Adsorption of 4,4″-Diamino-p-Terphenyl on Cu(001): A First-Principles Study


Abstract
1. Introduction
2. Computational Methods
3. Results and Discussion
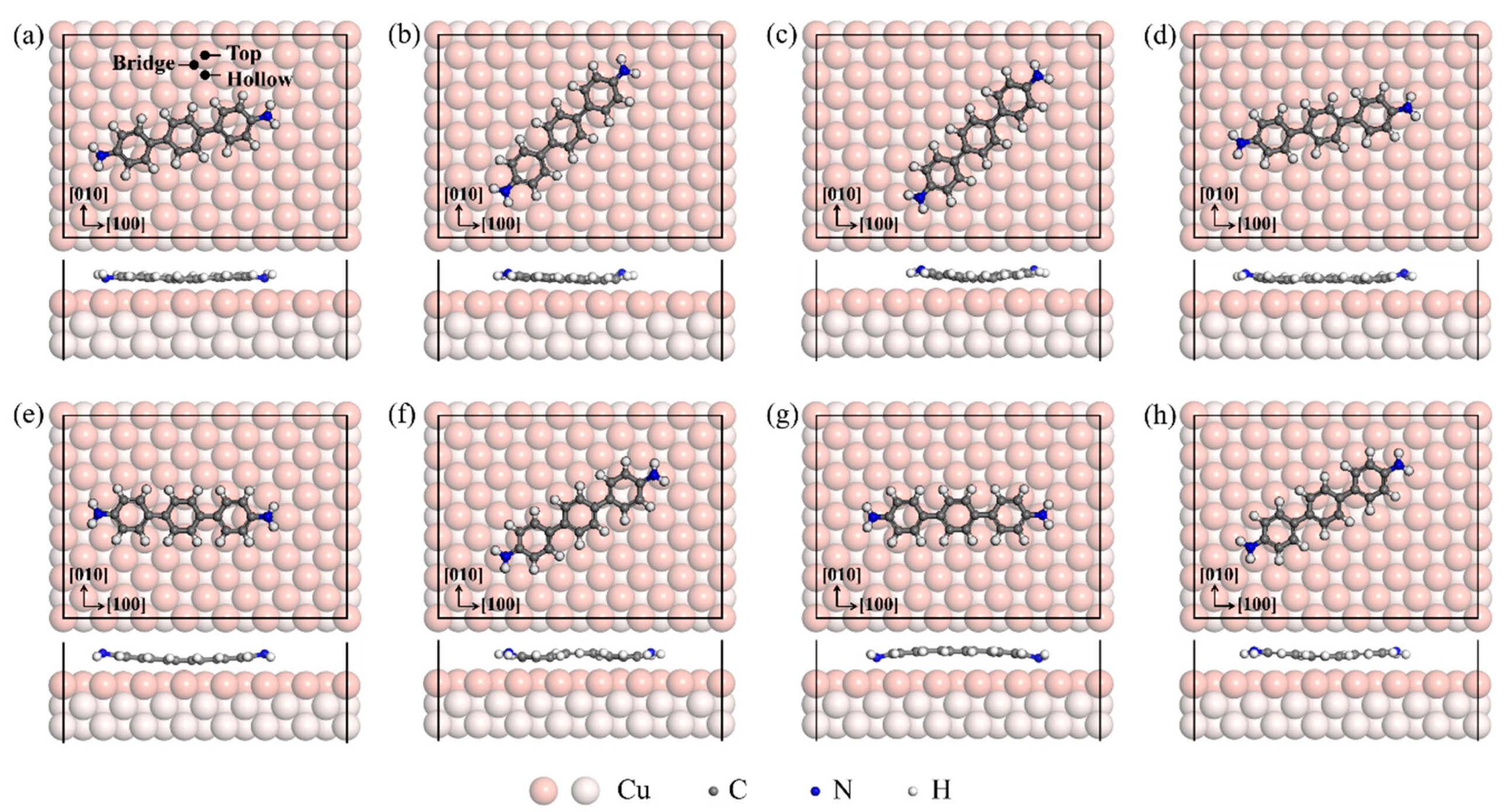
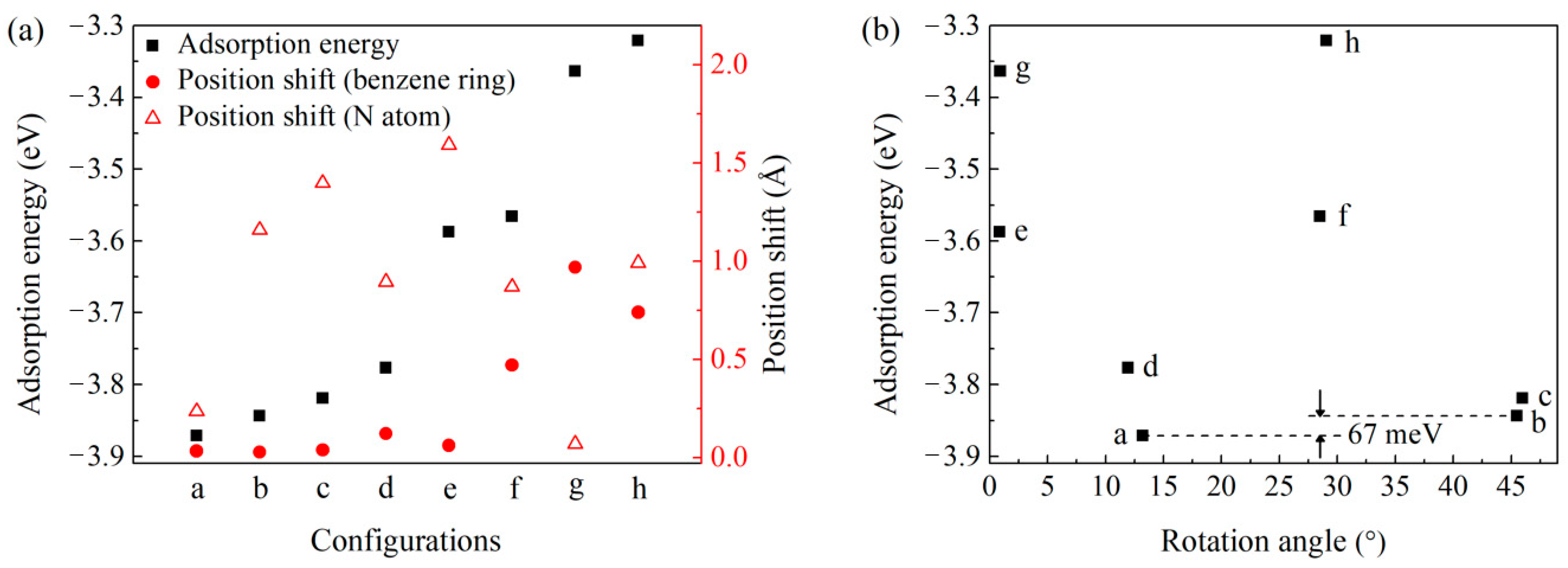
3.1. Adsorption Configurations
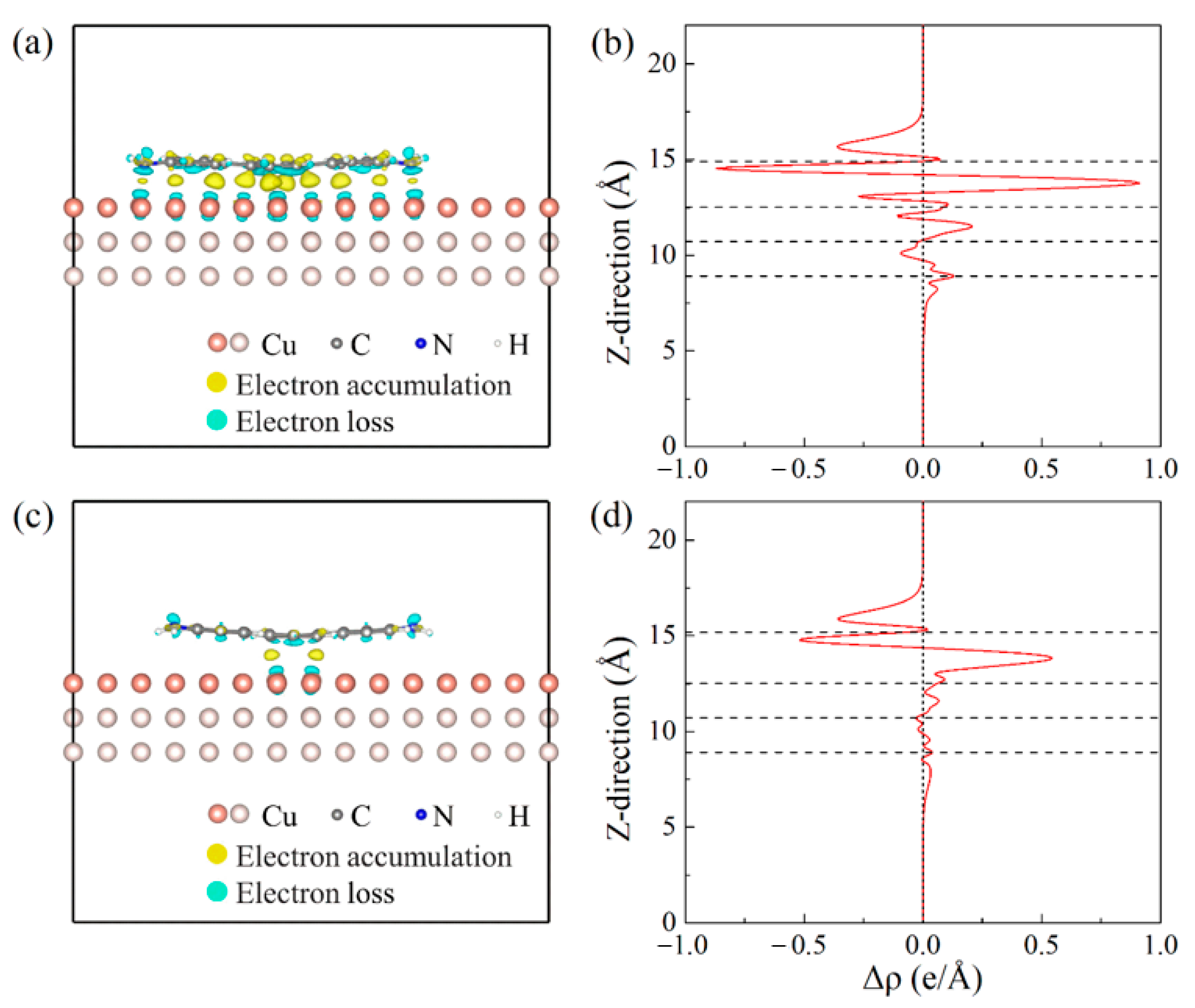
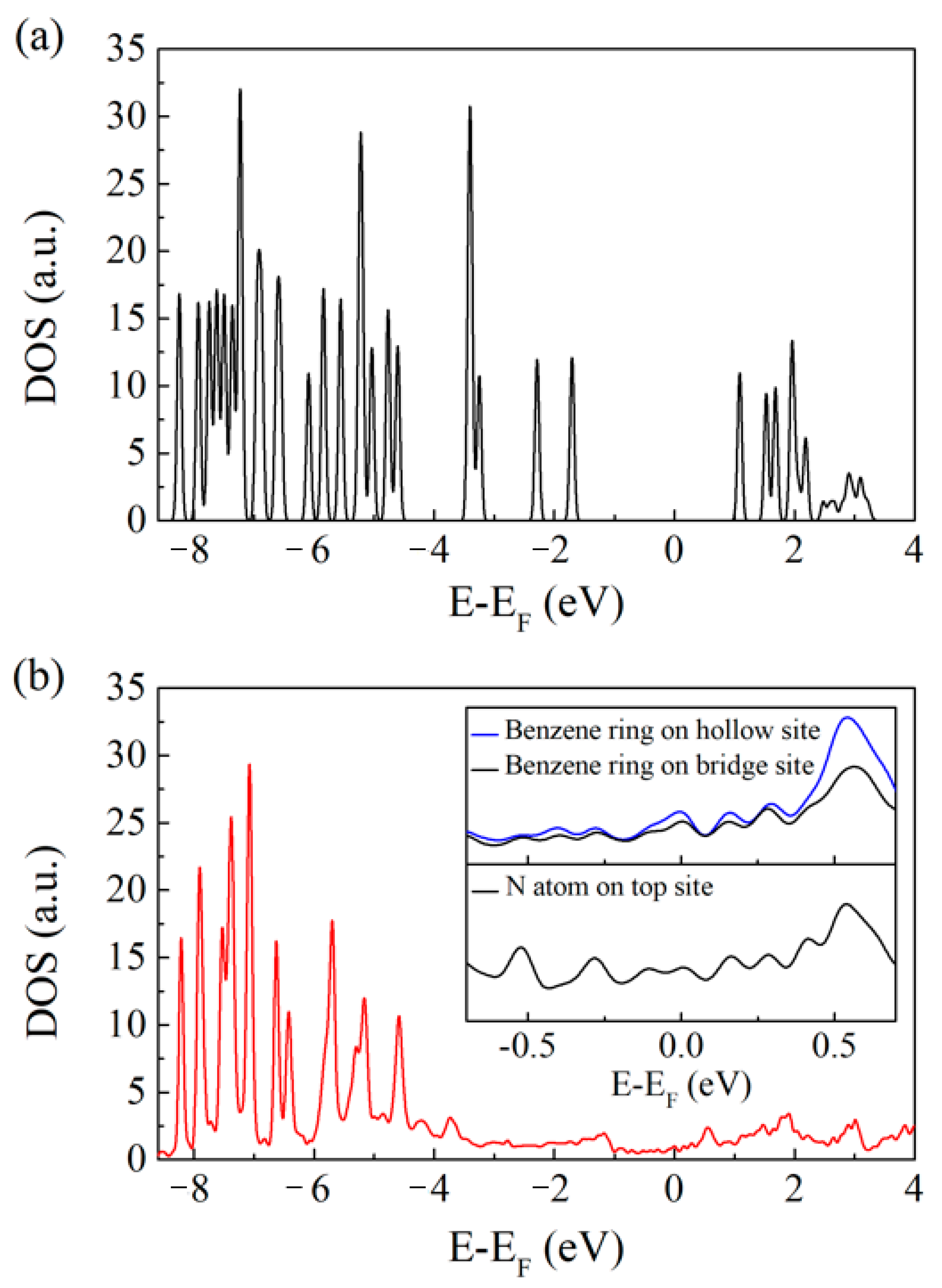
3.2. Electronic Properties
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Eremtchenko, M.; Schaefer, J.A.; Tautz, F.S. Understanding and tuning the epitaxy of large aromatic adsorbates by molecular design. Nature 2003, 425, 602–605. [Google Scholar] [CrossRef] [PubMed]
- Du, S.X.; Gao, H.J.; Seidel, C.; Tsetseris, L.; Ji, W.; Kopf, H.; Chi, L.F.; Fuchs, H.; Pennycook, S.J.; Pantelides, S.T. Selective Nontemplated Adsorption of Organic Molecules on Nanofacets and the Role of Bonding Patterns. Phys. Rev. Lett. 2006, 97, 156105. [Google Scholar] [CrossRef] [PubMed]
- Gao, H.J.; Gao, L. Scanning tunneling microscopy of functional nanostructures on solid surfaces: Manipulation, self-assembly, and applications. Prog. Surf. Sci. 2010, 85, 28–91. [Google Scholar] [CrossRef]
- Vuillaume, D. Molecular Nanoelectronics. Proc. IEEE 2010, 98, 2111–2123. [Google Scholar]
- Gao, L.; Liu, Q.; Zhang, Y.Y.; Jiang, N.; Zhang, H.G.; Cheng, Z.H.; Qiu, W.F.; Du, S.X.; Liu, Y.Q.; Hofer, W.A.; et al. Constructing an Array of Anchored Single-Molecule Rotors on Gold Surfaces. Phys. Rev. Lett. 2008, 101, 197209. [Google Scholar] [CrossRef]
