A Critical Review of the Physicochemical Properties of Lignosulfonates: Chemical Structure and Behavior in Aqueous Solution, at Surfaces and Interfaces


Abstract
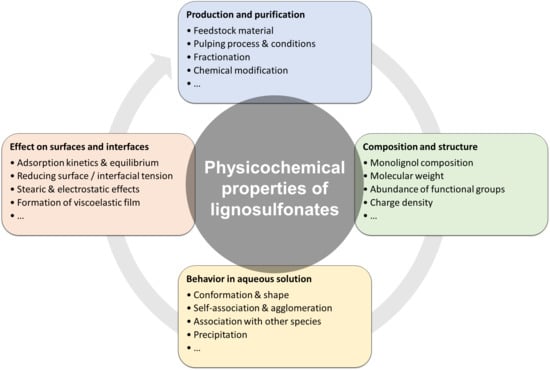
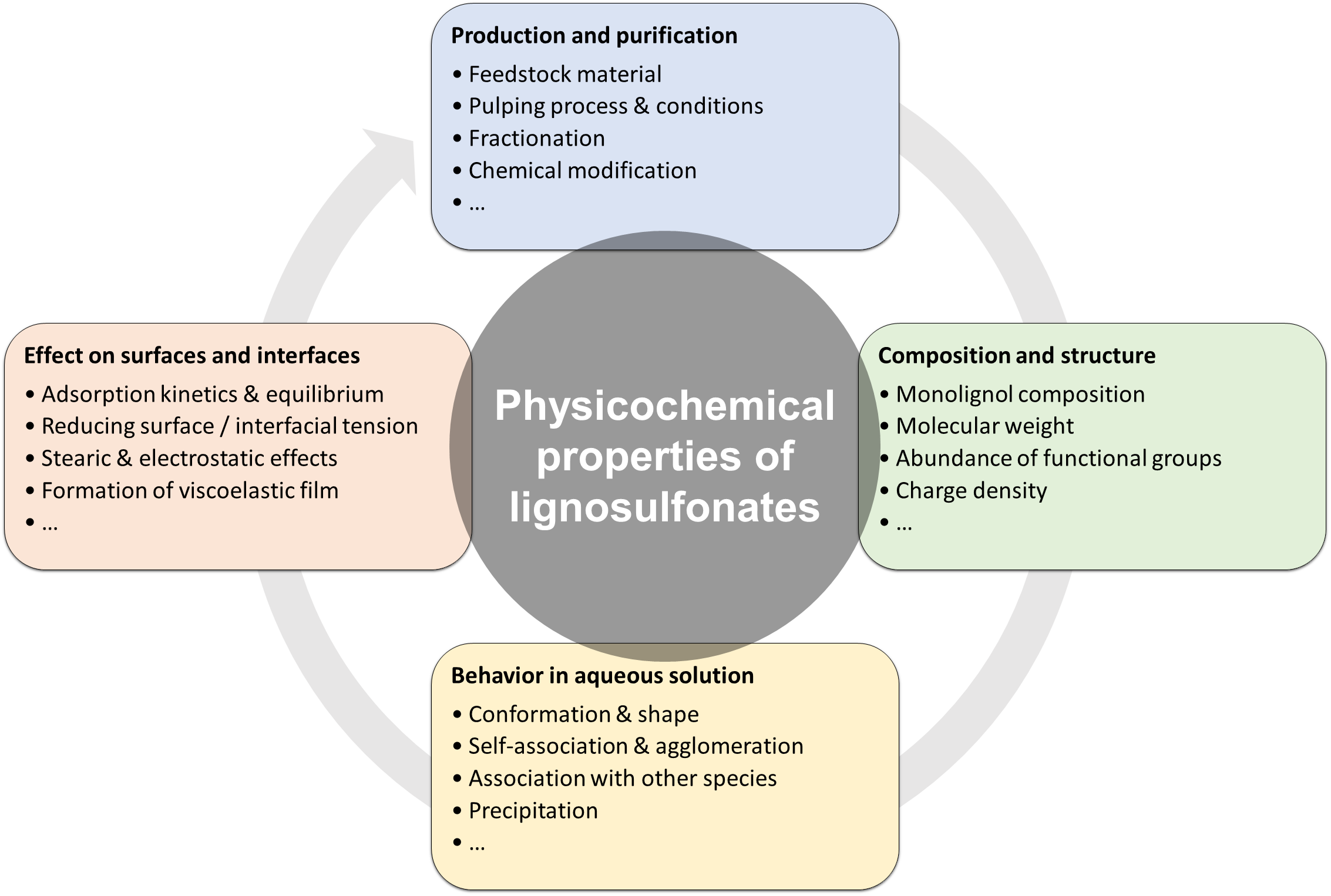
1. Introduction
2. Fundamentals
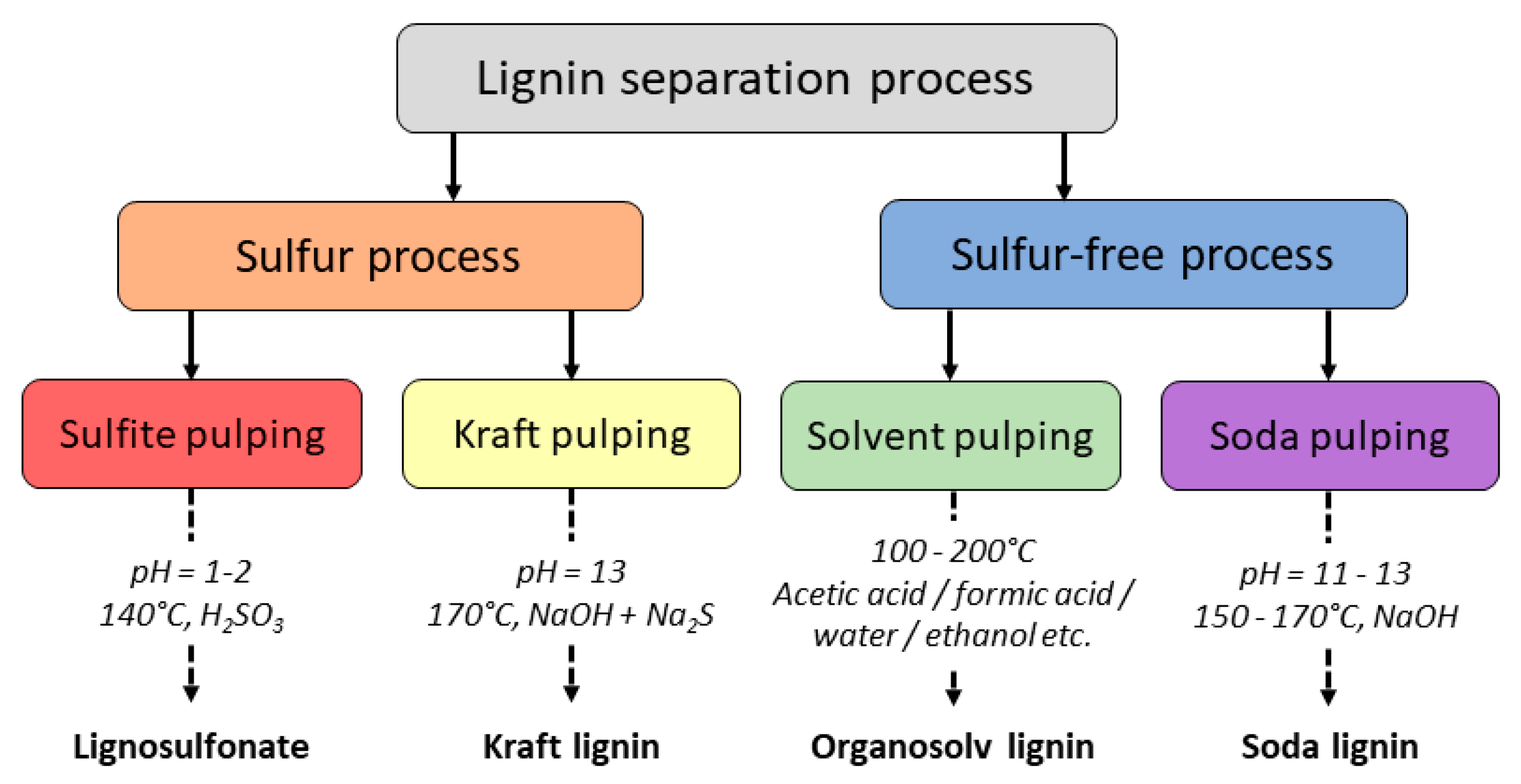
2.1. Definitions and Distinctions
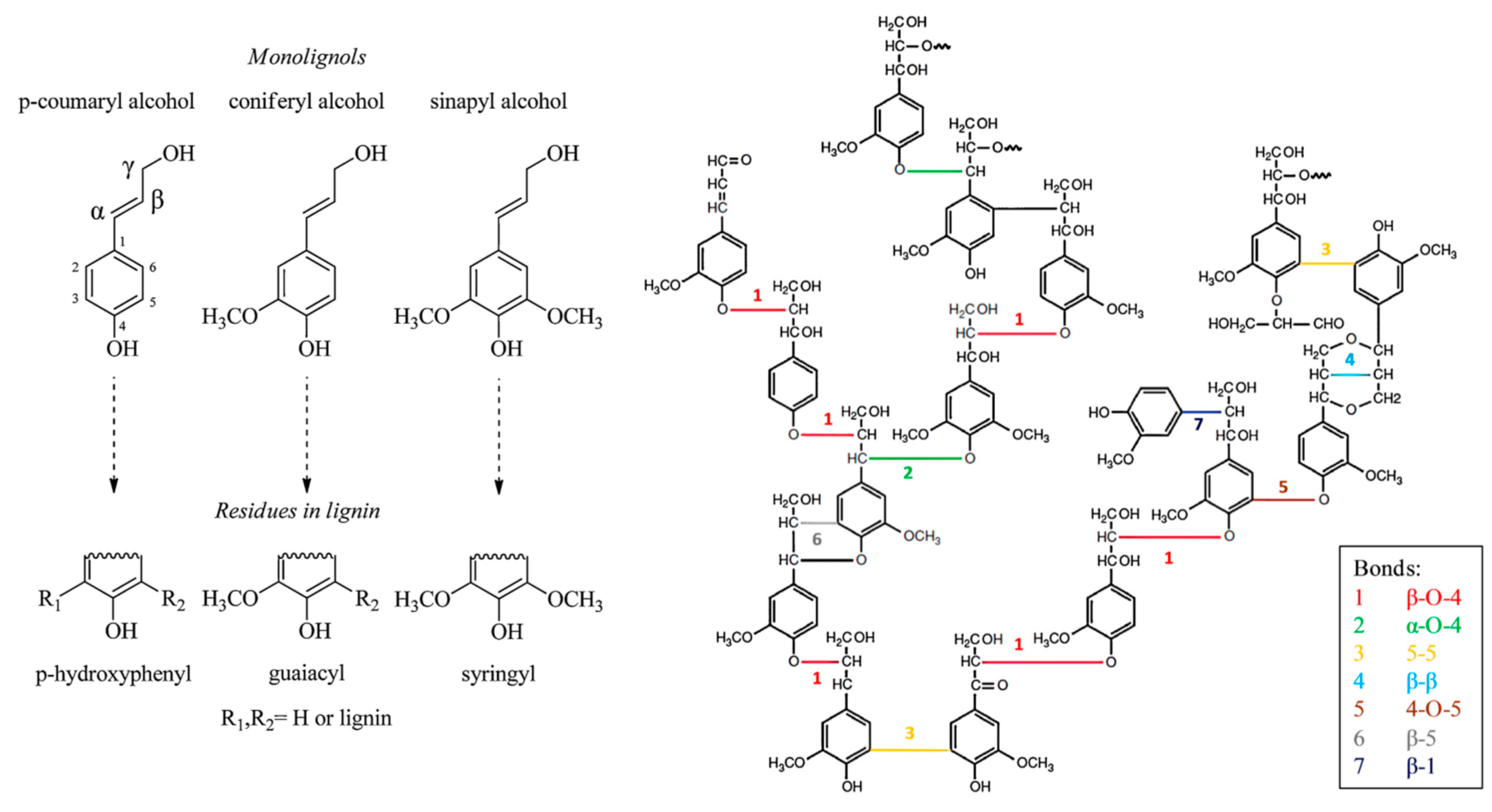
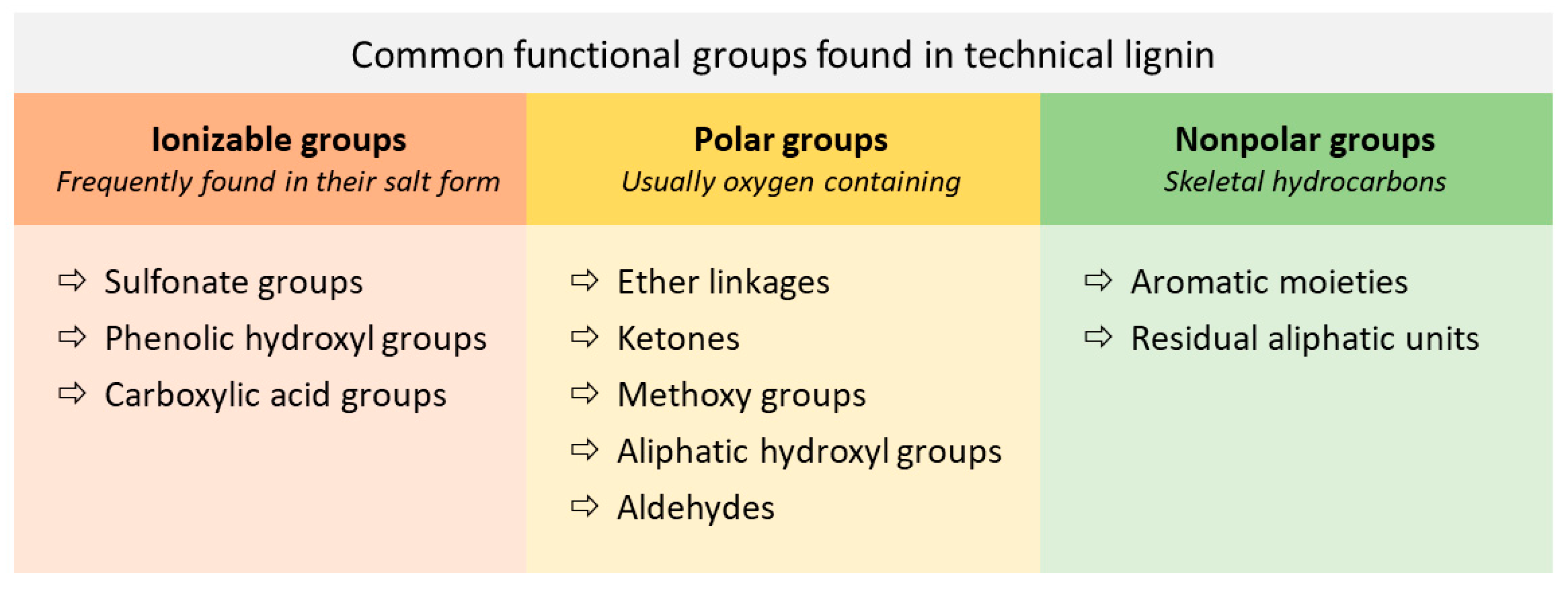
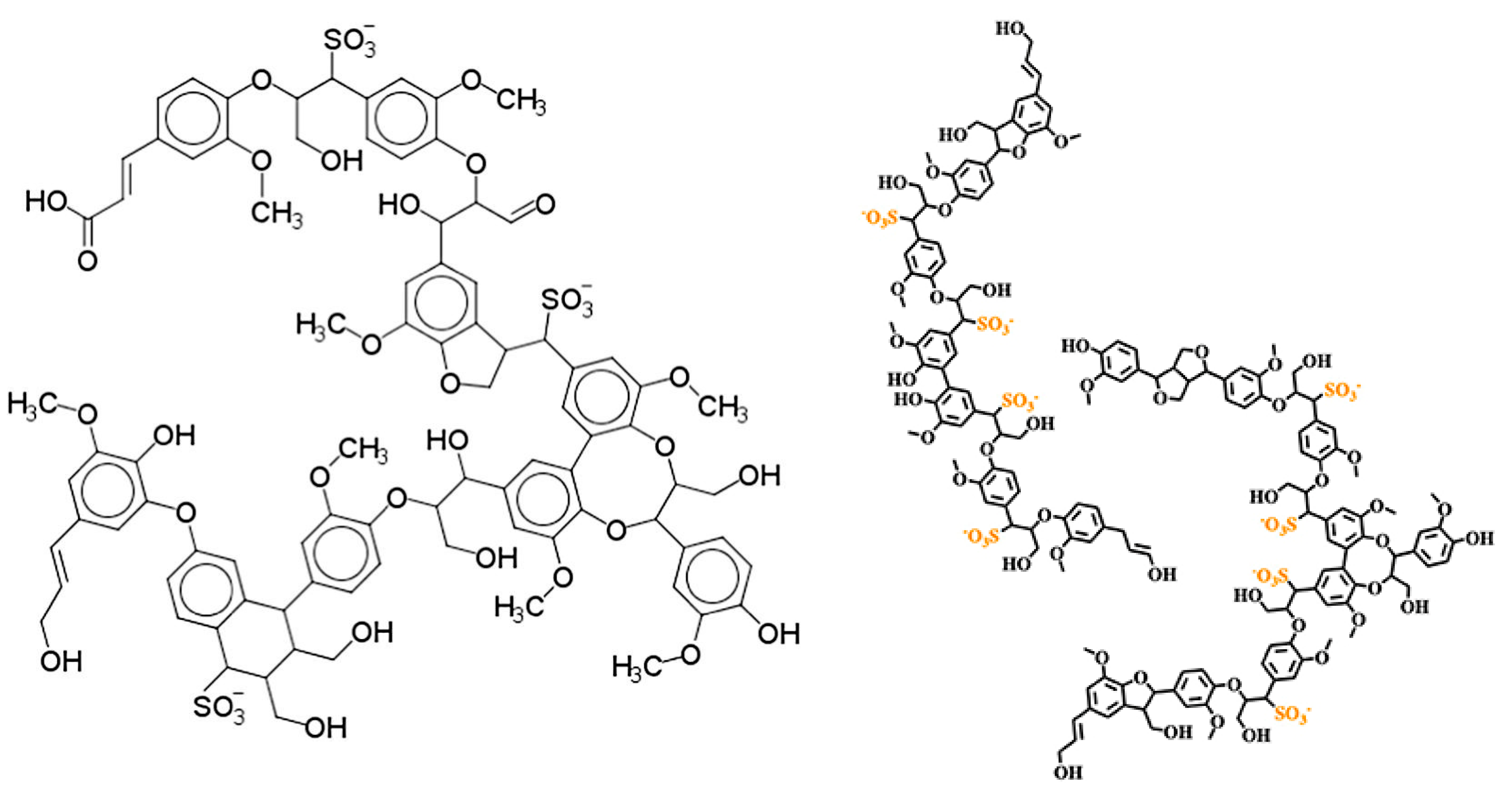
2.2. Chemical Composition and Structure
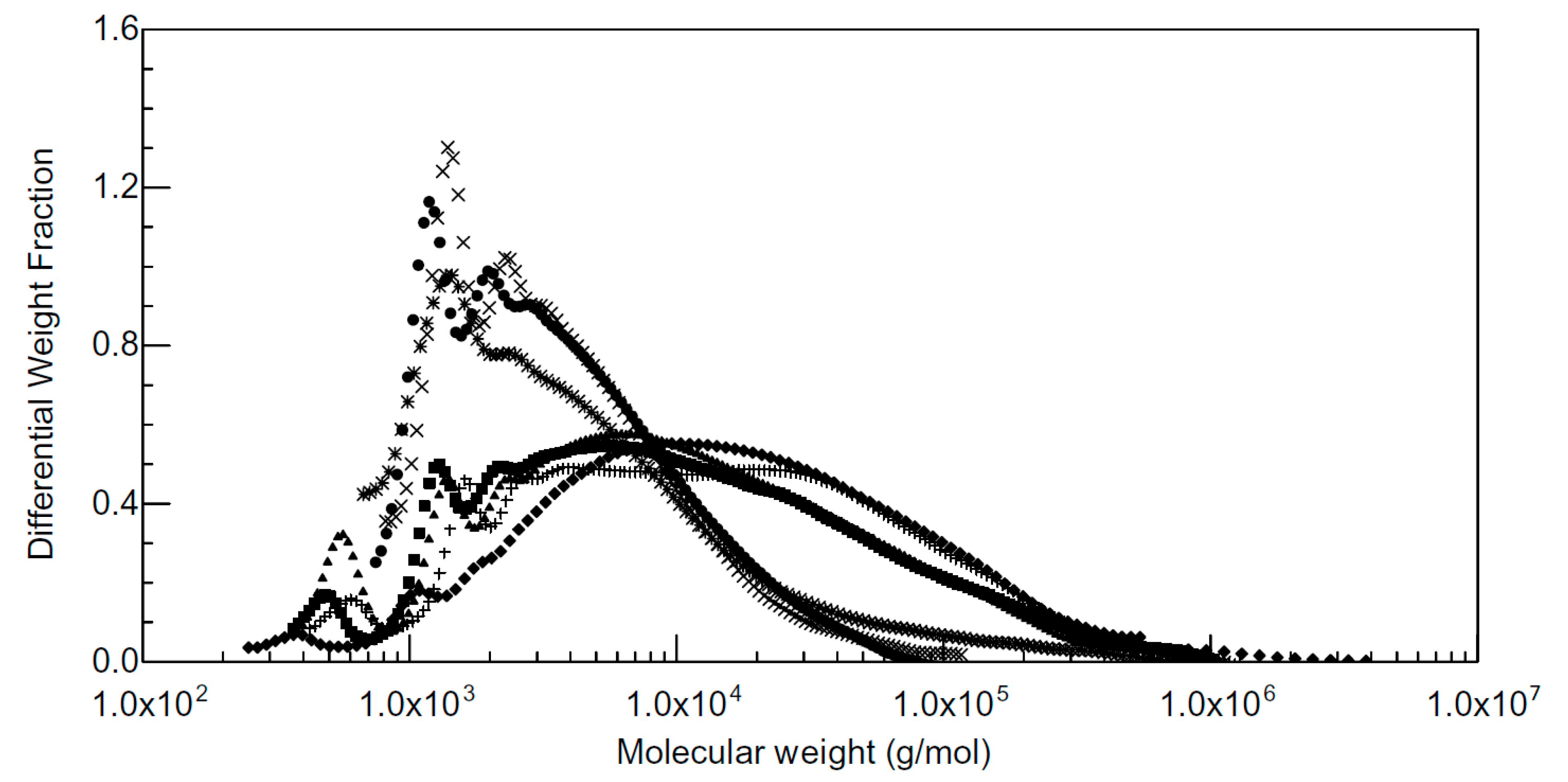
2.3. Analytical Techniques for Lignosulfonate Characterization
2.4. Fractionation
3. Physicochemical Properties of Lignosulfonates
3.1. Solubility in Different Solvents
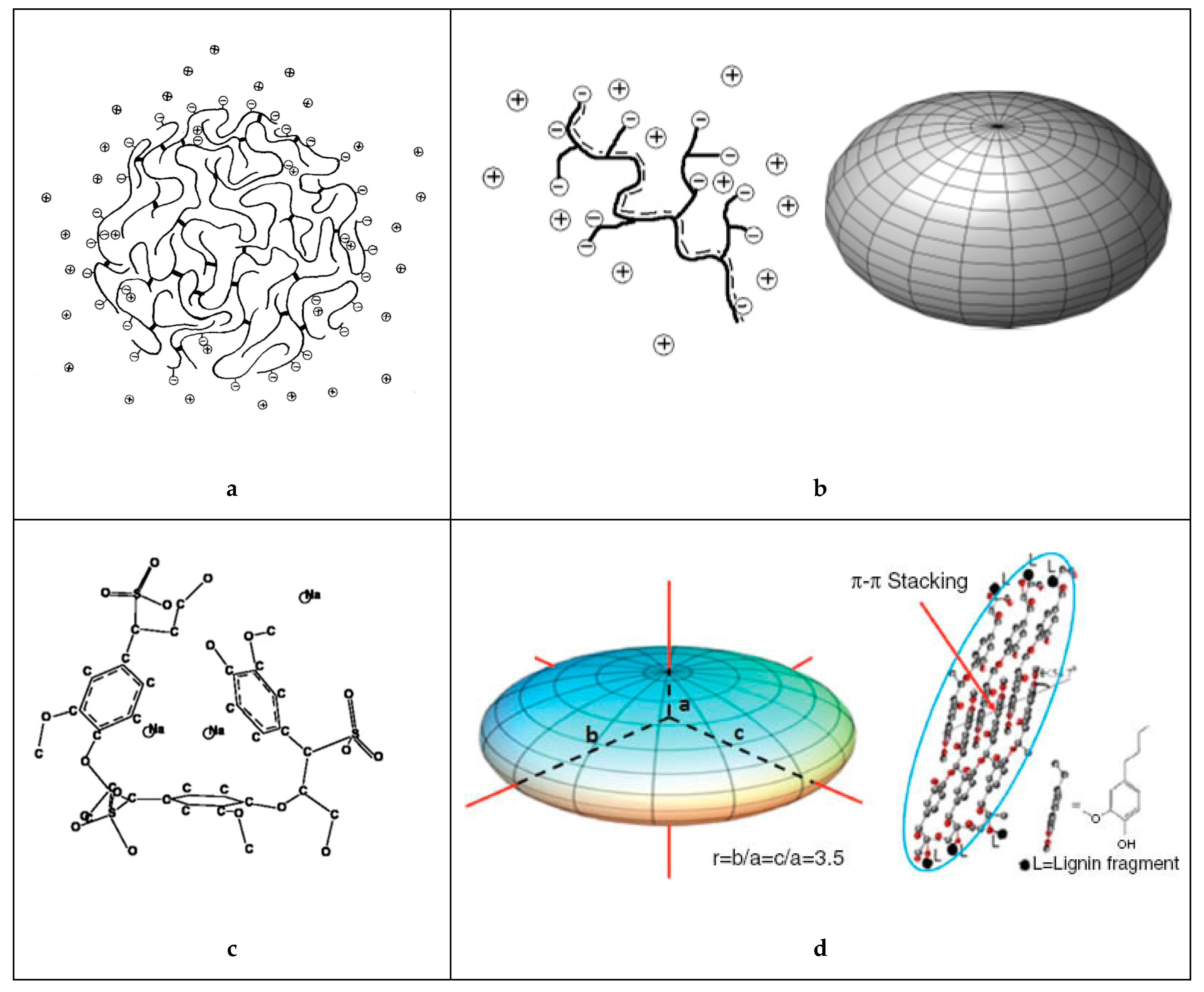
3.2. Conformation and Shape in Aqueous Solution
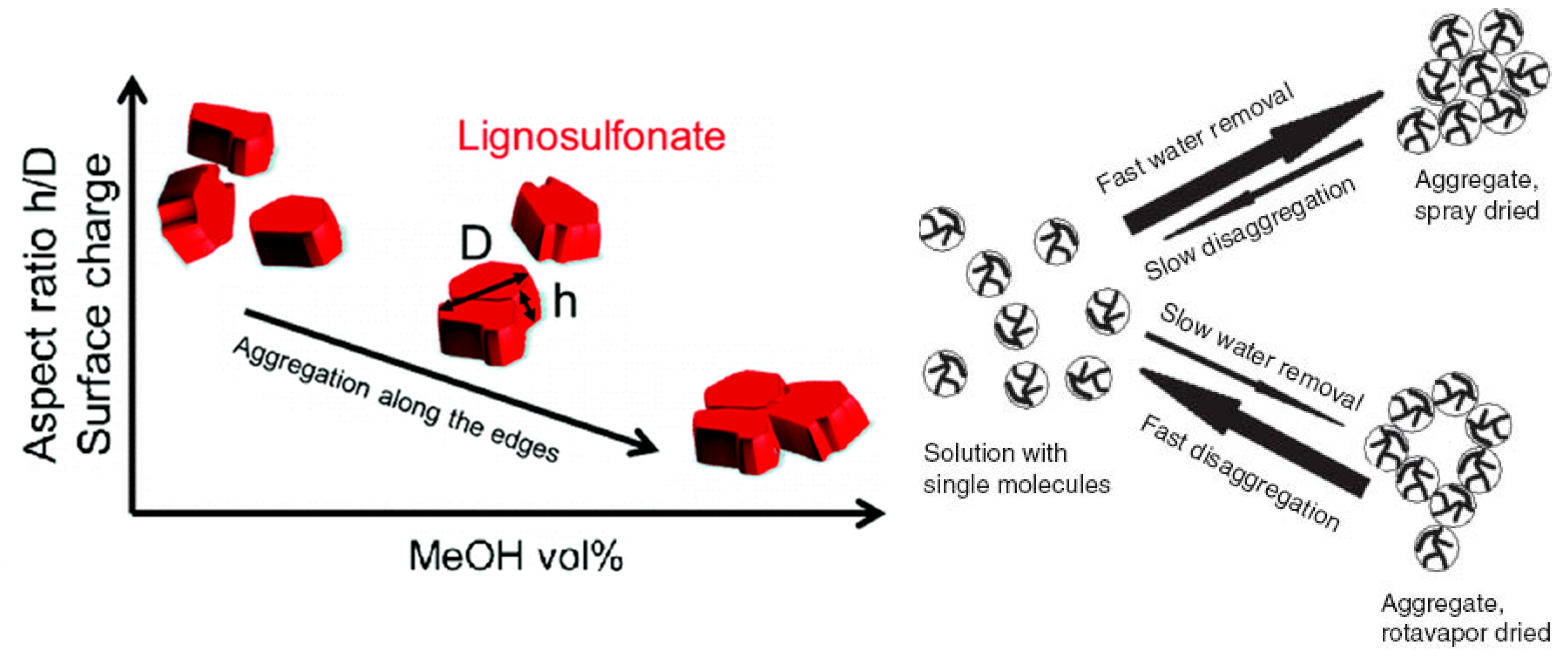
3.3. Self-Association and Agglomeration in Aqueous Solution
3.4. Precipitation and Gelling in Aqueous Solution
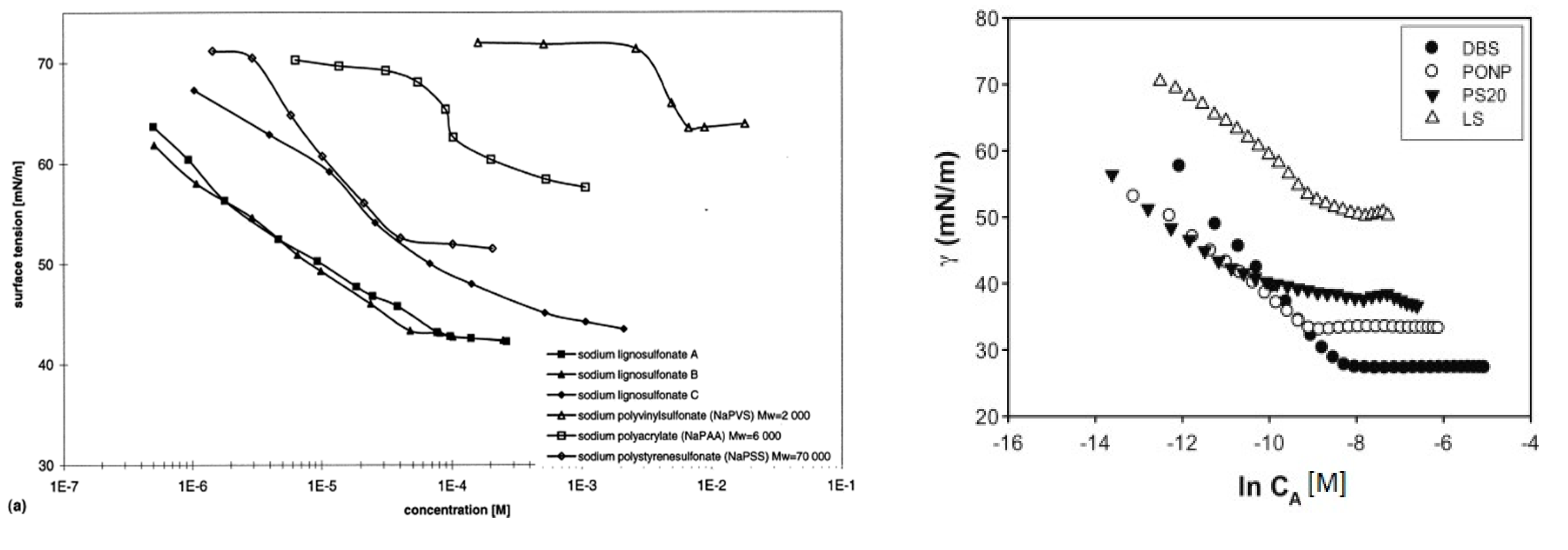
3.5. Adsorption at Surfaces and Interfaces
3.6. Interactions with other Components
3.7. Relationship between the Chemical Make-Up and the Physicochemical Behavior
3.8. Chemical Modification
- Modulation of the physicochemical properties, for example for improving the dispersant performance and compatibility. This can be done by addition or substitution of functional groups, but polymerization and depolymerization may be effective measures as well.
- Improving the use in and compatibility with polymer formulations, for example by altering the abundance and accessibility of certain functional groups. Lignosulfonate may be incorporated as a quasi-monomer it into a larger polymer matrix. Use as filler material by blending the modified lignin with thermoplastic materials has also been demonstrated.
4. Summary and Conclusions
Funding
Conflicts of Interest
References
- Aro, T.; Fatehi, P. Production and Application of Lignosulfonates and Sulfonated Lignin. ChemSusChem 2017, 10, 1861–1877. [Google Scholar] [CrossRef] [PubMed]
- Macfarlane, A.; Mai, M.; Kadla, J.F. Bio-based chemicals from biorefining: Lignin conversion and utilisation. In Advances in Biorefineries; Waldron, K., Ed.; Woodhead Publishing: Sawston, UK, 2014; pp. 659–692. [Google Scholar]
- Stern, T.; Schwarzbauer, P. Wood-based lignosulfonate versus synthetic polycarboxylate in concrete admixture systems: The perspective of a traditional pulping by-product competing with an oil-based substitute in a business-to-business market in central Europe. For. Prod. J. 2008, 58, 81–86. [Google Scholar]
