
Adsorption of Gadolinium Bisphthalocyanine on Atomically Flat Surfaces: Comparison of Graphene and Hexagonal Boron Nitride from DFT Calculations


Abstract

1. Introduction
2. Methods
3. Results and Discussion
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Candini, A.; Klyatskaya, S.; Ruben, M.; Wernsdorfer, W.; Affronte, M. Graphene spintronic devices with molecular nanomagnets. Nano Lett. 2011, 11, 2634–2639. [Google Scholar] [CrossRef] [PubMed]
- Gonidec, M.; Biagi, R.; Corradini, V.; Moro, F.; De Renzi, V.; del Pennino, U.; Summa, D.; Muccioli, L.; Zannoni, C.; Amabilino, D.B.; et al. Surface supramolecular organization of a terbium(III) double-decker complex on graphite and its single molecule magnet behavior. J. Am. Chem. Soc. 2011, 133, 6603–6612. [Google Scholar] [CrossRef] [PubMed]
- Klar, D.; Candini, A.; Joly, L.; Klyatskaya, S.; Krumme, B.; Ohresser, P.; Kappler, J.-P.; Ruben, M.; Wende, H. Hysteretic behaviour in a vacuum deposited submonolayer of single ion magnets. Dalton Trans. 2014, 43, 10686–10689. [Google Scholar] [CrossRef] [PubMed]
- Marocchi, S.; Candini, A.; Klar, D.; Van den Heuvel, W.; Huang, H.; Troiani, F.; Corradini, V.; Biagi, R.; De Renzi, V.; Klyatskaya, S.; et al. Relay-like exchange mechanism through a spin radical between TbPc2 molecules and graphene/Ni(111) substrates. ACS Nano 2016, 10, 9353–9360. [Google Scholar] [CrossRef] [PubMed]
- Corradini, V.; Candini, A.; Klar, D.; Biagi, R.; De Renzi, V.; Lodi Rizzini, A.; Cavani, N.; del Pennino, U.; Klyatskaya, S.; Ruben, M.; et al. Probing magnetic coupling between LnPc2 (Ln = Tb, Er) molecules and the graphene/Ni(111) substrate with and without Au-intercalation: Role of the dipolar field. Nanoscale 2018, 10, 277–283. [Google Scholar] [CrossRef] [PubMed]
