of Cr2O3 and CrOOH. Work on how to remove the SERS background is required in order to extract more information from the SER spectra about the structure and composition of the surface films.
- Comparison of in situ and ex situ SER spectra yielded mixed results. In some cases, air exposure caused significant change to the SER spectrum of surface films formed in PWR PW at 320 °C.
- SERS is most effective when used in combination with other tools for investigating surface films such as scanning electron microscopy, transmission electron microscopy, and electrochemical impedance spectroscopy.
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
Appendix A. Surface-Enhanced Raman Spectroscopy
Appendix B. Reversible Hydrogen Reference Electrode
Appendix C. SERS Background Subtraction
References
- Young, B.A.; Gao, X.; Srivatsan, T.S.; King, P.J. The Response of Alloy 690 Tubing in a Pressurized Water Reactor Environment. Mater. Des. 2007, 28, 373–379. [Google Scholar] [CrossRef]
- Grimmel, B.U.S. Plant Experience with Alloy 600 Cracking and Boric Acid Corrosion of Light-Water Reactor Pressure Vessel Materials; U.S. Nuclear Regulatory Commission Report NUREG1823; U.S. Nuclear Regulatory Commission: Washington, DC, USA, 2005.
- Jeon, S.-H.; Song, G.D.; Hur, D.H. Effects of Hydrogen Contents on Oxidation Behavior of Alloy 690TT and Associated Boron Accumulation within Oxides in High-Temperature Water. Scanning 2018, 2018, 7845176. [Google Scholar] [CrossRef] [PubMed]
- Scott, P.; Combrade, P. General Corrosion and Stress Corrosion Cracking of Alloy 600 in Lighht Water Reactor Primary Coolants. J. Nucl. Mater. 2019, 524, 340–375. [Google Scholar] [CrossRef]
- Machet, A.; Galtayries, A.; Marcus, P.; Combrade, P.; Scott, P. XPS Study of Oxides Formed on Nickel-Base Alloys in High-Temperature and High-Pressure Water. Surf. Interface Anal. 2002, 34, 197–200. [Google Scholar] [CrossRef]
- Jiang, Y.; Bustillo, K.C.; Devine, T.M. Investigation via Electron Microscopy and Electrochemical Impedance Spectroscopy of the Effect of Aqueous Zinc Ions on Passivity and the Surface Films of Alloy 600 in PWR PW at 320 °C. Corros. Mater. Degrad. 2023, 4, 54–89. [Google Scholar] [CrossRef]
- Ziemniak, S.E.; Hanson, M. Corrosion behavior of NiCrFe Alloy 600 in high temperature, hydrogenated water. Corros. Sci. 2006, 48, 498–521. [Google Scholar] [CrossRef]
- Terachi, T.; Totsuka, N.; Yamada, T.; Nakagawa, T.; Deguchi, H.; Horiuchi, M.; Oshitani, M. Influence of Dissolved Hydrogen on Structure of Oxide Film on Alloy 600 Formed in Primary Water of Pressurized Water Reactors. J. Nucl. Sci. Technol. 2003, 40, 509–516. [Google Scholar] [CrossRef]
- Panter, J.; Viguier, B.; Cloué, J.M.; Foucault, M.; Combrade, P.; Andrieu, E. Influence of oxide films on primary water stress corrosion cracking initiation of alloy 600. J. Nucl. Mater. 2006, 348, 213–221. [Google Scholar] [CrossRef]
