Abstract
In this review, we have assessed the possibility of metal–perovskite interfacial engineering to enhance the catalytic activity and selectivity in a range of heterogeneous catalytic reactions. We embarked on a literature screening of different perovskite material classes and reactions to show the versatility of the perovskite structures to induce the formation of such hetero-interfaces and the widespread nature of the phenomenon in catalytic research. There is almost no limitation on the chemical composition of the used perovskites and the nature of the catalyzed reaction, be it under reduction or oxidation conditions. We attempted to classify the perovskite materials, discuss the different strategies leading to the hetero-interfaces, and detail the synergistic action of the components of the respective interfaces. We also provide a critical assessment of the large body of data that is available in terms of a knowledge-based approach to the comparison of differently prepared interfaces with varying interfacial extent to gain a deeper understanding of the bi-functional operation of the interfaces and the urgent necessity to study and characterize such interfaces under realistic operation conditions.
1. Introduction: Simple Exsolution vs. Activity of the Metal–Perovskite Interface in Heterogeneous Catalysis
Perovskites are one of the fastest evolving materials classes in heterogeneous catalysis with promising catalytic properties in a wide field of research, including, but not limited to nitrogen, carbon, and oxygen (electro-) chemistry [1]. As such, perovskite materials are especially used for environmental and energy applications, such as hydrogen production, advanced oxidation processes, oxidative removal of air pollutants, wastewater treatment, degradation of volatile organic compounds, and photocatalytic applications, e.g., carbon dioxide reduction [2]. Comprehensive review articles, encompassing not only selected applications but also synthesis procedures and the influence of perovskite size and morphology, already exist in literature [1,2,3,4,5,6,7,8,9,10]. The main interest in the use of perovskite materials lies in the eventual replacement of noble metal-containing catalysts by various perovskite classes, thereby exploiting their unique feature of steering their properties by adjusting the chemical composition, redox properties, defect structure, oxygen vacancy concentration, or ion conductivity, likewise [1]. Perovskites have the general formula ABX3, where X is usually oxygen, a halid, sulfur, or nitrogen. A and B are (transition) bi- or trivalent metal cations, but depending on the doping level, higher oxidation states can also be accommodated to tune the physico-chemical and catalytic properties [1].
A unique feature inherent to perovskite materials, setting them apart from other classes of heterogeneous catalysts, is the potential gas-atmosphere-dependent instability of the perovskite structure [1,2,3,4,5,6,7,8,9,10]. Often viewed as a nuisance, it has been shown, especially for perovskites, that it can be used to tailor metal–oxide interfacial systems if the decomposition is carried out in a controlled way [11,12,13]. In this respect, the perovskite structure is used only as a precursor to prepare the catalytically active entity by steered breakdown of the perovskite structure. The result of such a preparation strategy usually is a metal-oxide system with a more-or-less defined interface if the perovskite is decomposed completely, or a metal–perovskite interface upon only partial decomposition. In any case, structurally and morphologically, a much more complex catalyst material evolves. The key factor in this decomposition process is exsolution of metal nanoparticles from the perovskite structure as a first step, followed by eventual decomposition of the entire perovskite structure. This exsolution process has been covered by review articles in detail, and many of its driving forces and the resulting structural consequences are already known [3,4,6,8,14,15]. Appreciating the complex exsolution as a delicate balance between thermodynamic and kinetic aspects of the different steps of the exsolution process, including phase transition, reduction, nucleation, and the diffusion of oxidized and reduced species (e.g., oxygen ions or cations), there has been a tremendous step forward in the understanding of the structural and morphological consequences of the exsolution process with respect to control composition, exsolved particle size, shape, or distribution. Pathways and synthesis strategies to tune the structure, morphology, and extent of the metal–oxide or perovskite–oxide interface, arising from controlled decomposition of the perovskite structure, exist [4,11,12,13].
However, as much as is known about the geometrical aspects of these interfaces and the associated structure tuning, a systematic discussion about the catalytic consequences is very much underrepresented, especially when it affects a direct bi-functional synergism between the supposedly catalytically active individual entities evolving from perovskite decomposition. With this review, we attempt to close this apparent knowledge gap and provide a comprehensive overview of the catalytic aspects of differently prepared metal–perovskite hetero-interfaces. As the structure–activity correlation concepts have, thus far, been mainly dominated by gas-phase catalytic approaches, we will focus our review on those studies and will not cover the large area of the electro-catalytic effects and aspects of such interfaces. As the geometrical aspects of interface formation and exsolution are vastly covered by review articles already, we leave these aspects also out [1,2,3,4,5,6,7,8,9,10].
This review is organized as follows. After a brief introduction into the concept of using perovskite in catalysis and the importance of the metal–perovskite interface, in Section 2, we will discuss the reactions in which selected perovskite materials form a metal–oxide or metal–perovskite interface and which enhancements effects are observed. We will cover the last 30 years for this overview with 100+ case studies and consider only those examples where authors indicate a clear interfacial activity/selectivity relationship. The simple appearance of a hetero-interface, e.g., as a result of exsolution, is not enough to be considered. While we cannot possibly include every study that has been published on this subject, we will include the vast majority of representative case studies. “Perovskite” materials, in this respect, include single perovskites, double perovskites, and Ruddlesden-Popper structures. As hetero-interfaces, we will count metal–perovskite interfaces prepared by simple support of metal particles on perovskite surfaces or those induced by partial decomposition of a perovskite precursor, as well as metal–oxide systems developing from the breakdown of the entire perovskite structure. This is necessary, as both strategies appear strongly entangled in the literature.
The summary of the overview of such interfaces in catalysis will lead to the strategies for the (in situ) steering of the extent of metal–perovskite interface and correlation to activity, discussed in Section 3. This will also include a classification of the literature-derived importance of different pathways for preparation of such interfaces and a discussion of the catalytic boost factor of those interfaces relative to reference catalysts without such interface present. We will therefore classify the literature-reported interfaces into four different strategies that we have identified as possible preparation strategies in the literature and our own research, including impregnation, exsolution, the combination of exsolution and impregnation, and deliberate over-doping. The discrimination between the four cases will not always be straightforward, but it helps in the appreciation of the different preparation strategies. The review will close in Section 4 with a balance of advantages and drawbacks of the preparation strategies and will present an outlook to pending questions.
2. Metal–Perovskite Interfaces in Heterogeneous Catalysis
2.1. Hydrocarbon Dry Reforming
Methane dry reforming is by far the most studied reaction with respect to perovskites and the potential formation and activity of the evolving metal–perovskite or metal–oxide interface upon decomposition. A very large body of all kinds of perovskite structures has been studied with respect to doping and cation exchange at the B or B’ site, but Ni-containing perovskites on nickelate basis are still in the vast majority [11,12,13,16,17,18,19,20,21,22,23,24,25,26,27,28,29,30,31,32,33,34,35,36].
2.1.1. Nickelates
Nickelates with La at the A-site are the archetypical materials, where the Ni exsolution and the formation of the active Ni/La2O3 interface has been observed at first. Almost all aspects of the mechanism and its structural and morphological prerequisites are known [11,12,13,16,17,18,19,20,21,22,23,24,25,26,27,28,29,30,31,32,33,34,35,36]. The structural decomposition pathway of LaNiO3 is known from in situ measurements and involves a sequence of structural transformations, the formation of oxygen-deficient perovskite structures, and the formation of a Ruddlesden-Popper structure La2NiO4 en route to full decomposition. The reversible formation of two polymorphs of La2O2CO3 oxy-carbonates has also been observed (Figure 1A) [11,12,13,16,17]. The Ni–La2NiO4 and Ni-La2O3 interfaces resulting from decomposition of La2NiO4 have been studied by Gallego et al., who revealed strong differences in the coking propensity [17]. Ni particles obtained from La2NiO4 decomposition, in contact with La2O3, are much less prone to carbon decomposition, owing to the efficient carbon reactivity at the Ni–La2O3 interface (Figure 1B) [17]. The effect of Cu-, Fe- and Ru-doping is also known to reduce the formation of coke, alongside steering the Ni crystallite size, the chemical composition of the exsolved Ni by Cu and Fe co-alloying and the reactivity of the oxy-carbonate structures [12,18,19,20,23,25,27,29,31,34,35]. Das et al. reported differences in the mechanistic pathway, switching between a La-oxy-carbonate-dominated pathway over Ni–La2O3 interfaces to an oxygen-vacancy-dominated pathway over a NiFe–La2O3 interface—both obtained via hydrogen reduction of La0.9Sr0.1NiO3 and La0.9Sr0.1Ni0.5Fe0.5O3, respectively (Figure 1C) [18]. An adverse effect of Ni–Fe alloying has also been reported on Fe-doped LaNiO3 perovskites, where formation of LaFeO3-encapsulated Ni particles with diminished reactivity was observed [23]. The role of reactivity of surface graphite has been found to be crucial for DRM activity over earth–alkaline-doped perovskite catalysts. The effect of Zn- and Ce-doping on LaNi1−xZnxO3 and La1−xCexNiO3 has also been studied: Zn stabilizes the LaNiO3 structure relative to pure LaNiO3 and, thus, allows steering the extent of Ni exsolution [31]. Instead, Ce provides activation of C–H bonds and increases the H2 selectivity [30]. Ce substitution in LaNiO3 has also been reported to act beneficial due to the formation of active Ce3+ cations in the Ni–CeO2–(LaCe)(NiFe)O3 interfacial mixture of the decomposed perovskite [32,33]. C–H bond activation by mobile lattice oxygen species was also reported over A-site doped Ni-M (M=Bi, Co, Cr, Cu, Fe) [26]. Addition of Ru to LaNi1-xRuxO3 perovskites enhances the stability of the evolving Ni–perovskite interface [27]. A similar observation has been made for a trimetallic LaNi0.34Co0.33Mn0.33O3 catalyst upon reduction [25].
2.1.2. Ferrites
Bimetallic Ni–Co catalysts supported on LaFeO3-La2O3 were prepared by steered decomposition of La(CoxNi1−x)0.5Fe0.5O3 precursors and revealed promotion of catalytic DRM activity and carbon removal by Co addition [37]. Oxygen vacancies in the LaFe0.9Ni0.1O3 perovskite were found to facilitate the activation/dissociation of CO2 in catalytic ethane dry reforming [38]. Effects of Fe partial substitution of La2NiO4 and LaNiO3 catalyst precursors have been studied by Song et al. and revealed a suppressed formation tendency of the Ni–La2O3 interface for the Fe-doped samples [24,35]. Provendier et al. studied LaNixFe1−xO3 structures in DRM and found that Ni–Fe alloys exsolved from the perovskite structures can prevent surface poisoning by deposited carbon [29].
2.1.3. Niobates
Perovskite-related mixed oxides of LaNiO3 and LaNbO4 were reported to form a Ni–La2O3-NbOx interface with increased metallic dispersion favoring the activity and stability of the catalysts [39].
2.1.4. Titanates
Ni doping into Ca0.8Sr0.2TiO3 and BaTiO3 yielded highly active DRM catalysts due to in situ formation of a Ni/perovskite interface and easy reverse spillover from the perovskite to the Ni particles, where it helps in oxidative removal of deposited carbon [21]. Joo et al. combined the exsolution process with atomic layer deposition (ALD) on La0.6Sr0.2Ti0.85Ni0.15O3−δ. This increased the number of exsolved nanoparticles, increased the catalytic activity, and prolonged the stability compared to the pristine catalyst in DRM [40].
2.1.5. Cuprates
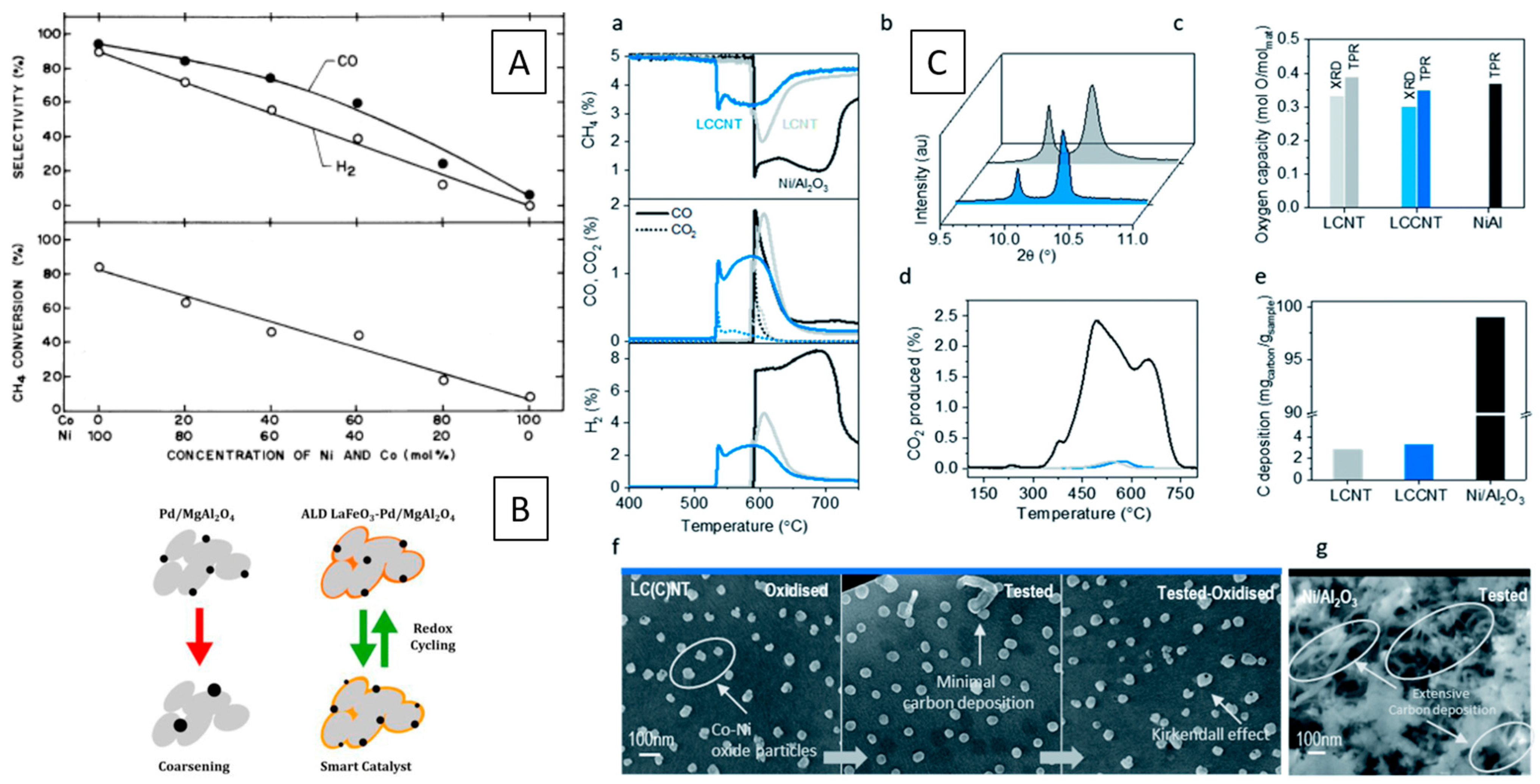
A Ni–LaCuO3 interface has been compared in DRM to a Ni–LaCuO3 interface developing from a LaCu0.53Ni0.47O3 perovskite and found to be more active, essentially due to a smaller Ni particle size as well as a strong interaction between NiO and the LaCuO3 support. (Figure 1D) [41].
2.1.6. Cobaltites
Osuzawa showed the excellent carbo-removal properties of an in situ-developed Co-Sm2O3 interface developing from a SmCoO3 perovskite upon DRM treatment [42].
2.1.7. Aluminates
Deactivation studies of impregnating Ni in different amounts on Ca-doped La-containing aluminate perovskites La0.9Ca0.1AlO2.95 yielded different deactivation mechanisms as a function of Ni particle size. Small Ni particles tended to deactivate via metal particle sintering, but for larger ones, deactivation by carbon deposition becomes more important [43]. A strongly reduced sintering of exsolved metallic Ni particles has been observed for LaNixAl−xO3 perovskites due to strong interaction of Ni with the perovskite [44]. Ni supported on LaAlO3 has been reported by Urasaki et al. to exhibit excellent carbon suppression properties due to oxygen migration from the perovskite to metallic Ni, outperforming LaFeO3, SrTiO3, BaTiO3, or a doped La–Ba ferrite perovskite [22]. Ma et al. reported the favorable anti-sintering and anti-carbon properties of a deliberately prepared Ni–LaAlO3 interface [45].
2.1.8. Zirconates
An array of MZr0.8Ni0.2O3 (M=Sr, Ba and Ca) perovskites has been investigated by Dama et al. to study the effect of basicity of the perovskite oxide, metal–support interaction, Ni crystallite size, surface hydroxyl groups, and oxygen defects, and revealed that, for CaZr0.8Zr0.2O3, the physicochemical properties are balanced in the most beneficial way to yield an excellent DRM catalyst [46].
2.1.9. Manganites
A comprehensive comparison of differently prepared Ni–LaMnO3 interfaces has been provided by Wei et al., revealing the smaller size distribution of exsolved Ni particles and a better attachment to the perovskite surface compared to Ni impregnated on the perovskite surface [47]. An atypical stability of exsolved Ni–Fe alloy particles anchored on an oxygen-excess PrBaMn1.6Ni0.3Fe0.1O5+δ perovskite under severe coking conditions was reported by Yao et al. [48].
2.1.10. Chromates
The importance of the Ni–LaCrO3 interface in providing strong exsolved Ni particle-support interaction, a high basicity, and a high oxygen vacancy concentration for improved methane dry reforming has been demonstrated on Sr- and Ni- co-doped chromate-based perovskites [49].
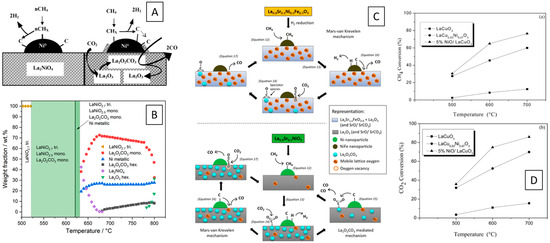
Figure 1.
Mechanistic details of methane dry reforming over metal–perovskite interfaces. (A) Difference in CO2 reforming between unreduced La2NiO4 with Ni metal decoration and decomposed La2NiO4 resulting in a Ni–La2O3 interface. Decomposed La2NiO4 facilitates CO2 adsorption sites via lanthanum oxy-carbonates, which can react on Ni-adsorbed carbon to form CO, while unreduced La2NiO4 leads to coking [17]. (B) Sequence of crystalline phases observed during in situ decomposition of LaNiO3 in a DRM mixture [11]. (C) Different pathways of methane dry reforming over Sr- and Fe-doped LaNiO3. Switching from a La-oxycarbonate-mediated pathway over Ni–La2O3 interfaces to a parallel Mars–van Krevelen mechanism over a NiFe–La2O3 interface resulting from hydrogen reduction which was observed by isotopic experiments and DRIFTS analysis [18]. (D) Catalytic methane and carbon dioxide conversions over LaCuO3, Cu-doped LaNiO3, and NiO-decorated LaCuO3 [41]. Reprinted with permission from Elsevier (A,D) [17,41] and American Chemical Society (B,C) [11,18].
Figure 1.
Mechanistic details of methane dry reforming over metal–perovskite interfaces. (A) Difference in CO2 reforming between unreduced La2NiO4 with Ni metal decoration and decomposed La2NiO4 resulting in a Ni–La2O3 interface. Decomposed La2NiO4 facilitates CO2 adsorption sites via lanthanum oxy-carbonates, which can react on Ni-adsorbed carbon to form CO, while unreduced La2NiO4 leads to coking [17]. (B) Sequence of crystalline phases observed during in situ decomposition of LaNiO3 in a DRM mixture [11]. (C) Different pathways of methane dry reforming over Sr- and Fe-doped LaNiO3. Switching from a La-oxycarbonate-mediated pathway over Ni–La2O3 interfaces to a parallel Mars–van Krevelen mechanism over a NiFe–La2O3 interface resulting from hydrogen reduction which was observed by isotopic experiments and DRIFTS analysis [18]. (D) Catalytic methane and carbon dioxide conversions over LaCuO3, Cu-doped LaNiO3, and NiO-decorated LaCuO3 [41]. Reprinted with permission from Elsevier (A,D) [17,41] and American Chemical Society (B,C) [11,18].
2.2. Methane (Partial) Oxidation
2.2.1. Nickelates
Pure and Sr/Ca/Co-doped LaNiO3 perovskites have been shown by Choudhary et al. to be active in the oxidative conversion of methane to syngas [50]. Doping significantly decreased both activity and selectivity, but pure LaNiO3 shows a very good performance in the simultaneous oxidative conversion and steam and CO2 reforming reactions due to the formation of a Ni–La2O3 interface in the initial stage of the reactions (Figure 2A). In a similar line of argumentation, Provendier et al. showed the capabilities of mixed LaNixFe1-xO3 materials, permitting to control the reversible migration of Ni from the bulk to the surface and the stabilization of Ni by Fe substitution [51]. Interestingly, this is a system where only partial decomposition of the perovskite structure has been observed after the catalytic tests, i.e., a structurally complex interface of still intact perovskite, La2O3 and Ni species, is formed. Clean LaNiO3 was studied by Nguyen et al. in the partial oxidation of methane, with formation of syngas prevailing over decomposed LaNiO3 (i.e., Ni and La2O3) [52].
