
Atmospheric Corrosion of Silver and Silver Nanoparticles

Abstract
:1. Introduction
- A thorough understanding of the corrosion mechanism can guide materials design to slow or prevent the corrosion process and expand the options for technological, medical, practical, or decorative applications.
- Knowledge about the speed and mechanism of the corrosion of silver nanoparticles can inform the extent to which we need to be concerned about their ecotoxic effect upon release to the environment.
- As both corrosion and antibacterial activity have a common link to the release of Ag+ ions, understanding the corrosion process can provide insight into antibacterial effectiveness.
- An understanding of the corrosion of silver has importance in the conservation and restoration of historical and cultural objects.
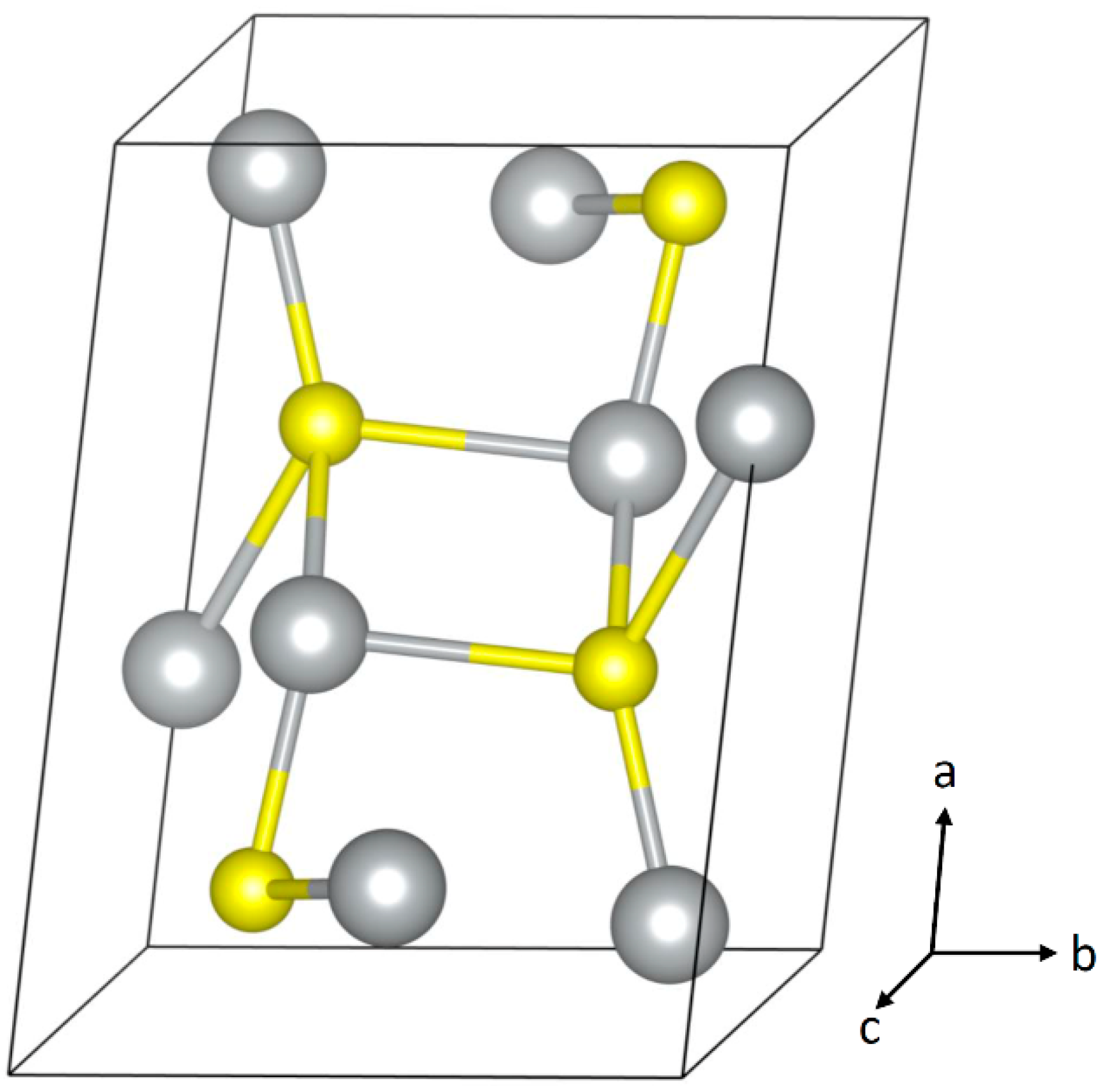
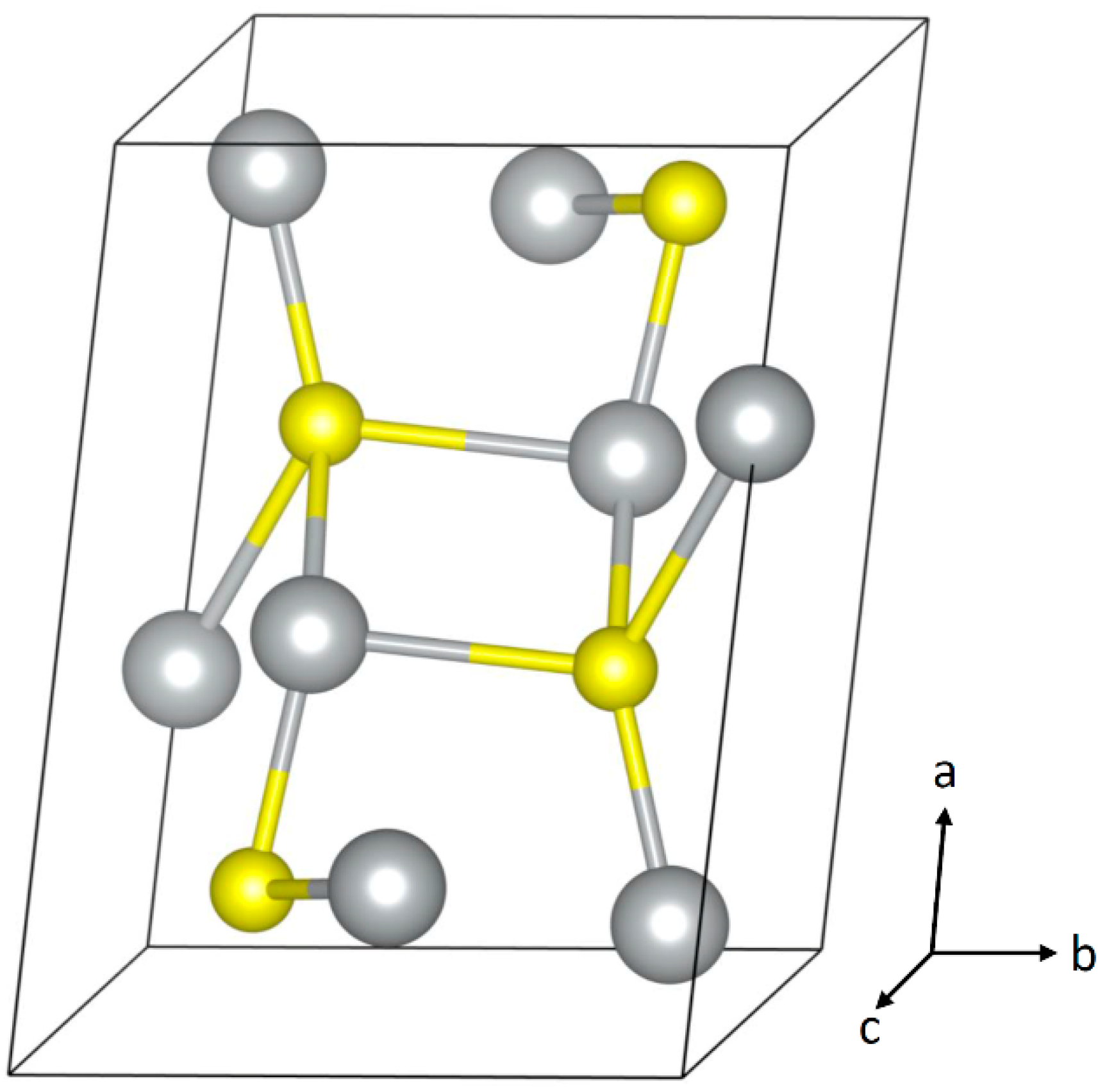
2. Corrosion of Bulk Silver

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Ag2S | Space Group: P21/c (14) | |||
|---|---|---|---|---|
| Unit Cell Parameters | ||||
| a = 4.231 Å | b = 6.93 Å | c = 9.526 Å | ||
| α = 90° | β = 125.48° | γ = 90° | ||
