Hydrogen Adsorption on Ru-Encapsulated, -Doped and -Supported Surfaces of C60



1
Department of Materials, Imperial College London, London SW7 2AZ, UK
2
Faculty of Engineering, Environment and Computing, Coventry University, Priory Street, Coventry CV1 5FB, UK
*
Author to whom correspondence should be addressed.
Surfaces 2020, 3(3), 408-422; https://doi.org/10.3390/surfaces3030030
Submission received: 5 June 2020
/
Revised: 25 July 2020
/
Accepted: 27 July 2020
/
Published: 19 August 2020
(This article belongs to the Special Issue Theory of Hetero-Interfaces and Surfaces: From Fundamentals to Applications)
Abstract
:Hydrogen is considered as one of the promising clean energy sources for future applications including transportation. Nevertheless, the development of materials for its storage is challenging particularly as a fuel in vehicular transport. In the present study, density functional theory simulations for hydrogen adsorption on the surfaces of pristine, Ru-encapsulated, -doped and -supported C60 are reported. The results show that adsorption on the pristine C60 is exoergic and there is an enhancement in the adsorption upon encapsulation of a single Ru atom. The Ru-doped surface also adsorbs H2 more strongly than the pristine surface, but its efficacy is slightly less than the Ru-encapsulated surface. The strongest adsorption is calculated for the C60 surface supported with Ru.

1. Introduction
Hydrogen is a promising alternative to the currently-used fossil fuels mainly due to its high energy efficiency and lower environmental burden in comparison with classical fuels such as gasoline [1,2,3,4]. Its technological applications are limited owing to its low volumetric energy density [5]. The storage of hydrogen in a suitable material with large quantities is a key challenge and considerable research activity has been devoted to this area both experimentally and theoretically [6,7,8,9].
A variety of materials including carbon nanomaterials [10,11,12], zeolites [13] and metal organic frameworks [14] have been examined for the adsorption of hydrogen. The requirements imposed on candidate host materials are cheap, non-toxic and thermodynamically, chemically and mechanically stable.
Buckyball structured fullerenes (C60) are candidate materials for the adsorption of hydrogen as they have open structures and are chemically inert [15,16]. Furthermore, they also have very high mechanical stability at higher pressures and temperatures [17]. The pristine surface of C60 cannot lead to the efficient adsorption of hydrogen as it is chemically less active. Surface modification is one of the strategies to improve the adsorption of atoms and molecules because such modification can alter the chemical nature of the surface. Surface modification via metal adsorption or doping has been considered both experimentally and theoretically for the adsorption of atoms and molecules in order to use the modified surface as a catalyst support [18,19,20,21,22,23,24,25,26,27,28]. A theoretical study by Shin et al. [29] demonstrates that Ni-dispersed fullerenes can store hydrogen exceeding the department of energy’s (DOE) target level of 6.5 wt % [30]. However, there is no experimental verification to validate this. Calcium was considered as a coating metal on the surface of the C60 fullerene in a theoretical study by Yoon et al. [31] and it was shown that a hydrogen uptake of > 8.4% is possible. Li-coated C60 was considered by Sun et al. [32] to show that about 120 hydrogen molecules can bind on the surface of Li12C60. Kaiser et al. [33] performed a combined quantum mechanical and experimental study to show that the Cs-doped C60 is a promising candidate and its adsorption efficacy is related to the significant charge transfer between Cs and C60. In an experimental study by Saha et al. [34], it is shown that there is an enhancement in the hydrogen adsorption on the surfaces of Pd-doped C60 (0.85 wt %) and Ru-doped C60 (0.69 wt %) compared to the adsorption on the surface of pristine C60 (0.3 wt %) though the department of energy (DOE) target for automobile application is 6.5 wt % [30]. There is no theoretical study reported to verify these experimental results on the enhancement upon doping. Furthermore, modification of the C60 surface via encapsulation and the deposition of metals is also worth investigating.
In this work, using density functional theory with dispersion correction (DFT+D), we examine the efficacy of pristine C60 and its modified forms (Ru-encapsulated, -doped and -supported) for the adsorption of hydrogen. The choice of Ru is based on its open-shell configuration (d7s1) that can interact with C60 via electron transfer and its catalytic activity in many useful reactions [35,36,37]. The current computational modelling technique provides insights into the nature of adsorption together with energy minimized structures and host–guest charge transfer.
2. Computational Methods
Electronic structure calculations were performed using DFT simulations as implemented in the VASP (Vienna ab initio simulation program) code [38] which solves standard Kohn–Sham (KS) equations using plane wave basis sets and projected augmented wave (PAW) potentials [39]. In all calculations, we used a plane wave basis set with a cut-off of 500 eV. The exchange correlation was described using the generalized gradient approximation (GGA) as parameterized by Perdew, Burke and Ernzerhof (PBE) [40]. Geometry optimizations were performed using a conjugate gradient algorithm [41]. Forces on the atoms were smaller than 0.001 eV/Å in relaxed configurations. A single k-point was used to optimize C60, its derivatives and molecular hydrogen (H2). Bulk ruthenium was optimized using a 4 × 4 × 8 Monk-horst [42] k-point mesh.
Here, C60, its derivatives and H2 were treated as molecules. A cubic supercell with a dimension of 15 Å × 15 Å × 15 Å was used to ensure that adjacent molecules do not interact each other. Dispersive interactions were modelled using a semi-empirical scheme (DFT+D3) as implemented by Grimme et al. [43].
Adsorption energy for a single H atom on the surface of C60 was calculated using the following equation.
where EH_C60 is the total energy of a single hydrogen atom adsorbed on the surface of C60, EC60 is the total energy of a C60 molecule and EH is the energy of an isolated gas phase hydrogen atom.
Eads = EH_C60 − EC60 − EH
Encapsulation energy of a single Ru atom within C60 was calculated using the following equation.
where ERu@C60 is the total energy of a single ruthenium encapsulated within C60, EC60 is the total energy of a C60 molecule and ERu is the energy of an isolated gas phase ruthenium atom.