- Lauten, R.A.; Myrvold, B.O.; Gundersen, S.A. New Developments in the Commercial Utilization of Lignosulfonates. Surfactants Renew. Resour. 2010, 269–283. [Google Scholar] [CrossRef]
- Xu, C.; Ferdosian, F. Utilization of Lignosulfonate as Dispersants or Surfactants. In Conversion of Lignin into Bio-Based Chemicals and Materials; Xu, C., Ferdosian, F., Eds.; Springer Berlin Heidelberg: Berlin/Heidelberg, Germany, 2017; pp. 81–90. [Google Scholar]
- Garguiak, J.D.; Lebo, S.E. Commercial Use of Lignin-Based Materials. In Lignin: Historical, Biological, and Materials Perspectives; American Chemical Society: Washington, DC, USA, 1999; Volume 742, pp. 304–320. [Google Scholar]
- Miretzky, P.; Cirelli, A.F. Cr(VI) and Cr(III) removal from aqueous solution by raw and modified lignocellulosic materials: A review. J. Hazard. Mater. 2010, 180, 1–19. [Google Scholar] [CrossRef]
- Alazigha, D.P.; Indraratna, B.; Vinod, J.S.; Heitor, A. Mechanisms of stabilization of expansive soil with lignosulfonate admixture. Transp. Geotech. 2018, 14, 81–92. [Google Scholar] [CrossRef]
- Ruwoldt, J.; Planque, J.; Øye, G. Lignosulfonate Salt Tolerance and the Effect on Emulsion Stability. ACS Omega 2020, 5, 15007–15015. [Google Scholar] [CrossRef]
- Ouyang, X.; Qiu, X.; Lou, H.; Yang, D. Corrosion and Scale Inhibition Properties of Sodium Lignosulfonate and Its Potential Application in Recirculating Cooling Water System. Ind. Eng. Chem. Res. 2006, 45, 5716–5721. [Google Scholar] [CrossRef]
- Blanck, G.; Cuisinier, O.; Masrouri, F. Soil treatment with organic non-traditional additives for the improvement of earthworks. Acta Geotech. 2014, 9, 1111–1122. [Google Scholar] [CrossRef]
- Tsau, J.-S.; Syahputra, A.E.; Yaghoobi, H.; Grigg, R.B. Use of Sacrificial Agents in CO2 Foam Flooding Application. In SPE Annual Technical Conference and Exhibition; Society of Petroleum Engineers: Houston, TX, USA, 1999; p. 9. [Google Scholar]
- Chiwetelu, C. Improving the Oil Recovery Efficacy of Lignosulfonate Solutions. J. Can. Pet. Technol. 1980, 19, 10. [Google Scholar] [CrossRef]
- Vebi, M.; Hassan, A.; Hubert, H.; Markus, B.; Ireen, G.; Robert, B.; Karin, F.; Antje, P.; Thomas, R. Lignosulfonate-based polyurethane materials via cyclic carbonates: Preparation and characterization. Holzforschung 2020, 74, 203–211. [Google Scholar]
- Hirose, S. Novel Epoxy Resins with Unsaturated Ester Chains Derived from Sodium Lignosulfonate. Macromol. Symp. 2015, 353, 31–38. [Google Scholar] [CrossRef]
- Hu, J.-P.; Guo, M.-H. Influence of ammonium lignosulfonate on the mechanical and dimensional properties of wood fiber biocomposites reinforced with polylactic acid. Ind. Crop. Prod. 2015, 78, 48–57. [Google Scholar] [CrossRef]