| Fractional Atom Coordinates (Wykoff site) | ||||
| Ag | 0.0438 | 0.0169 | 0.3075 | (4e) |
| Ag | 0.6465 | 0.3213 | 0.4362 | (4e) |
| S | 0.2612 | 0.2383 | 0.1306 | (4e) |
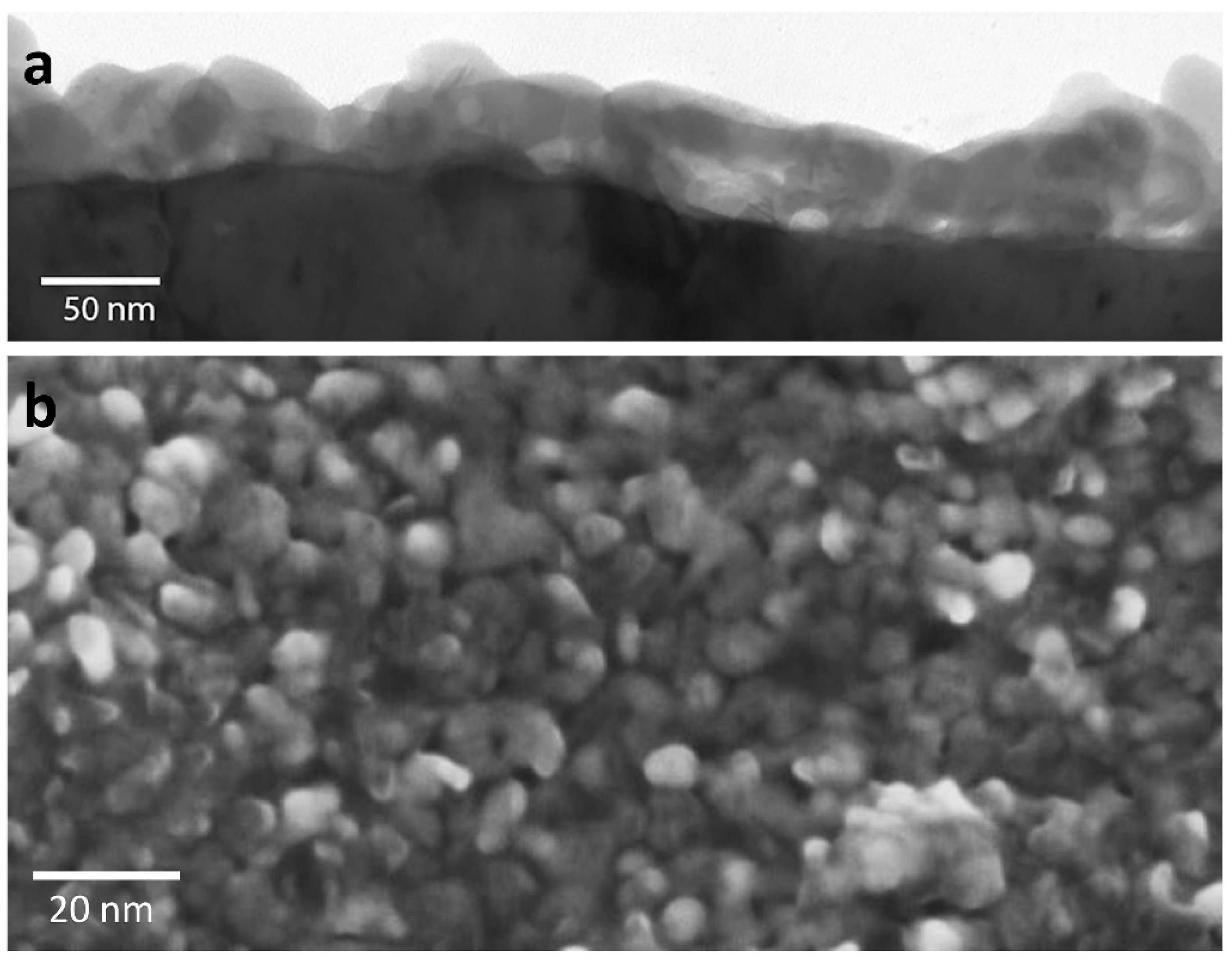
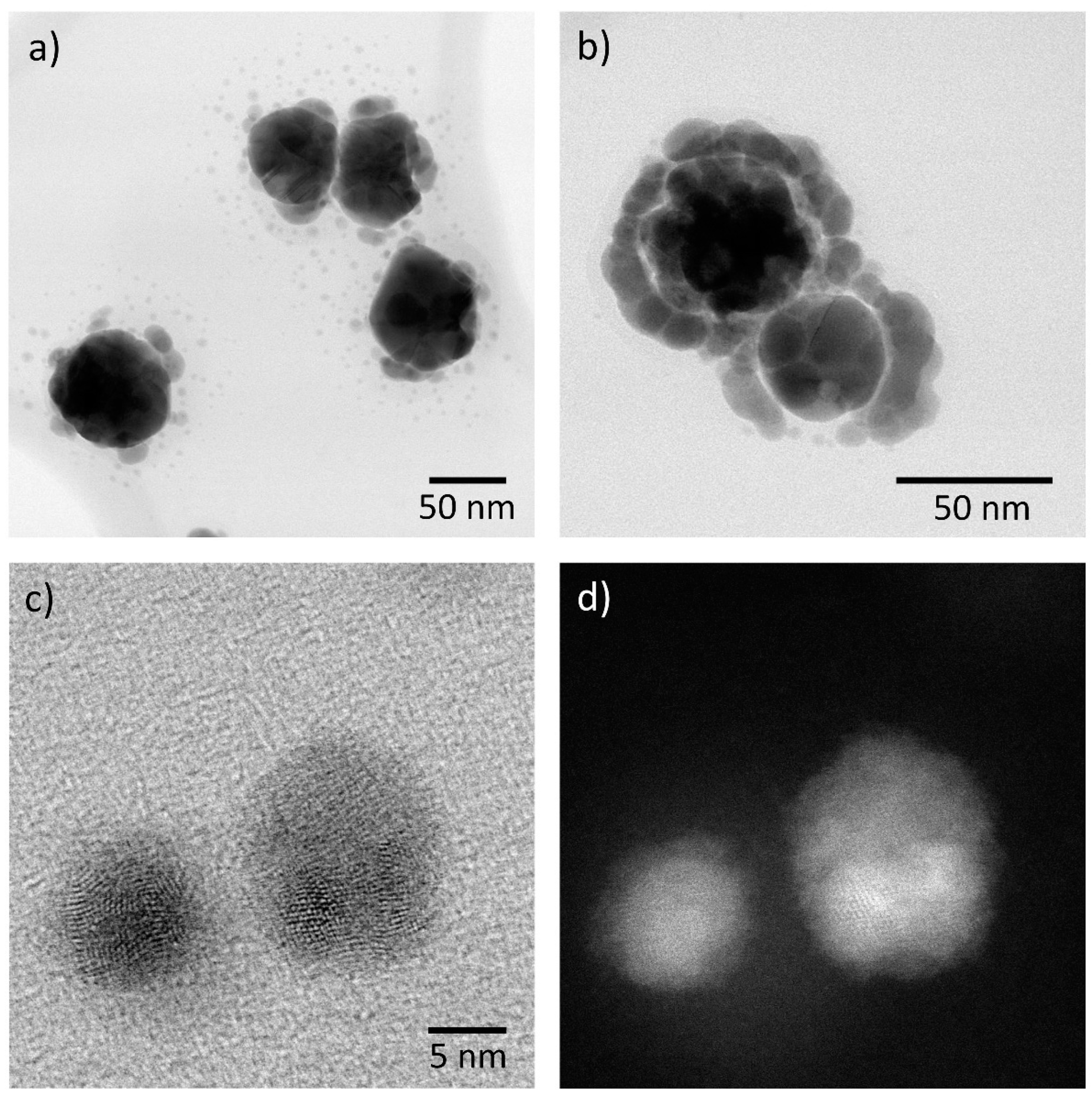
3. Corrosion of Nanoparticle Silver
4. Corrosion in Other Environments
5. Summary and Areas for Future Research
- direct conversion to Ag2S;
- oxidative dissolution of Ag to Ag+ followed by precipitation as Ag2S; and
- oxidative dissolution of Ag to Ag+ followed by precipitation as nanoparticulate Ag, then followed by conversion to Ag2S.