Eenc = ERu@C60 − EC60 − ERu
3. Results and Discussion
3.1. Validation of Computational Parameters
In this section, we discuss the efficacy of the pseudopotentials and basis sets used in this study by making comparisons between calculated and available experimental structural parameters for bulk Ru, C60 and the H2 molecule.
First, bulk ruthenium which crystalizes into a hexagonal closed pack structure with the space group P63/mmc (no. 194) [44], was relaxed under constant pressure. From Table 1 it is observed that there is an excellent agreement between the calculated and experimental lattice parameters.
Next, we considered the optimization of a C60 molecule. The calculated bond lengths of C–C and C=C (1.45 Å and 1.40 Å), were in excellent agreement with the corresponding experimental values (1.43 Å and 1.39 Å) [45].
Finally, molecular hydrogen was optimized and its bond length was compared with the experimental value reported by Olmsted et al. [46]. There is excellent agreement between the calculated (0.75 Å) and experimental values (0.74 Å).
3.2. Adsorption of Hydrogen on the Surface of Pristine C60
Here we discuss the adsorption of atomic and molecular hydrogen using on the surface of pristine C60. First the thermodynamic stability of atomic hydrogen on the pristine C60 surface was examined by considering five different initial sites as shown in the Figure 1. The Hydrogen atom occupying the top of the hexagonal ring, the pentagonal ring and the carbon atom on the surface are denoted by H, P and C, respectively. The sites 65 and 66 show the positions of the H atom on top of the bonds bridging hexagonal–pentagonal rings and hexagonal–hexagonal rings, respectively.
Table 2 reports the final configurations calculated for each initial configuration, adsorption energies calculated using atom and molecule as reference states, the Bader charge on the H atom and the shortest C–H bond distance in the optimized structures. The most favorable site is calculated for the site C. Adsorption energy is exothermic in all cases with respect to atomic hydrogen. This implies that isolated gas phase atomic hydrogen is unstable compared to its form adsorbed with C60. The adsorption energies are less negative or positive with respect to molecular hydrogen as the reference state. This is due to the energy penalty for dissociating a hydrogen molecule. The strong adsorption on the “C” site is due to the formation of strong C–H covalent bonds as shown in Figure 2. This is further confirmed by the shortest C–H bond distance of 1.11 Å and charge density plot. Furthermore, sp2 hybridization of carbon in the C60 molecule turns into sp3 hybridization as evidenced by the formation of the CC3H tetrahedral unit and elongation in the C–C bond lengths (refer to Figure 2b). The Bader charge analysis shows that there is no significant charge transfer from hydrogen. This is due to the formation of the C–H covalent bond in the CC3H tetrahedral unit. The configurations H and P are less stable by 2.06 eV than the configuration C. This is consistent with the weak van-der Waals interaction and this is supported by the long C–H distances (3.06 Å) (refer to Table 2).
Next, we considered the adsorption of two hydrogen atoms in three different possible configurations as shown Figure 3. In the first configuration (HH_66), hydrogen atoms are bonded to the carbon atoms bridged by two adjacent hexagonal rings. The second configuration (HH_65) has two hydrogen atoms bonded to the carbon atoms bridged by a hexagonal ring and a pentagonal ring that are next to each other. In the third configuration (HH_CC), hydrogen atoms are further away from each other.
The most favorable configuration is found to be HH_66. In C60, there are alternative single and double bonds present. The double bond bridges the two adjacent hexagonal rings (66). Addition of hydrogen is easier in double bond (C=C) than in single bond (C–C) explaining the preference of adsorption in 66 configurations. The second most stable configuration (HH_65) is 0.39 eV less energetically favorable than the HH_66 configuration. The stability of the third configuration is energetically less favorable by 0.80 eV than the first configuration. The preference of hydrogen adsorption on the bridge carbon atoms is also linked to the degree of C–C elongation in the tetrahedral unit because such elongation facilitates the formation of C–H bond. In the single hydrogen adsorption, the largest C–C bond distance is 1.536 Å (refer to Figure 2b). The second hydrogen adsorption elongates the C–C bond further by ~0.05 Å.
Table 3 reports the initial and final configurations, adsorption energies calculated with respect to the gas-phase hydrogen atom and diatomic molecule as reference states and Bader charges on the H. Exoergic adsorption energies are observed for all three cases with respect to atomic hydrogen implying that gaseous atomic hydrogen is not stable and prefers to interact with C60. The adsorption of H atom becomes stronger by 0.65 eV than that observed for the most stable single hydrogen adsorption (refer to Table 2). It clearly shows that the second atomic hydrogen prefers the carbon that is nearest neighbor to the carbon bonded with hydrogen.
Finally, molecular hydrogen was allowed to adsorb on the surface of pristine C60. Several configurations were considered. The first two most stable configurations are shown in Figure 4. In both cases, adsorption is non-covalent. Adsorption energies calculated with respect to atoms are −2.52 eV and −2.51 eV for configurations 1 (Figure 1a) and 2 (Figure 1d), respectively. Corresponding adsorption energies with respect to the hydrogen molecule as a reference are −0.22 eV and −0.21 eV, respectively. The high negative adsorption energy mainly consists of exothermic binding of hydrogen atoms to form dimers on the surface. According to the optimized configurations, there is no bonding between the H2 molecule and the surface. Negative adsorption energies calculated with respect to the molecule show that the molecule is stable on the surface with almost the same H–H bond distance (refer to Figure 4b,e). The Bader charge analysis shows that there is no electron lost or gained on the hydrogen molecule inferring the absence of charge transfer. The bond distance of the hydrogen molecule is almost the same as observed in the isolated hydrogen molecule. Furthermore, C–C bond distances in the C60 molecule do not show any significant difference compared to that calculated on the pristine C60 molecule. This further supports the physisorption of the hydrogen molecule on the surface.