- Combrade, P.; Scott, P.M.; Foucault, M.; Andrieu, E.; Marcus, P. Oxidation of Ni Base Alloys in PWR Water: Oxide Layers and Associated Damage to the Base Metal. In Proceedings of the 12th International Conference on Environmental Degradation of Materials in Nuclear Power System—Water Reactors, Salt Lake City, UT, USA, 14–18 August 2005; Allen, T.R., King, P.J., Nelson, L., Eds.; TMS (The Minerals, Metals & Materials Society): Pittsburgh, PA, USA; pp. 883–890. [Google Scholar]
- Morton, D.; Lewis, N.; Hanson, M.; Rice, S.; Sanders, P. Nickel Alloy Primary Water Bulk Surface and SCC Corrosion Film Analytical Characterization and SCC Mechanistic Implications; Lockheed-Martin Research Report, LM-07K022; Lockheed-Martin: Niskayuna, NY, USA, 2007. [Google Scholar]
- Delabrouille, F.; Legras, L.; Vaillant, F.; Scott, P.; Viguier, B.; Andrieu, E. Effect of the Chromium Content and Strain on the Corrosion of Nickel Based Alloys in Primary Water of Pressurized Water Reactors. In Proceedings of the 12th International Conference on Environmental Degradation of Materials in Nuclear Power System—Water Reactors, Salt Lake City, UT, USA, 14–18 August 2005; Allen, T.R., King, P.J., Nelson, L., Eds.; TMS (The Minerals, Metals & Materials Society): Pittsburgh, PA, USA; pp. 903–911. [Google Scholar]
- Kim, J.H.; Hwang, I.S. In-situ Raman Spectroscopic Study of Oxide Films on Alloy 600 in Simulated PWR Water. In Proceedings of the 11th International Conference on Environmental Degradation of Materials in Nuclear Systems—Water Reactors, Colorado Springs, CO, USA, 7–11 August 2011. [Google Scholar]
- James, R.O.; Healy, T.W. Adsorption of Hydrolyzable Metal Ions at the Oxide-Water Interface, Part I. J. Colloid Interface Sci. 1972, 40, 42–52. [Google Scholar] [CrossRef]
- James, R.O.; Healy, T.W. Adsorption of Hydrolyzable Metal Ions at the Oxide-Water Interface, Part II. J. Colloid Interface Sci. 1972, 40, 53–64. [Google Scholar] [CrossRef]
- James, R.O.; Healy, T.W. Adsorption of Hydrolyzable Metal Ions at the Oxide-Water Interface, Part III. J. Colloid Interface Sci. 1972, 40, 65–81. [Google Scholar] [CrossRef]
- Sten, P.; Puhakka, E.; Ikavalko, E.; Lehikoinen, J.; Olin, M.; Sirkia, P.; Kinnunen, P.; Laitinen, T. Adsorption Studies on Iron Oxides with Reference to the Oxide Films Formed on Materials Surfaces in Nuclear Power Plants; VTT Research Notes 2182; VTT Technical Research Center of Finland: Espoo, Finland, 2002. [Google Scholar]
- Radiation Protection Aspects of Primary Water Chemistry and Source Term Management, Radiological Protection, NEA/CRPPH/R(2014)2, July 2014. Available online: www.oecd-nea.org (accessed on 15 December 2024).
- Mintz, T.S. An In-Situ Investigation of the Passive Film on Stainless Steel and Inconel 600 in High Temperature, Pressurized, Impurity Containing Water. Ph.D. Thesis, University of California, Berkeley, CA, USA, 2003. [Google Scholar]
- Voyshnis, S.; Seyeux, A.; Zanna, S.; Martin-Cabanas, B.B.; Couvant, T.; Marcus, P. Oxide Layer Growth on Nickel-base Alloy Surfaces in High Temperature Water and in O2 Studied by ToF-SIMS with Isotropic Tracers. Corros. Sci. 2018, 145, 212–219. [Google Scholar] [CrossRef]