- Sixta, H. Handbook of Pulp; Wiley-vch: Weinheim, Germany, 2006. [Google Scholar]
- Konduri, M.K.R.; Fatehi, P. Production of Water-Soluble Hardwood Kraft Lignin via Sulfomethylation Using Formaldehyde and Sodium Sulfite. ACS Sustain. Chem. Eng. 2015, 3, 1172–1182. [Google Scholar] [CrossRef]
- Ouyang, X.; Ke, L.; Qiu, X.; Guo, Y.; Pang, Y. Sulfonation of Alkali Lignin and Its Potential Use in Dispersant for Cement. J. Dispers. Sci. Technol. 2009, 30, 1–6. [Google Scholar] [CrossRef]
- Huang, C.; Ma, J.; Zhang, W.; Huang, G.; Yong, Q. Preparation of Lignosulfonates from Biorefinery Lignins by Sulfomethylation and Their Application as a Water Reducer for Concrete. Polymers 2018, 10, 841. [Google Scholar] [CrossRef] [PubMed]
- Myrvold, B.O. A new model for the structure of lignosulphonates: Part 1. Behaviour in dilute solutions. Ind. Crop. Prod. 2008, 27, 214–219. [Google Scholar] [CrossRef]
- Li, T.; Takkellapati, S. The current and emerging sources of technical lignins and their applications. Biofuels, Bioprod. Biorefining 2018, 12, 756–787. [Google Scholar] [CrossRef]
- Upton, B.M.; Kasko, A.M. Strategies for the Conversion of Lignin to High-Value Polymeric Materials: Review and Perspective. Chem. Rev. 2016, 116, 2275–2306. [Google Scholar] [CrossRef]
- Chio, C.; Sain, M.; Qin, W. Lignin utilization: A review of lignin depolymerization from various aspects. Renew. Sustain. Energy Rev. 2019, 107, 232–249. [Google Scholar] [CrossRef]
- Szalaty, T.J.; Klapiszewski, Ł.; Jesionowski, T. Recent developments in modification of lignin using ionic liquids for the fabrication of advanced materials—A review. J. Mol. Liq. 2020, 301, 112417. [Google Scholar] [CrossRef]
- Laurichesse, S.; Avérous, L. Chemical modification of lignins: Towards biobased polymers. Prog. Polym. Sci. 2014, 39, 1266–1290. [Google Scholar] [CrossRef]
- Jacquet, N.; Maniet, G.; Vanderghem, C.; Delvigne, F.; Richel, A. Application of Steam Explosion as Pretreatment on Lignocellulosic Material: A Review. Ind. Eng. Chem. Res. 2015, 54, 2593–2598. [Google Scholar] [CrossRef]
- An, L.; Wang, G.; Jia, H.; Liu, C.; Sui, W.; Si, C. Fractionation of enzymatic hydrolysis lignin by sequential extraction for enhancing antioxidant performance. Int. J. Biol. Macromol. 2017, 99, 674–681. [Google Scholar] [CrossRef] [PubMed]
- Daintith, J. Surfactant; Oxford University Press: Oxford, UK, 2008. [Google Scholar]
- Paria, S.; Khilar, K.C. A review on experimental studies of surfactant adsorption at the hydrophilic solid–water interface. Adv. Colloid Interface Sci. 2004, 110, 75–95. [Google Scholar] [CrossRef] [PubMed]
