Spatially Resolved XPS Characterization of Electrochemical Surfaces

 ,
,  ,
,
Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
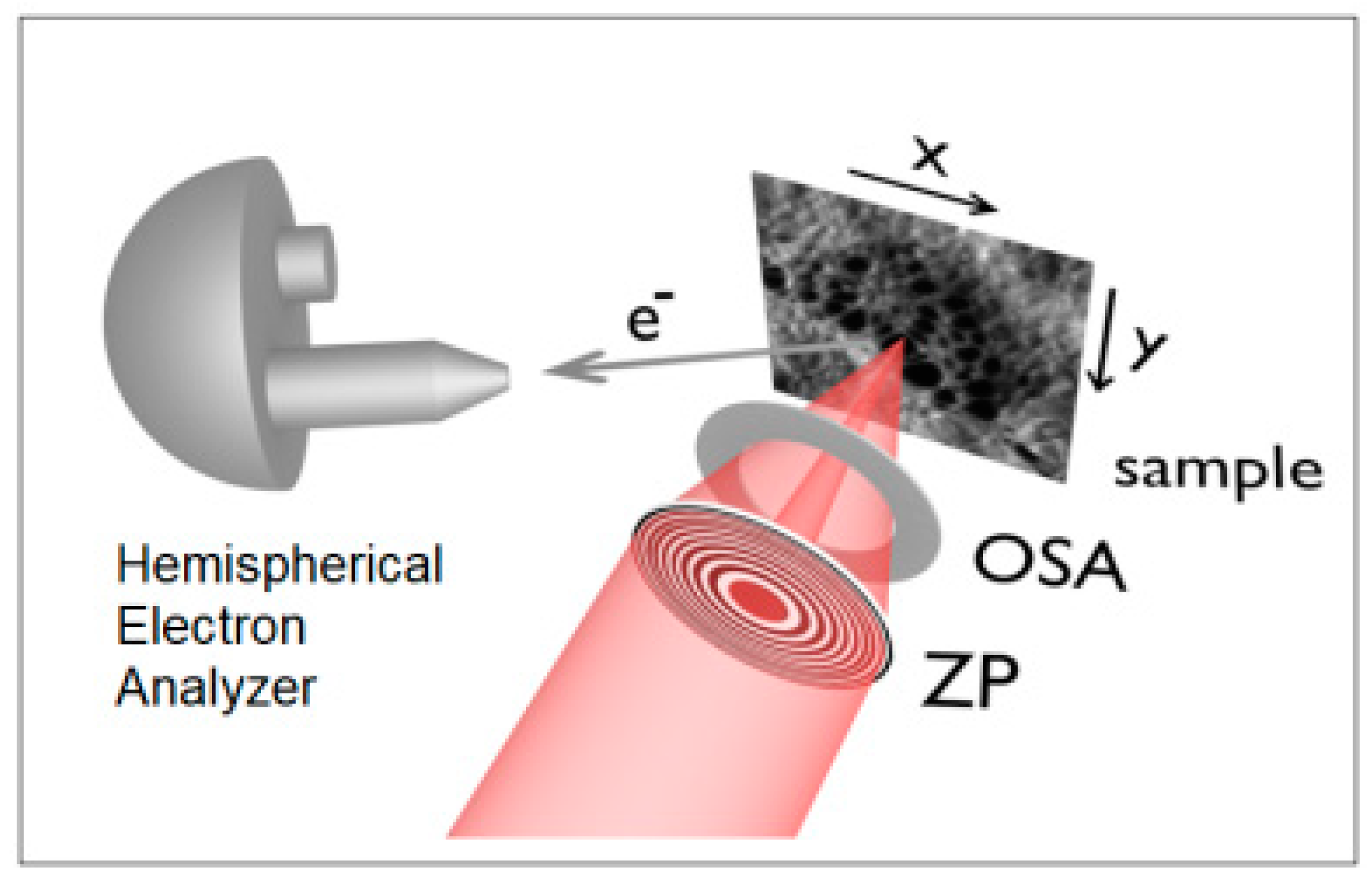