- Gui, J.; Devine, T.M. The Influence of Sulfate Ions on the Surface Enhanced Raman Spectra of Passive Films Formed on Iron. Corros. Sci. 1994, 36, 441–462. [Google Scholar] [CrossRef]
- Futamata, M.; Ishikura, M.; Iida, C.; Handa, S. The Critical Importance of Gap Modes in Surface Enhanced Raman Scattering. Faraday Discuss. 2015, 178, 203–220. [Google Scholar] [CrossRef]
- Karn-Orachai, K. Gap-Dependent Surface-Enhanced Raman Scattering (SERS) Enhancement Model of SERS Substrate–Probe Combination Using a Polyelectrolyte Nanodroplet as a Distance Controller. Langmuir 2021, 37, 10776–10785. [Google Scholar] [CrossRef] [PubMed]
- Schroeder, V.; Devine, T.M. SERS during the Galvanostatic Reduction of the Passive Film on Iron. J. Electrochem. Soc. 1999, 146, 4061–4070. [Google Scholar] [CrossRef]
- Porto, S.P.S.; Krishnan, R.S. Raman Effect of Corundum. J. Chem. Phys. 1967, 47, 1009–1012. [Google Scholar] [CrossRef]
- Oblonsky, L.J.; Devine, T.M. SERS from the Films Formed on Nickel in the Passive and Transpassive Regions. J. Electrochem. Soc. 1995, 142, 3677–3682. [Google Scholar] [CrossRef]
- Dietz, R.E.; Parisot, G.I.; Meixner, A.E. Infrared Absorption and Raman Scattering by Two-Magnon Processes in NiO. Phys. Rev. B Condens. Matter 1971, 4, 2302. [Google Scholar] [CrossRef]
- Maslar, J.E.; Hurst, W.S.; Bowers, W.J.; Hendricks, J.H., Jr.; Aquino, M.I. In Situ Raman Spectroscopic Investigation of Nickel Hydrothermal Corrosion. Corrosion 2002, 58, 225–231. [Google Scholar] [CrossRef]
- Melendres, C.A.; Xu, S. In situ Laser Raman Spectroscopic Study of Anodic Corrosion Films on Nickel and Cobalt. J. Electrochem. Soc. 1984, 131, 2239–2243. [Google Scholar] [CrossRef]
- Maslar, J.E.; Hurst, W.S.; Bowers, W.J.; Hendricks, J.H.; Aquina, M.J.; Levin, I. In-Situ Raman Spectroscopic Investigation of Chromium Surfaces under Hydrothermal Conditions. Appl. Surf. Sci. 2001, 180, 102–118. [Google Scholar] [CrossRef]
- Cubicciotti, D. Potential-pH Diagrams for Alloy-Water Systems. J. Nucl. Mater. 1993, 201, 176–183. [Google Scholar] [CrossRef]
- Hosterman, B.D. Raman Spectroscopic Study of Solid Solution Spinel Oxides. Ph.D. Thesis, Department of Physics, University of Nevada, Las Vegas, NV, USA, August 2011. [Google Scholar]
- Maslar, J.E.; Hurst, W.S.; Bowers, W.J.; Hendricks, J.H.; Windsor, E.S. Alloy 600 Aqueous Corrosion at Elevated Temperatures and Pressures: An In Situ Raman Spectroscopic Investigation. J. Electrochem. Soc. 2009, 156, C103–C113. [Google Scholar] [CrossRef]
- Yang, J.; Li, Y.; Macdonald, D.D. Effects of Temperature and pH on the Electrochemical Behavior of Alloy 600 in Simulated PWR Primary Water. J. Nucl. Mater. 2020, 528, 1–16. [Google Scholar] [CrossRef]
- Bojinov, M.; Kinnunen, P.; Sunduolm, G. Electrochemical Behavior of Nickel-Chromium Alloys in a High temperature Aqueous Electrolyte. Corrosion 2003, 59, 91–103. [Google Scholar] [CrossRef]