- Tao, L.; Zhang, Y.-Y.; Pantelides, S.T.; Du, S. Tuning the Catalytic Activity of a Quantum Nutcracker for Hydrogen Dissociation. Surfaces 2020, 3, 40–47. [Google Scholar] [CrossRef]
- Wu, R.; Yan, L.; Bao, D.-L.; Ren, J.; Du, S.; Wang, Y.; Huan, Q.; Gao, H.-J. Self-Assembly Evolution of Metal-Free Naphthalocyanine Molecules on Ag(111) at the Submonolayer Coverage. J. Phys. Chem. C 2019, 123, 7202–7208. [Google Scholar] [CrossRef]
- Lu, H.; Wenlong, E.; Ma, Z.; Yang, X. Organometallic polymers synthesized from prochiral molecules by a surface-assisted synthesis on Ag(111). Phys. Chem. Chem. Phys. 2020, 22, 8141–8145. [Google Scholar] [CrossRef]
- Martin-Jimenez, D.; Ahles, S.; Mollenhauer, D.; Wegner, H.A.; Schirmeisen, A.; Ebeling, D. Bond-Level Imaging of the 3D Conformation of Adsorbed Organic Molecules Using Atomic Force Microscopy with Simultaneous Tunneling Feedback. Phys. Rev. Lett. 2019, 122, 196101. [Google Scholar] [CrossRef]
- Ebeling, D.; Zhong, Q.; Schlöder, T.; Tschakert, J.; Henkel, P.; Ahles, S.; Chi, L.; Mollenhauer, D.; Wegner, H.A.; Schirmeisen, A. Adsorption Structure of Mono- and Diradicals on a Cu(111) Surface: Chemoselective Dehalogenation of 4-Bromo-3″-iodo-p-terphenyl. ACS Nano 2019, 13, 324–336. [Google Scholar] [CrossRef]