- Boerjan, W.; Ralph, J.; Baucher, M. Lignin Biosynthesis. Annu. Rev. Plant Biol. 2003, 54, 519–546. [Google Scholar] [CrossRef] [PubMed]
- Qiu, X.; Yan, M.; Yang, D.; Pang, Y.; Deng, Y. Effect of straight-chain alcohols on the physicochemical properties of calcium lignosulfonate. J. Colloid Interface Sci. 2009, 338, 151–155. [Google Scholar] [CrossRef] [PubMed]
- Ekeberg, D.; Gretland, K.S.; Gustafsson, J.; Bråten, S.M.; Fredheim, G.E. Characterisation of lignosulphonates and kraft lignin by hydrophobic interaction chromatography. Anal. Chim. Acta 2006, 565, 121–128. [Google Scholar] [CrossRef]
- Vishtal, A.G.; Kraslawski, A. Challenges in industrial applications of technical lignins. BioResources 2011, 6, 3547–3568. [Google Scholar]
- Fredheim, G.E.; Christensen, B.E.; Braaten, S.M. Comparison of Molecular Weight and Molecular Weight Distributions of Softwood and Hardwood Lignosulfonates. J. Wood Chem. Technol. 2003, 23, 197–215. [Google Scholar]
- Windeisen, E.; Wegener, G. 10.15—Lignin as Building Unit for Polymers. In Polymer Science: A Comprehensive Reference; Matyjaszewski, K., Möller, M., Eds.; Elsevier: Amsterdam, The Netherlands, 2012; pp. 255–265. [Google Scholar]
- Myrvold, B.O. Free radical gelling reactions of lignosulfonates. Holzforschung 2015, 69, 1089–1096. [Google Scholar] [CrossRef]
- Myrvold, B.O. Differences in solubility parameters and susceptibility to salting-out between softwood and hardwood lignosulfonates. Holzforschung 2016, 70, 1015–1021. [Google Scholar] [CrossRef]
- Heitner, C.; Dimmel, D.; Schmidt, J. Lignin and Lignans: Advances in Chemistry; CRC Press: Cleveland, OH, USA, 2016. [Google Scholar]
- Rovio, S.; Kuitunen, S.; Ohra-Aho, T.; Alakurtti, S.; Kalliola, A.; Tamminen, T. Lignin oxidation mechanisms under oxygen delignification conditions. Part 2: Advanced methods for the detailed characterization of lignin oxidation mechanisms. Holzforschung 2011, 65, 575. [Google Scholar] [CrossRef]
- Lachenal, D.; Fernandes, J.; Froment, P. Behaviour of residual lignin in kraft pulp during bleaching. J. Pulp Pap. Sci. 1995, 21, J173. [Google Scholar]
- Kazzaz, A.E.; Feizi, Z.H.; Fatehi, P. Grafting strategies for hydroxy groups of lignin for producing materials. Green Chem. 2019, 21, 5714–5752. [Google Scholar] [CrossRef]
- Myrvold, B.O. The Hansen solubility parameters of some lignosulfonates. World Acad. Sci. Eng. Technol. Trans. Energy Power Eng. 2014, 1, 261. [Google Scholar]
- Valencia, P.J.S.; Marinez, L.E.B.; Merchancano, S.T.P. Molecular Modeling of Ammonium, Calcium, Sulfur, and Sodium Lignosulphonates in Acid and Basic Aqueous Environments. Braz. J. Phys. 2015, 45, 567–574. [Google Scholar] [CrossRef]