- Yang, J.; Li, Y.; Xu, A.; Fekete, B.; Macdonald, D.D. The Electrochemical properties of Alloy 690 in Simulated Pressurized Water Reactor Primary Water: Effect of Temperature. J. Nucl. Mater. 2019, 518, 305–315. [Google Scholar] [CrossRef]
- Kaufman, L. Pourbaix Diagrams of Ni-Cr-Fe Ternary Alloys; EPRI Report C13751A; Electric Power Research Institute: Palo Alto, CA, USA, 2009. [Google Scholar]
- Pang, Y.S.; Hwang, H.J.; Kim, M.S. Reversible Temperature Dependence in Surface-Enhanced Raman Scattering of 1-Propanethiol Adsorbed on a Silver Island Film. J. Phys. Chem. B 1998, 102, 7203–7209. [Google Scholar] [CrossRef]
- Chiang, H.P.; Leung, P.T.; Tse, W.S. Remarks on the Substrate-Temperature Dependence of Surface-Enhanced Raman Scattering. J. Phys. Chem. B 2000, 104, 2348–2350. [Google Scholar] [CrossRef]
- Kwon, C.H.; Boo, D.W.; Hwang, H.J.; Kim, M.S. Temperature Dependence and Annealing Effects in Surface-Enhanced Raman Scattering on Chemically Prepared Silver Island Films. J. Phys. Chem. B 1999, 103, 9610–9615. [Google Scholar] [CrossRef]
- Leung, P.T.; Hider, M.H.; Sanchez, E.J. Surface-enhanced Raman Scattering at Elevated Temperatures. Phys. Rev. B 1996, 53, 12659–12662. [Google Scholar] [CrossRef] [PubMed]
- Galloway, C.M.; Le Ru, E.C.; Etchegoin, P.G. An Iterative Algorithm for Background Removal in Spectroscopy by Wavelet Transforms. Appl. Spectrosc. 2009, 63, 1370–1376. [Google Scholar] [CrossRef]
- Devine, T.M.; Adar, F. Raman Spectrsocopy of Solids. In Characterization of Materials; Wiley & Sons: Hoboken, NJ, USA, 2012; ISBN 9780471268826. [Google Scholar]
- Moskovits, M. Surface-enhanced Spectroscopy. Rev. Mod. Phys. 1985, 57, 783–829. [Google Scholar] [CrossRef]
- Moskovits, M. Surface Enhanced Raman Spectroscopy: A brief retrospective. J. Raman Spectrosc. 2005, 36, 485–496. [Google Scholar] [CrossRef]
- Gui, J.; Devine, T.M. Use of Surface Enhanced Raman Scattering as an In-Situ Probe of the Metal-Aqueous Solution Interface; Corrosion ’91, Paper #79; NACE: Houston, TX, USA, 1991. [Google Scholar]
- Kumai, C.S.; Devine, T.M. Oxidation of Iron in Oxygen-Containing Water at 288 °C. Corrosion 2005, 61, 201. [Google Scholar] [CrossRef]
- Kumai, C.S.; Devine, T.M. Influence of Oxygen Concentration of 288 °C Water and Alloy Composition on the Films Formed on Fe-Ni-Cr Alloys. Corrosion 2007, 63, 1101. [Google Scholar] [CrossRef]
- Kim, H.; Macdonald, D.D. Measurement of Steady-State Hydrogen Electrode Reactions on Alloy 600 and Alloy 690 Tubes. Corros. Sci. 2010, 52, 1139–1145. [Google Scholar] [CrossRef]
- Mahajan, S.; Cole, R.M.; Speed, J.D.; Pelfrey, S.H.; Russell, A.E.; Bartlett, P.N.; Barnett, S.M.; Baumberg, J.J. Understanding the Surface-Enhanced Raman Spectroscopy “Background”. J. Phys. Chem. C 2010, 114, 7242–7250. [Google Scholar] [CrossRef]
- Praveen, B.B.; Steuwe, C.; Mazilu, M.; Dholakia, K.; Mahajan, S. Wavelength modulated surface enhanced (resonance) Raman scattering for background-free detection. Analyst 2013, 138, 2816–2820. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Titanium Autoclave | |||
| Inner Layer | Outer Layer | Temperature and Time of Oxidation | Reference |