2. Materials and Methods: the Soft X-rays Scanning Photoemission Microscope of Elettra
2.1. The Standard UHV—HV Setup
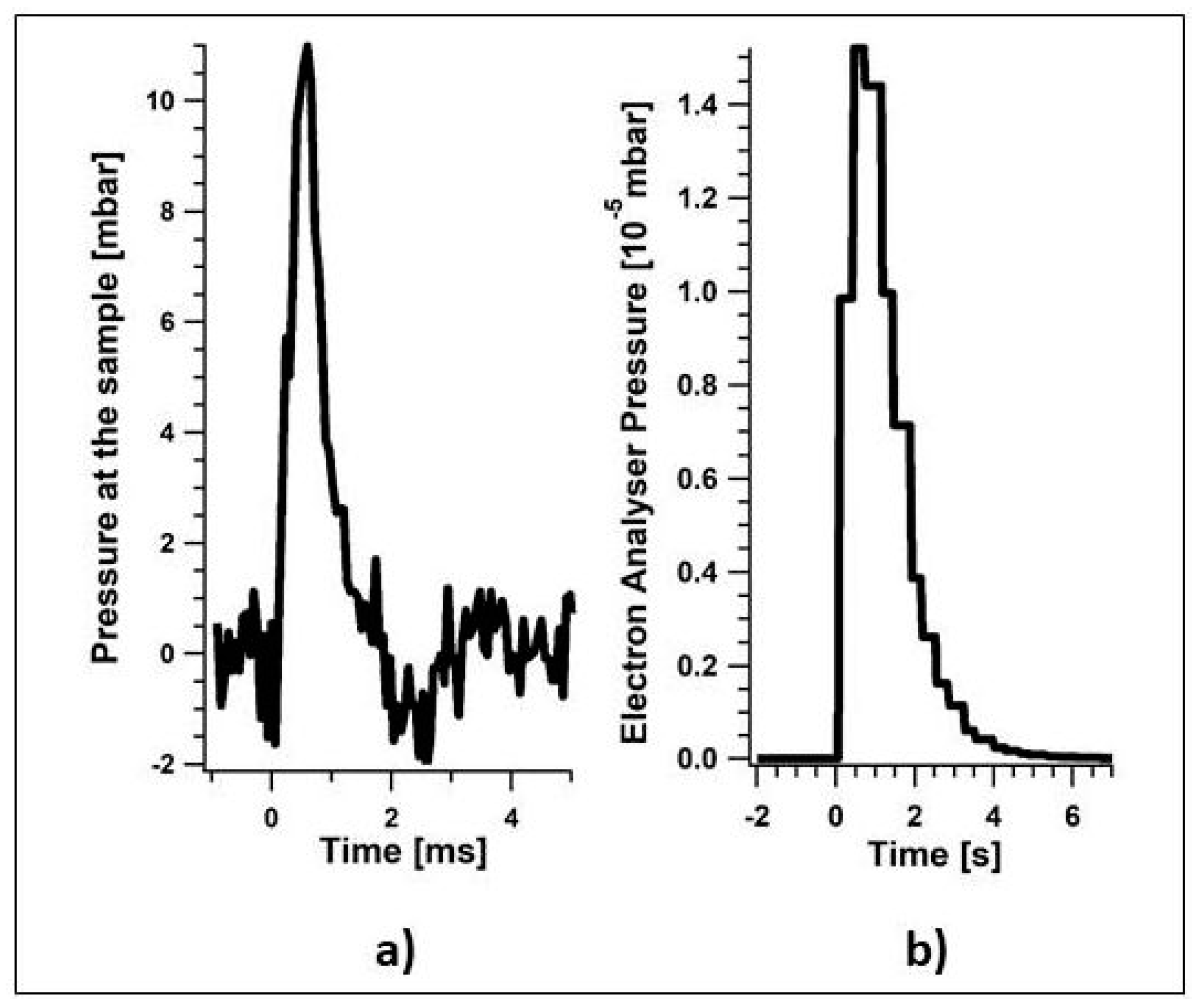