- Liu, J.; Chen, Q.; Cai, K.; Li, J.; Li, Y.; Yang, X.; Zhang, Y.; Wang, Y.; Tang, H.; Zhao, D.; et al. Stepwise on-surface dissymmetric reaction to construct binodal organometallic network. Nat. Commun. 2019, 10, 2545. [Google Scholar] [CrossRef] [PubMed]
- Telychko, M.; Su, J.; Gallardo, A.; Gu, Y.; Mendieta-Moreno, J.I.; Qi, D.; Tadich, A.; Song, S.; Lyu, P.; Qiu, Z.; et al. Strain-Induced Isomerization in One-Dimensional Metal–Organic Chains. Angew. Chem. Int. Ed. 2019, 58, 18591–18597. [Google Scholar] [CrossRef] [PubMed]
- Oreshkin, A.I.; Muzychenko, D.A.; Oreshkin, S.I.; Yakovlev, V.A.; Murugan, P.; Chandrasekaran, S.S.; Kumar, V.; Bakhtizin, R.Z. Real-Time decay of fluorinated fullerene molecules on Cu(001) surface controlled by initial coverage. Nano Res. 2018, 11, 2069–2082. [Google Scholar] [CrossRef]
- Ienaga, K.; Miyamachi, T.; Takahashi, Y.; Kawamura, N.; Komori, F. Enhanced periodic modulation of electronic states in a hexagonal iron-nitride monolayer on Cu(001) via interfacial interaction. Phys. Rev. B 2017, 96, 085439. [Google Scholar] [CrossRef]
- Bahlke, M.P.; Karolak, M.; Herrmann, C. Interplay between strong correlation and adsorption distances: Co on Cu(001). Phys. Rev. B 2018, 97, 035119. [Google Scholar] [CrossRef]
- Zhang, Y.; Zhao, Y.; Wang, C.; Wei, Z.; Yang, J.; Ma, J. Zn-Doped Cu(100) facet with efficient catalytic ability for the CO2 electroreduction to ethylene. Phys. Chem. Chem. Phys. 2019, 21, 21341–21348. [Google Scholar] [CrossRef]