| Cr2O3 | Ni(OH)2 | 325 °C/4–8 min | [5] * |
| Cr2O3/CrOOH | NiO whiskers | 320 °C/4 h | [6] ** |
| Stainless Steel Autoclaves | |||
| Inner layer | Outer Layer | Temperature and Time of Oxidation | Reference |
| (Ni0.7Fe 0.3)(Fe0.3Cr0.7)23O4 | (Ni0.9Fe0.1)(Fe0.85Cr0.15)2O4 | 260 °C 1000–10,000 h | [7] |
| Cr-rich oxide | (Ni,Cr,Fe)-spinel NiFe2O4 | 320 °C/1000 h | [8] |
| Cr-rich oxide | Ni(Cr,Fe)2O4 | 360 °C/300 h | [9] |
| Cr2O3/(Fe,Ni)Cr2O4 | NiFe2O4 | 360 °C/300 h | [10] |
| Ni(Cr,Fe)2O4 | NiFe2O4 | 338 °C/4000 h | [11] *** |
| Cr-rich oxide | NiFe2O4 | 360 °C/1000 h | [12] |
| Alloy 690 Autoclave: Alloy 600 Tubing | |||
| Inner layer | Outer Layer | Temperature and Time of Oxidation | Reference |
| CrOOH Cr-oxide | NiCr2O4 NiO | 70 h/350 °C | [13] |
| Compositions (wt.%) of Alloy 600 and Alloy 690 | ||||||||
|---|---|---|---|---|---|---|---|---|
| Alloy | Ni | Cr | Fe | Mn | C | Cu | Si | S |
| Alloy 600 | ≥72 | 14–17 | 6–10 | ≤1 | ≤0.15 | ≤0.5 | ≤0.5 | ≤0.015 |
| Alloy 690 | ≥58 | 27–31 | 7–11 | ≤0.5 | ≤0.05 | ≤0.5 | ≤0.5 | ≤0.015 |
| Ni | 538.0 cm−1 SD 2.7 cm−1 | ||
| Cr | 541.8cm−1 SD 2.9 cm−1 | 582.7 cm−1 SD 5.4 cm−1 | |
| Ni-10Cr * | 543.5 cm−1 ± 5.0 cm−1 | 590.2 cm−1 ± 0 cm−1 | |
| Ni-20Cr * | 543.2 cm−1 ± 1.3 cm−1 | 602.3 cm−1 ± 6.6 cm−1 | |
| Ni-5Cr-8Fe | 539.1 cm−1 SD 2.8 cm−1 | 618.0 cm−1 SD 5.2 cm−1 | 679.7 cm−1 ** |
| Ni-10Cr-8Fe | 539.3 cm−1 SD 3.3 cm−1 | 600.4 cm−1 SD 5.4 cm−1 | 659.3 cm−1 SD 4.7 cm−1 |
| Alloy 600 | 538.5 cm−1 SD 1.8 cm−1 | 611.5 cm−1 SD 4.9 cm−1 | 684.0 cm−1 SD 9.2 cm−1 |
| Alloy 690 | 539.0 cm−1 SD 2.9 cm−1 | 588.0 cm−1 SD 5.3 cm−1 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, F.; Devine, T.M. In Situ Surface-Enhanced Raman Spectroscopy Investigation of the Passive Films That Form on Alloy 600, Alloy 690, Unalloyed Cr and Ni, and Alloys of Ni-Cr and Ni-Cr-Fe in Pressurized Water Nuclear Reactor Primary Water. Corros. Mater. Degrad. 2025, 6, 16. https://doi.org/10.3390/cmd6020016
Wang F, Devine TM. In Situ Surface-Enhanced Raman Spectroscopy Investigation of the Passive Films That Form on Alloy 600, Alloy 690, Unalloyed Cr and Ni, and Alloys of Ni-Cr and Ni-Cr-Fe in Pressurized Water Nuclear Reactor Primary Water. Corrosion and Materials Degradation. 2025; 6(2):16. https://doi.org/10.3390/cmd6020016
Chicago/Turabian StyleWang, Feng, and Thomas M. Devine. 2025. "In Situ Surface-Enhanced Raman Spectroscopy Investigation of the Passive Films That Form on Alloy 600, Alloy 690, Unalloyed Cr and Ni, and Alloys of Ni-Cr and Ni-Cr-Fe in Pressurized Water Nuclear Reactor Primary Water" Corrosion and Materials Degradation 6, no. 2: 16. https://doi.org/10.3390/cmd6020016
APA StyleWang, F., & Devine, T. M. (2025). In Situ Surface-Enhanced Raman Spectroscopy Investigation of the Passive Films That Form on Alloy 600, Alloy 690, Unalloyed Cr and Ni, and Alloys of Ni-Cr and Ni-Cr-Fe in Pressurized Water Nuclear Reactor Primary Water. Corrosion and Materials Degradation, 6(2), 16. https://doi.org/10.3390/cmd6020016