- Laughlin, R.G. HLB, from a thermodynamic perspective. J. Soc. Cosmet. Chem. 1981, 32, 371–392. [Google Scholar]
- Ensing, B.; Tiwari, A.; Tros, M.; Hunger, J.; Domingos, S.R.; Pérez, C.; Smits, G.; Bonn, M.; Bonn, D.; Woutersen, S. On the origin of the extremely different solubilities of polyethers in water. Nat. Commun. 2019, 10, 1–8. [Google Scholar] [CrossRef]
- Holmberg, K. Natural surfactants. Curr. Opin. Colloid Interface Sci. 2001, 6, 148–159. [Google Scholar] [CrossRef]
- Brauns, F.E.; Brauns, D.A. The Chemistry of Lignin: Supplement Volume Covering the Literature for the Years 1949–1958, 1st ed.; Academic Press: New York, NY, USA, 1960. [Google Scholar]
- Kun, D.; Pukánszky, B. Polymer/lignin blends: Interactions, properties, applications. Eur. Polym. J. 2017, 93, 618–641. [Google Scholar] [CrossRef]
- Fiorani, G.; Crestini, C.; Selva, M.; Perosa, A. Advancements and Complexities in the Conversion of Lignocellulose into Chemicals and Materials. Front. Chem. 2020, 8, 797. [Google Scholar] [CrossRef] [PubMed]
- Fredheim, G.E.; Braaten, S.M.; Christensen, E.B. Molecular weight determination of lignosulfonates by size-exclusion chromatography and multi-angle laser light scattering. J. Chromatogr. A 2002, 942, 191–199. [Google Scholar] [CrossRef]
- Kontturi, A.-K. Diffusion coefficients and effective charge numbers of lignosulphonate. Influence of temperature. J. Chem. Soc. Faraday Trans. 1 Phys. Chem. Condens. Phases 1988, 84, 4043–4047. [Google Scholar] [CrossRef]
- Ruwoldt, J.; Simon, S.; Øye, G. Viscoelastic properties of interfacial lignosulfonate films and the effect of added electrolytes. Colloids Surfaces A: Physicochem. Eng. Asp. 2020, 606, 125478. [Google Scholar] [CrossRef]
- Tolbert, A.; Akinosho, H.; Khunsupat, R.; Naskar, A.K.; Ragauskas, A.J. Characterization and analysis of the molecular weight of lignin for biorefining studies. Biofuels Bioprod. Biorewfining 2014, 8, 836–856. [Google Scholar] [CrossRef]
- El Mansouri, N.-E.; Salvadó, J. Analytical methods for determining functional groups in various technical lignins. Ind. Crop. Prod. 2007, 26, 116–124. [Google Scholar] [CrossRef]
- El Mansouri, N.-E.; Salvadó, J. Structural characterization of technical lignins for the production of adhesives: Application to lignosulfonate, kraft, soda-anthraquinone, organosolv and ethanol process lignins. Ind. Crop. Prod. 2006, 24, 8–16. [Google Scholar] [CrossRef]
- Technical Association of the Pulp and Paper Industry (TAPPI) standard T 222 om-15. In Acid-Insoluble Lignin in Wood and Pulp; TAPPI Press: Atlanta, GA, USA, 2015.