2.2.2. Ferrites
A Pd–LaFeO3 interface was prepared by atomic layer deposition on a MgAl2O4 support by Onn et al. and exhibited a stable catalytic performance in methane oxidation after a high-temperature annealing treatment at 900 °C (Figure 2B) [53]. Noble metal (Pd, Pt and Rh) incorporated LaFeO3 perovskites were tested in the oxidative cracking of n-propane. Formation of a Rh/LaFeO3 and Pt/LaFeO3 interface was observed, which increased the redox properties of the systems and the number of oxygen moieties [54].
2.2.3. Titanates
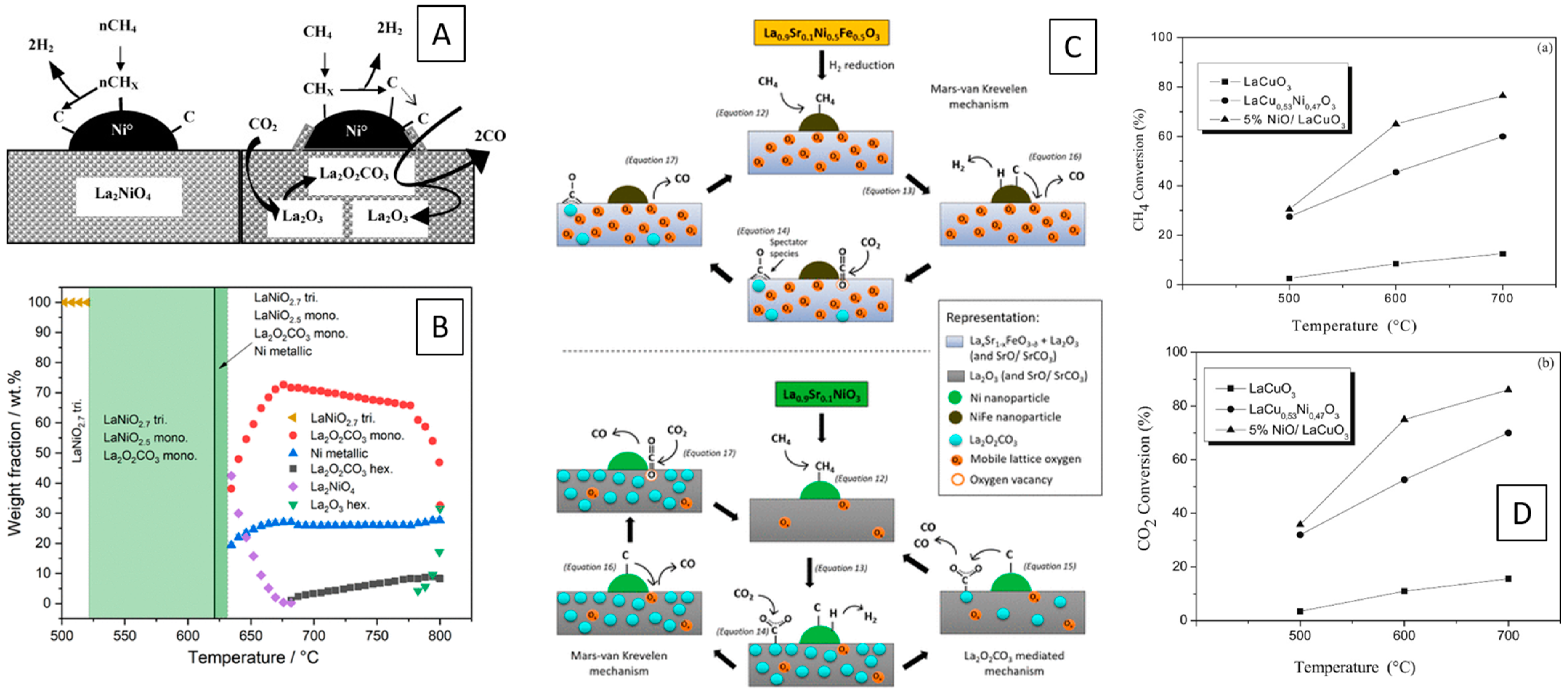
Chemical looping partial oxidation of methane was studied by Kousi et al. on a complex and especially tailored La0.7Ce0.1Co0.3Ni0.1Ti0.6O3−δ perovskite, where Co-doping of perovskite-supported exo/endo Ni particles at the interface allow to decrease the activation temperature of methane to as low as 450 °C (Figure 2C) [55].
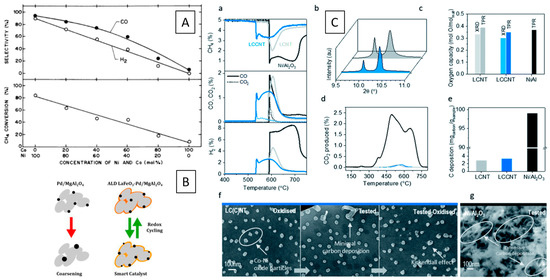
Figure 2.
Methane oxidation behavior of selected metal–perovskite materials. (A) Effect of Co-doping of LaNiO3 on the methane conversion and syngas selectivity [50]. (B) Improvement of the thermal stability and redox cycling ability by introducing a Pd–LaFeO3 interface to a MgAl2O4 spinel structure [53]. (C) Co-doping of La0.8Ce0.1Ni0.4Ti0.6O3−δ leading to an improvement of chemical looping partial oxidation of methane. In picture: (a) Conversion of methane to CO, CO2 and H2 for LCNT and LCCNT as a function of temperature. (b) X-ray diffraction patterns for the two samples (c). Calculated oxygen capacity for the catalysts in (a). (d) Temperature-programmed oxidation experiment after activity testing. (e) Carbon deposition calculated by integration of the curves in (d). (f) Representative SEM images for LCNT and LCCNT before testing, after testing, and after carbon oxidation experiment. (g) Ni/Al2O3 sample after testing displaying extensive carbon fiber growth all over the surface of the sample. [55] Reprinted with permission from Elsevier (A) [50], American Chemical Society (B) [53] and Royal Society of Chemistry (C) [55].
Figure 2.
Methane oxidation behavior of selected metal–perovskite materials. (A) Effect of Co-doping of LaNiO3 on the methane conversion and syngas selectivity [50]. (B) Improvement of the thermal stability and redox cycling ability by introducing a Pd–LaFeO3 interface to a MgAl2O4 spinel structure [53]. (C) Co-doping of La0.8Ce0.1Ni0.4Ti0.6O3−δ leading to an improvement of chemical looping partial oxidation of methane. In picture: (a) Conversion of methane to CO, CO2 and H2 for LCNT and LCCNT as a function of temperature. (b) X-ray diffraction patterns for the two samples (c). Calculated oxygen capacity for the catalysts in (a). (d) Temperature-programmed oxidation experiment after activity testing. (e) Carbon deposition calculated by integration of the curves in (d). (f) Representative SEM images for LCNT and LCCNT before testing, after testing, and after carbon oxidation experiment. (g) Ni/Al2O3 sample after testing displaying extensive carbon fiber growth all over the surface of the sample. [55] Reprinted with permission from Elsevier (A) [50], American Chemical Society (B) [53] and Royal Society of Chemistry (C) [55].
2.3. (Reverse) Water–Gas Shift Reaction
2.3.1. Ferrites
Ca-doped Nd-based ferrate perovskite structures have been studied by Lindenthal et al. to correlate the exsolution of metal particles with reverse water–gas shift activity. They identified a Mars–van Krevelen-type reaction mechanism with hydrogen activation at the exsolved metal, spillover of hydrogen to the interface for oxygen vacancy generation, and subsequent replenishment of the vacancies by CO2 activation (Figure 3A) [56,57]. A Ni/La0.9Fe0.1O3 perovskite was demonstrated by Huang et al. to exhibit a higher activity in the water–gas shift reaction than the corresponding perovskite-free materials. A combination of experiment and theory revealed the importance of water as the oxygen source for the water–gas shift reaction and highlighted the cooperative role of the perovskite as oxygen transmitter following water dissociation and the role of Ni in CO activation [58].
2.3.2. Nickelates
K-doped LaNiO3 has been studied as a promising catalyst for the high-temperature water-shift reaction. In that work, it was shown that the CO2 methanation could be effectively suppressed by K doping of the structure. It is inferred that this is due to the formation of hydroxylated K-OH species at the interface of the decomposed Ni–La2O3 interface. K-OH also promotes the formation of formate species, as a key intermediate for the water–gas shift reaction over reduced LaNiO3 [59]. Maluf et al. observed that the replacement of Co by Ni favored the reduction of the perovskite at lower temperatures (Figure 3B) [60] and accordingly boosted the water–gas shift activity over La1−ySryNixCo1−xO3 perovskites (Figure 3C) [61].
2.3.3. Titanates
A combined experimental and theoretical study of the water–gas shift reaction over Cu-modified SrTiO3 revealed an optimum Cu content of xCu = 0.2 in SrTi1−xCuxO3 materials with favorable interfacial dimensions to suppress Cu sintering and boosting the water–gas shift activity (Figure 3D,E) [61].
2.3.4. Cobaltites
Pt and Pd supported/incorporated LaCoO3 catalysts have been assessed in the water-gas shift reaction and it was found that they outperformed Pd and Pt particles supported on SiO2, indicating the importance of the metal–perovskite interface. The dominant reaction mechanism is of the Mars–van Krevelen type, including CO and steam (Figure 3F) [62]. In an additional study, Sekine revealed that the water–gas shift reactivity of those Pd– and Pt–LaCoO3 interfaces was not derived from Pt/Pd or the perovskite alone but, in fact, the interaction between the two entities promotes the water–gas shift reaction [63]. Carbon dioxide conversion by the reverse water–gas shift chemical looping over La1−xSrxCoO3−δ revealed the importance of metallic Co in contact with perovskite and La2O3 after hydrogen reduction to facilitate the role of the interface as an oxygen carrier in the reaction [64].
2.3.5. Yttrates
A Cu–Y2O3 interface derived by decomposition of a Cu2Y2O5 perovskite precursor was found to be active in the water–gas shift reaction due to facile water activation on differently exposed Cu facets [65].
2.3.6. Molybdates
Molybdates have essentially been used as double perovskite precursors in the reverse water–gas shift reaction, where exsolution of Ni–Fe alloys and the associated formation of a Ruddlesden-Popper structure in a core-shell structure has been found to be favorable for performance and stability [66].
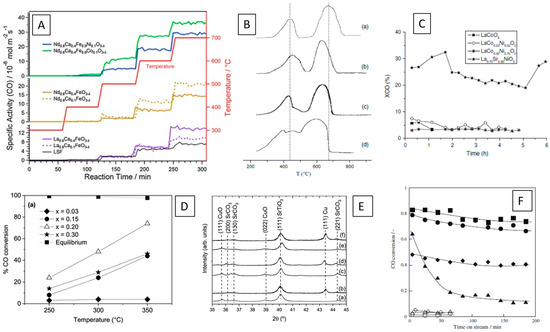
Figure 3.
(Reverse) water–gas shift reaction details over selected perovskite and metal–perovskite interfaces. (A) Comparison of the reverse water–gas shift reaction profiles over different Ni- and Co-doped ferrate perovskites [56]. (B,C) Correlation of reduction profiles with CO conversion in the water–gas shift reaction over La1−ySryNixCo1−xO3 [60]. (D,E) Influence of Cu doping SrTiO3 perovskites on CO conversion (D) and correlation to appearance and metallic Cu particles (E) [61]. (F) Catalytic water–gas shift activities over Pt- (diamonds) and Pd-doped (squares) LaCoO3 perovskites [62]. Reprinted with permission from Elsevier (A) [56], Springer (B,C,F) [60,62], and American Chemical Society (D,E) [61].
Figure 3.
(Reverse) water–gas shift reaction details over selected perovskite and metal–perovskite interfaces. (A) Comparison of the reverse water–gas shift reaction profiles over different Ni- and Co-doped ferrate perovskites [56]. (B,C) Correlation of reduction profiles with CO conversion in the water–gas shift reaction over La1−ySryNixCo1−xO3 [60]. (D,E) Influence of Cu doping SrTiO3 perovskites on CO conversion (D) and correlation to appearance and metallic Cu particles (E) [61]. (F) Catalytic water–gas shift activities over Pt- (diamonds) and Pd-doped (squares) LaCoO3 perovskites [62]. Reprinted with permission from Elsevier (A) [56], Springer (B,C,F) [60,62], and American Chemical Society (D,E) [61].
2.4. Alcohol and Hydrocarbon Steam Reforming
2.4.1. Ferrites
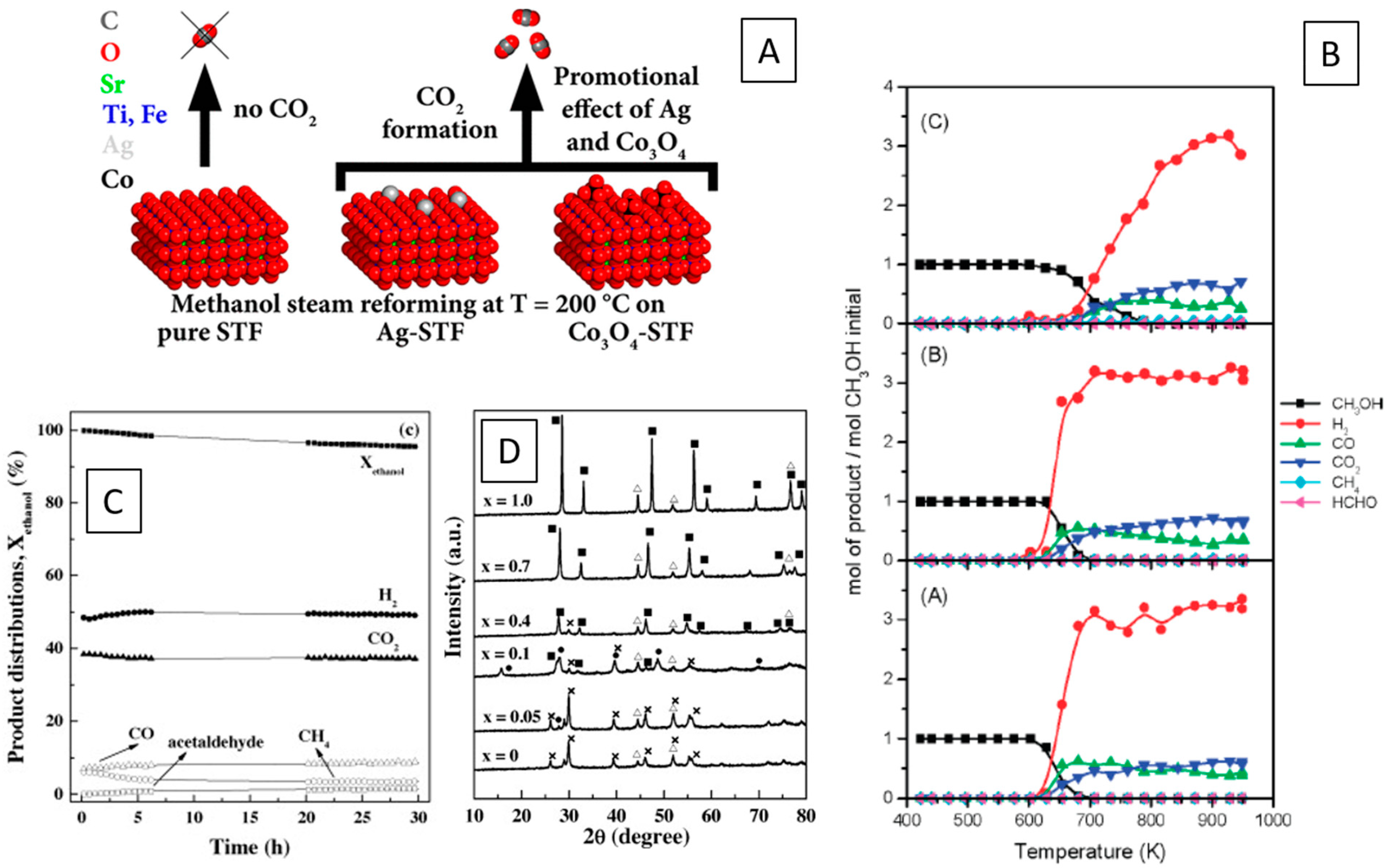
Ploner et al. have demonstrated the use of Co3O4- and Ag-impregnated SrTi0.7Fe0.3O3−δ (STF) as catalysts for methanol steam reforming. Ag and Co3O4 both promote the complex methanol chemistry with respect to pure STF. For Ag-STF, a clear effect of the Ag-STF heterointerface on the total oxidation of methanol to CO2 has been observed, ascribed to the fast and efficient transfer of lattice oxygen from STF to Ag via the extended metal–perovskite interface (Figure 4A) [67]. The effect of Ni doping on LaFeO3 perovskites has been studied by Provendier et al. Highly efficient catalysts for syngas production via steam reforming of methane were prepared, with the effect of Ni exsolution and the formation of a Ni/La2O3 by decomposition of LaFeO3 most pronounced for x > 0.4, if LaNixFe1−xO3 [68]. Chemical-looping steam reforming of methane over Ce-doped perovskites was shown by Zhang et al. to yield in situ-developed Fe0/La0.5Ce0.5)2O3 and Fe0/La0.5Ce0.5)2O2−x phases with improved water-splitting capabilities and superior reforming activity due to the amount of Ce3+ [69]. Ou et al. studied an array of Ni/ABO3 interfaces (A = Ca, Ba, Ce, Zn, Sr, Mg; B=Al, Fe, Mn), as well as different A-site doped materials, and highlighted the importance of the higher dispersion of Ni at the interface, in combination with a higher amount of surface oxygen for surface carbon removal and a stronger interaction between Ni and support. The Mg-doped aluminate perovskite–Ni interface showed the best performance [70]. Natile et al. provided a direct comparison of the methanol and ethanol steam-reforming performance of La0.6Sr0.4CoyFe1−yO3 catalysts and found that the differences in the activity, especially toward ethanol steam reforming, are attributable to a different stabilization of Co0 nanoparticles after the high-temperature reduction at 873 K, with particularly pronounced stability and high activity for La0.6Sr0.4Co0.5Fe0.5O3 (Figure 4B) [71].
2.4.2. Titanates
A series of Srn+1Tin−xNixO3n+1 Ruddlesden-Popper perovskite structures have been evaluated for methane, CO2, and bi-reforming of methane. For SrTi0.8Ni0.2O3−δ, the effect of the interface manifested itself in the spillover from labile oxygen vacancies from the bulk to the surface, assisting in the removal of surface-bound carbon [72].
2.4.3. Nickelates
Ce-doped La1−xCexNiO3 nickelate structures have been identified as promising catalysts for steam CO2 reforming of methane. For La0.9Ce0.1NiO3, a high degree of active Ni species (consequently forming a Ni/La2O3/CeO2/La2O2CO3 interface in the spent catalyst state) with promising activity and a high resistance to coke deposition, has been observed [73].
The ethanol steam-reforming reaction is a particularly well studied reaction in nickelate-type perovskites. Aguero et al. [74,75], Lima et al. [76], Lin et al. [77], and Zhao et al. [78] all studied different perovskite formulations on a lanthanum–nickelate basis; there is common agreement that a Ni–perovskite or Ni–La2O3 interface is present after reduction and that it exhibits beneficial properties with respect to stabilization of the Ni particles at the interface or the highest amount of oxygen vacancies, in correlation with high oxygen mobility and improved carbon clean-off performance. For example, Lima et al. identified a La0.9Ce0.1NiO3 composition exhibiting a very high ethanol conversion and particularly low amount of acetaldehyde. Structurally, this composition exhibits a high amount of exsolved Ni, alongside La2O3 and CeO2, after a hydrogen pre-treatment. The oxygen vacancy concentration is also highest for this composition, indicating a high carbon clean-off effectivity due to the high oxygen mobility (Figure 4C,D).
A Cu-decorated LaNi0.9Cu0.1O3 structure was also found to be active for low-temperature hydrogen production by steam reforming of glycerol. In the hydrogen-reduced state, a mix of Ni and Cu in contact with La2O3 was obtained, with consequently better resistance to coking [79]. Wu et al. showed the beneficial properties of smaller Ni particle sizes and a stronger metal–support interaction for reduced carbon deposition and higher stability in glycerol steam reforming [80]. A comparison of LaNiO3 and LaCoO3 by Aman et al. revealed the better glycerol steam-reforming performance of the latter due to the lower tendency of carbon deposition [81].