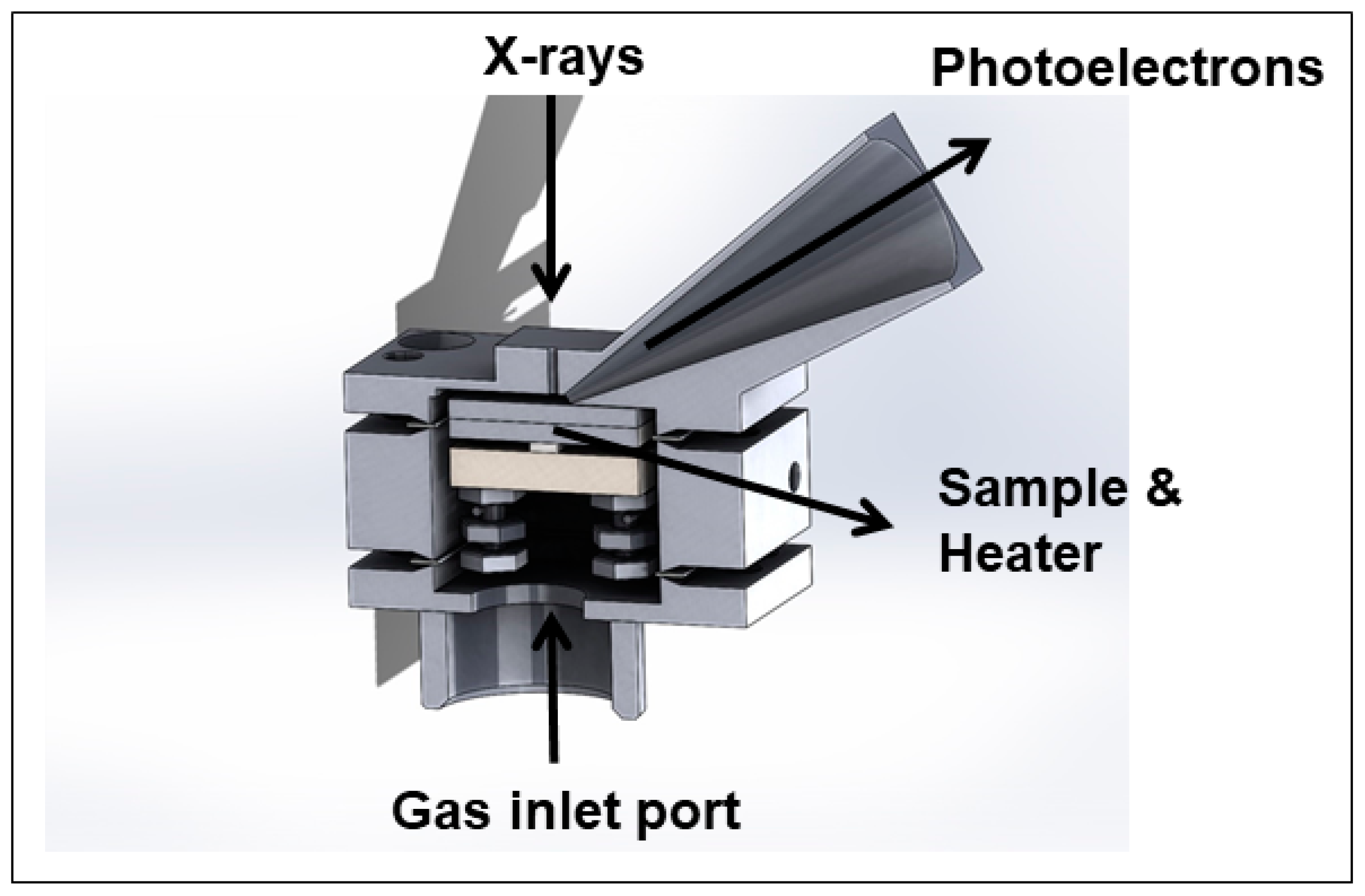
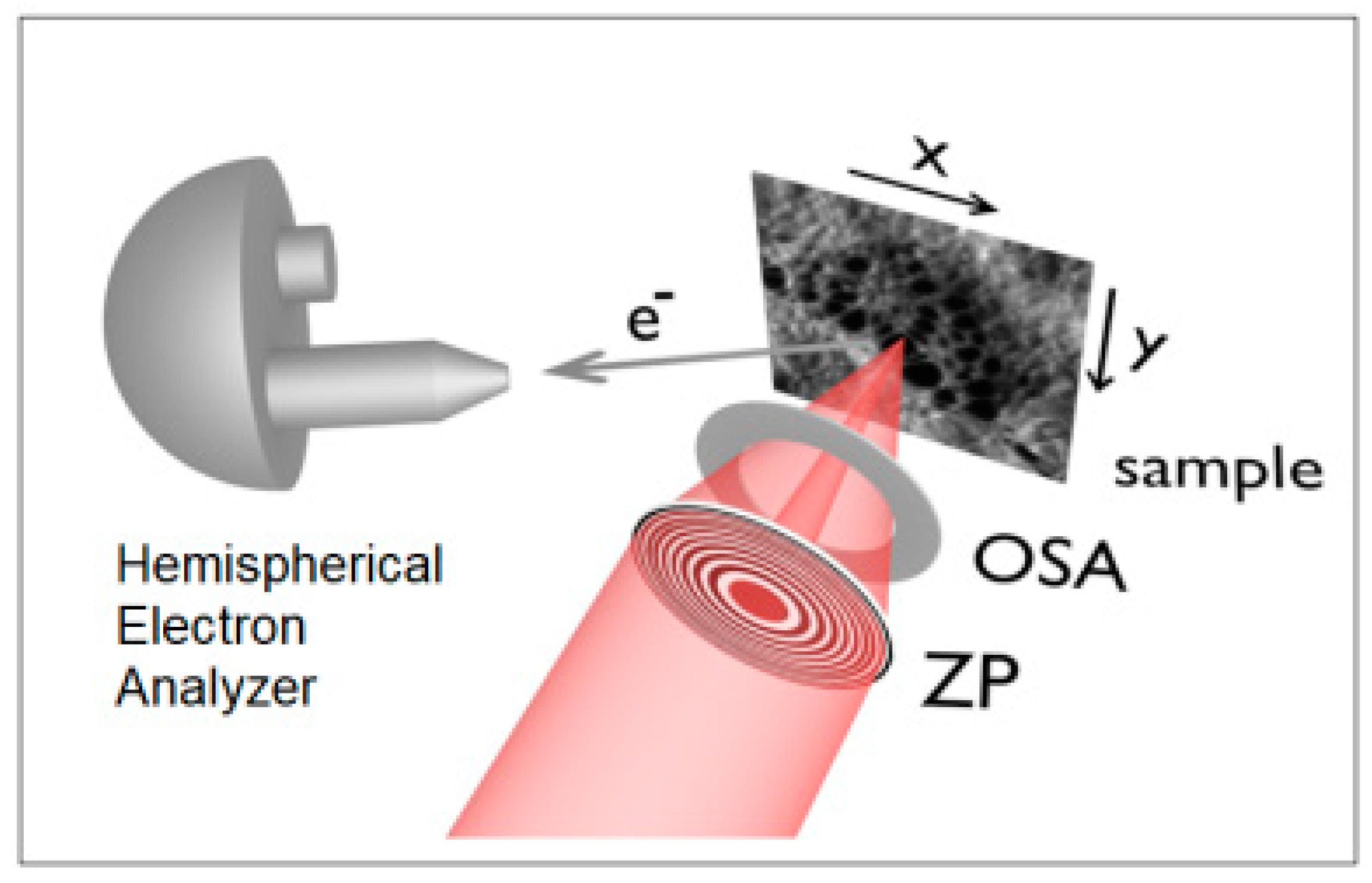
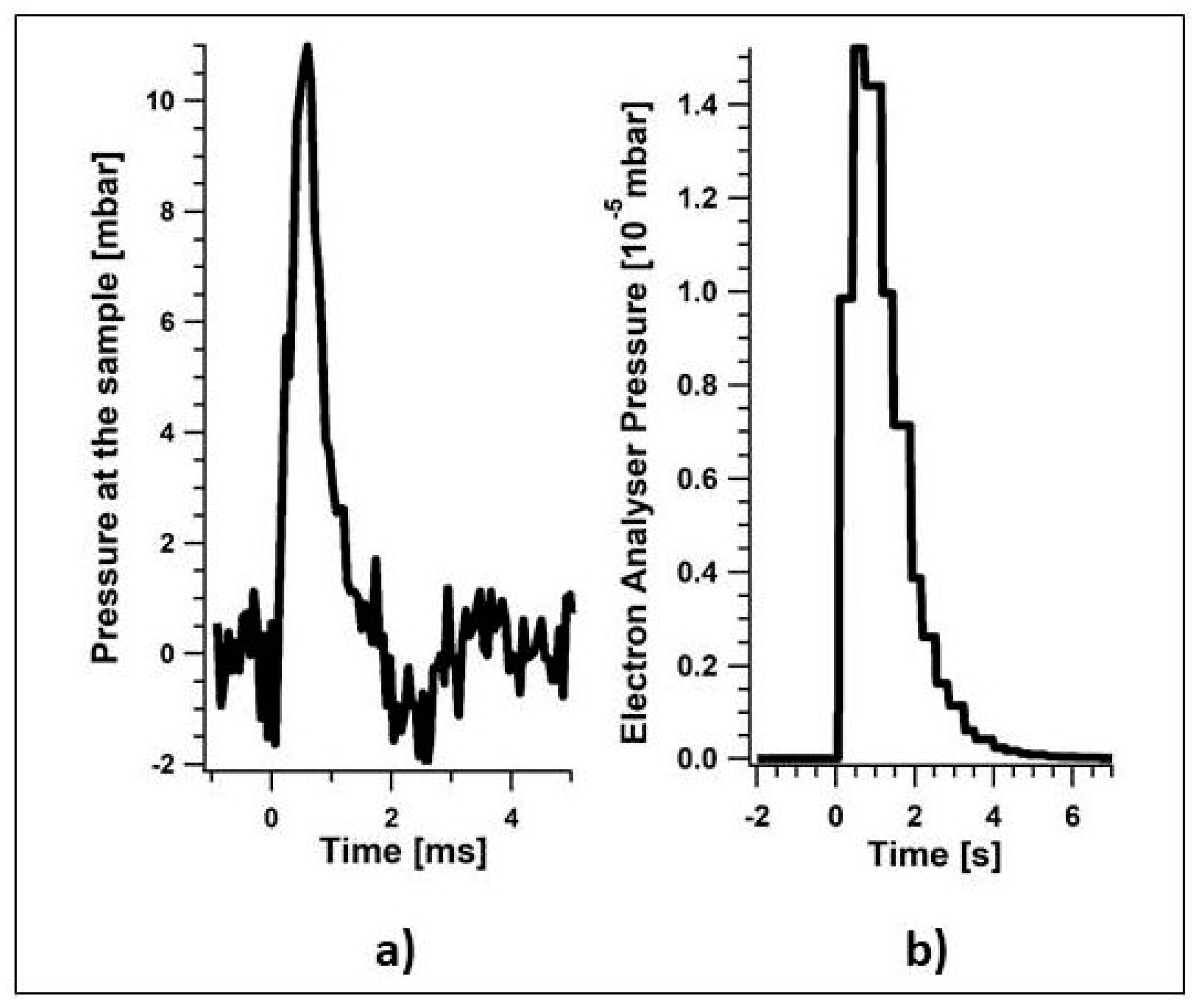
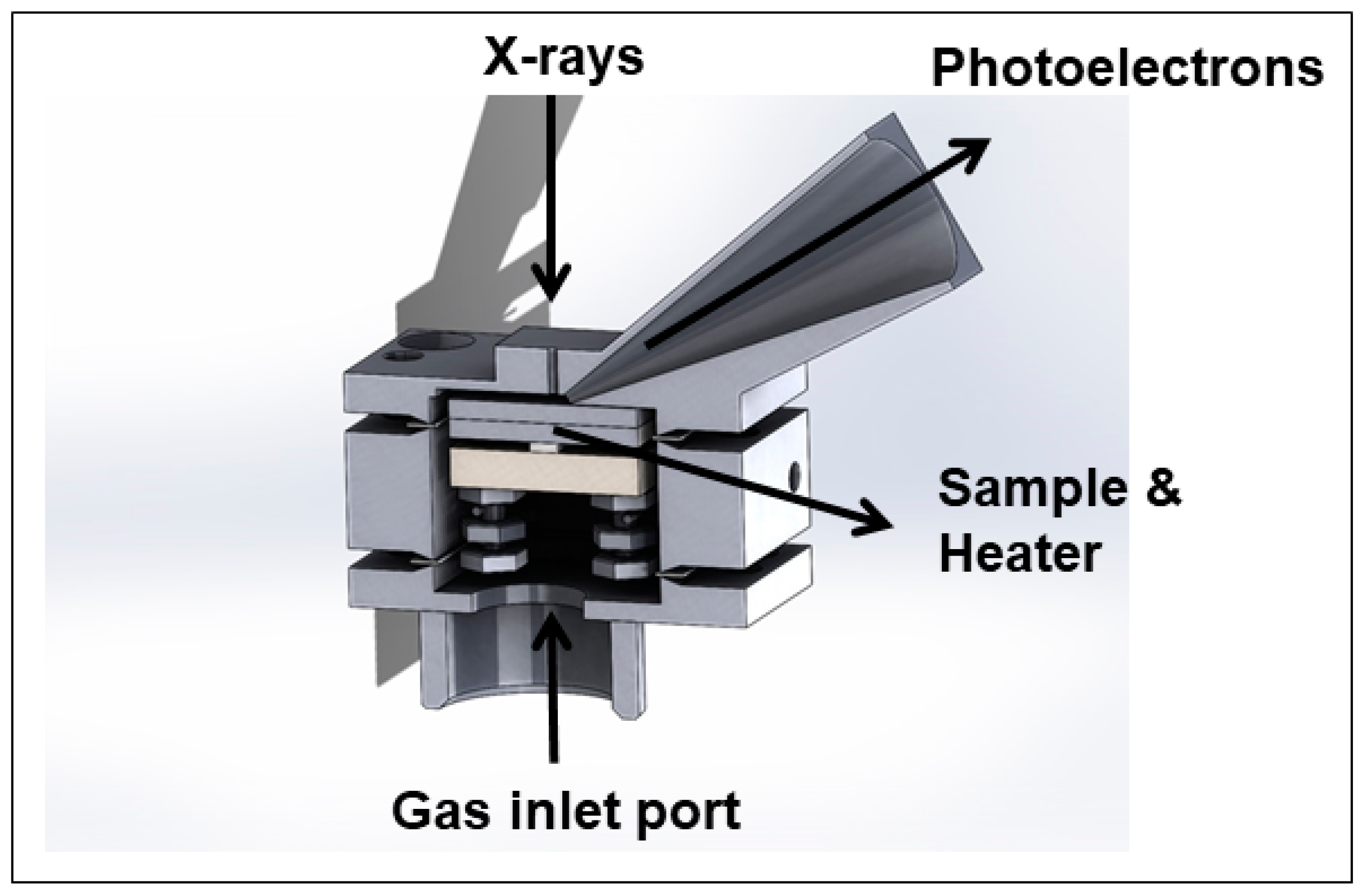