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Emsley, J. Nature’s Building Blocks: An A-Z Guide to the Elements, 2nd ed.; Oxford University Press: Oxford, UK, 2011. [Google Scholar]
- Rycenga, M.; Cobley, C.M.; Zeng, J.; Li, W.; Moran, C.H.; Zhang, Q.; Qin, D.; Xia, Y. Controlling the synthesis and assembly of silver nanostructures for plasmonic applications. Chem. Rev. 2011, 111, 3699–3712. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, H.; Peng, Y.; Yang, Y.; Li, Z.Y. Plasmon-enhanced light–matter interactions and applications. NPJ Comput. Mater. 2019, 5, 45. [Google Scholar] [CrossRef]
- Ardakani, L.S.; Surendar, A.; Thangavelu, L.; Mandal, T. Silver nanoparticles (Ag NPs) as catalyst in chemical reactions. Synth. Commun. 2021, 51, 1516–1536. [Google Scholar]
- Cortie, M.B.; Arnold, M.D.; Keast, V.J. The quest for zero loss: Unconventional materials for plasmonics. Adv. Mater. 2019, 32, 1904532. [Google Scholar] [CrossRef]
- Naik, G.V.; Shalaev, V.M.; Boltasseva, A. Alternative plasmonic materials: Beyond gold and silver. Adv. Mater. 2013, 25, 3264–3294. [Google Scholar] [CrossRef]
- Burdușel, A.C.; Gherasim, O.; Grumezescu, A.M.; Mogoantă, L.; Ficai, A.; Andronescu, E. Biomedical applications of silver nanoparticles: An up-to-date review. Nanomaterials 2018, 8, 681. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, J.; Sonshine, D.A.; Shervani, S.; Hurt, R.H. Controlled release of biologically active silver form nanosilver surfaces. ACS Nano 2010, 4, 6903–6913. [Google Scholar] [CrossRef] [Green Version]
- Lopez-Heras, M.; Theodorou, I.G.; Leo, B.F.; Ryan, M.P.; Porter, A.E. Towards understanding the antibacterial activity of Ag nanoparticles: Electron microscopy in the analysis of the materials-biology interface in the lung. Environ. Sci. Nano 2015, 2, 312–326. [Google Scholar] [CrossRef] [Green Version]
- Alexander, J.W. History of the medical use of silver. Surg. Infect. 2009, 10, 289–292. [Google Scholar] [CrossRef] [Green Version]
- Barillo, D.J.; Marx, D.E. Silver in medicine: A brief history BC 335 to present. Burns 2014, 40S, S3–S8. [Google Scholar] [CrossRef]
- Reidy, B.; Haase, A.; Luch, A.; Dawson, K.A.; Lynch, I. Mechanisms of silver nanoparticle release, transformation and toxicity: A critical review of current knowledge and recommendations for future studies and applications. Materials 2013, 6, 2295–2350. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marx, D.E.; Barillo, D.J. Silver in medicine: The basic science. Burns 2014, 405, S9–S18. [Google Scholar] [CrossRef] [PubMed]
- McQueen, R.H.; Keelan, M.; Xu, Y.; Mah, T. In vivo assessment of odour retention in an antimicrobial silver chloride-treated polyester textile. J. Text. Inst. 2013, 104, 108–117. [Google Scholar] [CrossRef]
- Toy, L.W.; Macera, L. Evidence-based review of silver dressing use on chronic wounds. J. Am. Acad. Nurse Pract. 2011, 23, 183–192. [Google Scholar] [CrossRef] [PubMed]
- Walter, N.; McQueen, R.H.; Keelan, M. In vivo assessment of antimicrobial treated textiles on skin microflora. Int. J. Cloth. Sci. Technol. 2014, 26, 330–342. [Google Scholar] [CrossRef]
- Tortella, G.R.; Rubilar, O.; Durán, N.; Diez, M.C.; Martinez, M.; Parada, J.; Seabra, A.B. Silver nanoparticles: Toxicity in model organisms as an overview of is hazard for human health and the environment. J. Hazard. Mater. 2020, 390, 121974. [Google Scholar] [CrossRef]
- Storme, P.; Schalm, O.; Wiesinger, R. The sulfidation process of sterling silver in different corrosive environments: Impact of the process on the surface films formed and consequences for the conservation-restoration community. Herit. Sci. 2015, 3, 25–39. [Google Scholar] [CrossRef] [Green Version]
- Tissot, I.; Monteiro, O.C.; Barreiros, M.A.; Correia, J.; Guerra, M.F. The influence of the constituent elements on the corrosion mechanisms of silver alloys in sulphide environments: The case of sterling silver. RSC Adv. 2017, 7, 28564–28572. [Google Scholar] [CrossRef] [Green Version]
- Phillips, V.A. Role of defects in evaporated silver films on the nucleation of sulfide “patches”. J. App. Phys. 1962, 33, 712–717. [Google Scholar] [CrossRef]
- Guan, R.; Yu, Y.D. A TEM study of Ag8S formed in the early stage of sulfidization of silver. Scripta Met. 1990, 24, 869–872. [Google Scholar] [CrossRef]