The promotional effect of Fe in LaNi1−xFexO3 catalysts for toluene steam reforming was highlighted by Oemar et al. and ascribed to the formation of Ni–Fe alloy particles at the bimetal–perovskite interface, also yielding a higher sintering stability [82].
2.4.4. Aluminates
Ni/LaAlO3 interfaces were studied by Sekine et al. also in the steam reforming of toluene. It was found that a Sr-doped La0.7Sr0.3AlO3−δ catalyst (prepared by a citrate complexing method) in contact with Ni exhibited the best bifunctional synergistic action due to the active oxygen species derived from the perovskite for coke suppression on the metallic Ni particles [83]. Urasaki et al. studied the production of hydrogen by ethanol steam reforming over Co and Ni particles supported on LaAlO3 in comparison to SrTiO3 and BaTiO3. LaAlO3 supported catalysts showed high catalytic activity and long-term stability with suppressed coke formation. It was concluded that the lattice oxygen plays a major role in suppressing coke formation and that the Co size has a great impact on the participation of lattice oxygen [84].
2.4.5. Cobaltites
Morales et al. studied a La0.6Sr0.4CoO3−δ precursor in the steam reforming of ethanol and revealed decomposition by hydrogen reduction into stabilized Co nanoparticles within a matrix of metal carbonates and metal oxides, leading to promising total ethanol conversion and high hydrogen selectivity [85].
2.4.6. Manganates
A Ni-doped La0.8Ce0.2Mn0.6Ni0.4O3 catalyst was studied with and without Cu addition for ethanol steam reforming by Wang et al. Hydrogen pre-reduction yielded Ni exsolution and accordingly, partial formation of a Ni/CeO2/perovskite interface, with complete ethanol conversion and a high syngas yield [86].
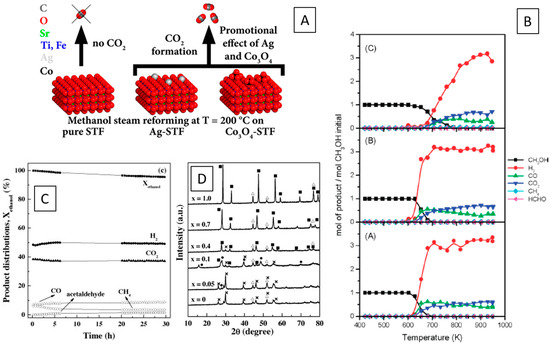
Figure 4.
Representative overview of the performance of different metal–perovskite interfaces in alcohol and hydrocarbon steam reforming reactions. (A) Display of methanol steam reforming over Ag- and Co3O4-decorated SrTi0.7Fe0.3O3−δ interfaces [67]. (B) Catalytic profiles of La0.6Sr0.4Co1−yFeyO3−δ perovskites in methanol steam reforming, with Co = (A) 0.8, (B) 0.5 and (C) 0.2 [71]. (C,D) Oxidative ethanol steam reforming performance of La0.9Ce0.1NiO3 (C) in correlation with the structural evolution of Ni particles and La2O3 during hydrogen reduction of La1−xCexNiO3 prior to reaction, that indicate a decreasing amount of oxygen vacancies with increasing ceria content [76]. Reprinted with permission from American Chemical Society (A,B) [67,71] and Elsevier (C,D) [76].
Figure 4.
Representative overview of the performance of different metal–perovskite interfaces in alcohol and hydrocarbon steam reforming reactions. (A) Display of methanol steam reforming over Ag- and Co3O4-decorated SrTi0.7Fe0.3O3−δ interfaces [67]. (B) Catalytic profiles of La0.6Sr0.4Co1−yFeyO3−δ perovskites in methanol steam reforming, with Co = (A) 0.8, (B) 0.5 and (C) 0.2 [71]. (C,D) Oxidative ethanol steam reforming performance of La0.9Ce0.1NiO3 (C) in correlation with the structural evolution of Ni particles and La2O3 during hydrogen reduction of La1−xCexNiO3 prior to reaction, that indicate a decreasing amount of oxygen vacancies with increasing ceria content [76]. Reprinted with permission from American Chemical Society (A,B) [67,71] and Elsevier (C,D) [76].
2.5. Methanation and Hydrogenation Reactions
2.5.1. Ferrites
Investigation of metal–support interaction effects on Ni- and Rh-decorated La0.6Sr0.4FeO3−δ perovskites have been investigated by Thalinger et al. [86,87]. These results highlighted the extreme variety in structure, extent, and electronic properties of the metal–perovskite interface induced upon hydrogen reduction. A clear correlation of deactivation with perovskite reducibility, as well as metal encapsulation effects and CO2 methanation, has been established (Figure 5A,B). Ni–LaFeO3 and Ni–Fe–LaFeO3 interfaces have also been identified to exhibit a superior resistance to coking in CO methanation. Comparison of Ni and Ni–Fe also allowed for deciphering the difference between metal–perovskite and alloy–perovskite interfaces, with the latter showing higher stability and better coking resistance. LaFeO3 perovskite supported Ni and Ni-Fe catalysts were studied by Wang et al. They revealed that NiFe@Ni/LaFeO3-La2O2CO3 and Ni/LaFeO3 formed after hydrogen reduction and syngas methanation. The former shows a core-shell structure with high stability and very good resistance to coking, while the latter is more active [88].
2.5.2. Titanates
Ni- and Rh-SrTi0.7Fe0.3O3−δ metal–perovskite interfaces prepared by impregnation have been assessed in CO and CO2 methanation in reference to a clean Ni/Al2O3 catalyst. Due to the small extent of reduction of SrTi0.7Fe0.3O3−δ, only a minor degree of Ni–Fe and Rh–Fe alloying has been observed and the deviation of the catalytic properties from the perovskite-free catalyst were equally less pronounced [86,87]. Cu-doped LaTi1−xCuxO3 perovskites have been found to be active for CO hydrogenation to methane, methanol, and CO2. XP spectra after pre-reduction indicated the presence of Cu0 species on the surface, which yielded the conclusion that the reaction proceeds at the interface via HxCO intermediates, followed by decomposition to HxC fragments and, subsequently, hydrocarbons [89]. Yan et al. provided evidence of a cooperative effect of a Rh–SrTiO3 interface in CO2 hydrogenation. The superior activity is assigned to a bi-functional cooperation between Rh (for efficient hydrogen dissociation) and oxygen vacancies on SrTiO3 for preferential adsorption and activation of carbon dioxide [90].
2.5.3. Aluminates
A three-dimensional porous LaAlO3 perovskite structure doped with Ni and Rh showed unique CO2 methanation capabilities, owing to the exsolution of Rh–Ni nanoalloys and the associated formation of a Ni–Rh/LaAlO3 interface. This interface shows that the low-temperature reducibility, rich-surface-adsorbed oxygen species, and associated basic sites have a distinct beneficial effect on CO2 methanation (Figure 5C) [91].
2.5.4. Rhodates and Platinates
CO hydrogenation has also been tested over BaMO3 structures (M=Rh, Ru, Pt, and Ir) and, in contrast to BaIrO3 and BaRuO3, crystalline BaRhO3 and BaPtO3 are both unstable in the synthesis gas mixture and an Rh (Pt)/BaCO3 interface was obtained in the spent catalyst state. Both perovskites have been found to exhibit a high oxygenate selectivity to methanol [92,93]. In a similar line of argumentation, LaRhO3 has been tested in both CO methanation and the hydroformylation of ethene after different hydrogen pre-treatments. After pre-reduction in hydrogen at 300 °C, the high selectivity to propionaldehyde and the observed chemical shift of the Rh 3d5/2 peak led to the conclusion of Rh0 species on the surface; thus, a Rh/LaRhO3 interface was most likely present [94].
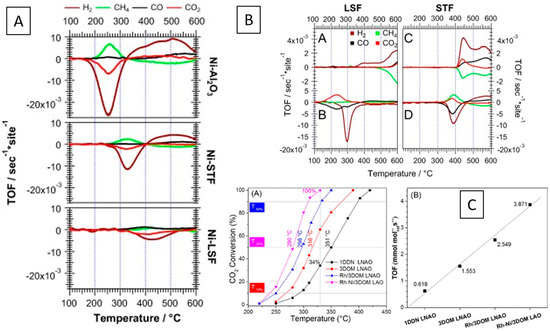
Figure 5.
Overview of the CO2 methanation capability of metal–perovskite interfaces derived via deliberate impregnation of metal particles on perovskite surfaces (A). Normalized methane steam reforming (A,C) as well as CO methanation reaction rates (B,D) for Ni–LSF and Ni–STF (B) [86]. Methanation capability of Rh–Ni alloys on a three dimensionally ordered microporous lanthanum aluminate perovskite catalyst prepared via exsolution (C) [91]. Reprinted with permission from Elsevier (A,B) [86] and American Chemical Society (C) [91].
Figure 5.
Overview of the CO2 methanation capability of metal–perovskite interfaces derived via deliberate impregnation of metal particles on perovskite surfaces (A). Normalized methane steam reforming (A,C) as well as CO methanation reaction rates (B,D) for Ni–LSF and Ni–STF (B) [86]. Methanation capability of Rh–Ni alloys on a three dimensionally ordered microporous lanthanum aluminate perovskite catalyst prepared via exsolution (C) [91]. Reprinted with permission from Elsevier (A,B) [86] and American Chemical Society (C) [91].
2.6. Car Exhaust Catalysis
2.6.1. Ferrites
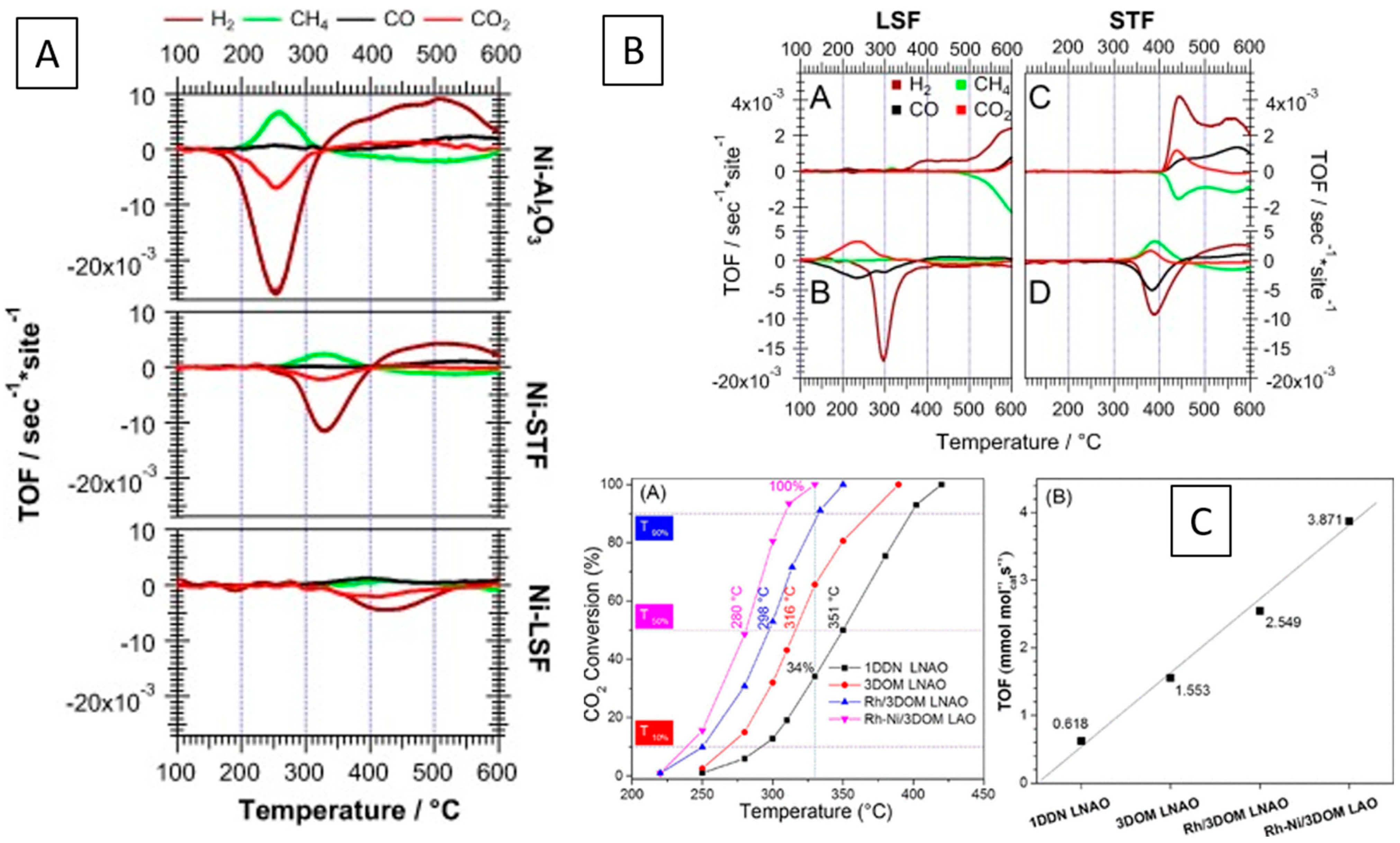
The archetypical perovskite system, where the exsolution of metal from the perovskite lattice and the reversible formation has been demonstrated in a systematic way, is the Pd-doped ferrite system LaFe0.57Co0.38Pd0.05O3 [95,96,97,98]. Oxidative and reductive cycling allows for reversible insertion and exsolution of Pd into and out from the lattice with improved long-term NO and CO conversion rates (Figure 6A).
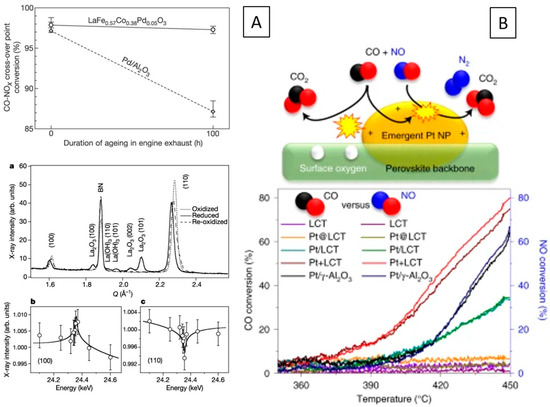
Figure 6.
Behavior of in situ-formed metal–perovskite interfaces in car exhaust catalysis. (A) Catalyst activity of LaFe0.57Co0.38Pd0.05O3 in NO reduction by CO in comparison to a Pd/Al2O3 reference catalyst (top) ascribed to reversible exsolution and incorporation of Pd out of and into the perovskite structure. XRD patterns are shown on the bottom (a), the energy dependency for Pd(100) (b) and Pd (110) (c) near the Pd K-edge indicates the occupation of the B-site in the perovskite structure in the oxidized state by Pd) [95]. (B) Reaction mechanism of Pt+LCT (top) and conversion rates of La0.4Ca0.3925Ba0.0075Pt0.005Ti0.995O3 under car exhaust NO+CO reduction conditions for different emergent Pt/La0.4Ca0.3925Ba0.0075Pt0.005Ti0.995O3 catalysts (bottom) [99]. Reprinted with permission from Wiley (A) [95] and Nature (B) [99]. Rights in the material are owned by a third party.
2.6.2. Titanates and Zirconates
In line with similar studies on the Pd–ferrite system discussed in the preceding section, also the respective earth–alkaline A-site perovskites in the titanate and zirconate systems have been evaluated, specifically CaTi.095Rh0.05O3, SrTi.095Rh0.05O3, BaTi.095Rh0.05O3, CaTi.095Pt0.05O3, and CaZr.095Rh0.05O3. Although the solid solution properties in the oxidized state are comparable and the formation of a metal–perovskite interface occurs for all systems, their oxidative regeneration properties are vastly different. The high stability of small Pt particles (1–3 nm) on the perovskite surface for CaTi.095Pt0.05O3 is particularly pronounced. Titanates in the composition La0.4Ca0.3925Ba0.0075Pt0.005Ti0.995O3 and La0.4Sr0.3925Ba0.0075Pt0.005Ti0.995O3 were also tested by Kothari et al. in various exhaust-related reactions, including diesel oxidation catalysis, CO and NO oxidation, and CO oxidation. Excellent integration of the Pt particles by a sophisticated “trojan horse” solid-state synthesis into the perovskite lattice enabled the controlled exsolution of well-anchored Pt particles on the surface during hydrogen reduction, in turn reaching high activity in the oxidation reactions (Figure 6B) [99].
2.6.3. Cobaltites
Glisenti et al. presented a Cu-doped LaCo1−xCux perovskite, which yielded surface-segregated Cu clusters and increased oxygen mobility due to the formation of vacancies after hydrogen reduction. Of particular interest is the LaCo0.5Cu0.5O3 composition, which yielded promising results in simple NO+CO, CO oxidation and complex exhaust-modelled gas mixtures under stoichiometric and lean oxygen conditions. Thus, a strong dependence on the amount of Cu and the associated extent of the Cu–perovskite interface has been reported [100].
2.7. CO Oxidation
2.7.1. Titanates
In addition to the cobaltite and titanate structures discussed in the previous section, also a Ir-doped SrIr0.005Ti0.995O3 perovskite showed Ir exsolution under hydrogen reduction and an associated high CO oxidation capability (Figure 7). The distinct socketing of the Ir particles with the perovskite support yields a high iridium stability. It has been concluded that metallic iridium is primarily responsible for the high CO oxidation activity, as the unreduced doped perovskite catalyst does not exhibit any activity [101].
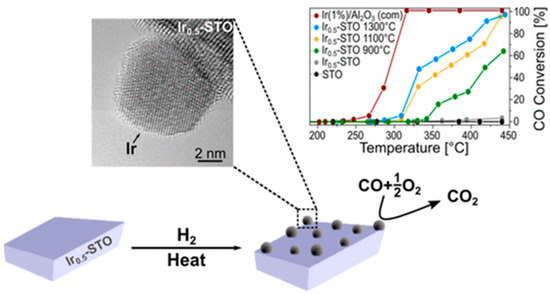
Figure 7.
CO oxidation profile of an in situ developed iridium-SrTiO3 interface via exsolution. Catalysts prepared at different exsolution temperatures gradually close the gap to a commercially available 1% Ir/Al2O3. Reprinted with permission from American Chemical Society [101].
2.7.2. Manganites
Differences in the CO oxidation behavior for Pt and Pd particles supported on thin-film LaMnO3 have been addressed at the high dispersion of Pd particles on LaMnO3 after several CO oxidation cycles associated with a low activation energy and a positive reaction order in CO [102].
2.8. deNOx NO+CO
2.8.1. Manganites
The role of Cu in model NO+CO, as well as realistic exhaust gas mixtures, has been assessed on Cu- and Pd-doped lanthanum manganite perovskites LaCuxMn1−xO3 to economize the use of noble metals by Thurner et al. [103,104]. It was shown that the Cu/reduced perovskite interface can indeed provide efficient NO activation sites in contact with in situ exsolved surface-bound monometallic Cu and bimetallic CuPd nanoparticles (Figure 8A,B). A similar result was obtained on Fe- and Pd-doped LaFe0.7Mn0.3O3 perovskites [105].
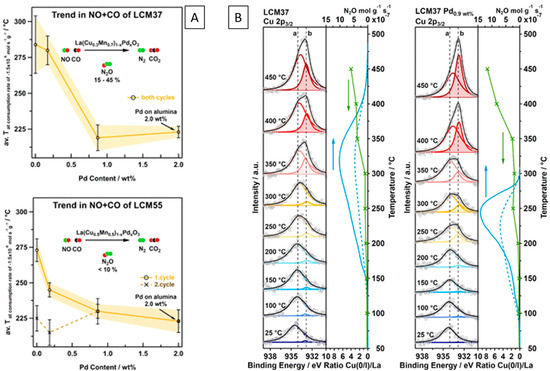
Figure 8.
NO reduction by CO over pure and Pd-doped LaCuxMn1−xO3 perovskite catalysts with in situ-developed Cu/perovskite and alloyed CuxPdy/perovskite interfaces. (A) Effect of perovskite chemical composition on the consumption temperature. (B) Correlation of in situ X-ray photoelectron spectra collected under catalytic NO+CO reaction conditions at different temperatures (left) with the La 3d5/2-normalized integral intensity of Cu(0/I) compared to the formation of N2O (right). Reprinted with permission from Elsevier [104].
2.8.2. Cobaltites
An adverse effect of NO+CO reactivity has been observed on a Co3O4–LaCoO3 interface: intermediate N2O decomposition has been seemingly blocked upon the presence of the Co3O4–LaCoO3 interface because of the decrease of active oxygen vacancy numbers. Part of this drawback could be overcome by doping the catalyst with potassium, rendering the catalyst a promising NO storage material at low temperatures [106].
2.8.3. Aluminates
A promotive effect of water on low-temperature NO reduction by CO has been observed over Pd/La0.9Ba0.9AlO3−δ. Steam addition promotes the desorption of surface-bound carbonates to accelerate the reaction of nitrate with CO. It has been stated that the mechanism specifically proceeds on the Pd–perovskite interface via participation of lattice oxygen and oxygen from La0.9Ba0.9AlO3−δ [107].