2.2. Novel Solutions for Operando Near-ambient Pressure Approaches
2.3. Sample Preparation
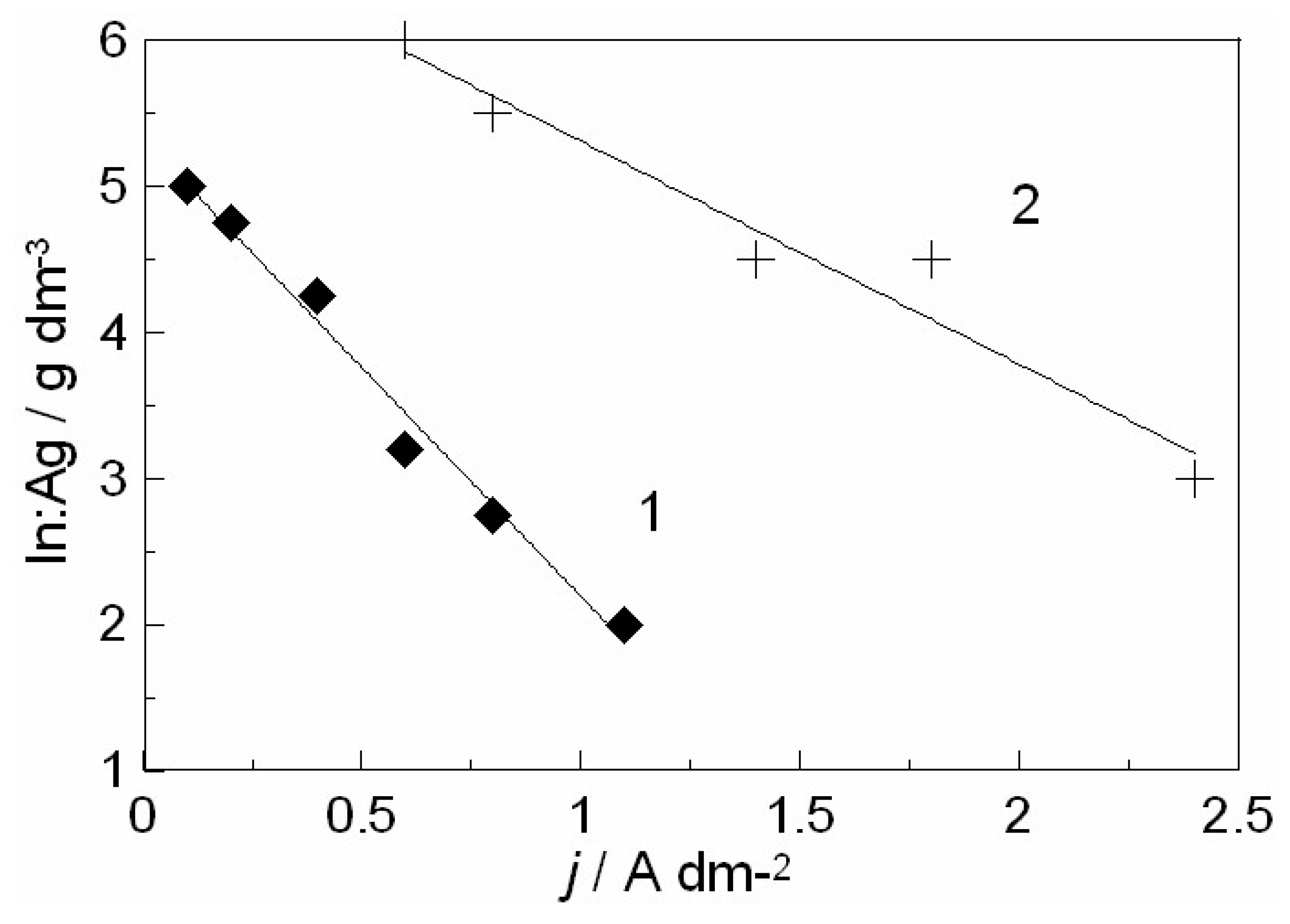
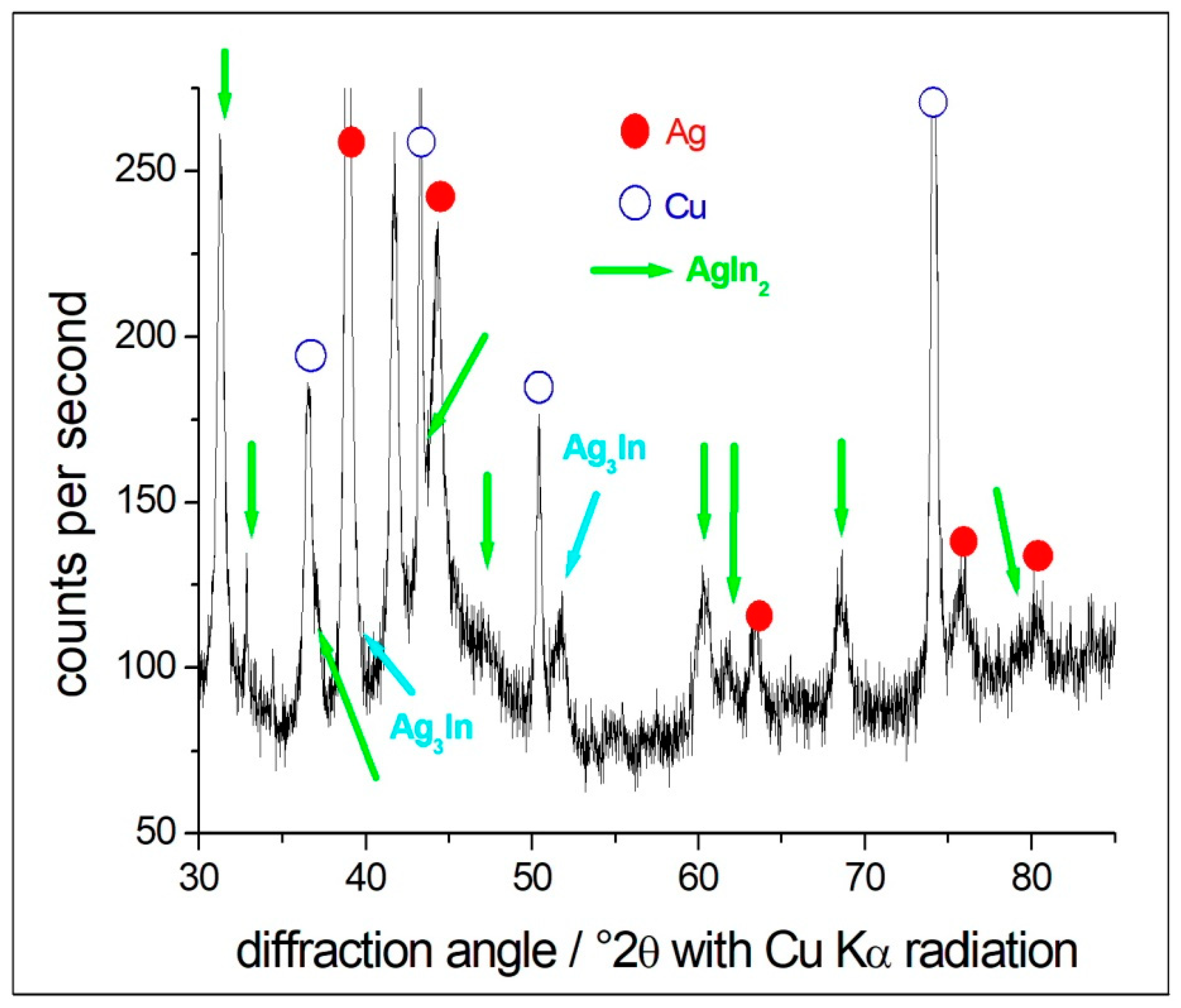