- Sluiter, A.; Hames, B.; Ruiz, R.; Scarlata, C.; Sluiter, J.; Templeton, D.; Crocker, D. Determination of structural carbohydrates and lignin in biomass. Lab. Anal. Proced. 2008, 1617, 1–16. [Google Scholar]
- Maekawa, E.; Ichizawa, T.; Koshijima, T. An Evaluation of the Acid-Soluble Lignin Determination in Analyses of Lignin by the Sulfuric Acid Method. J. Wood Chem. Technol. 1989, 9, 549–567. [Google Scholar] [CrossRef]
- Gosselink, R.J.A.; Abächerli, A.; Semke, H.; Malherbe, R.; Käuper, P.; Nadif, A.; Van Dam, J. Analytical protocols for characterisation of sulphur-free lignin. Ind. Crop. Prod. 2004, 19, 271–281. [Google Scholar] [CrossRef]
- Faix, O. Fourier Transform Infrared Spectroscopy. In Methods in Lignin Chemistry; Lin, S.Y., Dence, C.W., Eds.; Springer: Berlin/Heidelberg, Germany, 1992; pp. 83–109. [Google Scholar]
- Boeriu, C.G.; Bravo, D.; Gosselink, R.J.; Van Dam, J.E. Characterisation of structure-dependent functional properties of lignin with infrared spectroscopy. Ind. Crop. Prod. 2004, 20, 205–218. [Google Scholar] [CrossRef]
- Capanema, E.A.; Balakshin, A.M.Y.; Kadla, J.F. A Comprehensive Approach for Quantitative Lignin Characterization by NMR Spectroscopy. J. Agric. Food Chem. 2004, 52, 1850–1860. [Google Scholar] [CrossRef] [PubMed]
- Pu, Y.; Cao, S.; Ragauskas, A.J. Application of quantitative 31P NMR in biomass lignin and biofuel precursors characterization. Energy Environ. Sci. 2011, 4, 3154–3166. [Google Scholar] [CrossRef]
- Gupta, P.R.; McCarthy, J.L. Lignin. X.I.V. Gel Chromatography and the Distribution in Molecular Size of Lignin Sulfonates at Several Electrolyte Concentrations. Macromology 1968, 1, 236–244. [Google Scholar] [CrossRef]
- Siochi, E.J.; Ward, T.C.; Haney, M.A.; Mahn, B. The absolute molecular weight distribution of hydroxypropylated lignins. Macromology 1990, 23, 1420–1429. [Google Scholar] [CrossRef]
- Duval, A.; Molina-Boisseau, S.; Chirat, C. Fractionation of lignosulfonates: Comparison of ultrafiltration and ethanol solubility to obtain a set of fractions with distinct properties. Holzforschung 2015, 69, 127–134. [Google Scholar] [CrossRef]
- Evtuguin, D.; Domingues, P.; Amado, F.L.; Neto, C.P.; Correia, A.F. Electrospray Ionization Mass Spectrometry as a Tool for Lignins Molecular Weight and Structural Characterisation. Holzforschung 1999, 53, 525–528. [Google Scholar] [CrossRef]
- Qian, Y.; Deng, Y.; Guo, Y.; Yi, C.; Qiu, X. Determination of absolute molecular weight of sodium lignosulfonates (NaLS) by laser light scattering (LLS). Holzforschung 2013, 67, 265–271. [Google Scholar] [CrossRef]
- Shen, Q.; Fu, Z.; Li, R.; Wu, Y. A study on the pyrolysis mechanism of a β-O-4 lignin dimer model compound using DFT combined with Py-GC/MS. J. Therm. Anal. Calorim. 2020, 1–11. [Google Scholar] [CrossRef]
- Brudin, S.; Schoenmakers, P. Analytical methodology for sulfonated lignins. J. Sep. Sci. 2010, 33, 439–452. [Google Scholar] [CrossRef]
- Lebo, S.E.; Bråten, S.M.; Fredheim, G.E.; Lutnaes, B.F.; Lauten, R.A.; Myrvold, B.O.; McNally, T.J. Recent Advances in the Characterization of Lignosulfonates. In Characterization of Lignocellulosic Materials; Hu, T.Q., Ed.; Blackwell Publishing Ltd.: Hoboken, NJ, USA, 2008. [Google Scholar]
- Sulaeva, I.; Vejdovszky, P.; Henniges, U.; Mahler, A.K.; Rosenau, T.; Potthast, A. Molar Mass Characterization of Crude Lignosulfonates by Asymmetric Flow Field-Flow Fractionation. ACS Sustain. Chem. Eng. 2018, 7, 216–223. [Google Scholar] [CrossRef]
- Winowiski, T.; Lebo, S.; Gretland, K.; Gustafsson, J. Characterization of Sulfonated Lignin Dispersants by Hydrophobic Interactive Chromatography. J. ASTM Int. 2005, 2, 12915. [Google Scholar] [CrossRef]
- Musl, O.; Sulaeva, I.; Bacher, M.; Mahler, A.K.; Rosenau, T.; Potthast, A. Hydrophobic Interaction Chromatography (HIC) in 2D-LC Characterization of Lignosulfonates. ChemSusChem 2020, 13, 4595–4604. [Google Scholar] [CrossRef] [PubMed]
- Korntner, P.; Schedl, A.; Sumerskii, I.; Zweckmair, T.; Mahler, A.K.; Rosenau, T.; Potthast, A. Sulfonic Acid Group Determination in Lignosulfonates by Headspace Gas Chromatography. ACS Sustain. Chem. Eng. 2018, 6, 6240–6246. [Google Scholar] [CrossRef]
- Grigg, R.; Bai, B. Calcium lignosulfonate adsorption and desorption on Berea sandstone. J. Colloid Interface Sci. 2004, 279, 36–45. [Google Scholar] [CrossRef] [PubMed]
- Ratinac, K.R.; Standard, O.; Bryant, P. Lignosulfonate adsorption on and stabilization of lead zirconate titanate in aqueous suspension. J. Colloid Interface Sci. 2004, 273, 442–454. [Google Scholar] [CrossRef]
- Nanthakumar, B.; Arinaitwe, E.; Pawlik, M. Adsorption of sodium lignosulfonates on hematite. Adsorption 2010, 16, 447–455. [Google Scholar] [CrossRef]
- Ge, Y.; Li, Z.; Pang, Y.; Qiu, X. Influence of molecular mass of lignosulfonates on the resulting surface charges of solid particles. Int. J. Biol. Macromol. 2013, 52, 300–304. [Google Scholar] [CrossRef]
- Liu, D.; Peng, Y. Understanding different roles of lignosulfonate in dispersing clay minerals in coal flotation using deionised water and saline water. Fuel 2015, 142, 235–242. [Google Scholar] [CrossRef]
- Tang, Q.; Zhou, M.; Yang, D.; Qiu, X. Effects of pH on aggregation behavior of sodium lignosulfonate (NaLS) in concentrated solutions. J. Polym. Res. 2015, 22, 50. [Google Scholar] [CrossRef]
- Vainio, U.; Lauten, R.A.; Serimaa, R. Small-Angle X-ray Scattering and Rheological Characterization of Aqueous Lignosulfonate Solutions. Langmuir 2008, 24, 7735–7743. [Google Scholar] [CrossRef] [PubMed]
- Qian, Y.; Deng, Y.; Yi, C.; Yu, H.; Qiu, X. Solution Behaviors and Adsorption Characteristics of Sodium Lignosulfonate Under Different Ph Conditions. BioResources 2011, 6, 4686–4695. [Google Scholar]
- Vainio, U.; Lauten, R.A.; Haas, S.; Svedström, K.; Veiga, L.S.I.; Hoell, A.; Serimaa, R. Distribution of Counterions around Lignosulfonate Macromolecules in Different Polar Solvent Mixtures. Langmuir 2012, 28, 2465–2475. [Google Scholar] [CrossRef] [PubMed]
- Yan, M.; Yang, D.; Deng, Y.; Chen, P.; Zhou, H.; Qiu, X. Influence of pH on the behavior of lignosulfonate macromolecules in aqueous solution. Colloids Surf. A Physicochem. Eng. Asp. 2010, 371, 50–58. [Google Scholar] [CrossRef]
- Qin, Y.; Qiu, X.; Liang, W.; Yang, D. Investigation of Adsorption Characteristics of Sodium Lignosulfonate on the Surface of Disperse Dye Using a Quartz Crystal Microbalance with Dissipation. Ind. Eng. Chem. Res. 2015, 54, 12313–12319. [Google Scholar] [CrossRef]
- Wang, Y. Adsorption and Inhibition Behavior of Calcium Lignosulfonate on Steel in NaCl + Ca(OH)2 Solutions with Different pH Values. Int. J. Electrochem. Sci. 2016, 11, 6976–6992. [Google Scholar] [CrossRef]
- Deng, Y.; Zhang, W.; Wu, Y.; Yu, H.; Qiu, X. Effect of Molecular Weight on the Adsorption Characteristics of Lignosulfonates. J. Phys. Chem. B 2011, 115, 14866–14873. [Google Scholar] [CrossRef]
- Ge, Y.; Li, D.; Li, Z. Effects of Lignosulfonate Structure on the Surface Activity and Wettability to a Hydrophobic Powder. Bioresources 2014, 9, 9. [Google Scholar] [CrossRef]
- Pita, F.; Castilho, A.M. Plastics floatability: Effect of saponin and sodium lignosulfonate as wetting agents. Polímeros 2019, 29, 29. [Google Scholar] [CrossRef]
- Askvik, K.M. Complexation of Lignosulfonates with Multivalent Cations and Cationic Surfactants, and the Impact on Emulsion Stability. Ph.D. Thesis, University of Bergen, Bergen, Norway, 2000. [Google Scholar]
- Megiatto, J.D.; Cerrutti, B.M.; Frollini, E. Sodium lignosulfonate as a renewable stabilizing agent for aqueous alumina suspensions. Int. J. Biol. Macromol. 2016, 82, 927–932. [Google Scholar] [CrossRef]
- Askvik, K.M.; Gundersen, S.A.; Sjöblom, J.; Merta, J.; Stenius, P. Complexation between lignosulfonates and cationic surfactants and its influence on emulsion and foam stability. Colloids Surfaces A: Physicochem. Eng. Asp. 1999, 159, 89–101. [Google Scholar] [CrossRef]