2.8.4. Titanates
A LaTi0.5Mg0.5O3 catalyst with Pd added showed promising activity in the reduction of NO with CO and it was concluded that the distribution of Pd at the interface is responsible for the steering of the reaction between the reduction of NO by CO and direct NO decomposition [108].
Figure 9 intermediary summarizes the present chapter and, at the same time, provides the link to the following section on the strategies of preparing the metal–perovskite and metal–oxide interfaces derived from controlled synthesis or decomposition. Panel A reveals that nickelate perovskite systems represent by far the largest group of all perovskite compositions, owing to their widespread use in methane (dry) reforming reactions. Equally important are ferrite, cobaltite, titanate, and manganite materials. They are used in a wide variety of reactions, where oxygen vacancies and an associated redox couple, e.g., Mn3+/Mn4+, Fe2+/Fe3+ are important for charge stabilization. Aluminate perovskites also represent a large group, and are used in a variety of mostly steam-reforming reactions ranging from methane over toluene and glycerol to ethanol [109]. In terms of reactions, the overwhelming majority of reactions where the discussed hetero-interfaces play a dominant role is due to methane dry reforming. This is essentially due to the fact that the invoked bi-functional synergism is very pronounced, with job-sharing duties between the components for methane activation (mostly nickel), carbon dioxide activation (e.g., La2O3), and enhancing carbon reactivity (oxygen-deficient perovskite with oxygen spillover to the metal entity). The same is true for steam reforming reactions, where water activation usually takes place on the oxidic part, and NO reduction by CO, with shared duties for NO and CO activation. As a link to the subsequent chapter, Panel C highlights the overall synthesis strategies that are used to prepare the hetero-interfaces. The exsolution approach is by far the most used one, making up for more than 75% of all screened cases. Within this group, exsolution by hydrogen pre-reduction is strongly dominant, and only a small number of studies provide results on exsolution directly by decomposition in the reaction mixture. Characterization is strongly dominated by ex situ methods; if in situ characterization is provided, it is usually connected with monitoring the hydrogen pre-reduction.
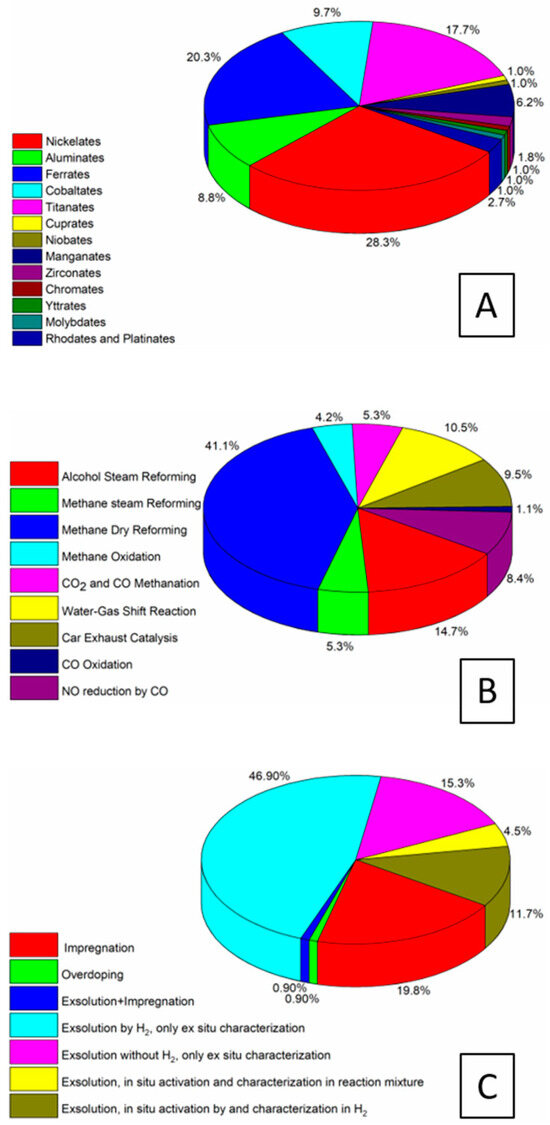
Figure 9.
Statistical analysis of key features of the studies of the formation of metal–perovskite and metal–oxide (from perovskite decomposition) interfaces. (A) Chemical composition across all studied reactions, (B) Appearance of hetero-interfaces among different reactions, and (C) Formation and characterization of the hetero-interfaces across all reactions.
A summary of the materials and the reactions is provided in Table 1.

Table 1.
Overview of the systems, reaction conditions, and observed structures after reduction and/or reaction and pathway of hetero-interface formation. * e/i indicates if the interface is obtained by exsolution or impregnation, respectively.
3. Strategies for the In Situ Steering of the Extent of Metal–Perovskite Interface and Correlation to Activity in Heterogeneous Catalysis
3.1. Formation by Metal Exsolution from Structure-Pure (Doped) Perovskites
The formation of a metal–perovskite interface by exsolution following a reduction treatment was first observed by Nishihata et al. upon reducing a Co-doped lanthanum ferrite structure modified with Pd, LaFe0.57Co0.38Pd0.05O3, in a hydrogen/nitrogen mixture. Following this treatment, the reversible partial decomposition of the parent perovskite lattice and the formation of La2O3, in addition to exsolution of Pd, has been observed. The active state under NO reduction by CO conditions relevant for automotive exhausts, hence, is a structurally extremely complex Pd/La2O3/reduced LaFe0.57Co0.38Pd0.05O3 heterointerface. The presence of the interface has been essentially inferred from XANES and EXAFS data [95]. Interestingly, direct observation of the Pd–perovskite interface has been only obtained by atomic-resolution electron microscopy on model Pd–-LaFeO3 compounds, mimicking the reduction conditions by Nishihata et al. Pd exsolution was indeed observed with the according formation of a Pd/reduced LaFeO3 interface, but the process was deemed to be very limited and localized, with a limited impact on catalysis [98]. (Figure 10)
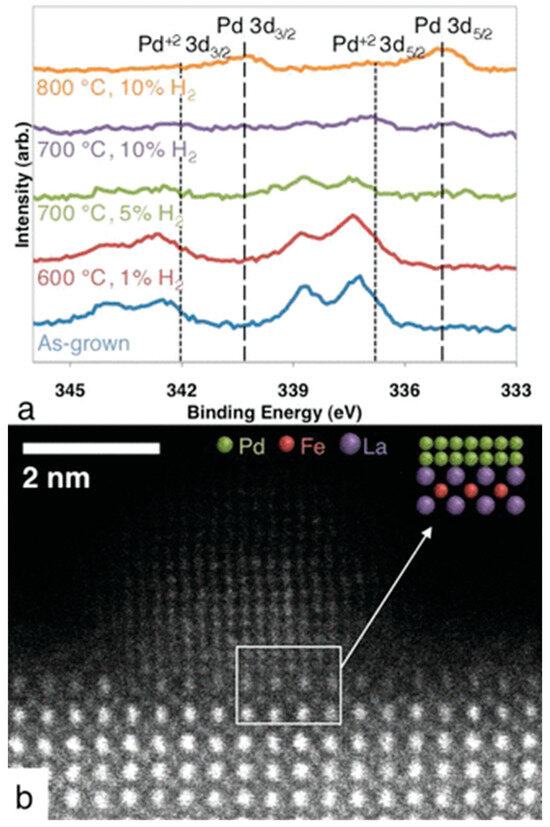
Figure 10.
Exsolution of Pd particles from a LaFe0.95Pd0.05O3−δ PLD thin film after various thermal treatments. XP spectra shown in (a), the high-angle annular dark-field image of a Pd particle found on the LaFe0.95Pd0.05O3−δ surface in (b). Reprinted with permission from Ref [98]. American Chemical Society.
This introductory example already underlines one of the core problems that occurs upon using exsolution as a preparation approach to metal–perovskite or metal–oxide interfaces: while the reduction process in hydrogen (or directly in the reaction mixture) can be steered by the experimental conditions, the resulting hetero-interface is, without exception, extremely complex and heterogeneous. In most cases, it is not a total decomposition but only a partial decomposition of the perovskite that is observed. In other words, in most cases, it is not a “metal–oxide” interface that is present but, rather, a “metal-reduced perovskite” or, even worse, a “metal–oxide-reduced perovskite” hetero-interface. This renders the establishment of the active site or a bi-functional synergism extremely complicated.
3.1.1. Methane Dry Reforming
Kühl et al. provided electron microscopy evidence of the formation of a Ni–La2O3 interface following hydrogen reduction of a Ru (2.5%)-doped LaNiO3 catalyst at 700 °C. The large, structurally homogeneous LaNiO3 particles were found to be transformed into aggregates of smaller La2O3 and Ni domains in random orientation. The authors also provided in situ reduction data by X-ray diffraction and were, thus, able to follow the structural decomposition of the perovskite through two distinct reduction steps associated with the formation of an oxygen-deficient LaNiO2.5 structure and then full decomposition at 475 °C [35]. In a similar line of argumentation, Bonmassar et al. studied the decomposition of LaNiO3 in hydrogen and within a dry reforming mixture in situ. Clear differences in the appearance and stability of transient structures have been observed, most importantly, a La2NiO4 Ruddlesden-Popper structure, which appears only in a carbon dioxide–methane mixture (Figure 11A,B) [11]. Similarly, the influence of A- and B-site doping of La2NiO4 has been studied by Bekheet et al. It was revealed that the differences in catalytic activity essentially arise due the formation of Ni–Cu alloy phases, the resulting Ni particle size and the inherently different reactivity of the present (oxy)carbonate phases. Nezhad et al. in turn focused on the formation, structure, and other aspects of the undoped La2NiO4 as a precursor material in contrast to the LaNiO3 structure. A much stronger coking and encapsulation propensity of Ni in contrast to LaNiO3 was observed, despite full decomposition of La2NiO4 [12]. Sr-doped samarium nickelate structures were also studied by Nezhad et al. in dry reforming of methane to reveal the role of earth–alkaline doping. It was found, that despite the often-reported beneficial role of the earth–alkaline metal in DRM catalysis, the detrimental role manifests itself in the sole formation of a SrCO3 phase and a generally destabilized Ni/Sm2O3 interface with pronounced Ni particle sintering [34]. The intermediate formation of La2NiO4 during DRM has also been observed by Batiot-Dupeyrat et al. at 700 °C. At higher reaction temperatures of 800 °C, La2NiO4 vanished in favor of full decomposition. The material switched from a Ni–La2NiO4 hetero-interface to Ni–La2O3 at higher temperatures [24].
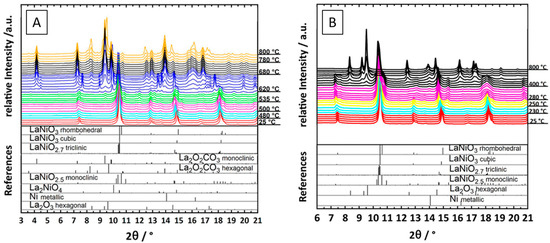
Figure 11.
Comparison of the development of the Ni–La2O3 interface during in situ heating of LaNiO3 in a CO2:CH4 (1:1) mixture (A) and in hydrogen (B) from room temperature to 800 °C. Reprinted with permission from American Chemical Society, Ref. [11].
Gao et. al. [36], Alvarez et al. [39], Su et al. [30], Wang et al. [33], Gallego et al. [17], Valderrama et al. [16], Rabelo-Neto et al. [32], and Moradi et al. [31] all reported full decomposition of La2NiO4/LaNiO3, Nb-, Cu-, or Ce-doped LaNiO3 into Ni and LaNiO3 upon hydrogen pre-reduction or by ex situ XRD characterization after the catalytic test. M-doped (M=Bi, Co, Cr, Cu, and Fe) La0.8Sr0.2Ni0.8M0.2O3 catalysts were found to yield very different metal–oxide interfaces after a dry reforming mixture: La2NiO4 was found in all structural mixtures after the catalytic test (alongside La oxy-carbonate structures and SrCO3) except for the Co-doped samples. Nevertheless, the analysis is severely hampered by the structurally impure starting materials containing at least SrCO3, NiO, SrO, La2O3, and Bi2O3 as parasitic structures [26]. Kim et al. also revealed full decomposition of Mn- and Co-doped LaNiO3, with clear differences in the formation propensity of the La-oxy-carbonate phases for the Co- and Mn-co-doped sample. While the manganese-free sample exhibited extensive coke formation due to the insertion of CO2 into the La2O3 support and thereby forming lanthanum oxycarbonate (La2O2CO3), this is not the case for the manganese-doped sample. MnO thereby strengthens the interaction of metal and support, leading to a smaller particle size (due to slower reduction) of the metallic particles initially and ascertains a constant particle size of Ni against their crystal growth during DRM. Additionally, MnO doping also provides more CO2 adsorption sites to supply oxygen species during the reaction to Ni, where the methane cracking takes place [25].
Stabilization of the perovskite–La2O2CO3-metallic Ni interface by Fe doping of La2NiO4 and LaNiO3 was reported by Song et al. [24]. In turn, the authors suggest that the interaction between Ni and the perovskite effectively suppresses the sintering of Ni particles alongside suppressing the coking propensity. The samples with equal nominal amounts of Ni and Fe are reported to be particularly promising.
In contrast, de Lima et al. studied Fe-doped LaNiO3 catalysts at varying stoichiometry and revealed the presence of a small amount of La2O3 in contact with LaFeO3 and metallic Ni particles. It was suggested that the presence of the oxidic phases gives rise to an oxidizing character, which destabilizes the carbonaceous species on the active Ni sites [28]. Tsoukalou also reported stabilization of Fe-doped LaNiO3 samples by in situ XRD measurements in a 1:1 = CO2:CH4 reaction mixture. While a Ni–LaFeO3-La2O2CO3 interface was present for this sample, the respective clean Ni and Co-doped samples shows full decomposition into Ni, La2O3, and La2O2CO3 [23]. Das et al. also pointed out that partial substitution of Ni by Fe in LaxSr1−xNiO3 perovskites increased the stability of the perovskite and the perovskite structure is obviously retained even under harsh reduction conditions (hydrogen, 700 °C). It was also reported that exposure of the hydrogen-reduced sample to DRM conditions at least partially regenerates the perovskite structure. This Mars–van Krevelen-type mechanism is attributed to this particular regeneration cycle and the associated high oxygen mobility and storage capability of the perovskite [18]. Different Fe-doped LaNiO3 materials have been characterized by Provendier et al. A clear dependence of the decomposition propensity as observed: higher Fe contents (0.2 ≤ x ≤ 0.7, x = Fe) lead to undecomposed LaFeO3, including the formation of exsolved Ni–Fe alloy plus La2O3. Hence, a complex Ni–Fe alloy, LaFeO3–La2O3, is present under DRM conditions [29]. Similar observations were made by Wang et al. on bimetallic Ni–Co catalysts supported on La2O3–LaFeO3 formed by Co–Fe-doped LaNiO3. It is suggested that the stable perovskite at the Ni–La2O3 interface affects the active oxygen species in the perovskite, which is highly beneficial for removal of deposited coke, boosting the catalytic DRM activity [37].
An illuminating example of the importance of the hetero-interface has been provided by Touahra et al. using surface-dispersed Ni particles in contact with LaCuO3 prepared by exsolution and by impregnation. Reduction in hydrogen, in all cases, resulted in the total decomposition into Ni and La2O3. After a subsequent DRM reaction, a La2O2CO3 structure in contact with metallic Ni and Cu was observed in both cases, and, interestingly, in the case of the impregnated sample, part of the initial LaCuO3 structure is restored during the DRM reaction [41].
Dama et al. focused on the structure and coking propensity of different Ni-doped zirconate structures and revealed that the particular stability of CaZr0.8Ni0.2O3 in comparison to the Sr- and Ba-doped variants is due to the suppressed sintering of the Ni particles resulting from strong interaction between Ni and the support. In addition, for the Ca-doped sample, only amorphous carbon was observed at the interface between Ni and perovskite, but no carbon filaments, which would cause deactivation of the catalyst (Figure 12A) [46].

Figure 12.
Transmission electron microscopy images of CaZr0.8Ni0.2O3-δ (a) SrZr0.8Ni0.2O3-d (b), and BaZr0.8Ni0.2O3-δ (c) after methane dry reforming at 900 °C (A) and Ni particle size, as well as coking propensity of La0.9Mn0.8Ni0.2O3 (a) in comparison with Ni impregnated on LaMnO3 in the spent catalyst state after methane dry reforming at 800 °C (b) (B). Reprinted with permission from Elsevier, adapted from Refs [46,47].
A structurally extremely stable Ni-LaMnO3 perovskite has been studied by Wei et al. Reduction and/or treatment in the DRM reaction up to 800 °C caused Ni exsolution, but the perovskite remained stable. If, compared to an impregnated Ni-LaMnO3 system, the coking propensity of the interface developed from exsolution is very much suppressed. It was concluded that the weak bonding of the Ni particles to the LaMnO3 surface for the impregnated catalyst results in accelerated sintering and a higher carbon filament formation rate (Figure 12B) [47].
Similar observations have been made by Zhao et al. in the dry reforming of ethane (DRE) with respect to LaFeO3 stability during DRE. In particular, the authors provided a direct structural comparison of the Ni–NiFe–La2O3 interface resulting from exsolution and Ni and NiFe deliberately supported on La2O3. The spent NiFe–La2O3 catalyst showed clear indications of partial formation of LaFeO3, alongside La2O2CO3 species. Interestingly, this hetero-interface turned out to be negatively affected by the interaction of LaFeO3 and NiFe particles. The interface developed during DRM by exsolution and perovskite decomposition was found to be more stable and active compared to the interfaces prepared via impregnation, due to the smaller Ni particle size and the higher number of oxygen vacancies (for CO2 dissociation) at the interface [38].
3.1.2. Methane Oxidation
Ni(Co) particles in contact with Ce- and Co-doped La0.8Ce0.1Ni0.4TiO6−δ were shown by Kousi et al. to be much more strongly coking-resistant due to the strong interaction of the exsolved Ni(Co) particles with the perovskite, in contrast to a Ni/Al2O3 reference catalyst. The peculiarity of this system is that it contains so-called exo and endo Ni(Co) particles, i.e., Ni(Co) particles on the surface and within the bulk of a porous perovskite material. The system therefore contains two interfaces, which synergistically act to lower the methane oxidation temperature to about 450 °C (Figure 13A) [55].

Figure 13.
SEM image of a cross section of La0.8Ce0.1Ni0.4TiO6−δ after reduction in forming gas at 900 °C highlighting the simultaneous presence of exo (i.e., surface) and endo (i.e., bulk) Ni particles (A). TEM image of 5 wt.-% K-doped LaNiO3 after reduction in hydrogen at 600 °C showing the Ni–La2O3-K2O interface (B). HR-TEM images of Cu particles exsolved from Cu2Y2O5 at 320 °C (left) and 500 °C (right) exposing 111 (a) and 110 (b) lattice fringes. Reprinted and adapted with permission from Royal Society of Chemistry (A), Elsevier (B) and MDPI (C) Refs. [55,59,65].
3.1.3. Water–Gas Shift Reaction
Formation of Ni, as well as Co–perovskite interfaces in the Ca-doped neodym–ferrite system has been studied by Lindenthal et al. in the reverse water–gas shift reaction [56,57]. It has been concluded that the combination of an easily reducible element (such as Co or Ni) in combination with a less reducible element (Fe, in this case) allows for the simple formation of a metal–perovskite interface, as the less reducible element acts as a stability factor for retaining the parent perovskite structure preventing full decomposition. Similar observations have been made by Huang et al. using La0.9Fe0.95Ni0.05O3 materials in the water–gas shift reaction. Ni particles were observed by reduction in hydrogen, but the L–FeO3 backbone was also retained.
Maneerung et al. provided evidence for the formation of a structurally complex Ni-K2O-La2O3 interface following hydrogen reduction of a 5 wt.-% K-doped LaNiO3 sample. Electron microscopy analysis evidenced the overall smaller Ni particle size compared to a conventionally impregnated Ni–La2O3 system and the formation of the Ni–K2O-La2O3 triple structure boundary, i.e., K2O formed after reduction is found in direct contact with both metallic Ni and La2O3. This subsequently gives rise to easier water activation at the interface by formation of K-OH species (Figure 13B) [59].
Daza et al. provided evidence for the dynamics of the metal–perovskite interface during several cycles of reduction and reverse water–gas shift reaction for Sr-doped La1−xSrxCoO3−d phases. Starting with a cubic perovskite phase, the initial perovskite was reduced to La2O3, Co, and SrCO3 and minor amounts of the parent perovskite. After the first water–gas shift cycle, Co and La2O3 were oxidized to CoO and a Sr-doped La2CoO4 structure. After yet another two reductions, CoO was partially reduced to Co and re-oxidized to CoO during the next two catalytic cycles [64].