- Serrano, G.; Velez-Fort, E.; Cimatti, I.; Cortigiani, B.; Malavolti, L.; Betto, D.; Ouerghi, A.; Brookes, N.B.; Mannini, M.; Sessoli, R. Magnetic bistability of a TbPc2 submonolayer on a graphene/SiC(0001) conductive electrode. Nanoscale 2018, 10, 2715–2720. [Google Scholar] [CrossRef] [PubMed]
- Berkley, R.S.; Hooshmand, Z.; Jiang, T.; Le, D.; Hebard, A.F.; Rahman, T.S. Characteristics of single-molecule magnet dimers ([Mn3]2) on graphene and h-BN. J. Phys. Chem. C 2020, 124, 28186–28200. [Google Scholar] [CrossRef]
- Yin, X.; Deng, L.; Ruan, L.; Wu, Y.; Luo, F.; Qin, G.; Han, X.; Zhang, X. Recent progress for single-molecule magnets based on rare earth elements. Materials 2023, 16, 3568. [Google Scholar] [CrossRef]
- Liu, S.; Zhu, Z.; Zhang, P.; Tang, J. Chemisorption of lanthanide single-molecule magnets on surfaces. Fundam. Res. 2023. [Google Scholar] [CrossRef]
- Gabarró-Riera, G.; Aromí, G.; Sañudo, E.C. Magnetic molecules on surfaces: SMMs and beyond. Coord. Chem. Rev. 2023, 475, 214858. [Google Scholar] [CrossRef]
- Auwärter, W. Hexagonal boron nitride monolayers on metal supports: Versatile templates for atoms, molecules and nanostructures. Surf. Sci. Rep. 2019, 74, 1–95. [Google Scholar]
- Wäckerlin, C.; Donati, F.; Singha, A.; Baltic, R.; Rusponi, S.; Diller, K.; Patthey, F.; Pivetta, M.; Lan, Y.; Klyatskaya, S.; et al. Giant hysteresis of single-molecule magnets adsorbed on a nonmagnetic insulator. Adv. Mater. 2016, 28, 5195–5199. [Google Scholar] [CrossRef]
- Trojan, K.L.; Kendall, J.L.; Kepler, K.D.; Hatfield, W.E. Strong exchange coupling between the lanthanide ions and the phthalocyaninato ligand radical in bis(phthalocyaninato)lanthanide sandwich compounds. Inorg. Chim. Acta 1992, 198–200, 795–803. [Google Scholar] [CrossRef]