3.3. Adsorption of Hydrogen on the Modified Surfaces C60
The improvement in the adsorption of hydrogen, particularly in the form of the molecule, was considered by modifying the surface of C60. Here we considered three different possible modifications namely encapsulation, doping and supporting. In the encapsulation process, a single Ru atom was encapsulated. Doping here is the substitutional replacement of a C atom by a Ru atom. In the process of supporting, C60 molecules were allowed to interact with a single Ru atom.
3.3.1. Ru-Encapsulated C60 (Ru@C60)
First, a single Ru atom was encapsulated. The relaxed structure is shown in Figure 5a. The C–C bond distances in the relaxed geometry and charge density plot are shown in Figure 1b,c, respectively. The encapsulated Ru atom occupies the center of the cage and the encapsulation energy is −1.48 eV, meaning that the Ru atom is stable inside the cage. The Bader charge analysis shows that the Ru atom transferred 0.32 e to the cage. A small change in the C–C bond distances is noted.
Next, we considered the adsorption of a single hydrogen atom on the surface of Ru@C60 at five different sites as discussed previously for the surface of pristine C60. Initial and final configurations, adsorption energies, Bader charges on the Ru and H atoms and C–H bond distances are tabulated in Table 4. The results show that the most favorable site is “C” as observed in the case of hydrogen on the pristine C60 surface. Adsorption is exothermic in all cases with respect to the hydrogen atom and endothermic for the sites “H and “P” with respect to molecular hydrogen. Interestingly, there is an enhancement in the adsorption of hydrogen upon Ru encapsulation. The charge transfer from the Ru atom to the cage increases with the adsorption of hydrogen particularly in the case of configuration “C” (refer to Table 4). The formation of a C–H single covalent bond has disturbed the electron delocalization on the ring and has reduced the electron density on the surface. The encapsulated Ru counteract the loss of the electron by donating some of its electrons. However, the donation is not significant due to its location, which is the center of the cage further away from the surface in the optimized structure.
Figure 6 shows the relaxed structure of the most stable configuration, C–C bond distances on the surface of C60 and the charge density plot showing the adsorption of hydrogen. The C–C bond distances in the tetrahedral unit differ slightly compared to those calculated on the surface of pristine C60.
Thereafter, the adsorptions of two hydrogen atoms were considered. As discussed earlier, three possible configurations were considered. The most favorable adsorption site is 66. As explained earlier, the preference for this site is due to its double bond character in which hydrogen can be easily reacted. Figure 7 shows the relaxed structure, bond distances in the tetrahedral units (CC3H) and charge density plot of the adsorbed configuration.
Encapsulation of Ru has increased the degree of hydrogen adsorption in all three configurations compared to the adsorption on the pristine C60 (refer to Table 5). Adsorption energies are exothermic in all cases with respect to both reference states. The positive Bader charges on the Ru infer the donation of electrons to the C60. This is due to the donation of π electrons from the ring to the C–H bond.
Next, we considered the adsorption of molecular hydrogen on the surface of Ru@C60. Two possible optimized configurations are shown in Figure 8. Encapsulation yields stronger adsorption than that observed on the surface of pristine C60 (refer to Table 6). A small amount of charge has been transferred from the Ru atom to the surface of C60. The C–C and H–H bond distances are not significantly affected.
3.3.2. Ru-Doped C60
The second modification was made by substituting a C atom with a single Ru atom. The relaxed structure is shown in Figure 9. The Ru atom forms a trigonal pyramid structure and displaces away from the surface. The Bader charge on the Ru is +1.01 meaning that one electron has been transferred from Ru to the adjacent carbons. The amount of charge transferred is almost equally distributed (refer to Figure 9b).
A single hydrogen atom was allowed to interact with the Ru-doped surface. The relaxed structure, bond distances and charge density plot are shown in Figure 10. Adsorption energies calculated using atom and molecular hydrogen as reference states are −1.94 eV and 0.36 eV, respectively. The strength of adsorption is lower than that observed on the surfaces of pristine and Ru-encapsulated C60. This is due to the different bonding feature between the Ru and hydrogen. A small charge transfer from Ru to hydrogen is observed. Hydrogen and Ru are negatively and positively charged, respectively. The formation of the Ru–H bond is due to the electrostatic attraction between the oppositely charges ions. A very small change in the bond distances on the Ru-doped surface is noted compared to Ru-doped C60 surface.
Molecular hydrogen was allowed to interact with Ru-doped C60. Figure 11 shows the relaxed structures of two possible configurations. Adsorption energies are negative with respect to both references meaning that both the atom and molecule would prefer to adsorb on the surface. The H–H bond distances are slightly elongated (refer to Table 7). The Bader charges on the Ru changes slightly. The adsorption of hydrogen by the Ru-doped C60 surface is less favorable than that observed on the Ru-encapsulated C60 surface and more favorable than that observed on the surface of pristine C60.
3.3.3. Ru-Supported C60
The last modification was made by interacting a single Ru atom on the surface of pristine C60. Five different positions were considered as discussed before. The lowest energy configuration was found to be “66”. Relaxed structure is shown in Figure 12. The adsorption energy was calculated to be −3.75 eV meaning that the formation is exoergic. The Bader charge on the Ru was +0.50.
A single hydrogen adsorption was first considered. Adsorption energies calculated with respect to hydrogen atom and molecular hydrogen were −2.48 eV and −0.18 eV, respectively. The strong adsorption is due to the charge transfer between the Ru and hydrogen. The Bader charge on the Ru in the Ru-supported structure is +0.50. Once the hydrogen is adsorbed, the Ru becomes more positively charged (+0.73) and hydrogen negatively charged (−0.39) (Figure 13). The Ru–H bond formation is driven by the electrostatic attraction between positively charged Ru and negatively charged hydrogen.