A structural dependence of Cu particles exsolved from a Cu2Y2O5 perovskite, yielding differently facetted Cu particles attached to Y2O3 as a result of total perovskite decomposition in a pre-reduction step in hydrogen, was provided by Wang et al. Most importantly, the faceting could be directly steered by the reduction temperature. Reduction at 320 °C produced more Cu(111) facets and higher reduction temperatures (500 °C) and yielded an increasing number of Cu(110) facets. It was shown that the so-resulting interface had different abilities to dissociate and activate water (Figure 13C) [65].
As a glimpse into the complexity of using double perovskite structures as precursor materials to form a metal–perovskite interface, Orsini et al. studied a Sr2FeMo0.6Ni0.4O6−d double perovskite in the reverse water–gas shift reaction and revealed that, depending on temperature and number of reduction cycles, at least four different structures were present: Ni–Fe alloy particles, Sr3MoO6, Sr3FeMoO7−d, and Sr2FeMoO6. Despite the structural complexity, the authors inferred a bi-functional synergism, especially between the Ni–Fe alloy (activation of reactants H2 and CO2) and the Sr3FeMoO7−δ structure (facilitating fast oxygen exchange) [66].
3.1.4. Steam Reforming of Hydrocarbons and Alcohols
Provendier et al. provided evidence for a composition-dependent formation and stability of a Ni–LaFeO3 interface during steam reforming of methane, arising from an initial LaNixFe1−xO3 structure. For values x ≤ 0.4, formation of small NiO particles on LaFeO3 have been observed, while higher x values lead to deconstruction of the perovskite lattice and the formation of a Ni–La2O3 interface. The system, therefore, switches from pure LaNixFe1−xO3 (a) over NiO-LaFeO3 (b) to Ni–La2O3 (c) as a function of Fe-doping (Figure 14) [68]. Similarly, Yang et. Studied the effect of Ce-doping on interface formation in the LaxCe1−xNiO3 system in the steam CO2 reforming of methane. Higher amounts of Ce also in the calcined initial samples cause formation of NiO–perovskite–CeO2 interfaces, which is, in due course, also reflected in the spent catalysts: Samples without Ce show the typical La-oxy-carbonate phases in contact with Ni in the spent catalyst state, but the Ce samples show a transition to a Ni–CeO2 interface with minor oxy-carbonate formation with increased Ce content [73]. A very similar observation has been made by Sarkari et al. in the case of oxidative steam reforming of ethanol [76].

Figure 14.
HR-TEM images of different Ni–oxide interfaces derived from treating LaNixFe1−xO3 in a methane steam reforming reaction (1:1 = H2O:CH4). Reprinted with permission from Elsevier from Ref. [68].
Ou et al. focused on the formation of a Ni–LaAlO3 interface also during methane dry reforming. Ni deposited on clean LaAlO3 was compared to Ni on La0.7Mg0.3AlO3−δ. While the interface for both samples appeared stable even after 35 h on stream, the coking propensity was very much suppressed for the Mg-doped sample due to the higher dispersion of Ni and the increased amount of surface oxygen [70].
Lin et al. reported that the behavior of the archetypical LaNiO3 in ethanol steam reforming, in fact, yields a very similar interfacial structure after reduction and reaction, with metallic Ni, La2O2CO3, and LaNiO3 still present [77]. A different approach to a metal–perovskite interface was taken by Zhao et al. by impregnating ZrO2 by a LaNi1−xCoxO3 perovskite, which decomposes into Ni–Co alloy particles highly dispersed on ZrO2 and La2O3. The anti-sintering and anti-coking propensity of this hetero-interface is pronounced. Reversible formation and deconstruction of an interface developing from La0.6Sr0.4CoO3 after reduction and ethanol steam reforming has been studied by Morales et al. After reaction at 600 °C, highly dispersed cobalt nanoparticles within a matrix of metal carbonates and metal oxides, including La2O3, La2O2CO3, SrO, and SrCO3, were present. Regeneration in air at 900 °C, in turn, caused re-formation of the initial perovskite [78].
The general behavior of Ca-, Co, and Co-doped LaNiO3 catalysts and the respective formation of a metal–oxide interface in glycerol steam reforming is the decomposition of the parent perovskite lattice into a mix of Ni, La2O2CO3, and CaCO3 [79,80,81].
For CO2 reduction, Yan et al. compared Rh–SrTiO3 interfaces developed either from the exsolution of Rh or from Rh-doped SrTiO3 with Rh particles supported on SrTiO3. The extent of the metal–perovskite interface was much larger for the exsolved sample after reduction, giving rise to enhanced adsorption and activation of CO2 (Figure 15A) [90]. SrTiO3 was also used by Cali et al. as a host for iridium (in a composition of SrIr0.005Ti0.995O3) and, in turn, to exsolve it during reduction under forming gas at and above 900 °C to form a Ir/SrTiO3 interface as a catalyst in CO oxidation. Ir particle sizes were found to be in the range of 2–4 nm (Figure 15B) [101]. In NO reduction by CO, titanates in composition of La0.4Ca0.3925Ba0.00775Pt0.05Ti0.995O3 and La0.4Sr0.3925Ba0.00775Pt0.05Ti0.995O3 have also been used by Kothari et al. to form an interface composed of Pt nanoparticles in contact with the perovskite backbone after reduction in forming gas at 700 °C (Figure 15C) [99].
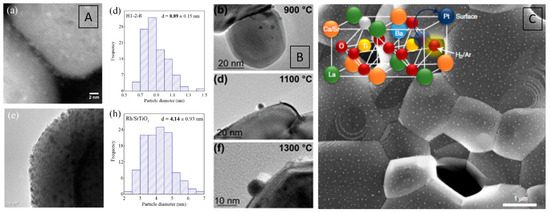
Figure 15.
Comparison of the Rh–SrTiO3 interface derived from exsolution of Rh-doped SrTiO3 samples and Rh impregnated on SrTiO3 with the respective Rh particle-size distributions during CO2 hydrogenation (A). Emerging Ir–SrTiO3 interface developing during a reduction process prior to CO oxidation as a function of reduction temperature (B). Pt particles exsolved from La0.4Ca0.3925Ba0.00775Pt0.05Ti0.995O3 during reduction in forming gas at 700 °C (C). Reprinted and adapted with permission from Elsevier (A), American Chemical Society (B), and Nature (C) from Refs. [90,99,101].
3.2. Impregnation Strategies
Impregnation of perovskites with metal and bimetals or alloys is the second most strategy to prepare metal (or bimetal)—perovskite hetero-interfaces. In some cases, the structure and the catalytic properties are compared to an interface obtained by an exsolution approach. Mohammadi et al. have studied a range of Pd-impregnated and Pd-incorporated lanthanum iron manganite compounds in the NO+CO reaction and revealed a strong dependence of CO adsorption and deNOx reactivity. Strong CO adsorption without a pronounced Pd-perovskite interface acts as an inhibitor for adsorption and desorption of NO, while samples with an extended interface show reduced CO poisoning. At the interface, the optimized use of lattice oxygen for CO oxidation and its replenishment from NO dissociation allows for the formation of sites for NO activation without CO poisoning. This directly translates into an increase in N2 selectivity [105].
Urasaki et al. studied an array of Ni-impregnated aluminate, ferrite, and titanate samples, including LaAlO3, LaFeO3, SrTiO3, and BaTiO3, in the methane steam-reforming reaction. Utilization of lattice oxygen at the Ni–perovskite interface appeared as the key parameter for promotion of adsorbed hydrocarbon fragments adsorbed on metallic Ni. A Mars–van Krevelen-type reaction mechanism was inferred, with the Ni/LaAlO3 and Ni/SrTiO3 interface performing best due to the easiest accessibility of lattice oxygen at the interface [84]. In a similar line of argumentation, Hayakawa inferred participation of lattice oxygen in the dry reforming methane reaction over Ni/BaTiO3 interfaces [21]. Similarly, Sekine et al. [63], Qi et al. [43] and Ma et al. [45] revealed that Ni–La0.9Ca0.1AlO2.95 and Ni/LaAlO3 hetero-interfaces performed outstandingly in methane dry reforming, due to the stabilization of small Ni particles at the interface and an enhanced metal–support interaction.
Onn et al. revealed the smart behavior of Pd particles supported on LaFeO3 for the beneficial redox cycling for CO oxidation and methane oxidation. The LaFeO3 film with a thickness of less than 1.5 nm was shown to be stable to at least 900 °C [53].
Sekine et al. and Watanabe et al. studied Pt– and Pd–LaCoO3 interfaces in the water–gas shift reaction. A Pt-LaCoO3 interface is present in the fresh catalyst, but after reduction (causing deactivation), the structure of the interface changes to Pt-Co-La2O3. Interestingly, similar reduction of a Pd-LaCoO3 causes the corresponding changes in the structure of the interface, but no deactivation [62]. Pt and Pd particles were also deposited on a LaMnO3 perovskite by Cao et al. as catalysts for CO oxidation. The authors report that, while the Pd–LaMnO3 interface is stable during a reductive pre-treatment, the Pt–LaMnO3 is not stable and decomposes into a Pt–Mn alloy, Mn2AlO4, and remains of LaMnO3 [102].
The importance of surface and bulk characterization in judgement of the structural properties of metal–perovskite interfaces were revealed by Ploner et al. on Ag- and Co3O4 impregnated SrTi0.7Fe0.3O3−d in methanol steam reforming. During catalytic pre-treatment, it was observed that Ag remained metallic in the bulk state during pre-oxidation, but XP spectra indicated a Ag2O shell around the Ag particles. After pre-reduction, the Ag2O shell turns metallic again. The interface of Co3O4 to SrTi0.7Fe0.3O3−d appears even more interesting, as it does not show up crystalline during any of the pre-treatments, but the surface—as monitored by XPS—followed the expected trends in Co chemistry during pre-oxidation and pre-reduction. Most importantly, highlighting the need for characterization in the reaction mixture, it was revealed, for both samples, that hydrogen formed during the methanol steam reforming reaction reduces Ag2O and Co3O4 to Ag and CoO, respectively. This underlines the sensitivity of the structure of the interface to the gas-phase environment [67].
Urasaki et al. revealed the dependence of the Co particle size in contact with BaTiO3 ans SrTiO3. The smaller Co particle size on the latter caused more frequent participation of lattice oxygen in the oxidation of intermediate species, causing a higher catalytic activity and stability [22].
Wang et al. studied Ni–LaFeO3 interfaces in syngas methanation following different preparation pathways. A strong dependence of the extent, stability, and chemical composition of the Ni–LaFeO3 interface has been observed. A NiO/LaFeO3 interface prepared by wet impregnation was revealed to be very stable during reaction, whereas preparation via a citrate complexing routine caused partial collapse of the Ni–LaFeO3 structure into Ni–Fe particles in contact with intact LaFeO3 and La2O2CO3 [88].
Ni and Rh interfaces with SrTi0.7Fe0.3O3-d and La0.6Fe0.4FeO3 were studied by Thalinger et al. in methane steam reforming and carbon dioxide methanation. Pre-reduction in hydrogen or catalytic gas mixtures induced a variety of different effects, including Fe exsolution, the formation of Ni–Fe (or Fe–Rh) alloy particles (Figure 16A). As La0.6Fe0.4FeO3 is more reducible than SrTi0.7Fe0.3O3−d, these effects are reported to be more pronounced on the former [86,87].
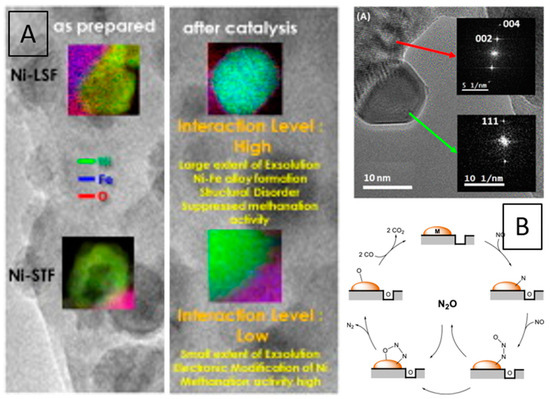
Figure 16.
EDX mapping of Ni–STF and Ni–LSF hetero-interfaces prepared by impregnation of the perovskites with Ni particles in the as prepared state and after CO2 methanation. Bullet points of the key parameters are also shown (A). Cu particles exsolved during NO reduction by CO from a LaCu0.3Mn0.7O3 catalyst alongside reaction mechanism observed at the interface (B). Reprinted with permission from Elsevier (A) and American chemical Society (B) from Refs. [86,103].
A stable Pd/LaAlO3 and Pd/La0.9Ba0.1AlO3 interface for low-temperature NO reduction by CO was prepared by Higo et al. by a citrate complexing method. As observed for other metal–perovskite interfaces, the key factor for activity is the promotion of surface carbonate species by perovskite lattice oxygen at the interface [107].
3.3. Deliberate Over-Doping
Over-doping of perovskite structures, thereby creating a structural mix even in initial catalyst entities, is somewhat common in perovskite research. In essence, this means that, even before any catalytic reaction, a normally ill-defined hetero-interface is present. While this should be generally avoided, it can indeed be helpful, if it is deliberately done to create a bi-functionally synergistically acting interface. Thurner et al. provided evidence, how copper can indeed substitute noble metals in the reduction of NO by CO, if a Cu-perovskite interface is initially prepared. Specifically, the Cu doping level of a LaCuxMn1-xO3 perovskite structure was deliberately exceeded at a composition of LaCu0.5Mn0.5O3 to yield a Cu/LaCu0.5Cu0.5MnO3 interface with increased Cu(0) surface loading, improved exsolution kinetics and a higher number of oxygen vacancies. It has been proven that this catalyst is similarly active and selective as a structure-pure 0.86 wt.-% Pd-doped LaCu0.3Mn0.7O3 catalyst [103].
3.4. Combination of Exsolution and Impregnation
Combining two standard preparation pathways to metal–perovskite interfaces, Thurner et al. highlighted how Pd impregnation of a LaCu0.5Mn0.5O3 catalyst in combination with enhanced Cu exsolution during the NO reduction by CO, can overcome the oxidative deactivation issues of Cu under realistic water-containing model exhaust gas mixtures. Minute impregnation with Pd (0.86 wt.-%) leads to formation of a Cu-Pd alloy, which essentially suppresses the oxidation of Cu to CuO under humid conditions (equivalent to 5 vol.% water in the NO+CO feed). This in turn enhances the N2O decomposition kinetics and improves the other main reaction branches of the deNOx network, including water–gas shift reaction and CO oxidation (Figure 16B) [104].
4. Further Development and Current Understanding of the Metal–Perovskite Interface in Heterogeneous Catalysis
In summary, we have shown that the formation of metal–perovskite or metal–oxide hetero-interfaces developing from decomposition of a perovskite precursor is a quite common phenomenon occurring for a vast amount of different perovskite classes and in many catalytic reactions. We have attempted to classify the perovskite materials, discuss the different strategies leading to the hetero-interfaces, and detail the synergistic action of the components of the interface. Despite the large body of data that is, in principle, available, we highlight four points that provide a combination of summary and outlook for further development and understanding.
- Knowledge-based steering of the metal–perovskite interface:
- Striking is the fact that, upon screening the literature, it is evident that, in the vast majority of cases, the formation of the interface is “by accident”, rather than the result of a knowledge-based approach. This is mostly the case if perovskite precursors are used. In this respect, one should be careful to discriminate between the understanding of the exsolution process itself and the interpretation of the synergistic action of the formed interface. While the former has been scrutinized in almost every aspect already, a deeper understanding of the latter is still mostly missing. To develop this understanding, a two-pronged approach is necessary: correlation of differently prepared interfaces, allowing for varying the extent of the interface, and in situ formation and analysis of the interfaces.
- Different approaches allow altering the extent of the metal–perovskite interface:
- There is a high need for detailed studies of hetero-interfaces with varying metal–perovskite or metal–oxide (if started from perovskite precursors) extent prepared by different approaches. This can for example achieved by varying the composition in the parent doped perovskite and steering the decomposition process, or by entirely different preparation approaches. The combination of impregnation, exsolution, and/or a combination of those appears particularly promising. A comparison of interfaces prepared by impregnation in comparison with exsolved ones is the absolute minority in the discussed cases studies, but it is still imperative for a deeper understanding.
- Mind the difference between a metal–perovskite and metal–oxide interface:
- Speaking about the knowledge-based approach to such interfaces, a clear discrimination must be made between the interfaces gained from a deliberate support of the metal particles on the perovskite surface, the metal–perovskite interface arising from a partial decomposition of a perovskite precursor and a metal–oxide interface developing from a total decomposition of such a perovskite precursor. While it is evident that, usually, only a comparison of such interfaces reveals the true mechanistic understanding, the literature-reported cases fail to a large extent in this respect. In connection with this deficiency, it has to be stated that, unfortunately, the bad habit of starting from structurally impure initial perovskite structures has wormed itself into research. While the appearance of parasitic stray phases might not have a strong effect on the decomposition process, it cannot be ruled out at will.
- Only in situ and operando analysis gives insight into the structure–activity correlation of metal–perovskite and metal–oxide interfaces derived from (partial) perovskite decomposition:
- A thorough understanding of the interface developing under operational reaction conditions is equally imperative. This is, sadly, still under-represented in most of the studies, despite the fact that such in situ and operando characterization exists for many methods nowadays. In addition, in the vast majority of the discussed cases, the interface is triggered by a pre-reduction step in hydrogen and not by the reaction mixture itself. This is a particular pity, as it has been shown, e.g., for methane dry reforming or the reduction of NO by CO, that the nature and reactivity in terms of, e.g., the size of exsolved metal particles (hence, extent of the interface), the structural pre-steps to the formation of the interface, and the reactivity of the interface itself, is strongly depending on the gas-phase chemical potential. It is the rule, rather than the exception, that the physico-chemical and catalytic properties of interfaces formed by reduction in hydrogen or in the reaction mixture are different. The archetypical material in methane dry reforming, LaNiO3, serves as the prime example: upon decomposition in the reaction mixture, the reactivity of the interface is governed by the intermediate appearance of the Ruddlesden-Popper structure La2NiO4. However, upon hydrogen reduction, this phase does not appear during perovskite decomposition. In consequence, the Ni particle size and the reactivity of the interface are different.
Author Contributions
Conceptualization and writing C.M. and S.P. All authors have read and agreed to the published version of the manuscript.
Funding
The research was funded by the FWF through project F 35770-N. The APC was funded in part by the Universitaet Innsbruck.
Data Availability Statement
We have created no additional data in this review. Permission for re-use of published material has been obtained from all publishers prior to submission.