- Salvado, N.; Buti, S.; Labrador, A.; Cinque, G.; Emerich, H.; Pradell, T. SR-XRD and SR-FTIR study of the alteration of silver foils in medieval paintings. Anal. Bioanal. Chem. 2011, 399, 3041–3052. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, Y.D.; Guan, R. Electron-microscope study of the structure of Ag8S formed in the initial stage of silver sulfidation. Acta Cryst. 1995, B51, 149–155. [Google Scholar] [CrossRef]
- Sadanaga, R.; Sueno, S. X-ray study on the α-β transition of Ag2S. Mineral. J. 1967, 5, 124–148. [Google Scholar] [CrossRef] [Green Version]
- Momma, K.; Izumi, F. VESTA 3 for three-dimensional visualization of crystal, volumetric and morphology data. J. Appl. Crystallogr. 2011, 44, 1272–1276. [Google Scholar] [CrossRef]
- Sanders, C.E.; Verreault, D.; Frankel, G.S.; Allen, H.C. The role of sulfur in the atmospheric corrosion of silver. J. Electrochem. Soc. 2015, 162, C630–C637. [Google Scholar] [CrossRef]
- Wan, Y.; Wang, X.; Wang, X.; Li, Y.; Sun, H.; Zhang, K. Determination and generation of the corrosion compounds in silver exposed to the atmospheres. Int. J. Electrochem. Sci. 2015, 10, 2336–2354. [Google Scholar]
- Lin, H.; Frankel, G.S.; Abbott, W.H. Analysis of Ag corrosion products. J. Electrochem. Soc. 2013, 160, C345–C355. [Google Scholar] [CrossRef] [Green Version]
- Wan, Y.; Macha, E.N.; Kelly, R.G. Modification of ASTM B117 salt spray corrosion test and its correlation to field measurements of silver corrosion. Corrosion 2012, 68, 036001. [Google Scholar] [CrossRef]
- Watanabe, M.; Hokazono, A.; Handa, T.; Ichino, T.; Kuwaki, N. Corrosion of copper and silver plates by volcanic gases. Corros. Sci. 2006, 48, 3759–3766. [Google Scholar] [CrossRef]
- Watanabe, M.; Shinozaki, S.; Toyoda, E.; Asakura, K.; Ichino, T.; Kuwaki, N.; Higashi, Y.; Tanaka, T. Corrosion products formed on silver after a one-month exposure to urban atmospheres. Corrosion 2006, 62, 243–250. [Google Scholar] [CrossRef]
- Lin, H.; Frankel, G.S. Accelerated atmospheric corrosion testing of Ag. Corrosion 2013, 69, 1060–1072. [Google Scholar] [CrossRef]
- Liang, D.; Allen, H.C.; Frankel, G.S.; Chen, Z.Y.; Kelly, R.G.; Wu, Y.; Wyslouzil, B.E. Effects of sodium chloride particles, ozone, UV and relative humidity on atmospheric corrosion of silver. J. Electrochem. Soc. 2010, 157, C146–C156. [Google Scholar] [CrossRef] [Green Version]
- Franey, J.P.; Kammlott, G.W.; Graedel, T.E. The corrosion of silver by atmospheric sulfur gases. Corros. Sci. 1985, 25, 133–143. [Google Scholar] [CrossRef]
- Graedel, T.E.; Franey, J.P.; Gualtieri, G.J.; Kammlott, G.W.; Malm, D.L. On the mechanism of silver and copper sulfidation by atmospheric H2S and OCS. Corros. Sci. 1985, 25, 1163–1180. [Google Scholar] [CrossRef]
- Graedel, T.E. Corrosion mechanisms for silver exposed to the atmosphere. J. Electrochem. Soc. 1992, 139, 1963–1970. [Google Scholar] [CrossRef]
- Pope, D.; Gibbens, H.R.; Moss, R.L. The tarnishing of Ag at naturally-occurring H2S and SO2 levels. Corros. Sci. 1968, 8, 883–887. [Google Scholar] [CrossRef]
- Rice, D.W.; Peterson, P.; Rigby, E.B.; Phipps, P.B.P.; Coppell, R.J.; Tremoureux, R. Atmospheric corrosion of copper and silver. J. Electrochem. Soc. 1981, 128, 275–284. [Google Scholar] [CrossRef]
- Martina, I.; Wiesinger, R.; Schreiner, M. Micro-Raman investigations of early stage silver corrosion products occurring in sulfur containing atmospheres. J. Raman Spectrosc. 2013, 44, 770–775. [Google Scholar] [CrossRef]
- Sinclair, J.D. Tarnishing of solver by organic sulfur vapours: Rates and film characteristics. Electrochem. Sci. Technol. 1982, 239, 33–40. [Google Scholar] [CrossRef]
- Liu, J.; Pennell, K.G.; Hurt, R.H. Kinetics and mechanisms of nanosilver oxysulfidation. Environ. Sci. Technol. 2011, 45, 7345–7353. [Google Scholar] [CrossRef] [Green Version]
- Bennett, H.E.; Peck, R.L.; Burge, D.K.; Bennett, J.M. Formation and growth of tarnish on evaporated silver films. J. Appl. Phys. 1969, 40, 3351–3360. [Google Scholar] [CrossRef]
- Wiesinger, R.; Grayburn, R.; Dowsett, M.; Sabbe, P.J.; Thompson, P.; Adriaens, A.; Schreiner, M. In situ time-lapse synchrotron radiation X-ray diffraction of silver corrosion. J. Anal. At. Spectrom. 2015, 30, 694–701. [Google Scholar] [CrossRef]
- Price, L.E.; Thomas, G.J. The tarnishing of silver and silver alloys and its prevention. J. Inst. Metals 1938, 63, 29–65. [Google Scholar]
- Chou, C.H. Hydrogen Sulfide: Human Health Aspects. In Concise International Chemical Assessment Document; World Health Organization: Geneva, Switzerland, 2003; Volume 53. [Google Scholar]
- Kleber, C.; Wiesinger, R.; Schnöller, J.; Hilfrich, U.; Hutter, H.; Schreiner, M. Initial oxidation of silver surfaces by S2− and S4+ species. Corros. Sci. 2008, 50, 1112–1121. [Google Scholar] [CrossRef]