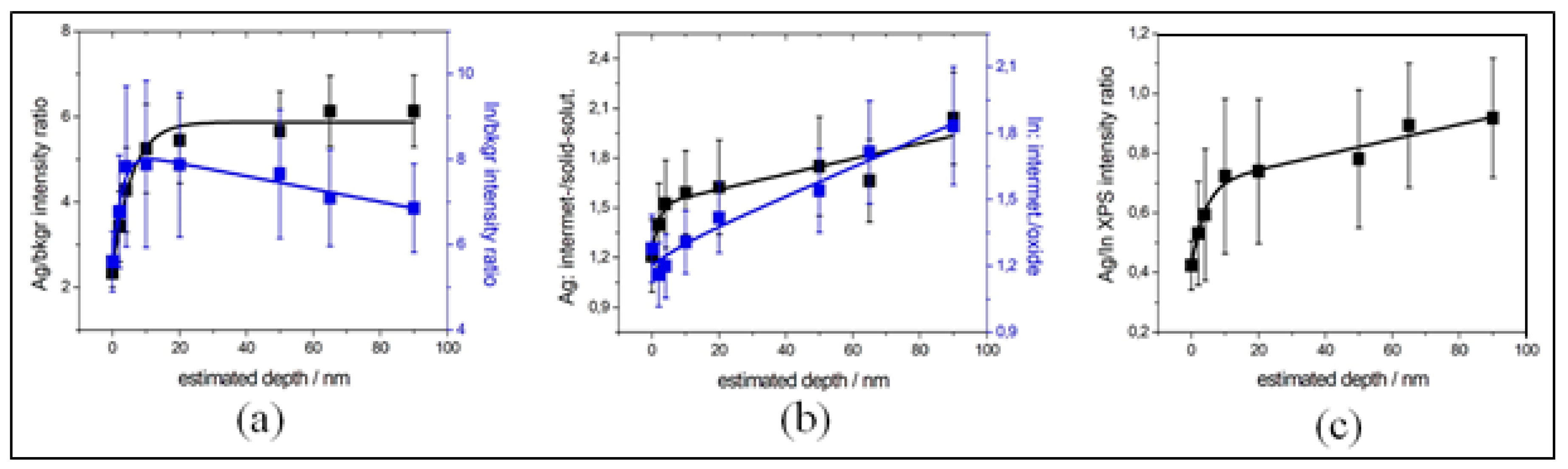
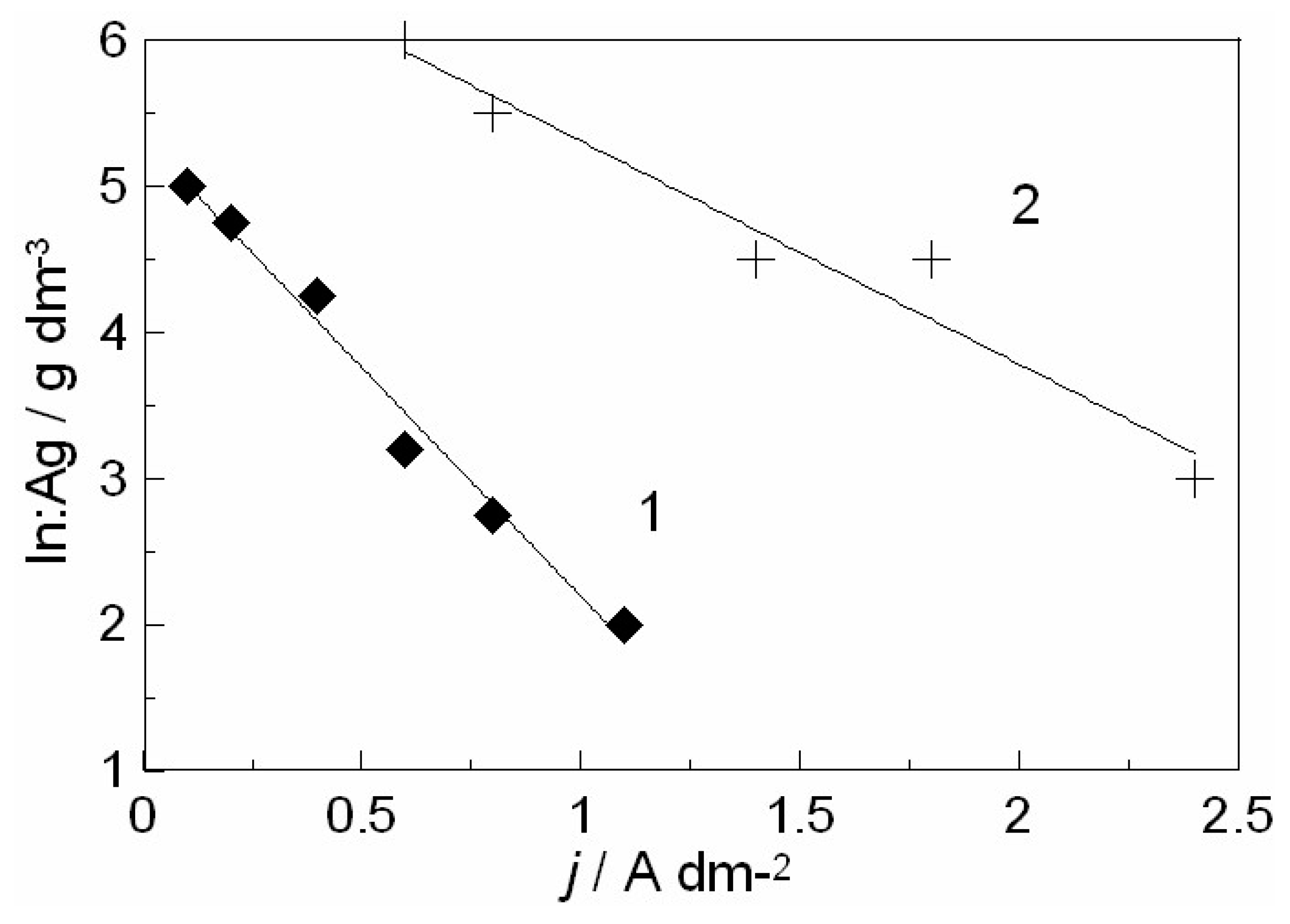
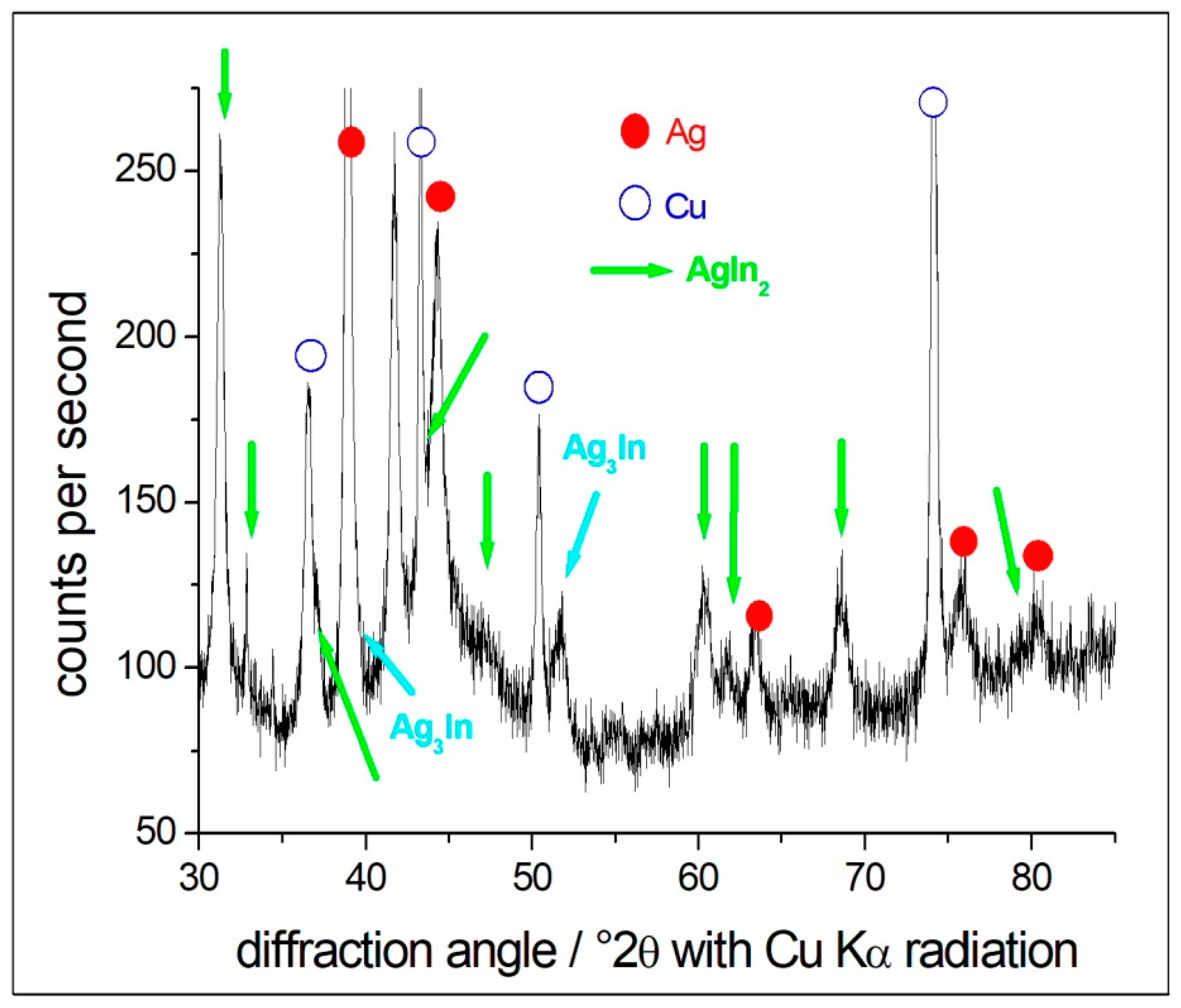
2.3.1. Ag-In Alloys