Next, molecular hydrogen was considered for the adsorption. Two different relaxed configurations are shown in Figure 14. Table 8 reports the adsorption energies, H-H bond distance and the Bader charges on the Ru and H atoms. In the first configuration, hydrogen atoms are completely dissociated and both hydrogen atoms are negatively charged. This is confirmed by the increase in the positive charge on the Ru atom compared to that observed in the Ru-supported C60. The adsorption energy is −3.20 eV with respect to the atom and −0.90 eV with respect to the molecular hydrogen showing the strongest adsorption of hydrogen.
4. Conclusions
The present computational study has considered the adsorption of hydrogen on the pristine and defective surfaces of C60 using density functional theory together with dispersion. The hydrogen is adsorbed exothermically on the pristine surface of C60. In order to increase the efficacy of adsorption, the surface of C60 was modified by encapsulating, doping and supporting a single Ru atom. The C60 encapsulated with Ru exhibited a strong adsorption. The enhancement in the adsorption was also noted for the doped-C60 but its degree of adsorption is less than that observed for the encapsulated complex. The C60 surface supported with a single Ru atom with the adsorption energy of −3.74 eV adsorbs hydrogen very strongly and this modified surface is identified as the best surface. The present model calculation results reported at the atomic scale can be tested experimentally for the scope of assembling storage materials for hydrogen.
Author Contributions
Computation, N.K.; writing of original draft preparation, N.K.; writing of review and editing, A.C. Both authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Acknowledgments
The High Performance Computing (HPC) Center at Imperial College London is acknowledged for providing computational facilities.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Moradi, R.; Groth, K.M. Hydrogen storage and delivery: Review of the state of the art technologies and risk and reliability analysis. Int. J. Hydrogen Energy 2019, 44, 12254–12269. [Google Scholar] [CrossRef]
- Kaur, M.; Pal, K. Review on hydrogen storage materials and methods from an electrochemical viewpoint. J. Energy Storage 2019, 23, 234–249. [Google Scholar] [CrossRef]
- Abdalla, A.M.; Hossain, S.; Nisfindy, O.B.; Azad, A.T.; Dawood, M.; Azad, A.K. Hydrogen production, storage, transportation and key challenges with applications: A review. Energy Convers. Manag. 2018, 165, 602–627. [Google Scholar] [CrossRef]
- Gnanapragasam, N.V.; Rosen, M.A. A review of hydrogen production using coal, biomass and other solid fuels. Biofuels 2017, 8, 725–745. [Google Scholar] [CrossRef]
- Graetz, J. New approaches to hydrogen storage. Chem. Soc. Rev. 2009, 38, 73–82. [Google Scholar] [CrossRef]
- Jena, P. Materials for Hydrogen Storage: Past, Present, and Future. J. Phys. Chem. Lett. 2011, 2, 206–211. [Google Scholar] [CrossRef]
- Nagar, R.; Vinayan, B.P.; Samantaray, S.S.; Ramaprabhu, S. Recent advances in hydrogen storage using catalytically and chemically modified graphene nanocomposites. J. Mater. Chem. A 2017, 5, 22897–22912. [Google Scholar] [CrossRef]
- Sugiyama, J. Study on Hydrogen Storage Materials. J. Phys. Soc. Jpn. 2016, 85, 091012. [Google Scholar] [CrossRef]
- Park, N.; Choi, K.; Hwang, J.; Kim, D.W.; Kim, D.O.; Ihm, J. Progress on first-principles-based materials design for hydrogen storage. Proc. Natl. Acad. Sci. USA 2012, 109, 19893–19899. [Google Scholar] [CrossRef] [Green Version]
- de la Casa-Lillo, M.A.; Lamari-Darkrim, F.; Cazorla-Amorós, D.; Linares-Solano, A. Hydrogen Storage in Activated Carbons and Activated Carbon Fibers. J. Phys. Chem. B 2002, 106, 10930–10934. [Google Scholar] [CrossRef]
- Cheng, H.-M.; Yang, Q.-H.; Liu, C. Hydrogen storage in carbon nanotubes. Carbon 2001, 39, 1447–1454. [Google Scholar] [CrossRef]
- Alonso, J.A.; Cabria, I.; López, M.J. Simulation of hydrogen storage in porous carbons. J. Mater. Res. 2012, 28, 589–604. [Google Scholar] [CrossRef]
- Anderson, P.A. 9—Storage of hydrogen in zeolites. In Solid-State Hydrogen Storage; Walker, G., Ed.; Woodhead Publishing: Cambridge, UK, 2008; pp. 223–260. [Google Scholar]
- Lin, X.; Jia, J.; Champness, N.R.; Hubberstey, P.; SchrÖDer, M. 11—Metal-organic framework materials for hydrogen storage. In Solid-State Hydrogen Storage; Walker, G., Ed.; Woodhead Publishing: Cambridge, UK, 2008; pp. 288–312. [Google Scholar]
- Kroto, H.W.; Heath, J.R.; O’Brien, S.C.; Curl, R.F.; Smalley, R.E. C60: Buckminsterfullerene. Nature 1985, 318, 162–163. [Google Scholar] [CrossRef]
- Kroto, H.W. C60: Buckminsterfullerene, the Celestial Sphere that Fell to Earth. Angew. Chem. Int. Ed. 1992, 31, 111–129. (In English) [Google Scholar] [CrossRef]
- Qiu, W.; Chowdhury, S.; Hammer, R.; Velisavljevic, N.; Baker, P.; Vohra, Y.K. Physical and mechanical properties of C60 under high pressures and high temperatures. High Press. Res. 2006, 26, 175–183. [Google Scholar] [CrossRef]
- Kuganathan, N.; Srikaran, R.; Chroneos, A. Stability of Coinage Metals Interacting with C60. Nanomaterials 2019, 9, 1484. [Google Scholar] [CrossRef] [Green Version]
- Kuganathan, N.; Arya, A.K.; Rushton, M.J.D.; Grimes, R.W. Trapping of volatile fission products by C60. Carbon 2018, 132, 477–485. [Google Scholar] [CrossRef]