- Rice, D.W.; Cappell, R.J.; Kinsolving, W.; Laskowski, J.J. Indoor corrosion of metals. J. Electrochem. Soc. 1980, 127, 891–901. [Google Scholar] [CrossRef]
- Ankersmit, H.A.; Tennant, N.H.; Watts, S.F. Hydrogen sulfide and carbonyl sulfide in the museum environment—Part I. Atmos. Environ. 2005, 39, 695–707. [Google Scholar] [CrossRef]
- Leygraf, C.; Wallinde, I.O.; Tidblad, J.; Graedel, T. Atmospheric Corrosion, 2nd ed.; John Wiley & Sons: Hoboken, NJ, USA, 2016. [Google Scholar]
- Volpe, L.; Peterson, P.J. The atmospheric sulfidation of silver in a tubular corrosion reactor. Corros. Sci. 1989, 29, 1179–1196. [Google Scholar] [CrossRef]
- Fischmeister, H.; Drott, J. Reaction rate and growth forms of reaction product in the system Ag-H2S. Acta Metall. 1959, 7, 777–781. [Google Scholar] [CrossRef]
- Lee, S.H.; Jun, B.H. Silver nanoparticles: Synthesis and application for nanomedicine. Int. J. Mol. Sci. 2019, 20, 865. [Google Scholar] [CrossRef] [Green Version]
- Yin, Y.; Li, Z.Y.; Zhong, Z.; Gates, B.; Xia, Y.; Venkateswaran, S. Synthesis and characterization of stable aqueous dispersions of silver nanoparticles through the Tollens process. J. Mater. Chem. 2002, 12, 522–527. [Google Scholar] [CrossRef]
- Allpress, J.G.; Sanders, J.V. The influence of surface structure on a tarnishing reaction. Philos. Mag. 1964, 10, 829–836. [Google Scholar] [CrossRef]
- Glover, R.D.; Miller, J.M.; Hutchinson, J.E. Generation of metal nanoparticles from silver and copper objects: Nanoparticle dynamics on surfaces and potential sources of nanoparticles in the environment. ACS Nano 2011, 5, 8950–8957. [Google Scholar] [CrossRef] [PubMed]
- Le Ouay, B.; Stellacci, F. Antibacterial activity of silver nanoparticles: A surface science insight. Nano Today 2015, 10, 339–354. [Google Scholar] [CrossRef] [Green Version]
- McMahon, M.D.; Lopez, R.; Meyer, H.M.; Feldmen, L.C.; Haglund, R.F., Jr. Rapid tarnishing of silver nanoparticles in ambient laboratory air. Appl. Phys. B 2005, 80, 915–921. [Google Scholar] [CrossRef]
- Cao, W.; Elsayed-Ali, H.E. Stability of Ag nanoparticles fabricated by electron beam lithography. Mater. Lett. 2009, 63, 2263–2266. [Google Scholar] [CrossRef]
- Elechiguerra, J.L.; Larios-Lopez, L.; Lui, C.; Garcia-Gutierrez, D.; Camacho-Bragado, A.; Yacaman, M.J. Corrosion at the nanoscale: The case of silver nanowires and nanoparticles. Chem. Mater. 2005, 17, 6042–6052. [Google Scholar] [CrossRef]
- Lu, W.; Yao, K.; Wang, J.; Yuan, J. Ionic liquids-water interfacial preparation of triangular Ag nanoplates and their shape-dependent antibacterial activity. J. Colloid Interface Sci. 2015, 437, 35–41. [Google Scholar] [CrossRef]
- Wang, L.; Xiong, W.; Nishijima, Y.; Yokota, Y.; Ueno, K.; Misawa, H.; Bi, G.; Qiu, J.R. Spectral properties and mechanism of instability of nanoengineered silver blocks. Opt. Express 2011, 19, 10640–10646. [Google Scholar] [CrossRef]
- Keast, V.J. Corrosion processes of silver nanoparticles. Appl. Nanosci. 2022, 12, 1859–1868. [Google Scholar] [CrossRef]
- Keast, V.J.; Myles, T.A.; Shahcheraghi, N.; Cortie, M.B. Corrosion processes of triangular silver nanoparticles compared to bulk silver. J. Nanopart. Res. 2016, 18, 45. [Google Scholar] [CrossRef]
- Scuderi, M.; Esposito, M.; Todisco, F.; Simeone, D.; Tatamtini, I.; De Marco, L.; De Giorgi, M.; Nicotra, G.; Carbone, L.; Sanvitto, D.; et al. Nanoscale study of the tarnishing process in electron beam lithography-fabricated silver nanoparticles for plasmonic applications. J. Phys. Chem. C 2016, 120, 24314–24323. [Google Scholar] [CrossRef]
- Trautmann, S.; Dathe, A.; Csáki, A.; Thiele, M.; Müller, R.; Fritzche, W.; Stranik, O. Time-resolved study of site-specific corrosion in a single crystalline silver nanoparticle. Nanoscale Res. Lett. 2019, 14, 240. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cai, W.; Zhong, H.; Zhang, L. Optical measurements of oxidation behavior of silver nanometer particle within pores of silica host. J. Appl. Phys. 1998, 83, 1705–1710. [Google Scholar] [CrossRef]
- Qi, H.; Alexson, D.; Glembocki, O.; Prokes, S.M. The effect of size and size distribution on the oxidation kinetics and plasmonics of nanoscale Ag particles. Nanotechnology 2010, 21, 215706. [Google Scholar] [CrossRef]
- Kuzma, A.; Weis, M.; Flickyngerova, S.; Jakabovic, J.; Satka, A.; Dobrocka, E.; Chlpik, J.; Cirak, J.; Donoval, M.; Telek, P.; et al. Influence of surface oxidation on plasmon resonance in monolayer of gold and silver nanoparticles. J. Appl. Phys. 2012, 112, 130531. [Google Scholar] [CrossRef]