- Korolev, V.V.; Lomova, T.N.; Ramazanova, A.G.; Korolev, D.V.; Mozhzhukhina, E.G. Phthalocyanine-based molecular paramagnets. Effect of double-decker structure on magnetothermal properties of gadolinium complexes. J. Organometal. Chem. 2016, 819, 209–215. [Google Scholar] [CrossRef]
- Kratochvílová, I.; Šebera, J.; Paruzel, B.; Pfleger, J.; Toman, P.; Marešová, E.; Havlová, Š.; Hubík, P.; Buryi, M.; Vrňata, M.; et al. Electronic functionality of Gd-bisphthalocyanine: Charge carrier concentration, charge mobility, and influence of local magnetic field. Synth. Met. 2018, 236, 68–78. [Google Scholar] [CrossRef]
- Taran, G.; Moreno-Pineda, E.; Schulze, M.; Bonet, E.; Ruben, M.; Wernsdorfer, W. Direct determination of high-order transverse ligand field parameters via μSQUID-EPR in a Et4N [160GdPc2] SMM. Nat. Commun. 2023, 14, 3361. [Google Scholar] [CrossRef] [PubMed]
- Martínez-Flores, C.; Bolivar-Pineda, L.M.; Basiuk, V.A. Lanthanide bisphthalocyanine single-molecule magnets: A DFT survey of their geometries and electronic properties from lanthanum to lutetium. Mater. Chem. Phys. 2022, 287, 126271. [Google Scholar] [CrossRef]
- Martínez-Flores, C.; Basiuk, V.A. Ln@C60 endohedral fullerenes: A DFT analysis for the complete series from lanthanum to lutetium. Comp. Theor. Chem. 2022, 1217, 113878. [Google Scholar] [CrossRef]
- Martínez-Flores, C.; Basiuk, V.A. DFT analysis of the electronic and structural properties of lanthanide nitride cluster fullerenes Ln3N@C80. Inorganics 2023, 11, 223. [Google Scholar] [CrossRef]