Acknowledgments
The work has been performed within the framework of the research focus “Functional Materials” at the University of Inssbruck and project P 35770-N of the Austrian Science Foundation. C.M. appreciates financial support within FWF project P 35770-N.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Royer, S.; Duprez, D.; Can, F.; Courtois, X.; Batiot-Dupeyrat, C.; Lassiri, S.; Alamdari, H. Perovskites as Substitutes of Noble Metals for Heterogeneous Catalysis: Dream or Reality. Chem. Rev. 2014, 114, 10292–10368. [Google Scholar] [CrossRef] [PubMed]
- Bhattar, S.; Abedin, M.A.; Kanitkar, S.; Spivey, J.J. A review on dry reforming of methane over perovskite derived catalysts. Catal. Today 2021, 365, 2–23. [Google Scholar] [CrossRef]
- Kim, Y.; Jeong, H.; Won, B.; Jeon, H.; Park, C.; Park, D.; Kim, Y.; Lee, S.; Myung, J. Nanoparticle exsolution on perovskite oxides: Insights into mechanism, chareristics and novel strategies. Nano Micro Lett. 2024, 16, 33. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Li, J.; Sun, Y. The metal/oxide heterointerface delivered by solid-based exsolution strategy: A review. Chem. Eng. J. 2022, 440, 135868. [Google Scholar] [CrossRef]
- Sharif, R.; Khalid, A.; Ahmad, S.; Rehman, A.; Qutab, H.; Akhtar, H.; Mahmood, K.; Afzal, S.; Saleem, F. A comprehensive review of the current progresses and material advances in perovskite solar cells. Nanoscale Adv. 2023, 5, 3803–3833. [Google Scholar] [CrossRef] [PubMed]
- Kwon, O.; Joo, S.; Choi, S.; Sengodan, S.; Kim, G. Review on exsolution and its driving forces in perovskites. J. Phys. Energy 2020, 2, 032001. [Google Scholar] [CrossRef]
- Sun, Z.; Hao, C.; Toan, S.; Zhang, R.; Li, H.; Wu, Y.; Liu, H.; Sun, Z. Recent advances in exsolved perovskite oxide construction: Exsolution theory, modulation, challenges, and prospects. J. Mater. Chem. A 2023, 11, 17961–19776. [Google Scholar] [CrossRef]
- Ruh, T.; Berkovec, D.; Schrenk, F.; Rameshan, C. Exsolution on perovskite oxides: Morphology and anchorage of nanoparticles. Chem. Commun. 2023, 59, 3948–3956. [Google Scholar] [CrossRef] [PubMed]
- Hwang, J.; Rao, R.; Giordano, L.; Katayama, Y.; Yu, Y.; Shao-Horn, Y. Perovskites in catalysis and electrocatalysis. Science 2017, 358, 7751–7756. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Yu, J.; Gong, Y.; Lin, N.; Yang, Q.; Zhang, X.; Wang, Y. Perovskite catalysts with different dimensionalities for environmental and energy applications: A review. Sep. Purif. Technol. 2023, 307, 122716. [Google Scholar] [CrossRef]
- Bonmassar, N.; Bekheet, M.F.; Schlicker, L.; Gili, A.; Gurlo, A.; Doran, A.; Gao, Y.; Heggen, M.; Bernardi, J.; Klötzer, B.; et al. In Situ-Determined Catalytically Active State of LaNiO3 in Methane Dry Reforming. ACS Catal. 2020, 10, 1102–1112. [Google Scholar] [CrossRef]
- Bekheet, M.F.; Nezhad, P.D.K.; Bonmassar, N.; Schlicker, L.; Gili, A.; Praetz, S.; Gurlo, A.; Doran, A.; Gao, Y.; Heggen, M.; et al. Steering the Methane Dry Reforming Reactivity of Ni/La2O3 Catalysts by Controlled In Situ Decomposition of Doped La2NiO4 Precursor Structures. ACS Catal. 2021, 11, 43–59. [Google Scholar] [CrossRef] [PubMed]
- Nezhad, P.D.K.; Bekheet, M.F.; Bonmassar, N.; Schlicker, L.; Gili, A.; Kamutzki, F.; Gurlo, A.; Doran, A.; Gao, Y.; Heggen, M.; et al. Mechanistic in situ insights into the formation, structural and catalytic aspects of the La2NiO4 intermediate phase in the dry reforming of methane over Ni-based perovskite catalysts. Appl. Catal. A 2021, 612, 117984. [Google Scholar] [CrossRef]
- Neagu, D.; Oh, T.; Miller, D.; Menard, H.; Bukhari, S.; Gamble, S.; Gorte, R.; Vohs, J.; Irvine, J.T.S. Nano-socketed nickel particles with enhanced coking resistance grown in situ by redox exsolution. Nat. Commun. 2015, 6, 8120. [Google Scholar] [CrossRef] [PubMed]
- Han, H.; Park, J.; Nam, S.; Kim, K.; Choi, G.; Parkin, S.; Jang, H.; Irvine, J.T.S. Lattice strain-enhanced exsolution of nanoparticles in thin films. Nat. Commun. 2019, 10, 1471. [Google Scholar] [CrossRef] [PubMed]
- Valderrama, G.; Goldwasser, M.; de Navarro, C.; Barrault, J.T.J.; Batiot-Dupeyrat, C.; Martinez, F. Dry reforming of methane over Ni perovskite type oxides. Catal. Today 2005, 107, 785–791. [Google Scholar] [CrossRef]
- Gallego, G.; Mondragon, F.; Tatibouet, J.; Barrault, J.; Batiot-Dupeyrat, C. Carbon dioxide reforming of methane over La2NiO4 as catalyst precursor-characterization of carbon deposition. Catal. Today 2008, 133, 200–209. [Google Scholar] [CrossRef]
- Das, S.; Bhattar, S.; Liu, L.; Wang, Z.; Xi, S.; Spivey, J.J.; Kawi, S. Effect of partial Fe substitution in La0.9Sr0.1NiO3 perovskite-derived catalysts on the reaction mechanism of methane dry reforming. ACS Catal. 2020, 10, 12466–12486. [Google Scholar] [CrossRef]
- Wang, M.; Zhao, T.; Li, M.; Wang, H. Perovskite La2(NiCu)O4 catalyst precursors for dry reforming of methane: Effects of Cu-substitution on carbon resistance. RSC Adv. 2017, 7, 41847. [Google Scholar] [CrossRef]
- Batiot-Dupeyrat, C.; Valderrama, G.; Meneses, A.; Martinez, F.; Barrault, J.; Tatibouet, J. Pulse study of CO2 reforming of methane over LaNiO3. Appl. Catal. A 2003, 248, 143–151. [Google Scholar] [CrossRef]
- Hayakawa, T.; Suzuki, S.; Nakamura, J.; Uchijima, T.; Hamakawa, S.; Suzuki, K.; Shishido, T.; Takehira, K. CO2 reforming of CH4 over Ni/perovskite catalysts prepared by solid phase crystallization method. Appl. Catal. A 1999, 183, 273–285. [Google Scholar] [CrossRef]
- Urasaki, K.; Sekine, Y.; Kawabe, S.; Kikuchi, E.; Matsukata, M. Catalytic activities and coking resistance of Ni/perovskites in steam reforming of methane. Appl. Catal. A 2005, 286, 23–29. [Google Scholar] [CrossRef]
- Tsoukalou, A.; Imtiaz, Q.; Kim, S.; Abdala, P.; Yoon, S.; Müller, C.R. Dry reforming of methane over bimetallic Ni-N/La2O3 (M=Co, Fe): The effect of the rate of La2O2CO3 formation and phase stability on the catalytic activity and stability. J. Catal. 2016, 343, 208–214. [Google Scholar] [CrossRef]
- Song, X.; Dong, X.; Yin, S.; Weng, M.; Li, M.; Wang, H. Effects of Fe partial substitution of La2NiO4/LaNiO3 catalyst precursors prepared by wet impregnation method for the dry reforming of methane. Appl. Catal. A 2016, 526, 132–138. [Google Scholar] [CrossRef]
- Kim, W.; Jang, J.; Ra, E.; Kim, K.; Kim, E.; Lee, J. Reduced perovskite LaNiO3 catalysts modified with Co and Mn for low coke formation in dry reforming of methane. Appl. Catal. A 2019, 575, 198–203. [Google Scholar] [CrossRef]
- Sutthiumporn, K.; Maneerung, T.; Kathiraser, Y.; Kawi, S. CO2 dry reforming of methane over La0.8Sr0.2Ni0.8M0.2O3 perovskite (M=Bi, Co, Cr, Cu, Fe): Roles of lattice oxygen on C-H activation and carbon suppression. Int. J. Hydrogen Energy 2012, 37, 11195–11207. [Google Scholar] [CrossRef]
- Araujo, G.; Lima, S.; Assaf, J.; Pena, M.; Fierro, J.; Rangel, M. Catalytic evaluation of perovskite-type oxide LaNi1-xRuxO3 in methane dry reforming. Catal. Today 2008, 133, 129–135. [Google Scholar] [CrossRef]
- Lima, S.; Assaf, J. Ni-Fe catalysts based on perovskite-type oxides for dry reforming of methane to syngas. Catal. Lett. 2006, 108, 63–70. [Google Scholar] [CrossRef]
- Provendier, H.; Petit, C.; Estournes, C.; Kiennemann, A. Dry reforming of methane. Interest of La-Ni-Fe solid solutions compared to LaNiO3 and LaFeO3, Natural Gas conversions V. Stud. Surf. Sci. Catal. 1998, 119, 741–746. [Google Scholar]
- Su, Y.; Pan, K.; Chang, M. Modifying perovskite-type oxide catalyst LaNiO3 with Ce for carbon dioxide reforming of methane. Int. J. Hydrogen Energy 2014, 39, 4917–4925. [Google Scholar] [CrossRef]
- Moradi, G.R.; Rahmanzadeh, M.; Khosravian, F. The effects of pertial substitution of Ni by Zn in LaNiO3 perovskite catalyst for methane dry reforming. J. CO2 Util. 2014, 6, 7–11. [Google Scholar] [CrossRef]
- Rabelo-Nieto, R.C.; Sales, H.; Inocencio, C.; Varga, E.; Osko, A.; Erdohelyi, A.; Noronha, F.; Mattos, L. CO2 reforming of methane over supported LaNiO3 perovskite-type oxides. Appl. Catal. B 2018, 221, 349–361. [Google Scholar] [CrossRef]
- Wang, M.; Zhao, T.; Dong, X.; Li, M.; Wang, H. Effect of Ce substitution at the A-site of LaNi0.5Fe0.5O3 perovskite on the enhanced catalytic activity for dry reforming of methane. Appl. Catal. B 2018, 224, 214–221. [Google Scholar] [CrossRef]
- Nezhad, P.D.K.; Bekheet, M.F.; Bonmassar, N.; Gili, A.; Kamutzki, F.; Gurlo, A.; Doran, A.; Schwarz, S.; Bernardi, J.; Praetz, S.; et al. Elucidating the role of earth alkaline doping in perovskite-based methane dry reforming catalysts. Catal. Sci. Technol. 2022, 12, 1229–1244. [Google Scholar] [CrossRef]
- Kühl, S.; Düdder, H.; Girgsdies, F.; Kähler, K.; Muhler, M.; Behrens, M. Perovskites as precursors for Ni/La2O3 catalysts in the dry reforming of methane: Synthesis by constant pH co-precipitation, reduction mechanism and effect of Ru-doping. Z. Für Anorg. Und Allg. Chem. 2017, 643, 1088–1095. [Google Scholar] [CrossRef]
- Gao, J.; Jia, L.; Fang, W.; Li, Q.; Song, H. Methanation of carbon dioxide over the LaNiO3 perovskite catalysts activaed under the reactant stream. J. Fuel Chem. Technol. 2009, 37, 573–577. [Google Scholar] [CrossRef]
- Wang, H.; Dong, X.; Zhao, T.; Yu, H.; Li, M. Dry reforming of methane over bimetallic Ni-Co catalyst prepared from La(CoxNi1-x)0.5Fe0.5O3 perovskite precursor: Catalytic activity and coking resistance. Appl. Catal. B 2019, 245, 302–313. [Google Scholar] [CrossRef]