- Yang, D.; Qiu, X.; Pang, Y.; Zhou, M. Physicochemical Properties of Calcium Lignosulfonate with Different Molecular Weights as Dispersant in Aqueous Suspension. J. Dispers. Sci. Technol. 2008, 29, 1296–1303. [Google Scholar] [CrossRef]
- Rana, D.; Neale, G.; Hornof, V. Surface tension of mixed surfactant systems: Lignosulfonate and sodium dodecyl sulfate. Colloid Polym. Sci. 2002, 280, 775–778. [Google Scholar] [CrossRef]
- Park, S.; Lee, E.S.; Sulaiman, W.R.W. Adsorption behaviors of surfactants for chemical flooding in enhanced oil recovery. J. Ind. Eng. Chem. 2015, 21, 1239–1245. [Google Scholar] [CrossRef]
- Ruwoldt, J.; Øye, G. Effect of Low-Molecular-Weight Alcohols on Emulsion Stabilization with Lignosulfonates. ACS Omega 2020, 5, 30168–30175. [Google Scholar] [CrossRef]
- Gundersen, S. Langmuir surface and interface films of lignosulfonates and Kraft lignins in the presence of electrolyte and asphaltenes: Correlation to emulsion stability. Colloids Surf. A Physicochem. Eng. Asp. 2001, 182, 199–218. [Google Scholar] [CrossRef]
- Okko, R.; Bodo, S.; Ralph, L. Isolation and fractionation of lignosulfonates by amine extraction and ultrafiltration: A comparative study. Holzforschung 2005, 59, 405–412. [Google Scholar]
- Kienberger, M.; Demmelmayer, P.; Weißl, M.; Zankl, A.; Spirk, S. Biobased Support Layers for the Fractionation and Selective Extraction of Lignosulfonates. Solvent Extr. Ion Exch. 2019, 38, 132–141. [Google Scholar] [CrossRef]
- Qian, C.; Fang, H.; Cui, P.; Cai, F.; Gao, X.; He, H.; Hu, X. Rapid determination of lignosulfonate depolymerization products by advanced polymer chromatography. J. Sep. Sci. 2019, 42, 2289–2297. [Google Scholar] [CrossRef]
- Ouyang, X.P.; Zhang, P.; Tan, C.M.; Deng, Y.; Yang, D.J.; Qiu, X. Isolation of lignosulfonate with low polydispersity index. Chin. Chem. Lett. 2010, 21, 1479–1481. [Google Scholar] [CrossRef]
- Ouyang, X.; Zhang, P.; Qiu, X.; Deng, Y.; Chen, P. Lignosulfonate Separation Using Preparative Column Chromatography. Ind. Eng. Chem. Res. 2011, 50, 10792–10799. [Google Scholar] [CrossRef]
- Humpert, D.; Ebrahimi, M.; Czermak, P. Membrane Technology for the Recovery of Lignin: A Review. Membranes 2016, 6, 42. [Google Scholar] [CrossRef] [PubMed]
- Felicetta, V.F.; Ahola, A.; McCarthy, J.L. Lignin. VII. Distribution in Molecular Weight of Certain Lignin Sulfonates1a. J. Am. Chem. Soc. 1956, 78, 1899–1904. [Google Scholar] [CrossRef]
- Leger, C.A.; Chan, F.D.; Schneider, M.H. Fractionation and characterisation of technical ammonium lignosulphonate. BioResources 2010, 5, 2239–2247. [Google Scholar]
- Kontturi, A.-K.; Sundholm, G.; Nielsen, K.M.; Zingales, R.; Vikholm, I.; Urso, F.; Weidlein, J.; Zingaro, R.A. The Extraction and Fractionation of Lignosulfonates with Long Chain Aliphatic Amines. Acta Chem. Scand. 1986, 40, 121–125. [Google Scholar] [CrossRef]
- Kontturi, A.-K.; Kontturi, K.; Niinikoski, P.; Sundholm, G.; Euranto, E.K.; Pettersson, T. Extraction of a Polyelectrolyte Using a Supported Liquid Membrane. I. Choice of a Suitable Carrier--Solvent System. Acta Chem. Scand. 1990, 44, 879–882. [Google Scholar] [CrossRef][Green Version]
- Kontturi, A.-K.; Kontturi, K.; Niinikoski, P.; Sundholm, G. Extraction of a Polyelectrolyte Using a Supported Liquid Membrane, II. Extraction and Fractionation of Lignosulfonate. Acta Chem. Scand. 1990, 44, 883–891. [Google Scholar] [CrossRef]
- Rojas, O.; Salager, J.-L. Surface activity of bagasse lignin derivatives found in the spent liquor of soda pulping plants. Tappi J. 1994, 77, 169–174. [Google Scholar]
- Myrvold, B.O. Salting-out and salting-in experiments with lignosulfonates (LSs). Holzforschung 2013, 67, 549–557. [Google Scholar] [CrossRef]
- Glas, D.; Van Doorslaer, C.; Depuydt, D.; Liebner, F.; Rosenau, T.; Binnemans, K.; De Vos, D.E. Lignin solubility in non-imidazolium ionic liquids. J. Chem. Technol. Biotechnol. 2014, 90, 1821–1826. [Google Scholar] [CrossRef]
- Rezanowich, A.; Goring, D. Polyelectrolyte expansion of a lignin sulfonate microgel. J. Colloid Sci. 1960, 15, 452–471. [Google Scholar] [CrossRef]
- Qian, Y.; Deng, Y.; Guo, Y.; Li, H.; Qiu, X. Light scattering characterization of lignosulfonate structure in saline solutions. Holzforschung 2015, 69, 377–383. [Google Scholar] [CrossRef]
- Myrvold, B.O. The polyelectrolyte behavior of randomly branched lignosulfonates. Tappi J. 2007, 6, 10–14. [Google Scholar]
- Deng, Y.; Wu, Y.; Qian, Y.; Ouyang, X.; Yang, D.; Qiu, X. Adsorption and desorption behaviors of lignosulfonate during the self-assembly of multilayers. BioResources 2010, 5, 1178–1196. [Google Scholar]
- Li, H.; Deng, Y.; Ye, H.; Xiao, L.; Qiu, X. Effect of Temperature on Polyelectrolyte Expansion of Lignosulfonate. Bioresources 2014, 10, 575–587. [Google Scholar] [CrossRef][Green Version]
- Rezanowich, A.; Yean, W.Q.; Goring, D.A.I. High resolution electron microscopy of sodium lignin sulfonate. J. Appl. Polym. Sci. 1964, 8, 1801–1812. [Google Scholar] [CrossRef]
- Deng, Y.; Feng, X.; Yang, D.; Yi, C.; Qiu, X. Pi-Pi stacking of the aromatic groups in lignosulfonates. BioResources 2012, 7, 1145–1156. [Google Scholar]
- Qiu, X.; Kong, Q.; Zhou, M.; Yang, D. Aggregation Behavior of Sodium Lignosulfonate in Water Solution. J. Phys. Chem. B 2010, 114, 15857–15861. [Google Scholar] [CrossRef]
- Myrvold, B.O. Evidence for a very slow disaggregation of lignosulfonates. Holzforschung 2015, 69, 9–16. [Google Scholar] [CrossRef]
- Qian, Y.; Deng, Y.; Qiu, X.; Huang, J.; Yang, D. Aggregation of sodium lignosulfonate above a critical temperature. Holzforschung 2014, 68, 641–647. [Google Scholar] [CrossRef]
- Li, B.; Ouyang, X.P. Structure and properties of Lignosulfonate with different molecular weight isolated by gel column chromatography. Adv. Mater. Res. Trans Tech. Publ. 2012, 554, 2024–2030. [Google Scholar] [CrossRef]
- Zhukova, O.V.; Medvedeva, V.V.; Semchikov, Y.D. Gelation in sodium lignosulfonate solutions in the presence of a hexavalent chromium salt. Russ. J. Appl. Chem. 2008, 81, 2162–2165. [Google Scholar] [CrossRef]
- Felber, B.J. Factors Affecting the Kinetics of Gelation of Lignosulfonate and the Measurement of the Heats of Association of Transition Metal Complexes of Ethylene-Maleic Acid. PhD Thesis, Oklahoma State University, Stillwater, OK, USA, 1980. [Google Scholar]
- Narron, R.; Wolken, G.; Gargulak, J. Accelerated Polymerization of Ammonium Lignosulfonate from Loblolly Pine. For. Prod. J. 2020, 70, 134–142. [Google Scholar]
- Zulfikar, M.A.; Wahyuningrum, D.; Lestari, S. Adsorption of Lignosulfonate Compound from Aqueous Solution onto Chitosan-Silica Beads. Sep. Sci. Technol. 2013, 48, 1391–1401. [Google Scholar] [CrossRef]
- Bai, B.; Grigg, R.B. Kinetics and Equilibria of Calcium Lignosulfonate Adsorption and Desorption onto Limestone. In SPE International Symposium on Oilfield Chemistry; Society of Petroleum Engineers: The Woodlands, TX, USA, 2005; p. 11. [Google Scholar]
- Pang, Y.-X.; Qiu, X.; Yang, D.; Lou, H. Influence of oxidation, hydroxymethylation and sulfomethylation on the physicochemical properties of calcium lignosulfonate. Colloids Surf. A Physicochem. Eng. Asp. 2008, 312, 154–159. [Google Scholar] [CrossRef]
- Yan, M.; Yang, N. Adsorption Mechanism of Lignosulfonate at the Air/Liquid Interface. J. Braz. Chem. Soc. 2015, 26, 555–561. [Google Scholar] [CrossRef]
- Simon, S.; Saadat, M.; Ruwoldt, J.; Dudek, M.; Ellis, R.J.; Øye, G. Lignosulfonates in Crude Oil Processing: Interactions with Asphaltenes at the Oil/Water Interface and Screening of Potential Applications. ACS Omega 2020, 5, 30189–30200. [Google Scholar] [CrossRef]
- Zaki, N.N.; Ahmed, N.S.; Nassar, A.M. Sodium Lignin Sulfonate to Stabilize Heavy Crude Oil-In-Water Emulsions for Pipeline Transportation. Pet. Sci. Technol. 2000, 18, 1175–1193. [Google Scholar] [CrossRef]