- Basiuk, V.A.; Acevedo-Guzmán, D.A.; Meza-Laguna, V.; Álvarez-Zauco, E.; Huerta, L.; Serrano, M.; Kakazey, M.; Basiuk, E.V. High-energy ball-milling preparation and characterization of Ln2O3−graphite nanocomposites. Mater. Today Commun. 2021, 26, 102030. [Google Scholar] [CrossRef]
- Basiuk, E.V.; Prezhdo, O.V.; Basiuk, V.A. Strong bending distortion of supercoronene graphene model upon adsorption of lanthanide atoms. J. Phys. Chem. Lett. 2023, 14, 2910–2916. [Google Scholar] [CrossRef] [PubMed]
- Basiuk, V.A.; Prezhdo, O.V.; Basiuk, E.V. Adsorption of lanthanide atoms on graphene: Similar, yet different. J. Phys. Chem. Lett. 2022, 13, 6042–6047. [Google Scholar] [CrossRef] [PubMed]
- Bolivar-Pineda, L.M.; Mendoza-Domínguez, C.U.; Basiuk, V.A. Adsorption of lanthanide double-decker phthalocyanines on single-walled carbon nanotubes: Structural changes and electronic properties as studied by density functional theory. J. Mol. Model. 2023, 29, 158. [Google Scholar] [CrossRef] [PubMed]
- Mendoza-Domínguez, C.U.; Bolivar-Pineda, L.M.; Basiuk, V.A. Effect of structural defects in graphene on the geometry and electronic properties of adsorbed lanthanide bisphthalocyanines: A DFT analysis. Comp. Theor. Chem. 2023, 1225, 114152. [Google Scholar] [CrossRef]
- Delley, B.; Ellis, D.E.; Freeman, A.J.; Baerends, E.J.; Post, D. Binding energy and electronic structure of small copper particles. Phys. Rev. B 1983, 27, 2132–2144. [Google Scholar] [CrossRef]
- Delley, B. An all-electron numerical method for solving the local density functional for polyatomic molecules. J. Chem. Phys. 1990, 92, 508–517. [Google Scholar] [CrossRef]
- Delley, B. Fast calculation of electrostatics in crystals and large molecules. J. Phys. Chem. 1996, 100, 6107–6110. [Google Scholar] [CrossRef]