- Kuganathan, N.; Selvanantharajah, N.; Iyngaran, P.; Abiman, P.; Chroneos, A. Cadmium trapping by C60 and B-, Si-, and N-doped C60. J. Appl. Phys. 2019, 125, 054302. [Google Scholar] [CrossRef]
- Šuligoj, A.; Arčon, I.; Mazaj, M.; Dražić, G.; Arčon, D.; Cool, P.; Štangar, U.L.; Tušar, N.N. Surface modified titanium dioxide using transition metals: Nickel as a winning transition metal for solar light photocatalysis. J. Mater. Chem. A 2018, 6, 9882–9892. [Google Scholar] [CrossRef] [Green Version]
- You, B.; Liu, X.; Hu, G.; Gul, S.; Yano, J.; Jiang, D.-E.; Sun, Y. Universal Surface Engineering of Transition Metals for Superior Electrocatalytic Hydrogen Evolution in Neutral Water. J. Am. Chem. Soc. 2017, 139, 12283–12290. [Google Scholar] [CrossRef] [Green Version]
- Friend, C.M.; Xu, X. Reactions on Transition Metal Surfaces. Annu. Rev. Phys. Chem. 1991, 42, 251–278. [Google Scholar] [CrossRef]
- Saha, D.; Deng, S. Hydrogen Adsorption on Ordered Mesoporous Carbons Doped with Pd, Pt, Ni, and Ru. Langmuir 2009, 25, 12550–12560. [Google Scholar] [CrossRef] [PubMed]
- Lueking, A.; Yang, R.T. Hydrogen Spillover from a Metal Oxide Catalyst onto Carbon Nanotubes—Implications for Hydrogen Storage. J. Catal. 2002, 206, 165–168. [Google Scholar] [CrossRef]
- Wang, L.; Yang, R.T. Hydrogen Storage Properties of Carbons Doped with Ruthenium, Platinum, and Nickel Nanoparticles. J. Phys. Chem. C 2008, 112, 12486–12494. [Google Scholar] [CrossRef]
- Zieliński, M.; Wojcieszak, R.; Monteverdi, S.; Mercy, M.; Bettahar, M.M. Hydrogen storage in nickel catalysts supported on activated carbon. Int. J. Hydrogen Energy 2007, 32, 1024–1032. [Google Scholar] [CrossRef]
- Yang, R.T.; Wang, Y. Catalyzed Hydrogen Spillover for Hydrogen Storage. J. Am. Chem. Soc. 2009, 131, 4224–4226. [Google Scholar] [CrossRef]
- Shin, W.H.; Yang, S.H.; Goddard, W.A.; Kang, J.K. Ni-dispersed fullerenes: Hydrogen storage and desorption properties. Appl. Phys. Lett. 2006, 88, 053111. [Google Scholar] [CrossRef] [Green Version]
- Target Explanation Document: Onboard Hydrogen Storage for Light-Duty Fuel Cell Vehicles. Available online: https://www.energy.gov/sites/prod/files/2017/05/f34/fcto_targets_onboard_hydro_storage_explanation.pdf (accessed on 28 July 2020).
- Yoon, M.; Yang, S.; Hicke, C.; Wang, E.; Geohegan, D.; Zhang, Z. Calcium as the Superior Coating Metal in Functionalization of Carbon Fullerenes for High-Capacity Hydrogen Storage. Phys. Rev. Lett. 2008, 100, 206806. [Google Scholar] [CrossRef] [Green Version]
- Sun, Q.; Jena, P.; Wang, Q.; Marquez, M. First-Principles Study of Hydrogen Storage on Li12C60. J. Am. Chem. Soc. 2006, 128, 9741–9745. [Google Scholar] [CrossRef]
- Kaiser, A.; Renzler, M.; Kranabetter, L.; Schwärzler, M.; Parajuli, R.; Echt, O.; Scheier, P. On enhanced hydrogen adsorption on alkali (cesium) doped C60 and effects of the quantum nature of the H2 molecule on physisorption energies. Int. J. Hydrogen Energy 2017, 42, 3078–3086. [Google Scholar] [CrossRef] [Green Version]
- Saha, D.; Deng, S. Hydrogen Adsorption on Pd- and Ru-Doped C60 Fullerene at an Ambient Temperature. Langmuir 2011, 27, 6780–6786. [Google Scholar] [CrossRef] [PubMed]
- Kitano, M.; Kanbara, S.; Inoue, Y.; Kuganathan, N.; Sushko, P.V.; Yokoyama, T.; Hara, M.; Hosono, H. Electride support boosts nitrogen dissociation over ruthenium catalyst and shifts the bottleneck in ammonia synthesis. Nat. Commun. 2015, 6, 6731. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kuganathan, N.; Hosono, H.; Shluger, A.L.; Sushko, P.V. Enhanced N2 Dissociation on Ru-Loaded Inorganic Electride. J. Am. Chem. Soc. 2014, 136, 2216–2219. [Google Scholar] [CrossRef]
- Pagliaro, M.; Campestrini, S.; Ciriminna, R. Ru-based oxidation catalysis. Chem. Soc. Rev. 2005, 34, 837–845. [Google Scholar] [CrossRef]
- Kresse, G.; Furthmüller, J. Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set. Phys. Rev. B 1996, 54, 11169–11186. [Google Scholar] [CrossRef]
- Blöchl, P.E. Projector augmented-wave method. Phys. Rev. B 1994, 50, 17953–17979. [Google Scholar] [CrossRef] [Green Version]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized Gradient Approximation Made Simple. Phys. Rev. Lett. 1996, 77, 3865–3868. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Press, W.H.; Teukolsky, S.A.; Vetterling, W.T.; Flannery, B.P. The art of scientific computing. In Numerical Recipes in C; Cambridge University Press: Cambridge, UK, 1992. [Google Scholar]
- Monkhorst, H.J.; Pack, J.D. Special points for Brillouin-zone integrations. Phys. Rev. B 1976, 13, 5188–5192. [Google Scholar] [CrossRef]
- Grimme, S.; Antony, J.; Ehrlich, S.; Krieg, H. A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu. J. Chem. Phys. 2010, 132, 154104. [Google Scholar] [CrossRef] [Green Version]
- Urashima, Y.; Wakabayashi, T.; Masaki, T.; Terasaki, Y. Ruthenium, a new mineral from Horokanai, Hokkaido, Japan. Mineral. J. 1974, 7, 438–444. [Google Scholar] [CrossRef] [Green Version]
- Hawkins, J.M. Osmylation of C60: Proof and characterization of the soccer-ball framework. Acc. Chem. Res. 1992, 25, 150–156. [Google Scholar] [CrossRef]
- Olmsted, J.M.H.; Williams, G.M.; Friedman, D.G. Chemistry: The Molecular Science; Brown Publishers: Dubuque, IA, USA, 1997. [Google Scholar]
Figure 1.