- Verruto, V.J.; Le, R.K.; Kilpatrick, P.K. Adsorption and Molecular Rearrangement of Amphoteric Species at Oil−Water Interfaces. J. Phys. Chem. B 2009, 113, 13788–13799. [Google Scholar] [CrossRef]
- Freer, E.M.; Radke, C.J. Relaxation of Asphaltenes at the Toluene/Water Interface: Diffusion Exchange and Surface Rearrangement. J. Adhes. 2004, 80, 481–496. [Google Scholar] [CrossRef]
- Keleşoğlu, S.; Ponce, A.B.; Sørland, G.H.; Simon, S.; Paso, K.; Sjöblom, J. Rheological properties of highly concentrated dense packed layer emulsions (w/o) stabilized by asphaltene. J. Pet. Sci. Eng. 2015, 126, 1–10. [Google Scholar] [CrossRef]
- Ouyang, X.; Deng, Y.; Qian, Y.; Zhang, P.; Qiu, X. Adsorption Characteristics of Lignosulfonates in Salt-Free and Salt-Added Aqueous Solutions. Biomacromolecules 2011, 12, 3313–3320. [Google Scholar] [CrossRef] [PubMed]
- Bai, B.; Wu, Y.; Grigg, R.B. Adsorption and Desorption Kinetics and Equilibrium of Calcium Lignosulfonate on Dolomite Porous Media. J. Phys. Chem. C 2009, 113, 13772–13779. [Google Scholar] [CrossRef]
- Zulfikar, M.A.; Setiyanto, H.; Djajanti, S.D. Effect of temperature and kinetic modelling of lignosulfonate adsorption onto powdered eggshell in batch systems. Songklanakarin J. Sci. Technol. 2013, 35, 1–31. [Google Scholar]
- Klapiszewski, Ł.; Zdarta, J.; Szatkowski, T.; Wysokowski, M.; Nowacka, M.; Szwarc-Rzepka, K.; Bartczak, P.; Siwińska-Stefańska, K.; Ehrlich, H.; Jesionowski, T. Silica/lignosulfonate hybrid materials: Preparation and characterization. Open Chem. 2014, 12, 719–735. [Google Scholar] [CrossRef]
- Li, H.; Huang, G.; An, C.; Zhang, W.-X. Kinetic and equilibrium studies on the adsorption of calcium lignosulfonate from aqueous solution by coal fly ash. Chem. Eng. J. 2012, 200, 275–282. [Google Scholar] [CrossRef]
- Li, J.; Li, H.; Yuan, Z.; Fang, J.; Chang, L.; Zhang, H.; Li, C. Role of sulfonation in lignin-based material for adsorption removal of cationic dyes. Int. J. Biol. Macromol. 2019, 135, 1171–1181. [Google Scholar] [CrossRef]
- Banfill, P.F.G.; Bowen, P.; Flatt, R.; Galmiche, L.; Houst, Y.; Kauppi, A.; Lafuma, F.; Livesey, P.; Mäder, U.; Myrvold, B. Improved superplasticisers for high performance concrete: The Superplast project. In Proceedings of the 12th International Congress on the Chemistry of Cement, Conseil National de Recherches du Canada, Montreal, QC, Canada, 8–13 July 2007; p. fin00344. [Google Scholar]
- Gundersen, S.A. Lignosulfonates and Kraft Lignins as Oil-in-Water Emulsion Stabilizers; Department of Chemistry, University of Bergen: Bergen, Norway, 2000. [Google Scholar]
- Ström, G.; Stenius, P. Formation of complexes, colloids and precipitates in aqueous mixtures of lignin sulphonate and some cationic polymers. Colloids Surf. 1981, 2, 357–371. [Google Scholar] [CrossRef]
- Fredheim, G.E.; Christensen, B.E. Polyelectrolyte Complexes: Interactions between Lignosulfonate and Chitosan. Biomacromolecules 2003, 4, 232–239. [Google Scholar] [CrossRef]
- Luo, H.; Shen, Q.; Ye, F.; Cheng, Y.-F.; Mezgebe, M.; Qin, R.-J. Structure and properties of layer-by-layer self-assembled chitosan/lignosulfonate multilayer film. Mater. Sci. Eng. C 2012, 32, 2001–2006. [Google Scholar] [CrossRef]
- Askvik, K.M.; Hetlesæther, S.; Sjöblom, J.; Stenius, P. Properties of the lignosulfonate–surfactant complex phase. Colloids Surf. A Physicochem. Eng. Asp. 2001, 182, 175–189. [Google Scholar] [CrossRef]
- Manasrah, K.; Neale, G.; Hornof, V. Properties of mixed surfactant solutions containing petroleum sulfonates and lignosulfonates. Cellul. Chem. Technol. 1985, 19, 291–299. [Google Scholar]
- Hornof, V.; Neale, G.; Margeson, J.; Chiwetelu, C. Lignosulfonate-based mixed surfactants for low interfacial tension. Cellul. Chem. Technol. 1984, 18, 297–303. [Google Scholar]
- Hong, S.; Bae, J.; Lewis, G. An Evaluation of Lignosulfonate as a Sacrificial Adsorbate in Surfactant Flooding. SPE Reserv. Eng. 1987, 2, 17–27. [Google Scholar] [CrossRef]
- Hong, S.; Bae, J. Field Experiment of Lignosulfonate Preflushing for Surfactant Adsorption Reduction. SPE Reserv. Eng. 1990, 5, 467–474. [Google Scholar] [CrossRef]
- Nyman, V.; Rose, G.; Ralston, J. The colloidal behaviour of kraft lignin and lignosulfonates. Colloids Surfaces 1986, 21, 125–147. [Google Scholar] [CrossRef]
- De Oliveira, F.; Ramires, E.C.; Frollini, E.; Belgacem, M.N. Lignopolyurethanic materials based on oxypropylated sodium lignosulfonate and castor oil blends. Ind. Crop. Prod. 2015, 72, 77–86. [Google Scholar] [CrossRef]
- Qiu, X.; Zeng, W.; Liang, W.; Xue, Y.; Hong, N.; Li, Y. Sulfobutylated Lignosulfonate with Ultrahigh Sulfonation Degree and Its Dispersion Property in Low-Rank Coal-Water Slurry. J. Dispers. Sci. Technol. 2015, 37, 472–478. [Google Scholar] [CrossRef]
- Matsushita, Y. Conversion of technical lignins to functional materials with retained polymeric properties. J. Wood Sci. 2015, 61, 230–250. [Google Scholar] [CrossRef]
- Wang, Y.-Y.; Meng, X.; Pu, Y.; Ragauskas, A.J. Recent Advances in the Application of Functionalized Lignin in Value-Added Polymeric Materials. Polymers 2020, 12, 2277. [Google Scholar] [CrossRef]
- Thielemans, W.; Wool, R.P. Lignin Esters for Use in Unsaturated Thermosets: Lignin Modification and Solubility Modeling. Biomacromolecules 2005, 6, 1895–1905. [Google Scholar] [CrossRef] [PubMed]
- Gao, W.; Inwood, J.P.W.; Fatehi, P. Sulfonation of Phenolated Kraft Lignin to Produce Water Soluble Products. J. Wood Chem. Technol. 2019, 39, 225–241. [Google Scholar] [CrossRef]
- Konduri, M.K.; Fatehi, P. Alteration in interfacial properties and stability of coal water slurry by lignosulfonate. Powder Technol. 2019, 356, 920–929. [Google Scholar] [CrossRef]
- Tang, Y.; Lin, T.; Ai, S.; Li, Y.; Zhou, R.; Peng, Y. Super and selective adsorption of cationic dyes using carboxylate-modified lignosulfonate by environmentally friendly solvent-free esterification. Int. J. Biol. Macromol. 2020, 159, 98–107. [Google Scholar] [CrossRef] [PubMed]
- Mahmood, N.; Yuan, Z.; Schmidt, J.; Souzanchi, S. Depolymerization of lignins and their applications for the preparation of polyols and rigid polyurethane foams: A review. Renew. Sustain. Energy Rev. 2016, 60, 317–329. [Google Scholar] [CrossRef]
- Yanhua, J.; Weihong, Q.; Zongshi, L.; Lubai, C. A Study on the Modified Lignosulfonate from Lignin. Energy Sources 2004, 26, 409–414. [Google Scholar] [CrossRef]
- Zhou, H.; Yang, D.; Wu, X.; Deng, Y.; Qiu, X. Physicochemical properties of sodium lignosulfonates (NaLS) modified by laccase. Holzforschung 2012, 66, 825–832. [Google Scholar] [CrossRef]
- Zhou, H.; Yang, D.; Qiu, X.; Wu, X.; Li, Y. A novel and efficient polymerization of lignosulfonates by horseradish peroxidase/H2O2 incubation. Appl. Microbiol. Biotechnol. 2013, 97, 10309–10320. [Google Scholar] [CrossRef]
- Kalliola, A.; Asikainen, M.; Talja, R.; Tamminen, T. Experiences of Kraft Lignin Functionalization by Enzymatic and Chemical Oxidation. Bioresources 2014, 9, 7336–7351. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ruwoldt, J. A Critical Review of the Physicochemical Properties of Lignosulfonates: Chemical Structure and Behavior in Aqueous Solution, at Surfaces and Interfaces. Surfaces 2020, 3, 622-648. https://doi.org/10.3390/surfaces3040042
Ruwoldt J. A Critical Review of the Physicochemical Properties of Lignosulfonates: Chemical Structure and Behavior in Aqueous Solution, at Surfaces and Interfaces. Surfaces. 2020; 3(4):622-648. https://doi.org/10.3390/surfaces3040042
Chicago/Turabian StyleRuwoldt, Jost. 2020. "A Critical Review of the Physicochemical Properties of Lignosulfonates: Chemical Structure and Behavior in Aqueous Solution, at Surfaces and Interfaces" Surfaces 3, no. 4: 622-648. https://doi.org/10.3390/surfaces3040042
APA StyleRuwoldt, J. (2020). A Critical Review of the Physicochemical Properties of Lignosulfonates: Chemical Structure and Behavior in Aqueous Solution, at Surfaces and Interfaces. Surfaces, 3(4), 622-648. https://doi.org/10.3390/surfaces3040042