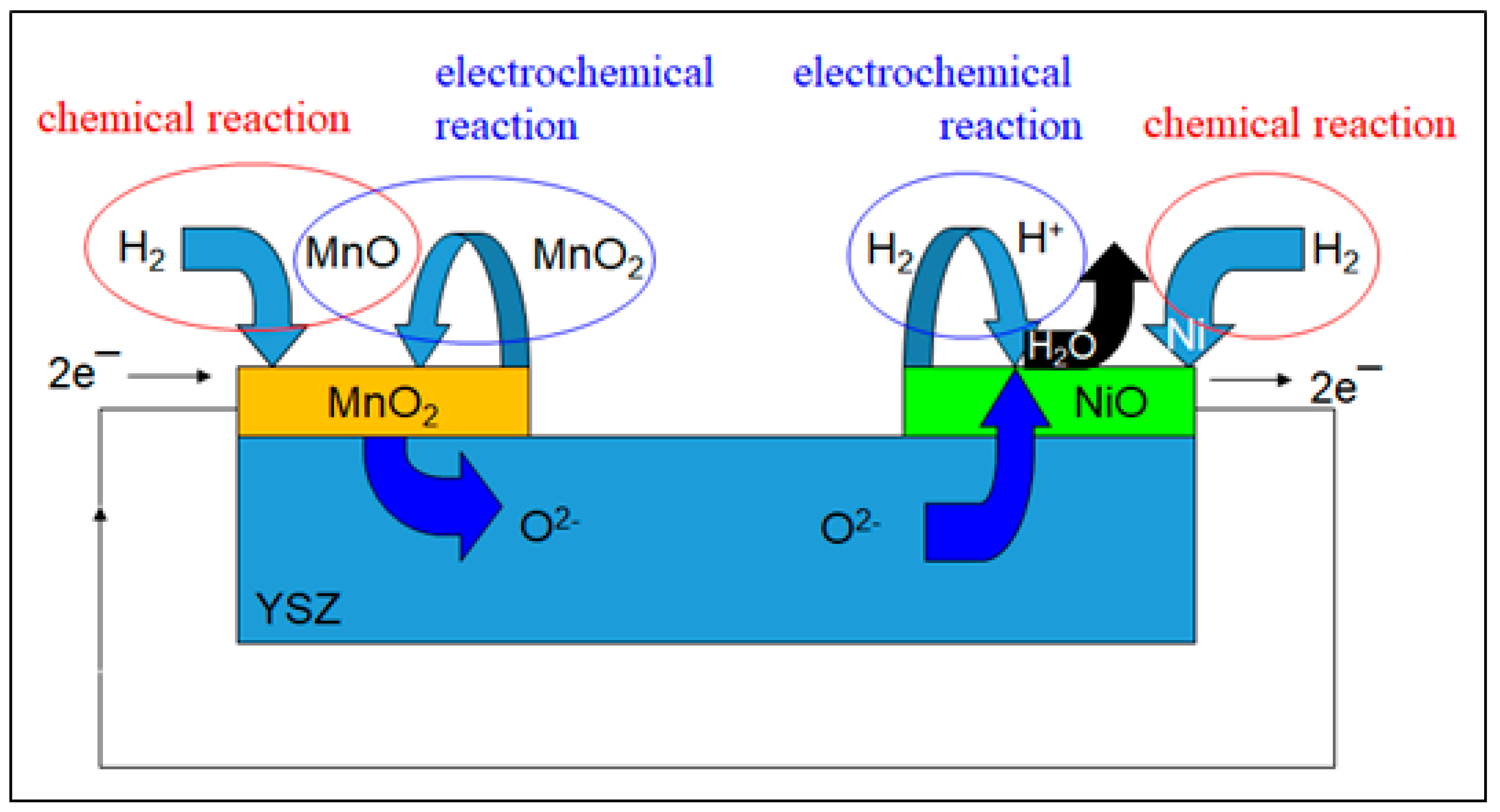
2.3.2. Mn-Ni SOFC at High Vacuum
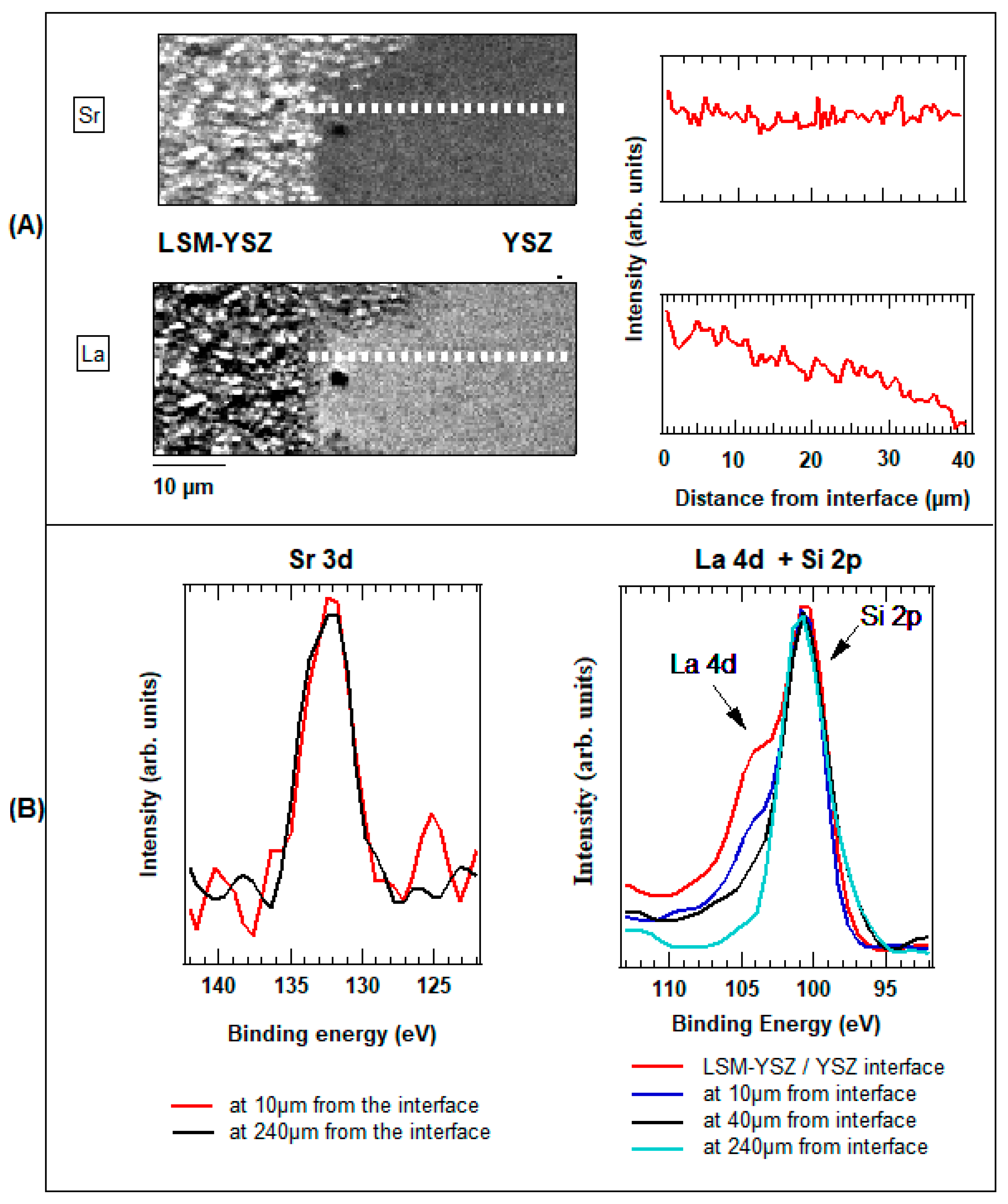
2.3.3. LSM-NiO Single Chamber SOFC
3. Results
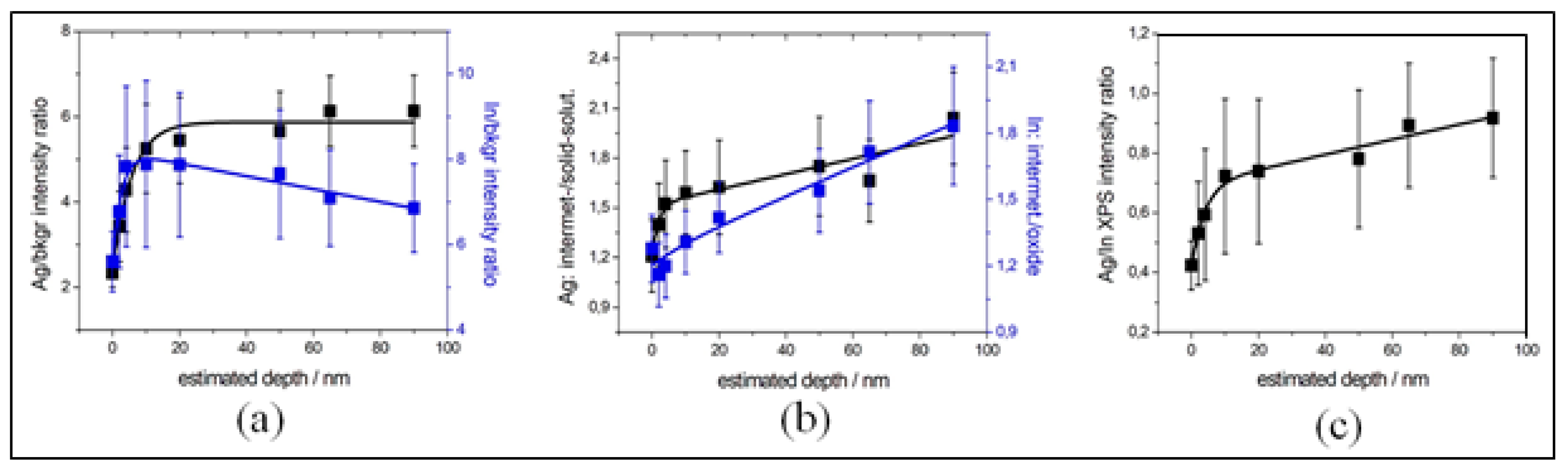