- Zhao, B.; Yan, B.; Yao, S.; Xie, Z.; Wu, Q.; Ran, R.; Weng, D.; Zhang, C.; Chen, J. LaFe0.9Ni0.1O3 perovskite catalyst with enhanced activity and coke-resistance for dry reforming of ethane. J. Catal. 2018, 358, 168–178. [Google Scholar] [CrossRef]
- Alvarez, J.; Valderrama, G.; Pietri, E.; Perez-Zurita, M.; Navarro, C.; Sousa-Aguiar, E.; Goldwasser, M. Ni-Nb-based mixed oxide precursors for the dry refroming of methane. Top. Catal. 2011, 54, 170–178. [Google Scholar] [CrossRef]
- Joo, S.; Seong, A.; Kwon, O.; Kim, K.; Lee, J.H.; Gorte, R.J.; Vohs, J.M.; Han, J.W.; Kim, G. Highly active dry methane reforming catalysts with boosted in situ grown Ni-Fe nanoparticles on perovskite via atomic layer deposition. Sci. Adv. 2020, 6, 2375–2548. [Google Scholar] [CrossRef] [PubMed]
- Touahra, F.; Rabahi, A.; Chebout, R.; Boudjama, A.; Lerari, D.; Sehailia, M.; Halliche, D.; Bachari, K. Enhanced catalytic behaviour of surfae dispersed nickel on LaCuO3 perovskite in the production of syngas: An expedient approach to carbon resistance during CO2 reforming of methane. Int. J. Hydrogen Energy 2016, 41, 2477–2486. [Google Scholar] [CrossRef]
- Osuzawa, O.; Setiabudi, H.; Rasid, R.; Cheng, C. Syngas production via methane dry reforming: A novel application of SmCoO3 perovskite catalyst. J. Nat. Gas Sci. Eng. 2017, 27, 435–448. [Google Scholar] [CrossRef]
- Qi, R.; Jin, R.; An, L.; Bai, X.; Wang, Z. A Ni/perovskite catalyst with low metal content for CO2 reforming of methane. Catal. Commun. 2022, 163, 106419. [Google Scholar] [CrossRef]
- Sagar, T.; Padmakar, D.; Lingaiah, N.; Rao, K.S.R.; Reddy, I.; Prasad, P. Syngas production by CO2 reforming of methane on LaNixAl-xO3 perovskite catalysts: Influence of method of preparation. J. Chem. Sci. 2017, 129, 1787–1794. [Google Scholar] [CrossRef]
- Ma, Y.; Su, P.; Ge, Y.; Wang, F.; Xue, R.; Wang, Z.; Li, Y. A novel LaAlO3 perovskite with large surface area supported Ni-based catalyst for methane dry reforming. Catal. Lett. 2022, 152, 2993–3003. [Google Scholar] [CrossRef]
- Dama, S.; Godhke, S.; Bobade, R.; Gurav, H.R.; Chilukuri, S. Active and durable alkaline earth metal substituted perovskite catalysts for dry reforming of methane. Appl. Catal. B 2018, 224, 146–158. [Google Scholar] [CrossRef]
- Wei, T.; Chia, L.; Zheng, H.; Chi, B.; Pu, J.; Li, J. LaMnO3-based perovskite with in situ exsolved Ni nanoparticles: A highly active, performance stable and coking resistant catalyst for CO2 dry reforming of CH4. Appl. Catal. A 2018, 564, 199–207. [Google Scholar] [CrossRef]
- Yao, X.; Cheng, Q.; Attada, Y.; Ould-Chikh, S.; Ramírez, A.; Bai, X.; Mohamed, H.O.; Li, G.; Shterk, G.; Zheng, L.; et al. Atypical stability of exsolved Ni-Fe alloy nanoparticles on double layered perovskite for CO2 dry reforming og methane. Appl. Catal. B 2023, 328, 122479. [Google Scholar] [CrossRef]
- Wei, T.; Jia, L.; Luo, J.; Chi, B.; Pu, J.; Li, J. CO2 dry reforming of methane with Sr and Ni co-doped LaCrO3 perovskite. Appl. Surf. Sci. 2020, 506, 144699. [Google Scholar] [CrossRef]
- Choudhary, V.R.; Uphade, B.S.; Belhekar, A.A. Oxidative coupling of methane to syngas over LaNiO3 perovskite with and without simultaneous steam and CO2 reforming. J. Catal. 1996, 163, 312–318. [Google Scholar] [CrossRef]
- Provendier, H.; Petit, C.; Estournes, C.; Libs, S.; Kiennemann, A. Stabilisation of active nickel catalysts in partial oxidation of methane to synthesis gas by iron addition. Appl. Catal. A 1999, 180, 163–173. [Google Scholar] [CrossRef]
- Nguyen, T.; Lamacz, A.; Beaunier, P.; Czajkowska, S.; Domanski, M.; Krzton, A.; Le, T.; Djega-Mariadassou, G. Partial oxidation of methane over bifunctional catalyst I. In situ formation of Ni0/La2O3 during temperature-programmed POM reaction over LaNiO3 perovskite. Appl. Catal. B 2014, 152, 360. [Google Scholar] [CrossRef]
- Onn, T.; Monai, M.; Dai, S.; Fonda, E.; Montini, T.; Pan, X.; Graham, G.; Fornasiero, P.; Gorte, R. Smart Pd catalyst with improved thermal stability supported on high-surface area LaFeO3 prepared by atomic layer deposition. J. Am. Chem. Soc. 2018, 140, 4841–4848. [Google Scholar] [CrossRef] [PubMed]
- Basahel, S.; Medkhali, A.; Mokhtar, M.; Narasimharao, K. Noble metal (Pd, Pt and Rh) incorporated LaFeO3 perovskite oxides for catalytic oxidative cracking of n-propane. Catal. Today 2022, 397, 81–93. [Google Scholar] [CrossRef]
- Kousi, K.; Neagu, D.; Bekris, L.; Cali, E.; Kerherve, G.; Papaioannou, E.; Payne, D.; Metcalfe, I. Low-temperature methane conversion with perovskite-supported exo/endo-particles. J. Mater. Chem. A 2020, 8, 12406–12417. [Google Scholar] [CrossRef]
- Lindenthal, L.; Popovic, J.; Rameshan, R.; Huber, J.; Schrenk, F.; Ruh, T.; Nenning, A.; Löffler, S.; Opitz, A.K.; Rameshan, C. Novel perovskite catalysts for CO2 utilization—Exsolution enhanced reverse water-gas shift activity. Appl. Catal. B 2021, 292, 120183. [Google Scholar] [CrossRef]
- Lindenthal, L.; Huber, J.; Drexler, H.; Ruh, T.; Rameshan, R.; Schrenk, F.; Löffler, S.; Rameshan, C. In situ growth of exsolved nanoparticles under varying rWGS reaction conditions—A catalysis and near ambient pressure-XPS study. Catalysts 2021, 11, 1484. [Google Scholar] [CrossRef]
- Huang, R.; Lim, C.; Jang, M.; Hwang, J.; Han, J. Exsolved metal-boosted active perovskite catalyst for water-gas shift reaction. J. Catal. 2021, 400, 148–159. [Google Scholar] [CrossRef]
- Maneerung, T.; Hidajat, K.; Kawi, S. K-doped LaNiO3 perovskite for high.temperature water-gas shift of reformate gas: Role of potassium on suppressing methanation. Int. J. Hydrogen Energy 2017, 42, 9840–9857. [Google Scholar] [CrossRef]
- Maluf, S.S.; Tanabe, E.Y.; Nascente, P.A.P.; Assaf, E.M. Study of water-gas shift reaction over La1-ySryNixCo1-xO3. Top. Catal. 2011, 54, 210–218. [Google Scholar] [CrossRef]
- Coletta, V.C.; Gonçalves, R.V.; Bernardi, M.I.B.; Hanaor, D.A.H.; Assadi, M.H.N.; Marcos, F.C.F.; Nogueira, F.G.E.; Assaf, E.M.; Mastelaro, V.R. Cu-modified SrTiO3 perovskites toward enhanced water-gas shift catalysis: A combined experimental and computational study. ACS Appl. Energy Mater. 2021, 4, 452–461. [Google Scholar] [CrossRef]
- Watanabe, R.; Sekine, Y.; Takamatsu, H.; Samaoto, Y.; Aramaki, S.; Matsukata, M.; Kikuchi, E. Pt and/or Pd-supported/incorporated catalyst on perovskite-type oxide for water gas shift reaction. Top. Catal. 2010, 53, 621–628. [Google Scholar] [CrossRef]
- Sekine, Y.; Takamatsu, H.; Aramaki, S.; Ichishima, K.; Takada, M.; Matsukata, M.; Kikuchi, E. Synergistic effect of Pt or Pd and perovskite oxide for water gas shift reaction. Appl. Catal. A 2009, 352, 214–222. [Google Scholar] [CrossRef]
- Daza, Y.; Kent, R.; Yung, M.; Kuhn, J. Carbon dioxide conversion by reverse water-gas shift chemical looping on perovskite-type oxides. Ind. Eng. Chem. Res. 2014, 53, 5828–5837. [Google Scholar] [CrossRef]
- Wang, Z.; Luo, F.; Wang, N.; Li, X. Cu-Y2O3 catalyst derived from Cu2Y2O5 perovskite for water gas shift reaction: The effect of reduction temperature. Catalysts 2022, 12, 481. [Google Scholar] [CrossRef]
- Orsini, F.; Ferrero, D.; Cannone, S.F.; Santarelli, M.; Felli, A.; Boaro, M.; de Leitenburg, C.; Trovarelli, A.; Llorca, J.; Dimitrakopoulos, G.; et al. Exsolution-enhanced reverse water-gas shift chemical looping activity of Sr2FeMo0.6Ni0.4O6-d. Chem. Eng. J. 2023, 475, 146083. [Google Scholar] [CrossRef]
- Ploner, K.; Götsch, T.; Kogler, G.; Thalinger, R.; Bernardi, J.; Zhao, Q.; Zhuo, C.; Klötzer, B.; Penner, S. Structural and catalytic properties of Ag- and Co3O4-impregnated strontium titanium ferrite SrTi0.7Fe0.3O3-d in methanol steam reforming. Ind. Eng. Chem. Res. 2017, 56, 13654–13662. [Google Scholar] [CrossRef]
- Provendier, H.; Petit, C.; Kiennemann, A. Steam reforming of methane on LaNixFe1-xO3 (0 ≤ x ≤ 1) perovkites. Reactivity and characterisation after test. Comptes Rendus L’académie Des Sci. Ser. IIC-Chem. 2001, 4, 57–66. [Google Scholar]
- Zhang, X.; Su, Y.; Pei, C.; Zhao, Z.; Liu, R.; Gong, J. Chemical Looping steam reforming of methane over Ce-doped perovskites. Chem. Eng. J. 2020, 223, 115707. [Google Scholar]
- Ou, Z.; Zhang, Z.; Qin, C.; Xia, H.; Deng, T.; Niu, J.; Ran, J.; Wu, C. Highly active and stable Ni/perovskite catalysts in steam methane reforming for hydrogen production. Sustain. Energy Fuels 2021, 5, 1845. [Google Scholar] [CrossRef]
- Natile, M.; Poletto, F.; Galenda, A.; Glisenti, A.; Montini, T.; De Rogatis, L.; Fornasiero, P. La0.6Sr0.4CoyFe1-yO3 perovskites: Influence of the Co/Fe atomic ratio on properties and catalytic activity toward alcohol steam reforming. Chem. Mater. 2008, 20, 2314–2327. [Google Scholar] [CrossRef]
- Dama, S.; Ghudke, S.; Bobade, R.; Gurav, H.; Chilukuri, S. Tuning the dimensionality of layered Srn+1Tin-xNixO3n+1 perovskite structures for improved activity in syngas generation. J. Catal. 2018, 360, 27–39. [Google Scholar] [CrossRef]
- Yang, E.; Kim, N.; Noh, Y.; Lim, S.; Jung, J.; Lee, J.; Hong, G.; Moon, D. Steam reforming of methane over La1-xCexNiO3 perovskite catalysts. Int. J. Hydrogen Energy 2015, 40, 11831–11839. [Google Scholar] [CrossRef]