- Delley, B. From molecules to solids with the DMol3 approach. J. Chem. Phys. 2000, 113, 7756–7764. [Google Scholar] [CrossRef]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized gradient approximation made simple. Phys. Rev. Lett. 1996, 77, 3865–3868. [Google Scholar] [CrossRef]
- Grimme, S. Semiempirical GGA-type density functional constructed with a long-range dispersion correction. J. Comput. Chem. 2006, 27, 1787–1799. [Google Scholar] [CrossRef]
- Basiuk, V.A. Interaction of tetraaza[14]annulenes with single-walled carbon nanotubes: A DFT study. J. Phys. Chem. B 2004, 108, 19990–19994. [Google Scholar] [CrossRef]
- Michelini, M.C.; Pis Diez, R.; Jubert, A.H. A density functional study of small nickel clusters. Int. J. Quantum Chem. 1998, 70, 693–701. [Google Scholar] [CrossRef]
- Migliore, A.; Sit, P.H.-L.; Klein, M.L. Evaluation of electronic coupling in transition-metal systems using DFT: Application to the hexa-aquo ferric−ferrous redox couple. J. Chem. Theory Comput. 2009, 5, 307–323. [Google Scholar] [CrossRef] [PubMed]
- Basiuk, V.A.; Prezhdo, O.V.; Basiuk, E.V. Thermal smearing in DFT calculations: How small is really small? A case of La and Lu atoms adsorbed on graphene. Mater. Today Commun. 2020, 25, 101595. [Google Scholar] [CrossRef]
- Slanina, Z.; Uhlík, F.; Akasaka, T.; Lu, X.; Adamowicz, L. Computational modeling of the Ce@C82 metallofullerene isomeric composition. ECS J. Solid State Sci. Technol. 2019, 8, M118–M121. [Google Scholar] [CrossRef]
- Slanina, Z.; Uhlík, F.; Akasaka, T.; Lu, X.; Adamowicz, L. Calculated relative thermodynamic stabilities of the Gd@C82 isomers. ECS J. Solid State Sci. Technol. 2021, 10, 071013. [Google Scholar] [CrossRef]