3.1. In-Depth Analysis of Ag-In Ex Situ Electrodeposited Alloys Exhibiting a Dynamic Pattern Formation Mechanism
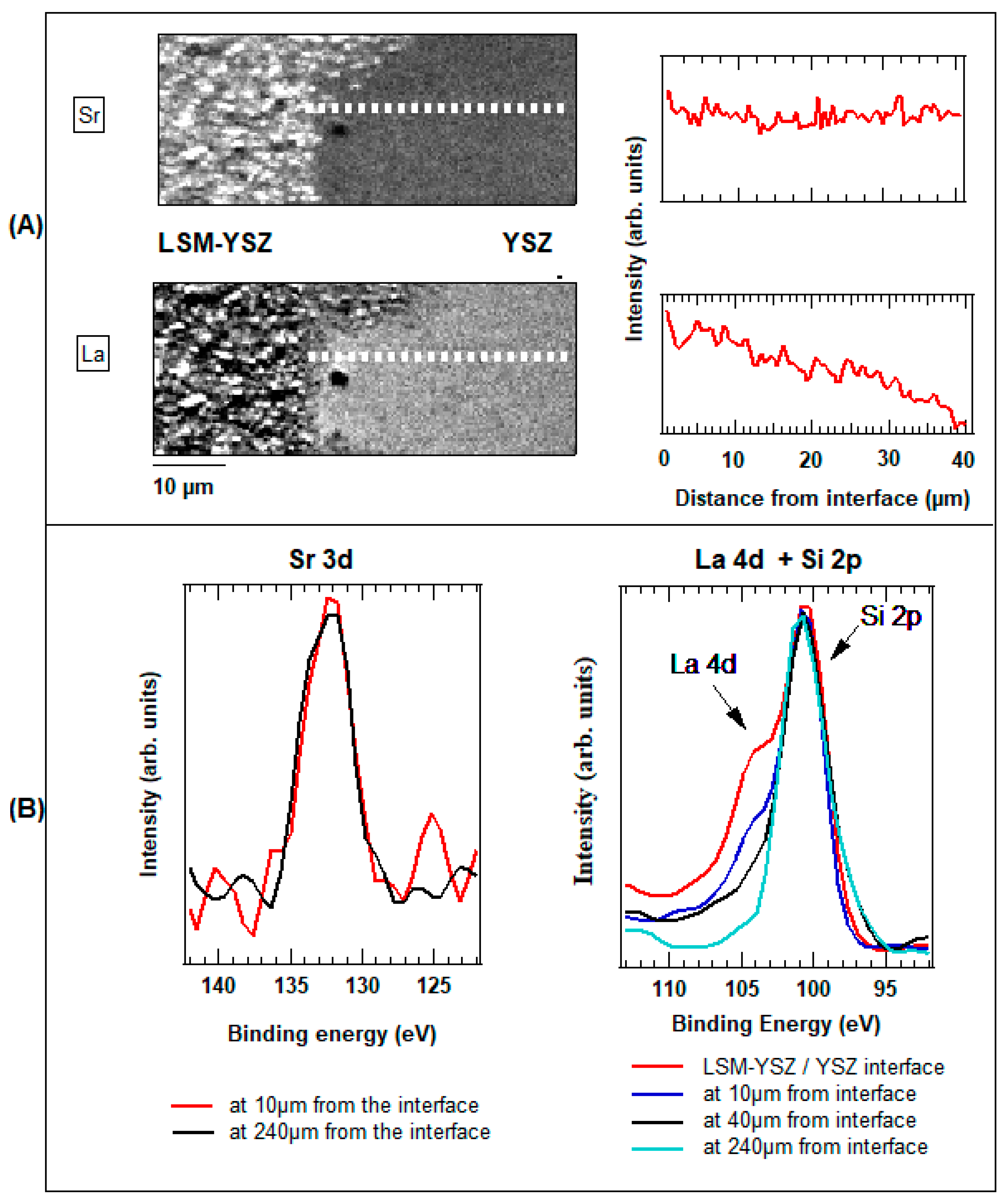