- Dokukin, S.A.; Kolesnikov, S.V.; Saletsky, A.M.; Klavsyuk, A.L. Diffusion-Mediated processes in Pt/Cu(001) surface alloy. Surf. Sci. 2020, 692, 121515. [Google Scholar] [CrossRef]
- Dou, W.-D.; Zhang, H.-J.; Bao, S.-N. Scanning tunneling microscopy study of surface reconstruction induced by N adsorption on Cu (100) surface. Chin. Phys. B 2010, 19, 026803. [Google Scholar]
- Benlattar, M.; Elkoraychy, E.; Sbiaai, K.; Mazroui, M.; Boughaleb, Y. Ehrlich-Schwöbel barriers and adsorption of Au, Cu and Ag stepped (100) surfaces. Mod. Phys. Lett. B 2017, 31, 1750037. [Google Scholar] [CrossRef]
- Chen, S.; Sun, S.; Lian, B.; Ma, Y.; Yan, Y.; Hu, S. The adsorption and dissociation of H2S on Cu(100) surface: A DTF study. Surf. Sci. 2014, 620, 51–58. [Google Scholar] [CrossRef]
- Robledo, M.; Díaz-Tendero, S. Exploring the Adsorption and the Potential Energy Surface of Acrylonitrile on Cu(100) and Cu(100) Coated with NaCl Layers. J. Phys. Chem. C 2015, 119, 15125–15136. [Google Scholar] [CrossRef]
- Guo, Q.; Qin, Z.; Huang, M.; Mantsevich, V.N.; Cao, G. Image potential states mediated STM imaging of cobalt phthalocyanine on NaCl/Cu(100). Chin. Phys. B 2016, 25, 036801. [Google Scholar] [CrossRef]
- Eren, B.; Weatherup, R.S.; Liakakos, N.; Somorjai, G.A.; Salmeron, M. Dissociative Carbon Dioxide Adsorption and Morphological Changes on Cu(100) and Cu(111) at Ambient Pressures. J. Am. Chem. Soc. 2016, 138, 8207–8211. [Google Scholar] [CrossRef] [PubMed]