Initial configurations of a single hydrogen atom adsorbed on five different sites on the surface of pristine C60.
Figure 1.
Initial configurations of a single hydrogen atom adsorbed on five different sites on the surface of pristine C60.

Figure 2.
(a) Optimized structure of a single hydrogen atom adsorbed on the “C” site, (b) bond distances and (c) charge density plot showing the strong bonding nature of hydrogen with C atom on the surface.
Figure 2.
(a) Optimized structure of a single hydrogen atom adsorbed on the “C” site, (b) bond distances and (c) charge density plot showing the strong bonding nature of hydrogen with C atom on the surface.

Figure 3.
(a–c) Three different initial configurations considered for the adsorption of two hydrogen atoms, (d) relaxed structure of the most stable configuration (HH_66), (e) bond distances near the adsorbed atoms and (f) charge density plot associated with the interaction of hydrogen atoms with carbon atoms on the surface.
Figure 3.
(a–c) Three different initial configurations considered for the adsorption of two hydrogen atoms, (d) relaxed structure of the most stable configuration (HH_66), (e) bond distances near the adsorbed atoms and (f) charge density plot associated with the interaction of hydrogen atoms with carbon atoms on the surface.

Figure 4.
(a) Relaxed configuration of molecular hydrogen adsorbed on the surface of pristine C60, (b) bond distances (H–H and C–C) in the relaxed configuration and (c) charge density plot showing the non-covalent interaction. Similar figures (d–f) are shown for configuration 2.
Figure 4.
(a) Relaxed configuration of molecular hydrogen adsorbed on the surface of pristine C60, (b) bond distances (H–H and C–C) in the relaxed configuration and (c) charge density plot showing the non-covalent interaction. Similar figures (d–f) are shown for configuration 2.

Figure 5.
(a) Optimized structure of Ru@C60, (b) bond distances and (c) charge density plot of encapsulated configuration.
Figure 5.
(a) Optimized structure of Ru@C60, (b) bond distances and (c) charge density plot of encapsulated configuration.

Figure 6.
(a) Energy minimized structure of a single hydrogen atom adsorbed on the most stable site (C) of Ru@C60, (b) bond distances in the relaxed configuration and (c) charge density plot.
Figure 6.
(a) Energy minimized structure of a single hydrogen atom adsorbed on the most stable site (C) of Ru@C60, (b) bond distances in the relaxed configuration and (c) charge density plot.

Figure 7.
(a) Energy minimized structure of two hydrogen atoms adsorbed on the most stable site (66) of Ru@C60, (b) bond distances in the relaxed configuration and (c) charge density plot.
Figure 7.
(a) Energy minimized structure of two hydrogen atoms adsorbed on the most stable site (66) of Ru@C60, (b) bond distances in the relaxed configuration and (c) charge density plot.

Figure 8.
(a) Relaxed configuration (configuration 1) of molecular hydrogen adsorbed on the surface of Ru@C60, (b) bond distances (H–H and C–C) in the relaxed configuration and (c) charge density plot showing the non-covalent interaction. Similar figures (d–f) are shown for configuration 2.
Figure 8.
(a) Relaxed configuration (configuration 1) of molecular hydrogen adsorbed on the surface of Ru@C60, (b) bond distances (H–H and C–C) in the relaxed configuration and (c) charge density plot showing the non-covalent interaction. Similar figures (d–f) are shown for configuration 2.

Figure 9.
(a) Relaxed structure of Ru-doped C60, (b) bond distances and Bader charges on the Ru and its adjacent C atoms and (c) charge density plot showing the interaction of Ru with the surface.
Figure 9.
(a) Relaxed structure of Ru-doped C60, (b) bond distances and Bader charges on the Ru and its adjacent C atoms and (c) charge density plot showing the interaction of Ru with the surface.

Figure 10.
(a) Relaxed structure of a single hydrogen adsorbed on the Ru-doped C60 surface, (b) bond distances and Bader charges on the Ru and its adjacent C atoms and (c) charge density plot showing the interaction between a single hydrogen atom and Ru-doped C60 surface.
Figure 10.
(a) Relaxed structure of a single hydrogen adsorbed on the Ru-doped C60 surface, (b) bond distances and Bader charges on the Ru and its adjacent C atoms and (c) charge density plot showing the interaction between a single hydrogen atom and Ru-doped C60 surface.