- Uhlík, F.; Slanina, Z.; Bao, L.; Akasaka, T.; Lu, X.; Adamowicz, L. Eu@C88 isomers: Calculated relative populations. ECS J. Solid State Sci. Technol. 2022, 11, 101008. [Google Scholar] [CrossRef]
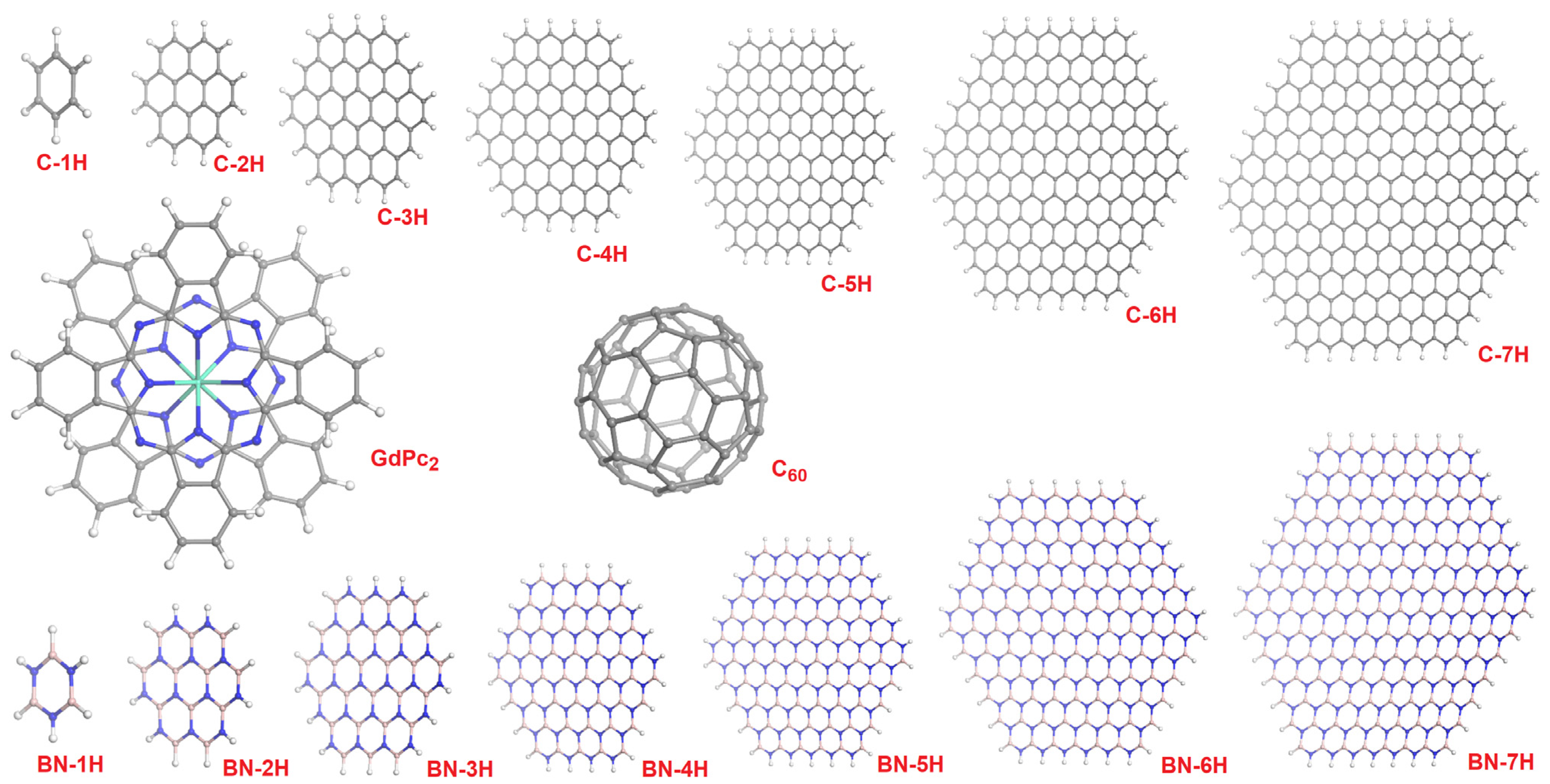
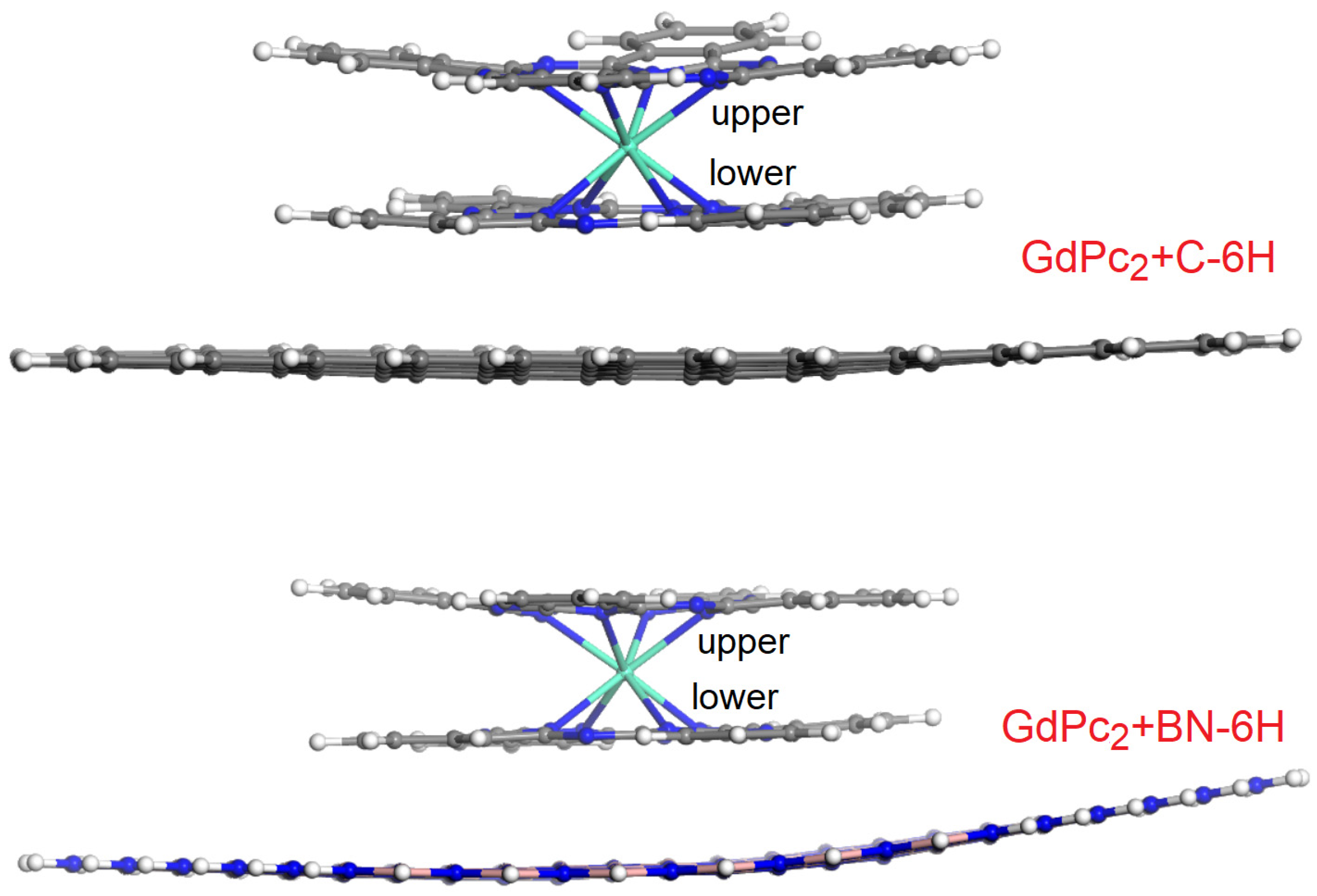

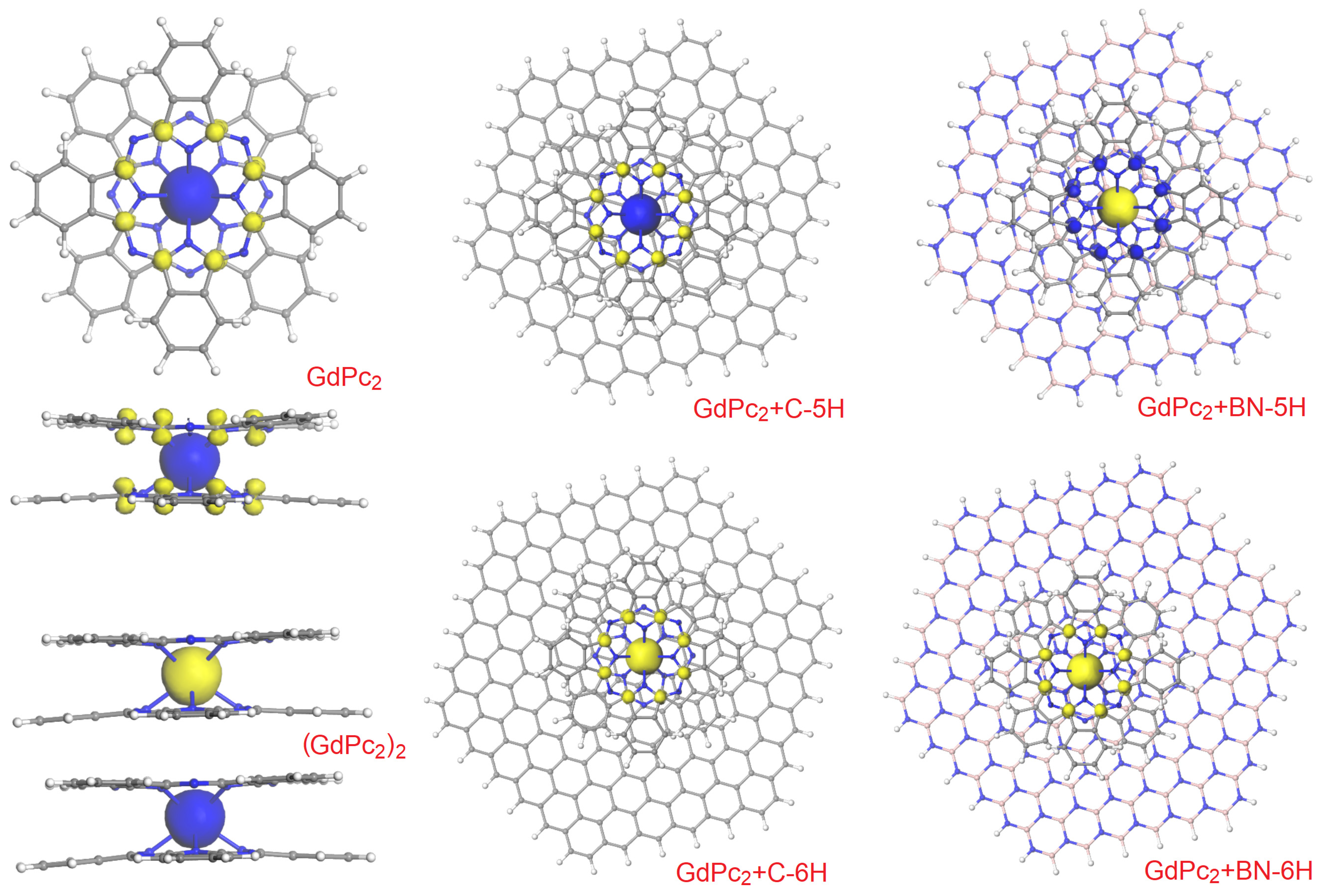

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Etotal (Ha) | ΔE (kcal/mol) | Gd-Nlower (Å) a | Gd-Nupper (Å) | Gd Charge (e) | Gd Spin (e) | GdPc2 Spin (e) | |
|---|---|---|---|---|---|---|---|
| GdPc2 | −3528.7468834 | 2.450 | 2.450 | 1.300 | 7.007 | 6.011 | |
| C-1H | −232.0208765 | ||||||
| C-2H | −921.0808194 | ||||||
| C-3H | −2067.1815845 | ||||||
| C-4H | −3670.3260780 | ||||||
| C-5H | −5730.5170328 | ||||||
| C-6H | −8247.7564282 | ||||||
| C-7H | −11,222.0439594 | ||||||
| C60 | −2284.4334844 | ||||||
| BN-1H | −242.4167720 | ||||||
| BN-2H | −962.7988404 | ||||||
| BN-3H | −2161.1563381 | ||||||
| BN-4H | −3837.4911698 | ||||||
| BN-5H | −5991.8039867 | ||||||
| BN-6H | −8624.0947660 | ||||||
| BN-7H | −11,734.3621054 | ||||||
| GdPc2 + C-1H | −3760.7904162 | −14.2 | 2.447 | 2.453 | 1.367 | −7.006 | −6.010 |
| GdPc2 + C-2H | −4449.8723439 | −28.0 | 2.487 | 2.473 | 1.364 | −7.000 | −6.096 |
| GdPc2 + C-3H | −5596.0080928 | −50.0 | 2.429 | 2.439 | 1.344 | 7.005 | 6.038 |
| GdPc2 + C-4H | −7199.1737356 | −63.2 | 2.439 | 2.426 | 1.353 | −7.004 | −6.065 |
| GdPc2 + C-5H | −9259.3781214 | −71.7 | 2.427 | 2.445 | 1.324 | 7.004 | 6.041 |
| GdPc2 + C-6H | −11,776.6182309 | −72.1 | 2.420 | 2.420 | 1.307 | −7.002 | −7.974 |
| GdPc2 + C-7H | −14,750.9094551 | −74.4 | 2.429 | 2.446 | 1.320 | 7.004 | 6.034 |
| GdPc2 + C60 | −5813.2211513 | −25.6 | 2.424 | 2.440 | 1.333 | 7.008 | 6.014 |