- Itabashi, A.; Fukushima, M.; Murata, H. Multi-Layer Polymer Light-Emitting Diodes Prepared by Vapor Deposition Polymerization of Polyazomethine Thin Film. Jpn. J. Appl. Phys. 2008, 47, 1271–1275. [Google Scholar] [CrossRef]
- Zhong, Q.; Ebeling, D.; Tschakert, J.; Gao, Y.; Bao, D.; Du, S.; Li, C.; Chi, L.; Schirmeisen, A. Symmetry breakdown of 4,4″-diamino-p-terphenyl on a Cu(111) surface by lattice mismatch. Nat. Commun. 2018, 9, 3277. [Google Scholar] [CrossRef] [PubMed]
- Nishimura, T.; Itabashi, A.; Sasahara, A.; Murata, H.; Arai, T.; Tomitori, M. Adsorption State of 4,4″-Diamino-p-terphenyl through an Amino Group Bound to Si(111)-7 × 7 Surface Examined by X-ray Photoelectron Spectroscopy and Scanning Tunneling Microscopy. J. Phys. Chem. C 2010, 114, 11109–11114. [Google Scholar] [CrossRef]
- Nishimura, T.; Sasahara, A.; Murata, H.; Arai, T.; Tomitori, M. Thermal Transformation of 4,4″-Diamino-p-terphenyl on a Si(111)-7 × 7 Surface Analyzed by X-ray Photoemission Spectroscopy and Scanning Tunneling Microscopy. J. Phys. Chem. C 2014, 118, 25104–25109. [Google Scholar] [CrossRef]
- Hassan, A.M.A.; Nishimura, T.; Sasahara, A.; Murata, H.; Tomitori, M. Stable alignment of 4,4″-diamino-p-terphenyl chemically adsorbed on a Si(001)-(2 × 1) surface observed by scanning tunneling microscopy. Surf. Sci. 2014, 630, 96–100. [Google Scholar] [CrossRef]