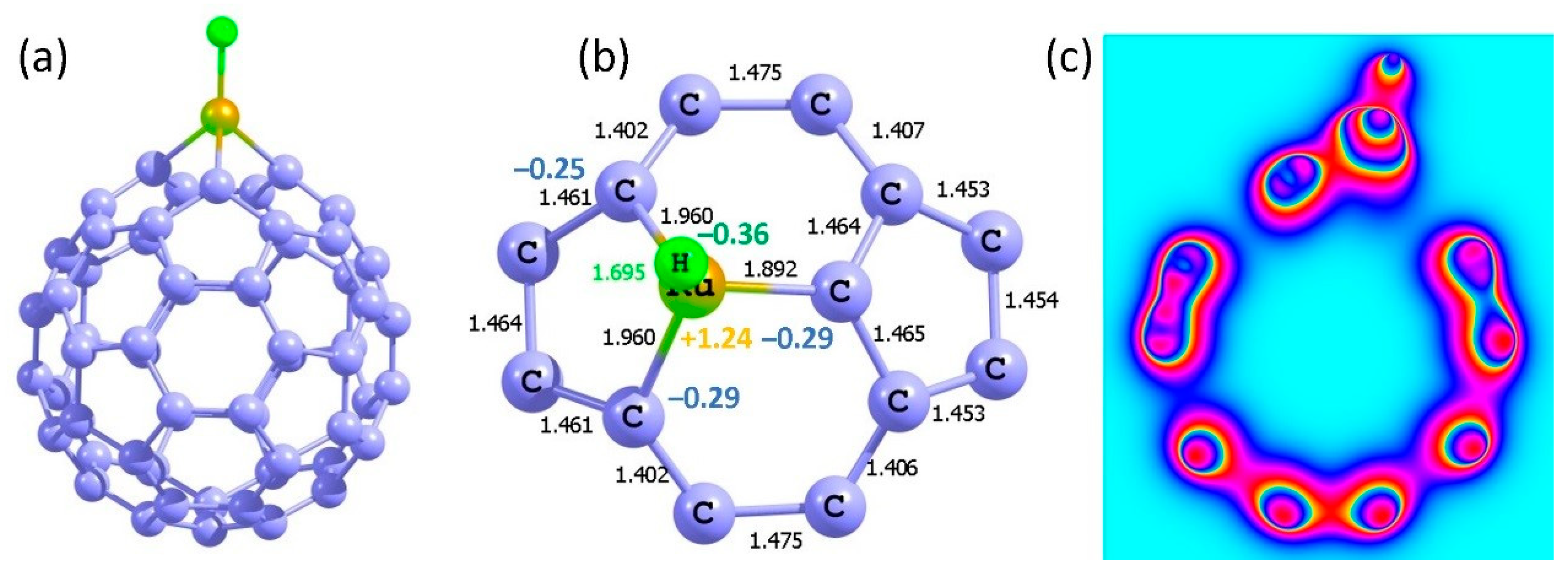
Figure 11.
(a) Relaxed configuration (configuration 1) of molecular hydrogen adsorbed on the surface of Ru-doped C60, (b) bond distances (H–H and C–C) in the relaxed configuration and (c) charge density plot showing the interaction of molecular hydrogen with Ru-doped C60. Similar figures (d–f) are shown for configuration 2.
Figure 11.
(a) Relaxed configuration (configuration 1) of molecular hydrogen adsorbed on the surface of Ru-doped C60, (b) bond distances (H–H and C–C) in the relaxed configuration and (c) charge density plot showing the interaction of molecular hydrogen with Ru-doped C60. Similar figures (d–f) are shown for configuration 2.

Figure 12.
(a) Relaxed structure of Ru-supported C60, (b) bond distances and Bader charges on the Ru and its adjacent C atoms and (c) charge density plot showing the interaction of Ru with the surface.
Figure 12.
(a) Relaxed structure of Ru-supported C60, (b) bond distances and Bader charges on the Ru and its adjacent C atoms and (c) charge density plot showing the interaction of Ru with the surface.

Figure 13.
(a) Relaxed structure of a single hydrogen atom adsorbed on the Ru-supported C60 surface (b) bond distances and Bader charges on the Ru and its adjacent C atoms and (c) charge density plot showing the interaction of hydrogen atom with Ru-supportedC60 surface.
Figure 13.
(a) Relaxed structure of a single hydrogen atom adsorbed on the Ru-supported C60 surface (b) bond distances and Bader charges on the Ru and its adjacent C atoms and (c) charge density plot showing the interaction of hydrogen atom with Ru-supportedC60 surface.

Figure 14.
(a) Relaxed configuration (configuration 1) of molecular hydrogen adsorbed on the surface of Ru-doped C60, (b) bond distances (H–H and C–C) in the relaxed configuration and (c) charge density plot showing the non-covalent interaction. Similar figures (d–f) are shown for configuration 2.
Figure 14.
(a) Relaxed configuration (configuration 1) of molecular hydrogen adsorbed on the surface of Ru-doped C60, (b) bond distances (H–H and C–C) in the relaxed configuration and (c) charge density plot showing the non-covalent interaction. Similar figures (d–f) are shown for configuration 2.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Table 1.
Calculated and experimental lattice parameters of hexagonal (P63/mmc) Ru.
| Parameter | Calc | Expt [44] | |∆| (%) |
|---|---|---|---|
| a = b (Å) | 2.72 | 2.71 | 0.37 |
| c (Å) | 4.30 | 4.28 | 0.47 |
| c/a | 1.58 | 1.58 | 0.00 |
| α = β (°) | 90.0 | 90.0 | 0.00 |
| γ (°) | 120.0 | 120.0 | 0.00 |
Table 2.
Initial and final configurations of a single H atom adsorbed on the surface of pristine C60, adsorption energies calculated with respect to gas phase hydrogen atom and diatomic molecule as reference states, Bader charges on the H atoms and shortest C–H bond in the optimized configurations.
Table 2.
Initial and final configurations of a single H atom adsorbed on the surface of pristine C60, adsorption energies calculated with respect to gas phase hydrogen atom and diatomic molecule as reference states, Bader charges on the H atoms and shortest C–H bond in the optimized configurations.
| Initial Configuration | Final Configuration | Adsorption Energy (eV) | Bader Charge on H (|e|) | C–H (Å) | |
|---|---|---|---|---|---|
| Ref: H | Ref: ½ H2 | ||||
| H | H | −0.26 | 2.04 | +0.01 | 3.06 |
| P | P | −0.26 | 2.04 | +0.01 | 3.05 |
| C | C | −2.32 | −0.02 | +0.08 | 1.11 |
| 66 | C | −2.32 | −0.02 | +0.03 | 1.11 |
| 65 | C | −2.32 | −0.02 | +0.04 | 111 |
Table 3.