- Grillet, N.; Manchon, D.; Cottancin, E.; Bertorelle, F.; Bonnet, C.; Broyer, M.; Lerme, J.; Pellarin, M. Photo-oxidation of individual silver nanoparticles: A real-time tracking of optical and morphological changes. J. Phys. Chem. C 2013, 117, 2274–2282. [Google Scholar] [CrossRef]
- Han, Y.; Lupitsky, R.; Chou, T.M.; Stafford, C.M.; Du, H.; Sukhishvila, S. Effect of oxidation on surface-enhanced Raman scattering activity of silver nanoparticles: A quantitative correlation. Anal. Chem. 2011, 83, 5873–5880. [Google Scholar] [CrossRef]
- Liu, J.; Hurt, R.H. Ion release kinetics and particle persistence in aqueous nano-silver colloids. Environ. Sci. Technol. 2010, 44, 2169–2175. [Google Scholar] [CrossRef]
- Levard, C.; Reinsch, B.C.; Michel, F.M.; Oumahi, C.; Lowry, G.V.; Brown, G.E., Jr. Sulfidation processes of PVP-coated silver nanoparticles in aqueous solution: Impact on dissolution rate. Environ. Sci. Technol. 2011, 45, 5260–5266. [Google Scholar] [CrossRef]
- Liu, B.; Ma, Z. Synthesis of Ag2S-Ag nanoprisms and their use as DNA hybridization probes. Small 2011, 7, 1587–1592. [Google Scholar] [CrossRef]
- Zeng, J.; Tao, J.; Su, D.; Zhu, Y.; Qin, D.; Xia, Y. Selective sulfuration at the corner sites of silver nanocrystal and it’s use in stabilization of the shape. Nano Lett. 2011, 11, 3010–3015. [Google Scholar] [CrossRef] [PubMed]
- Park, G.; Lee, C.; Seo, D.; Song, H. Full-color tuning of surface plasmon resonance by compositional variation of Ay@Ag core-shell nanocubes with sulfides. Langmuir 2012, 28, 9003–9009. [Google Scholar] [CrossRef] [PubMed]
- Huang, H.; Li, Q.; Wang, J.; Li, Z.; Yu, X.F.; Chu, P.K. Sensitive and robust colorimetric sensing of sulfide anion by plasmonic nanosensors based on quick crystal growth. Plasmonics 2014, 9, 11–16. [Google Scholar] [CrossRef]
- Yu, S.; Yin, Y.; Chao, J.; Shen, M.; Liu, J. Highly dynamic PVP-coated silver nanoparticles in aquatic environments: Chemical and morphology change induced by oxidation of Ag0 and reduction of Ag+. Environ. Sci. Technol. 2014, 48, 403–411. [Google Scholar] [CrossRef] [PubMed]
- Chen, R.; Morris, H.R.; Whitmore, P.M. Fast detection of hydrogen sulfide gas in the ppm range with silver nanoparticle films at ambient conditions. Sens. Actuators B Chem. 2013, 186, 431–438. [Google Scholar] [CrossRef]
- Davidson, R.A.; Anderson, D.S.; Van Winkle, L.S.; Pinkerton, K.E.; Guo, T. Evolution of silver nanoparticles in the rat lung investigated by X-ray absorption spectroscopy. J. Phys. Chem. A 2014, 119, 281–289. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, X.; Santschi, C.; Martin, O.J.F. Strong improvement of long-Term chemical and thermal stability of plasmonic silver nanoantennas and films. Small 2017, 13, 1700044. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mayousse, C.; Celle, C.; Fraczkiewicz, A.; Simonato, J.P. Stability of silver nanowire based electrodes under environmental and electrical stresses. Nanoscale 2015, 7, 2107–2115. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Theodorou, I.G.; Goode, A.E.; Gow, A.; Schwander, S.; Zhang, J.; Chung, K.F.; Tetley, T.D.; Shaffer, M.S.; Ryan, M.P.; et al. High-resolution analytical electron microscopy reveals cell culture media-induced changes to the chemistry of silver nanowires. Environ. Sci. Technol. 2013, 47, 13813–13821. [Google Scholar] [CrossRef] [Green Version]
- Impellitteri, C.A.; Harmon, S.; Silva, R.G.; Miller, B.W.; Scheckel, K.G.; Luxton, T.P.; Schupp, D.; Panguluri, S. Transformation of silver nanoparticles in fresh, ages and incinerated biosolids. Water Res. 2013, 47, 3678–3886. [Google Scholar] [CrossRef]
- Geng, H.; Poologasundarampillai, G.; Todd, N.; Devlin-Mullin, A.; Moore, K.L.; Golrokhi, Z.; Gilchrist, J.B.; Jones, E.; Potter, R.J.; Sutcliffe, C.; et al. Biotransformation of silver released from nanoparticle coated titanium implants revealed in regenerating bone. Appl. Mater. Interfaces 2017, 9, 21169–21180. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, S.; Goode, A.E.; Sweeney, S.; Theodorou, I.G.; Thorley, A.J.; Ruenraroengsak, P.; Chang, Y.; Gow, A.; Schwander, S.; Skepper, J.; et al. Sulfidation of silver nanowires inside human alveolar epithelial cells: A potential detoxification mechanism. Nanoscale 2013, 5, 9839–9847. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Theodorou, I.G.; Botelho, D.; Schwander, S.; Zhang, J.; Chung, K.F.; Tetley, T.D.; Shaffer, M.S.P.; Gow, A.; Ryan, M.P.; Porter, A.E. Static and dynamic microscopy of the chemical stability and aggregation state of silver nanowires in components of Murine pulmonary surfactant. Environ. Sci. Technol. 2015, 49, 8048–8056. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, S.; Goode, A.E.; Skepper, J.N.; Thorley, A.J.; Seiffert, J.M.; Chung, K.F.; Tetley, T.D.; Shaffer, M.S.P.; Ryan, M.P.; Porter, A.E. Avoiding artefacts during electron microscopy of silver nanomaterials exposed to biological environments. J. Microsc. 2015, 261, 157–166. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaegi, R.; Voegelin, A.; Ort, C.; Sinnet, B.; Thalmann, B.; Krismer, J.; Hagendorfer, H.; Elumela, M.; Mueller, E. Fate and transformation of silver nanoparticles in urban wastewater systems. Water Res. 2013, 47, 3866–3877. [Google Scholar] [CrossRef]
- Kaegi, R.; Voegelin, A.; Sinnet, B.; Zuleeg, S.; Hagendorfer, H.; Burkhardt, M.; Siegrist, H. Behavior of metallic silver nanoparticles in a pilot wastewater treatment plant. Environ. Sci. Technol. 2011, 45, 3902–3908. [Google Scholar] [CrossRef]
- Kent, R.D.; Oser, J.G.; Vikesland, P.J. Controlled evaluation of silver nanoparticle sulfidation in a full-scale wastewater treatment plant. Environ. Sci. Technol. 2014, 48, 8564–8572. [Google Scholar] [CrossRef]
- Lombi, E.; Donner, E.; Taheri, S.; Tavakkoli, E.; Jamting, A.K.; McClure, S.; Naidu, R.; Miller, B.W.; Scheckel, K.G.; Vasilev, K. Transformation of four silver/silver chloride nanoparticles during anaerobic treatment of wastewater and post-processing of sewage sludge. Environ. Pollut. 2013, 176, 193–197. [Google Scholar] [CrossRef]
- Ma, R.; Levard, C.; Judy, J.D.; Unrine, J.M.; Durenkamp, M.; Martin, B.; Jefferson, B.; Lowry, G.V. Fate of zinc oxide and silver nanoparticles in a pilot wastewater treatment plant and in processed biosolids. Environ. Sci. Technol. 2013, 48, 104–112. [Google Scholar] [CrossRef]
- Lorenz, C.; Windler, L.; von Goetz, N.; Lehman, R.P.; Schuppler, M.; Hungerbuhler, K.; Heuberger, M.; Nowack, B. Characterization of silver release from commercially available functional (nano)textiles. Chemosphere 2012, 89, 817–824. [Google Scholar] [CrossRef]
- Mitrano, D.M.; Rimmele, E.; Wischer, A.; Erni, R.; Height, M.; Nowack, B. Presence of nanoparticles in wash water from conventional silver and nano-silver textiles. ACS Nano 2014, 8, 7208–7219. [Google Scholar] [CrossRef] [PubMed]
- Impellitteri, C.A.; Tolaymat, T.M.; Scheckel, K.G. The speciation of silver nanoparticles in antimicrobial fabric before and after exposure to a hypochlorite/detergent solution. J. Environ. Qual. 2009, 38, 1528–1530. [Google Scholar] [CrossRef] [PubMed]
- Lowry, G.V.; Epinasse, B.P.; Badireddy, A.R.; Richardson, C.J.; Reinsch, B.C.; Bryant, L.D.; Bone, A.J.; Deonarine, A.; Chae, S.; Therezin, M.; et al. Long-term transformation and fate of manufactured Ag nanoparticles in a simulated large scale freshwater emergent wetland. Environ. Sci. Technol. 2012, 46, 7027–7036. [Google Scholar] [CrossRef] [PubMed]
- Coutris, C.; Joner, E.J.; Oughton, D.H. Aging and soil organic matter content affect the fate of silver nanoparticles in soil. Sci. Total Environ. 2012, 420, 327–333. [Google Scholar] [CrossRef] [PubMed]
- Khaksar, M.; Jolley, D.E.; Sekine, R.; Vasilev, K.; Johannessen, B.; Donner, E.; Lombi, E. In situ chemical transformations of silver nanoparticles along the water-sediment continuum. Environ. Sci. Technol. 2015, 49, 318–325. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hashimoto, Y.; Takeuchi, S.; Mitsunobu, S.; Ok, Y.S. Chemical speciation of silver (Ag) in soils under aerobic and anaerobic conditions: Ag nanoparticles vs. ionic Ag. J. Hazard. Mater. 2015, 322, 318–324. [Google Scholar] [CrossRef]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Keast, V.J. Atmospheric Corrosion of Silver and Silver Nanoparticles. Corros. Mater. Degrad. 2022, 3, 221-234. https://doi.org/10.3390/cmd3020013
Keast VJ. Atmospheric Corrosion of Silver and Silver Nanoparticles. Corrosion and Materials Degradation. 2022; 3(2):221-234. https://doi.org/10.3390/cmd3020013
Chicago/Turabian StyleKeast, Vicki J. 2022. "Atmospheric Corrosion of Silver and Silver Nanoparticles" Corrosion and Materials Degradation 3, no. 2: 221-234. https://doi.org/10.3390/cmd3020013
APA StyleKeast, V. J. (2022). Atmospheric Corrosion of Silver and Silver Nanoparticles. Corrosion and Materials Degradation, 3(2), 221-234. https://doi.org/10.3390/cmd3020013