| GdPc2 + BN-1H | −3771.1790578 | −9.7 | 2.492 | 2.493 | 1.408 | 7.003 | 6.008 |
| GdPc2 + BN-2H | −4491.6012679 | −34.9 | 2.452 | 2.462 | 1.350 | 7.002 | 6.020 |
| GdPc2 + BN-3H | −5689.9869344 | −52.5 | 2.443 | 2.427 | 1.328 | 7.005 | 6.019 |
| GdPc2 + BN-4H | −7366.3531079 | −72.2 | 2.467 | 2.432 | 1.335 | −7.004 | −6.028 |
| GdPc2 + BN-5H | −9520.6731475 | −76.7 | 2.429 | 2.424 | 1.316 | −7.005 | −6.019 |
| GdPc2 + BN-6H | −12,152.9688187 | −79.8 | 2.415 | 2.433 | 1.307 | −7.002 | −7.982 |
| GdPc2 + BN-7H | −15,263.2324587 | −77.5 | 2.434 | 2.410 | 1.303 | −7.007 | −6.024 |
| (GdPc2)2 | −7057.5896494 | −60.2 | 2.427 | 2.429 | 1.337, 1.337 | 7.004, −7.004 | 7.006, −7.004 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Basiuk, V.A.; Basiuk, E.V. Adsorption of Gadolinium Bisphthalocyanine on Atomically Flat Surfaces: Comparison of Graphene and Hexagonal Boron Nitride from DFT Calculations. Surfaces 2024, 7, 404-413. https://doi.org/10.3390/surfaces7020025
Basiuk VA, Basiuk EV. Adsorption of Gadolinium Bisphthalocyanine on Atomically Flat Surfaces: Comparison of Graphene and Hexagonal Boron Nitride from DFT Calculations. Surfaces. 2024; 7(2):404-413. https://doi.org/10.3390/surfaces7020025
Chicago/Turabian StyleBasiuk, Vladimir A., and Elena V. Basiuk. 2024. "Adsorption of Gadolinium Bisphthalocyanine on Atomically Flat Surfaces: Comparison of Graphene and Hexagonal Boron Nitride from DFT Calculations" Surfaces 7, no. 2: 404-413. https://doi.org/10.3390/surfaces7020025
APA StyleBasiuk, V. A., & Basiuk, E. V. (2024). Adsorption of Gadolinium Bisphthalocyanine on Atomically Flat Surfaces: Comparison of Graphene and Hexagonal Boron Nitride from DFT Calculations. Surfaces, 7(2), 404-413. https://doi.org/10.3390/surfaces7020025