- Ren, J.; Bao, D.-L.; Dong, L.; Gao, L.; Wu, R.; Yan, L.; Wang, A.; Yan, J.; Wang, Y.; Du, S.-X.; et al. Thermo-controllable self-assembled structures of single-layer 4, 4″-diamino-p-terphenyl molecules on Au(110). Chin. Phys. B 2017, 26, 086801. [Google Scholar] [CrossRef]
- Nguyen, M.-T.; Pignedoli, C.A.; Treier, M.; Fasel, R.; Passerone, D. The role of van der Waals interactions in surface-supported supramolecular networks. Phys. Chem. Chem. Phys. 2010, 12, 992–999. [Google Scholar] [CrossRef]
- Treier, M.; Nguyen, M.-T.; Richardson, N.V.; Pignedoli, C.; Passerone, D.; Fasel, R. Tailoring Low-Dimensional Organic Semiconductor Nanostructures. Nano Lett. 2009, 9, 126–131. [Google Scholar] [CrossRef] [PubMed]
- Cao, N.; Ding, J.; Yang, B.; Zhang, J.; Peng, C.; Lin, H.; Zhang, H.; Li, Q.; Chi, L. Deprotonation-Induced Phase Evolutions in Co-Assembled Molecular Structures. Langmuir 2018, 34, 7852–7858. [Google Scholar] [CrossRef] [PubMed]
- Rauls, E.; Blankenburg, S.; Schmidt, W.G. Chemical reactivity on surfaces: Modeling the imide synthesis from DATP and PTCDA on Au(111). Phys. Rev. B 2010, 81, 125401. [Google Scholar] [CrossRef]
- Blöchl, P.E. Projector augmented-wave method. Phys. Rev. B 1994, 50, 17953–17979. [Google Scholar] [CrossRef] [PubMed]