- Aguero, F.; Morales, M.; Larregola, S.; Izurieta, E.; Lopez, E.L.; Cadus, E. La1-xCaxAl1-yNiyO3 perovskites used as catalyst precursors of nickel-based catalysts for ethanaol steam reforming. Int. J. Hydrogen Energy 2015, 40, 15510–15520. [Google Scholar] [CrossRef]
- Aguero, F.; Alonso, J.; Fernandez-Diaz, M.; Cadus, L. Ni-based catalysts obtained from perovskite oxides for ethaol steam reforming. J. Fuel Chem. Technol. 2018, 46, 1332–1341. [Google Scholar] [CrossRef]
- da Lima, S.; da Silva, A.; da Costa, L.; Assaf, J.; Mattos, L.; Sarkari, R.; Venugopal, A.; Noronha, R. Hydrogen production through oxidative steam reforming of ethanol over Ni-based catalysts derived from La1-xCexNiO3. Appl. Catal. B 2012, 121, 1–9. [Google Scholar] [CrossRef]
- Lin, K.; Wang, C.; Chien, S. Catalytic performance of steam reforming of ethanol at low temperature over LaNiO3 perovskite. Int. J. Hydrogen Energy 2013, 38, 3226–3232. [Google Scholar] [CrossRef]
- Zhao, L.; Han, T.; Wang, H.; Zhang, L.; Liu, Y. Ni-Co alloy catalysts from La1-xNixCoO3 perovskite supported on zirconia for steam reforming of ethanol. Appl. Catal. B 2016, 187, 19–29. [Google Scholar] [CrossRef]
- Ramesh, S.; Yang, E.; Jung, J.; Moon, D. Copper decorated perovskite an efficient catalyst for low temperature hydroen production by steam reforming of glycerol. Int. J. Hydrogen Energy 2015, 40, 11428–11435. [Google Scholar] [CrossRef]
- Wu, G.; Li, S.; Zhang, C.; Wang, T.; Gong, J. Glycerol steam reforming over perovskite-derived nickel-based catalysts. Appl. Catal. B 2014, 144, 277–285. [Google Scholar] [CrossRef]
- Aman, D.; Radwan, D.; Ebaid, M.; Mikhail, S.; van Steen, E. Comparing nickel and cobalt perovskites for steam reforming of glycerol. Mol. Catal. 2018, 452, 60–67. [Google Scholar] [CrossRef]
- Oemar, U.; Ang, P.S.; Hidajat, K.; Kawi, S. Promotional effect of Fe on perovskite LaNixFe1-xO3 catalyst for hydrogen production via steam reforming of toluene. Int. J. Hydrogen Energy 2013, 38, 5525–5534. [Google Scholar] [CrossRef]
- Sekine, Y.; Mukai, D.; Murai, Y.; Tochiya, S.; Izutsu, Y.; Sekiguchi, K.; Hosomura, N.; Arai, H.; Kikuchi, E.; Sugiura, Y. Steam reforming of toluene over perovskite-supported Ni catalysts. Appl. Catal. A 2013, 451, 160–167. [Google Scholar] [CrossRef]
- Urasaki, K.; Tokunaga, K.; Sekine, Y.; Matsukata, M.; Kikuchi, E. Production of hydrogen by steam reforming of ethanol over cobalt and nickel catalysts supported on perovskite.type catalysts. Catal. Commun. 2008, 9, 600–604. [Google Scholar] [CrossRef]
- Morales, M.; Segarra, M. Steam reforming and oxidate steam reforming of ethanol over La0.6Sr0.4CoO3-d perovskite as catalyst precursor for hydrogen production. Appl. Catal. A 2015, 502, 305–311. [Google Scholar] [CrossRef]
- Thalinger, R.; Gocyla, M.; Heggen, M.; Dunin-Borkowski, R.; Grünbacher, M.; Stöger-Pollach, M.; Schmidmair, D.; Klötzer, B.; Penner, S. Ni-perovskite interaction and ist structural and catalytic consequences in methane steam reforming and methanation reactions. J. Catal. 2016, 337, 26–35. [Google Scholar] [CrossRef]
- Thalinger, R.; Götsch, T.; Zhuo, C.; Hetaba, W.; Wallisch, W.; Stöger-Pollach, M.; Schmidmair, D.; Klötzer, B.; Penner, S. Rhodium-catalyzed methanation and methane steam reforming reactions on Rhodium-perovskite systems: Metal-support interaction. ChemCatChem 2016, 8, 2057–2067. [Google Scholar] [CrossRef]
- Wang, H.; Fang, Y.; Liu, Y.; Bai, X. Perovskite LaFeO3 supported bi.metal catalyst for syngas methanation. J. Nat. Gas Chem. 2012, 21, 745–752. [Google Scholar] [CrossRef]
- Dokuchits, E.V.; Larina, T.V.; Gerasimov, E.Y.; Pochtar, A.A.; Minyukova, T.P. Syngas conversion over perovskite-like LaCuxTi1-xO3/KIT-6 catalysts. Appl. Catal. A 2020, 608, 117834. [Google Scholar] [CrossRef]
- Yan, B.; Wu, Q.; Cen, J.; Timoshenko, J.; Frenkel, A.; Su, D.; Chen, X.; Parise, J.; Stach, A.; Orlev, A.; et al. Highly active subnanmoeter Rh clusters derived from Rh-doped SrTiO3 for CO2 redcution. Appl. Catal. B 2018, 237, 1003–1011. [Google Scholar] [CrossRef]
- Aryandiyan, H.; Wang, Y.; Scott, J.; Mesgari, S.; Dai, H.; Amal, R. In situ exsolution of bimetallic Rh-Ni nanoalloys: A highly efficient catalyst for CO2 methanation. ACS Appl. Mater. Interfaces 2018, 10, 16352–16357. [Google Scholar] [CrossRef] [PubMed]
- Fierro, J.L.G. Hydrogenation of carbon oxides over perovskite-type oxides. Catal. Rev. 1992, 34, 321–336. [Google Scholar] [CrossRef]
- Gisling, H.J.; Monnier, J.R.; Apai, G. Synthesis, characterization and catalytic activity of LaRhO3. J. Catal. 1987, 103, 407. [Google Scholar] [CrossRef]
- Broussard, J.A.; Wade, L.E. Catalytic conversion of syngas with perovskites. Am. Chem. Soc. Div. Fuel Chem. 1986, 31, 75. [Google Scholar]
- Nishihata, Y.; Mizuki, J.; Akao, T.; Tanaka, H.; Uenishi, M.; Kimura, M.; Okamoto, T.; Hamada, H. Self-generation of a Pd-perovskite catalyst for automotive emissions control. Nature 2002, 418, 165–167. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, H.; Taniguchi, M.; Uenishi, M.; Kajita, N.; Tan, I.; Nishihata, Y.; Mizuki, J.; Narita, K.; Kimura, M.; Kaneko, K. Self-generating Rh- and Pt-based perovskite catalysts for automotive emissions control. Angew. Chem. Int. Ed. 2006, 45, 5998–6002. [Google Scholar] [CrossRef]
- Tanaka, H.; Tan, I.; Uenishi, M.; Taniguchi, M.; Kimura, M.; Nishihata, Y.; Mizuki, J. LaFePdO3 perovskite automotive catalyst having a self-regenerative function. J. Alloys Compd. 2006, 408, 1071–1077. [Google Scholar] [CrossRef]
- Katz, M.; Graham, G.; Duan, Y.; Liu, H.; Adamo, C.; Schlom, D.; Pan, X. Self-regeration of Pd-LaFeO3 catalysts: New insights from atomic-resolution electron microscopy. J. Am. Chem. Soc. 2011, 133, 18090–18093. [Google Scholar] [CrossRef]
- Kothari, M.; Jeon, Y.; Miller, D.; Pascui, A.; Kilmartin, J.; Wails, D.; Ramos, S.; Chadwick, A.; Irvine, J.T.S. Platinum incorporation into titanate perovskites to deliver emergent active and stable platinum nanparticles. Nat. Chem. 2021, 13, 677–682. [Google Scholar] [CrossRef]
- Glisenti, A.; Pacella, M.; Guiotto, M.; Natile, M.M.; Canu, P. Largely Cu-doped LaCo1-xCux perovskites for TWC: Toward new PGM-free catalysts. Appl. Catal. B 2016, 180, 94–105. [Google Scholar] [CrossRef]
- Calì, E.; Kerherve, G.; Naufal, F.; Kousi, K.; Neagu, D.; Papaioannou, E.I.; Thomas, M.P.; Guiton, B.S.; Metcalfe, I.S.; Irvine, J.T.S.; et al. Exsolution of catalytically active iridium nanparticles from strontium titanate. ACS Appl. Energy Mater. 2020, 12, 37444–37453. [Google Scholar] [CrossRef] [PubMed]
- Cao, T.; Wang, C.; Shen, K.; Vohs, J.; Gorte, R. Investigation into support effects for Pt and Pd on LaMnO3. Appl. Catal. A 2022, 646, 1188773. [Google Scholar] [CrossRef]
- Thurner, C.W.; Bonmassar, N.; Winkler, D.; Haug, L.; Ploner, K.; Nezhad, P.D.K.; Drexler, X.; Mohammadi, A.; van Aken, P.A.; Kunze-Liebhäuser, J.; et al. Who does the job? How copper can replace noble metals in sustainable catalysis by the formation of copper-mixed oxide interfaces. ACS Catal. 2022, 12, 7696–7708. [Google Scholar] [CrossRef] [PubMed]
- Thurner, C.; Drexler, X.; Haug, L.; Winkler, D.; Kunze-Liebhäuser, J.; Bernardi, J.; Klötzer, B.; Penner, S. When copper is not enough: Advantages and drawbacks of using copper in deNOx reactions over lanthanum manganite perovskite structures. Appl. Catal. B 2023, 331, 122693. [Google Scholar] [CrossRef]
- Mohammadi, A.; Farzi, A.; Thurner, C.; Klötzer, B.; Schwarz, S.; Bernardi, J.; Niaei, A.; Penner, S. Tailoring the metal-perovskite interface for promotional steering of the catalytic NO reduction by CO in the presence of H2O on Pd-lanthanum iron manganite composites. Appl. Catal. B 2022, 307, 121160. [Google Scholar] [CrossRef]
- Simonot, L.; Garin, F.; Maire, G. A comparative study of LaCoO3, Co3O4 and a mix of LaCoO3-Co3O4 II. Catalytic properties for the CO+NO reaction. Appl. Catal. B 1997, 11, 181–191. [Google Scholar] [CrossRef]
- Higo, T.; Omori, Y.; Shigemoto, A.; Ueno, K.; Ogo, S.; Sekine, Y. Promotive effect of H2O on low-temperature NO reduction by CO over Pd/La0.9Ba0.9AlO3-δ. Catal. Today 2020, 352, 192–197. [Google Scholar] [CrossRef]
- Khristova, M.; Petrovic, S.; Terlecki-Baricevic, A.; Mehandjiev, D. Catalytic reduction of NO by CO over Pd–doped Perovskite-type catalysts. Cent. Eur. J. Chem. 2009, 7, 857–863. [Google Scholar]
- Munoz, H.; Korili, S.; Gil, A. Progress and recent strategies in the synthesis and catalytic applications of perovskites based on lanthanum and aluminum. Materials 2002, 15, 3288. [Google Scholar] [CrossRef] [PubMed]
- Kapokova, L.; Pavlova, S.; Bunina, R.; Alikina, G.; Krieger, T.; Ishchenko, A.; Rogov, V.; Sadykov, V. Dry reforming of methane over LnFe0.7Ni0.3O3-δ perovskites: Influence of Ln nature. Catal. Today 2011, 164, 227–233. [Google Scholar] [CrossRef]
- Wang, M.; Yang, J.; Chi, B.; Pu, J.; Li, J. High performance Ni exsolved and Cu added La0.8Ce0.2Mn0.6Ni0.4O3-based perovskites for ethanol steam reforming. Int. J. Hydrogen Energy 2020, 45, 16458–16468. [Google Scholar] [CrossRef]
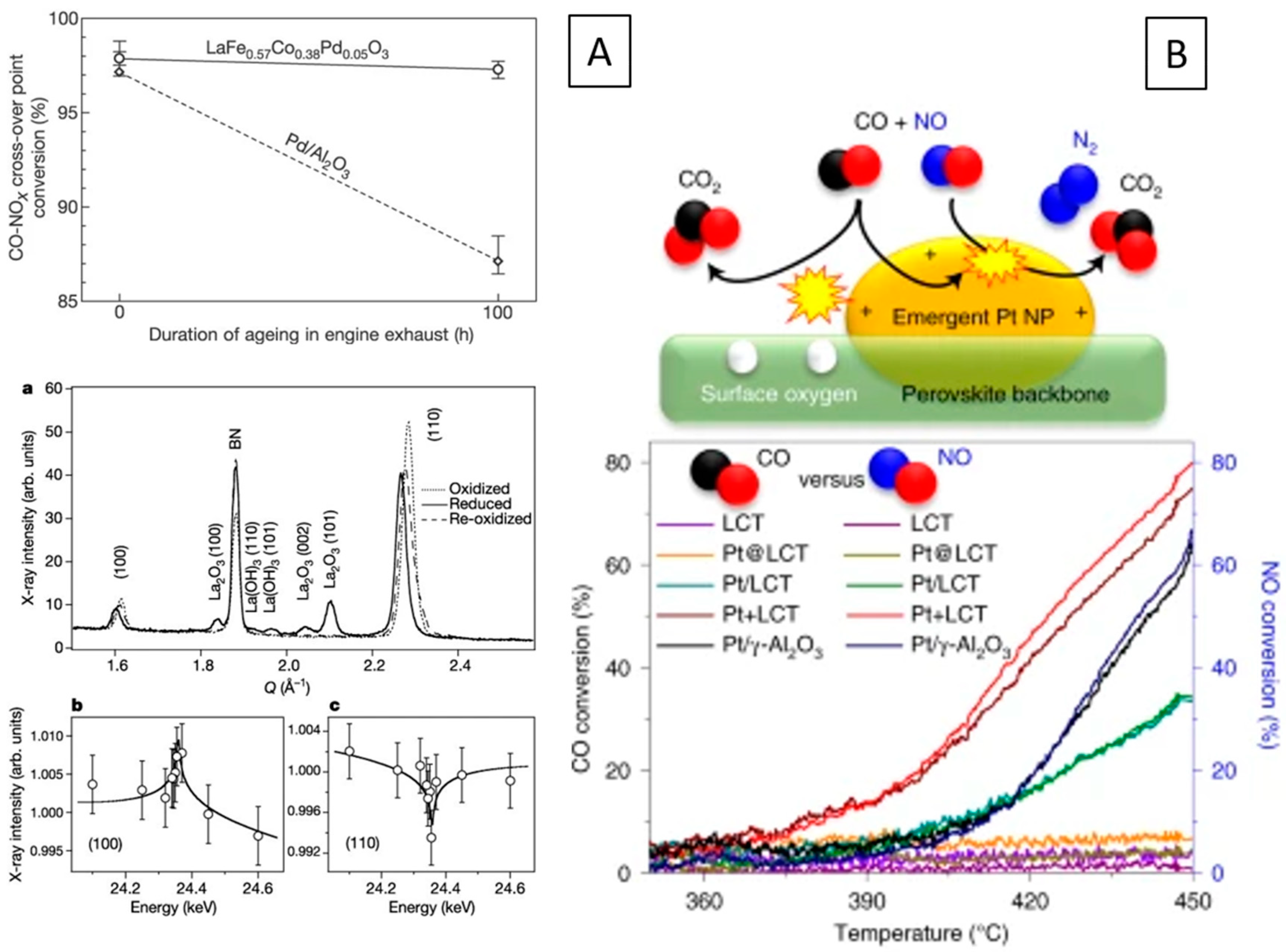
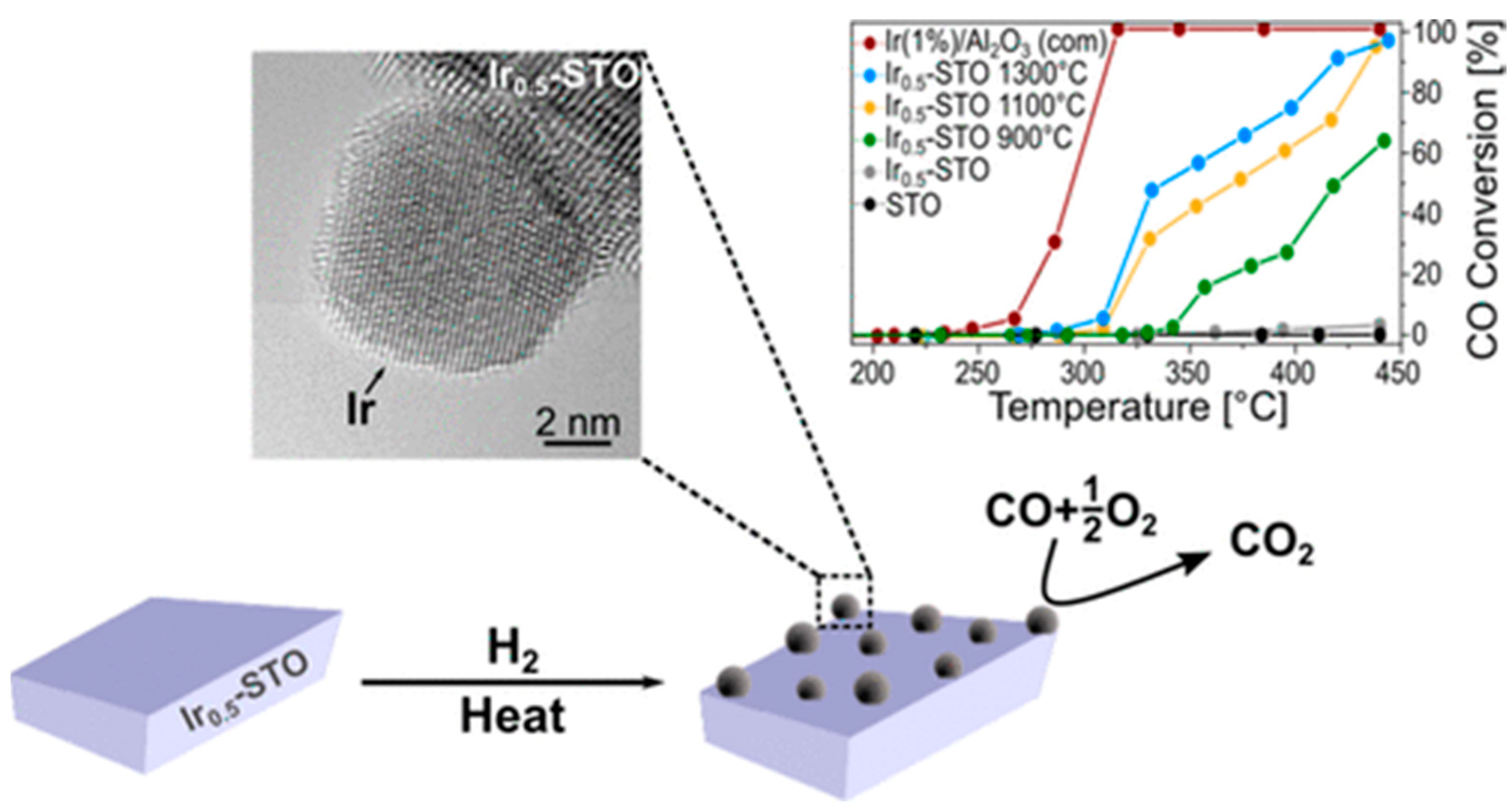
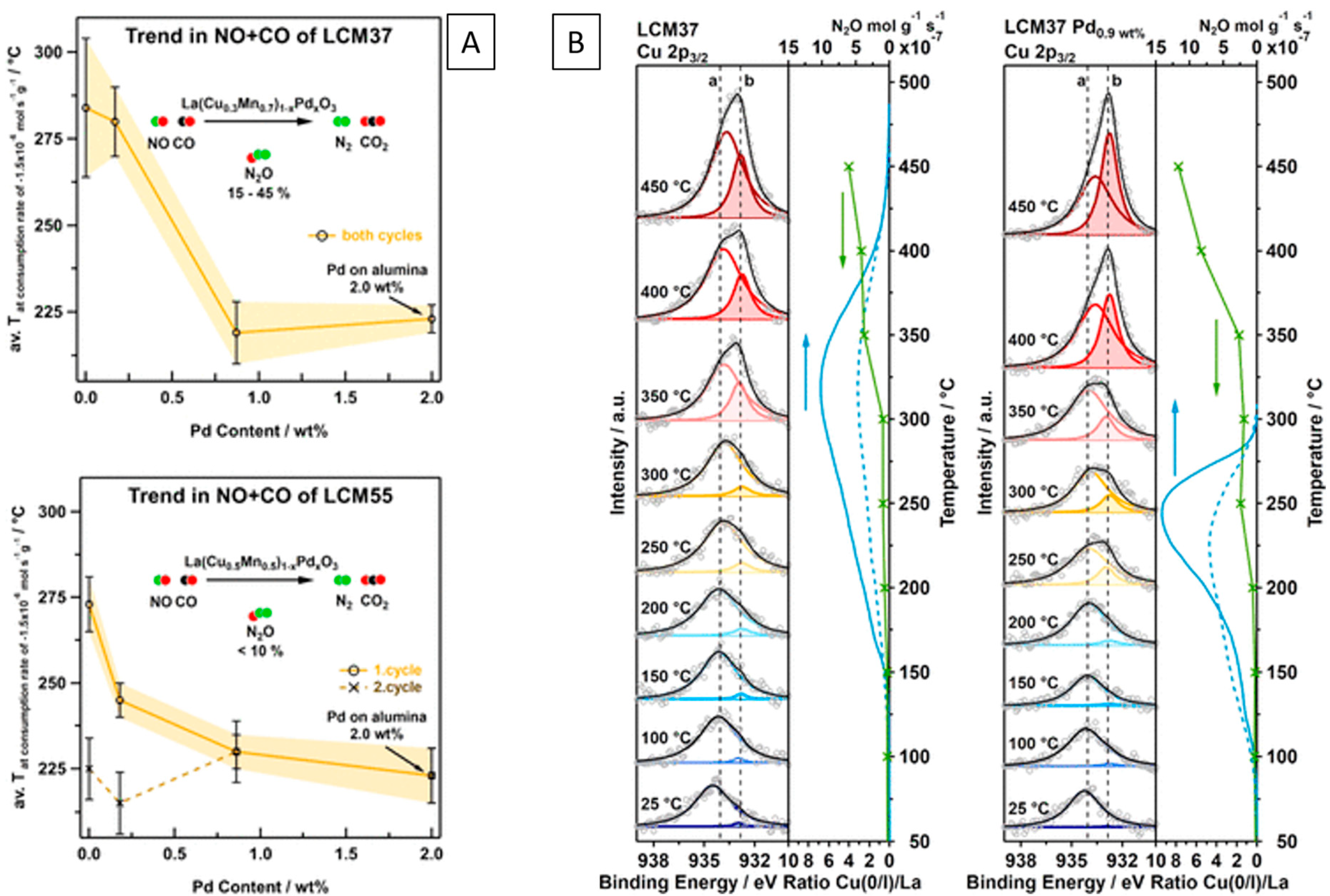
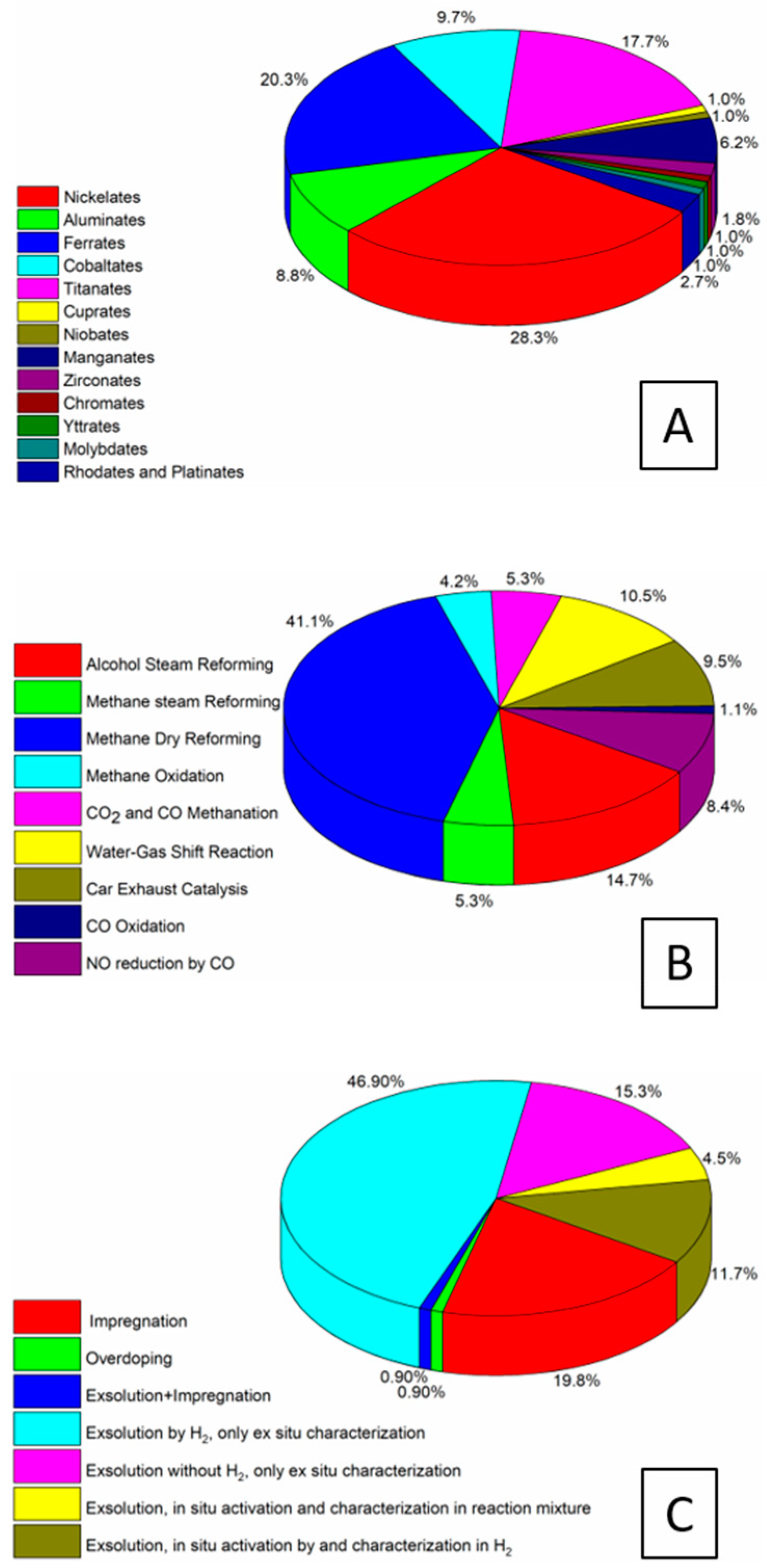
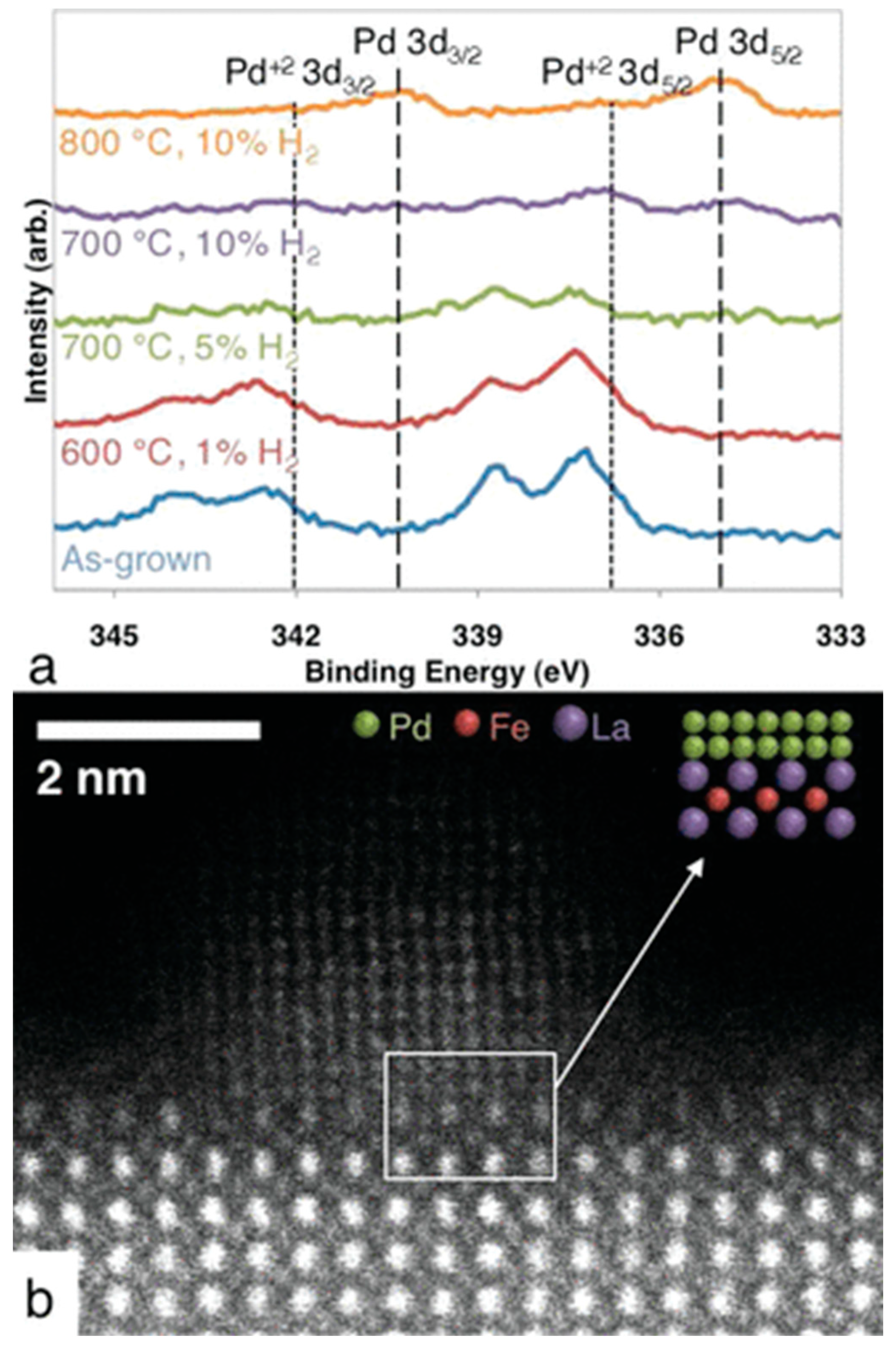
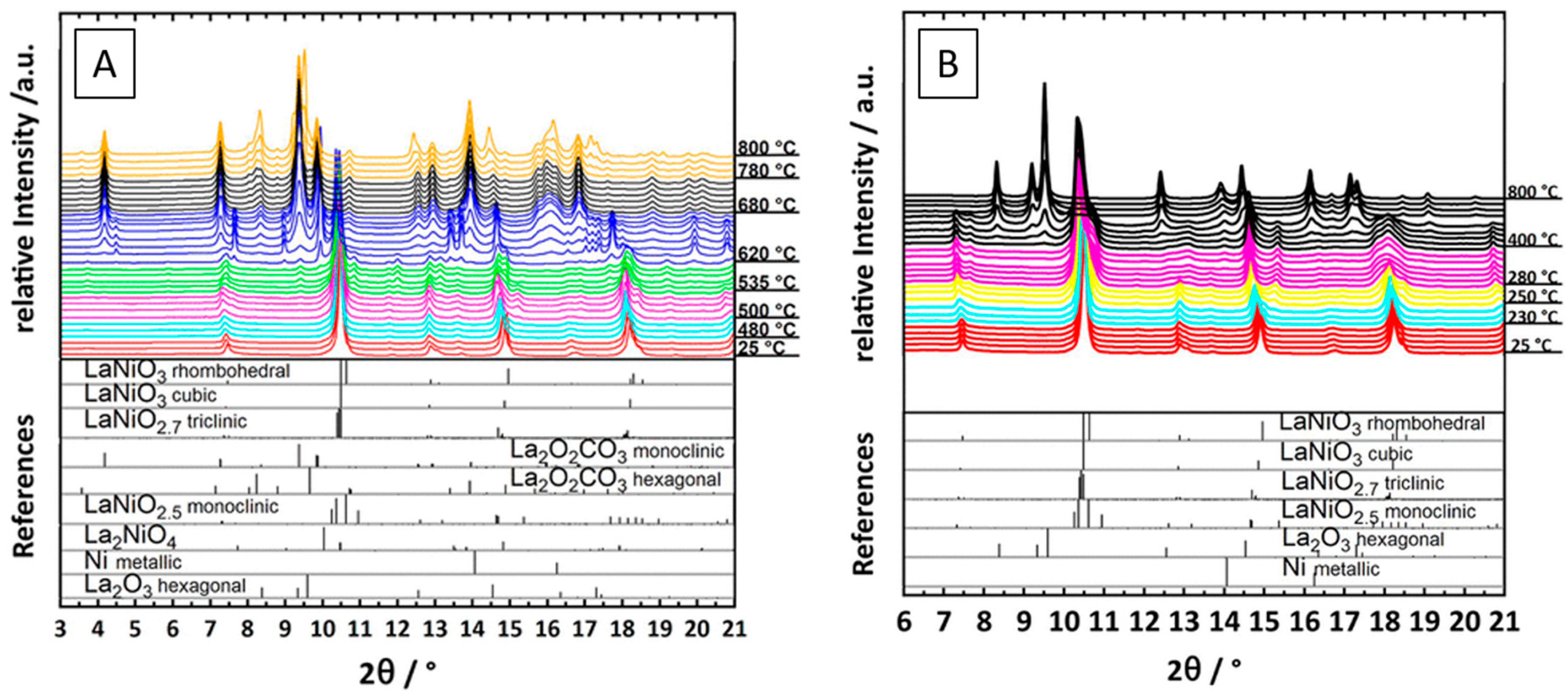



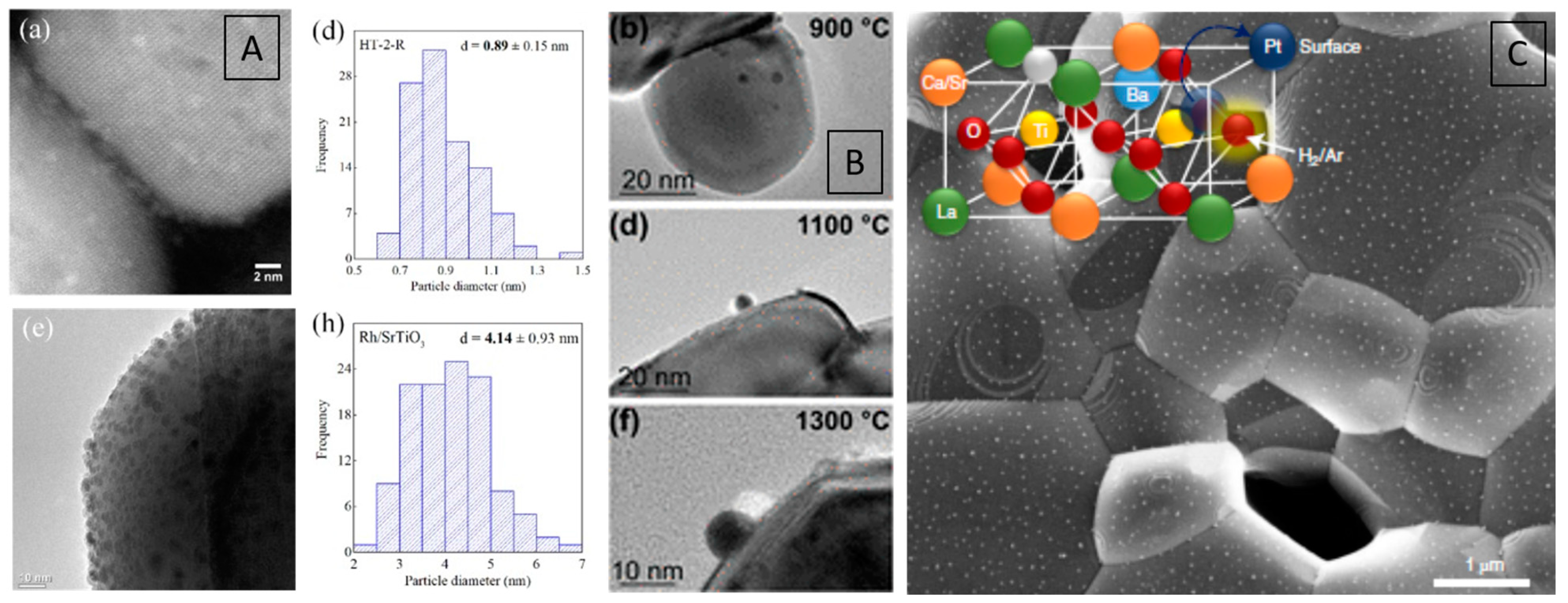
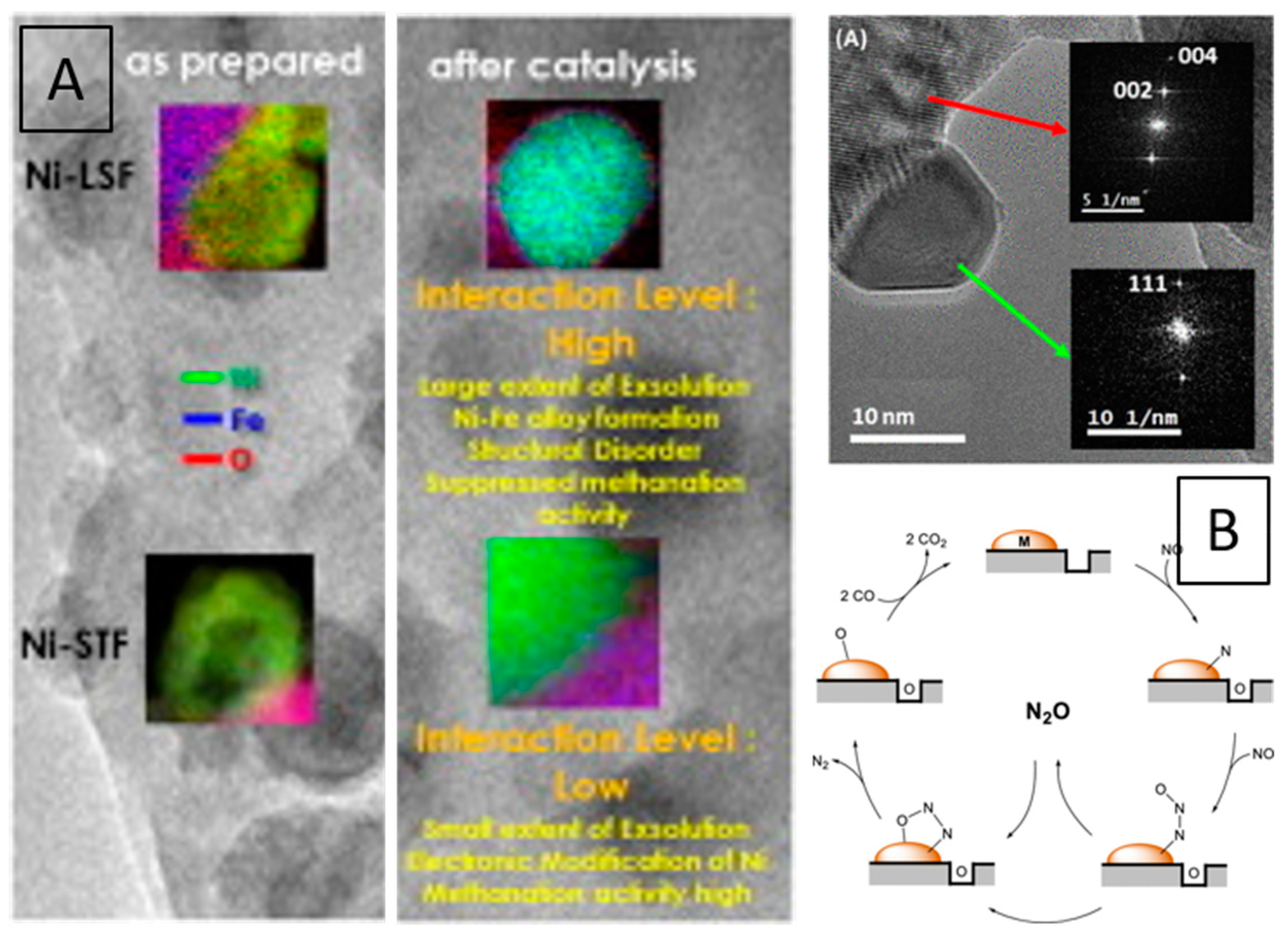
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).