Initial and final configurations of two single H atoms adsorbed on the surface of pristine C60, adsorption energies with respect to gas phase hydrogen atom and Bader charges on the H atoms.
Table 3.
Initial and final configurations of two single H atoms adsorbed on the surface of pristine C60, adsorption energies with respect to gas phase hydrogen atom and Bader charges on the H atoms.
| Initial Configuration | Final Configuration | Adsorption Energy (eV)/Atom | Bader Charge on H (|e|) | |
|---|---|---|---|---|
| Ref: H | Ref: H2 | |||
| HH_65 | HH_65 | −2.58 | −0.28 | +0.01, +0.06 |
| HH_66 | HH_66 | −2.97 | −0.67 | +0.03, +0.04 |
| HH_CC | HH_CC | −2.17 | +0.14 | +0.08, +0.04 |
Table 4.
Final configurations of a single H atom adsorbed on the surface of Ru-encapsulated C60, adsorption energies with respect to gas phase hydrogen atom and diatomic molecule, Bader charges on the Ru and H atoms and shortest C–H bond distances in the relaxed configurations.
Table 4.
Final configurations of a single H atom adsorbed on the surface of Ru-encapsulated C60, adsorption energies with respect to gas phase hydrogen atom and diatomic molecule, Bader charges on the Ru and H atoms and shortest C–H bond distances in the relaxed configurations.
| Initial Configuration | Final Configuration | Adsorption Energy (eV) | Bader Charge (|e|) | C–H (Å) | ||
|---|---|---|---|---|---|---|
| Ref: H | Ref: ½ H2 | H | Ru | |||
| H | H | −0.68 | 1.62 | 0.00 | +0.28 | 3.05 |
| P | P | −0.68 | 1.62 | +0.01 | +0.28 | 3.04 |
| C | C | −2.93 | −0.62 | +0.07 | +0.51 | 1.11 |
| 66 | C | −2.93 | −0.62 | +0.02 | +0.52 | 1.11 |
| 65 | C | −2.93 | −0.62 | +0.04 | +0.52 | 1.11 |
Table 5.
Final configurations of two single H atoms adsorbed on the surface of Ru@C60, adsorption energies with respect to gas phase hydrogen atom and Bader charges on the Ru and H atoms.
Table 5.
Final configurations of two single H atoms adsorbed on the surface of Ru@C60, adsorption energies with respect to gas phase hydrogen atom and Bader charges on the Ru and H atoms.
| Initial Configuration | Final Configuration | Adsorption Energy (eV)/Atom | Bader Charge (|e|) | ||
|---|---|---|---|---|---|
| Ref: H | Ref: H2 | H | Ru | ||
| HH_65 | HH_65 | −2.87 | −0.57 | +0.01, +0.05 | +0.45 |
| HH_66 | HH_66 | −3.20 | −0.90 | +0.03, +0.04 | +0.29 |
| HH_CC | HH_CC | −2.56 | −0.26 | +0.03, +0.08 | +0.49 |
Table 6.
Calculated adsorption energies with respect to gas phase molecular hydrogen, H–H bond distances and Bader charges on the Ru and H atoms.
Table 6.
Calculated adsorption energies with respect to gas phase molecular hydrogen, H–H bond distances and Bader charges on the Ru and H atoms.
| Configuration | Adsorption Energy (eV) | H–H (Å) | Bader Charge (|e|) | ||
|---|---|---|---|---|---|
| Ref: H | Ref: H2 | H | Ru | ||
| 1 | −2.60 | −0.31 | 0.749 | +0.02, −0.03 | +0.32 |
| 2 | −2.73 | −0.43 | 0.750 | +0.02, −0.03 | +0.28 |
Table 7.
Calculated adsorption energies with respect to gas phase molecular hydrogen, H–H bond distances and Bader charges on the Ru and H atoms.
Table 7.
Calculated adsorption energies with respect to gas phase molecular hydrogen, H–H bond distances and Bader charges on the Ru and H atoms.
| Configuration | Adsorption Energy (eV)/Atom | H–H (Å) | Bader Charge (|e|) | ||
|---|---|---|---|---|---|
| Ref: H | Ref: H2 | H | Ru | ||
| 1 | −2.69 | −0.39 | 0.807 | +0.02, −0.05 | +1.05 |
| 2 | −2.65 | −0.35 | 0.787 | +0.02, −0.03 | +1.04 |
Table 8.
Calculated adsorption energies with respect to gas phase molecular hydrogen, H-H bond distances and Bader charges on the Ru and H atoms.
Table 8.
Calculated adsorption energies with respect to gas phase molecular hydrogen, H-H bond distances and Bader charges on the Ru and H atoms.
| Configuration | Adsorption Energy (eV)/Atom | H–H (Å) | Bader Charge (|e|) | ||
|---|---|---|---|---|---|
| Ref: H | Ref: H2 | H | Ru | ||
| 1 | −3.20 | −0.90 | 1.889 | −0.20, −0.20 | +0.73 |
| 2 | −3.02 | −0.72 | 0.899 | +1.00, −1.15 | +0.61 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Kuganathan, N.; Chroneos, A. Hydrogen Adsorption on Ru-Encapsulated, -Doped and -Supported Surfaces of C60. Surfaces 2020, 3, 408-422. https://doi.org/10.3390/surfaces3030030
AMA Style
Kuganathan N, Chroneos A. Hydrogen Adsorption on Ru-Encapsulated, -Doped and -Supported Surfaces of C60. Surfaces. 2020; 3(3):408-422. https://doi.org/10.3390/surfaces3030030
Chicago/Turabian StyleKuganathan, Navaratnarajah, and Alexander Chroneos. 2020. "Hydrogen Adsorption on Ru-Encapsulated, -Doped and -Supported Surfaces of C60" Surfaces 3, no. 3: 408-422. https://doi.org/10.3390/surfaces3030030