- Kresse, G.; Furthmüller, J. Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set. Phys. Rev. B 1996, 54, 11169–11186. [Google Scholar] [CrossRef]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized Gradient Approximation Made Simple. Phys. Rev. Lett. 1996, 77, 3865–3868. [Google Scholar] [CrossRef]
- Grimme, S. Semiempirical GGA-type density functional constructed with a long-range dispersion correction. J. Comput. Chem. 2006, 27, 1787–1799. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Structure | a | b | c | d | e | f | g | h |
|---|---|---|---|---|---|---|---|---|
| Eads (eV/molecule) | −3.871 | −3.844 | −3.819 | −3.777 | −3.587 | −3.566 | −3.364 | −3.321 |
| Angle (°) | 13.2 | 45.5 | 46.0 | 11.9 | 0.9 | 28.5 | 0.9 | 29.0 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, C.-T.; Zhang, Y.-F.; Du, S. Adsorption of 4,4″-Diamino-p-Terphenyl on Cu(001): A First-Principles Study. Surfaces 2021, 4, 31-38. https://doi.org/10.3390/surfaces4010005
Wang C-T, Zhang Y-F, Du S. Adsorption of 4,4″-Diamino-p-Terphenyl on Cu(001): A First-Principles Study. Surfaces. 2021; 4(1):31-38. https://doi.org/10.3390/surfaces4010005
Chicago/Turabian StyleWang, Chang-Tian, Yan-Fang Zhang, and Shixuan Du. 2021. "Adsorption of 4,4″-Diamino-p-Terphenyl on Cu(001): A First-Principles Study" Surfaces 4, no. 1: 31-38. https://doi.org/10.3390/surfaces4010005
APA StyleWang, C.-T., Zhang, Y.-F., & Du, S. (2021). Adsorption of 4,4″-Diamino-p-Terphenyl on Cu(001): A First-Principles Study. Surfaces, 4(1), 31-38. https://doi.org/10.3390/surfaces4010005