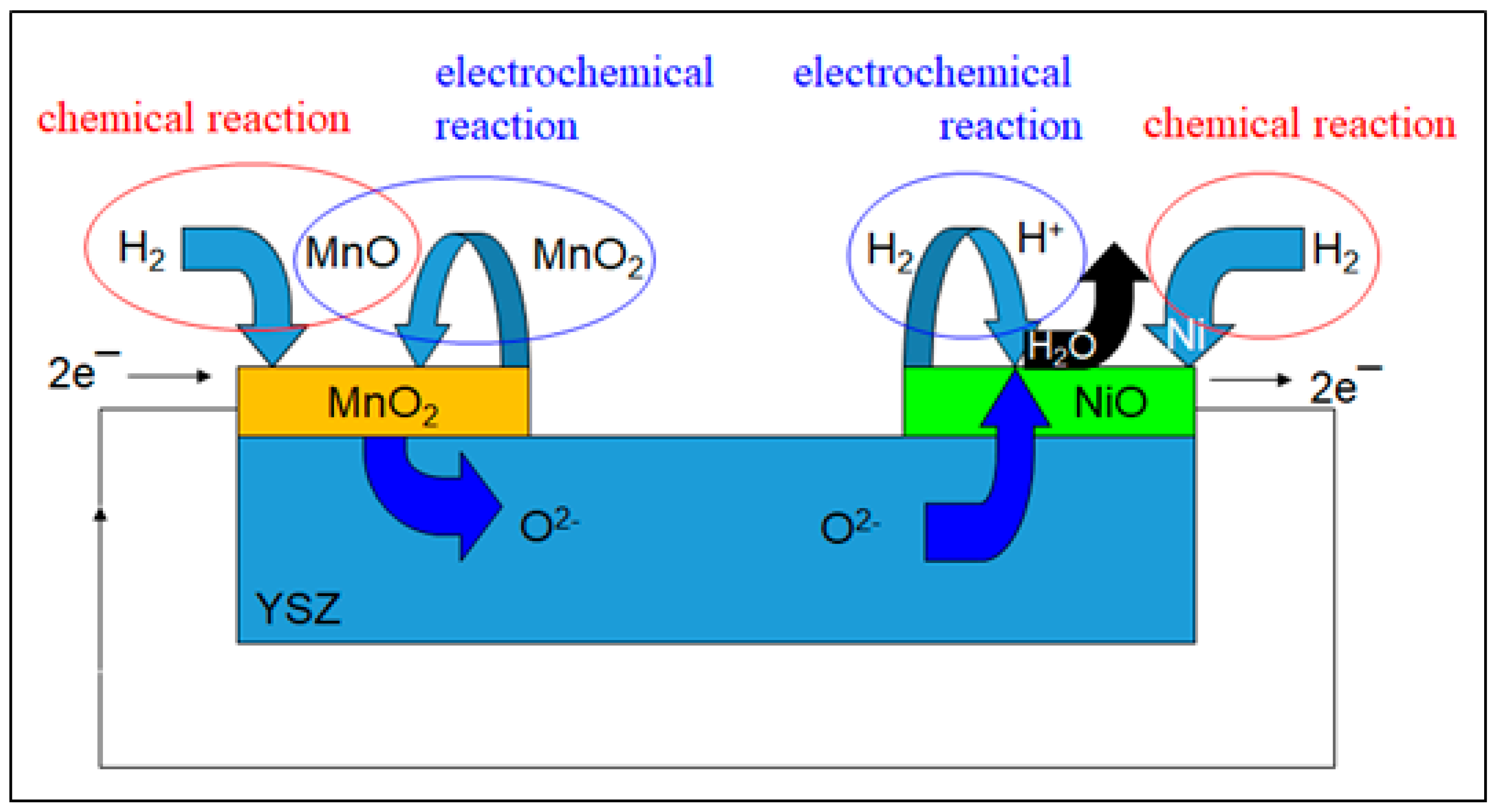
3.2. Spectromicroscopy Analysis of Electrochemical Processes at the Electrodes of a Self-driven Solid Oxide Fuel Cell
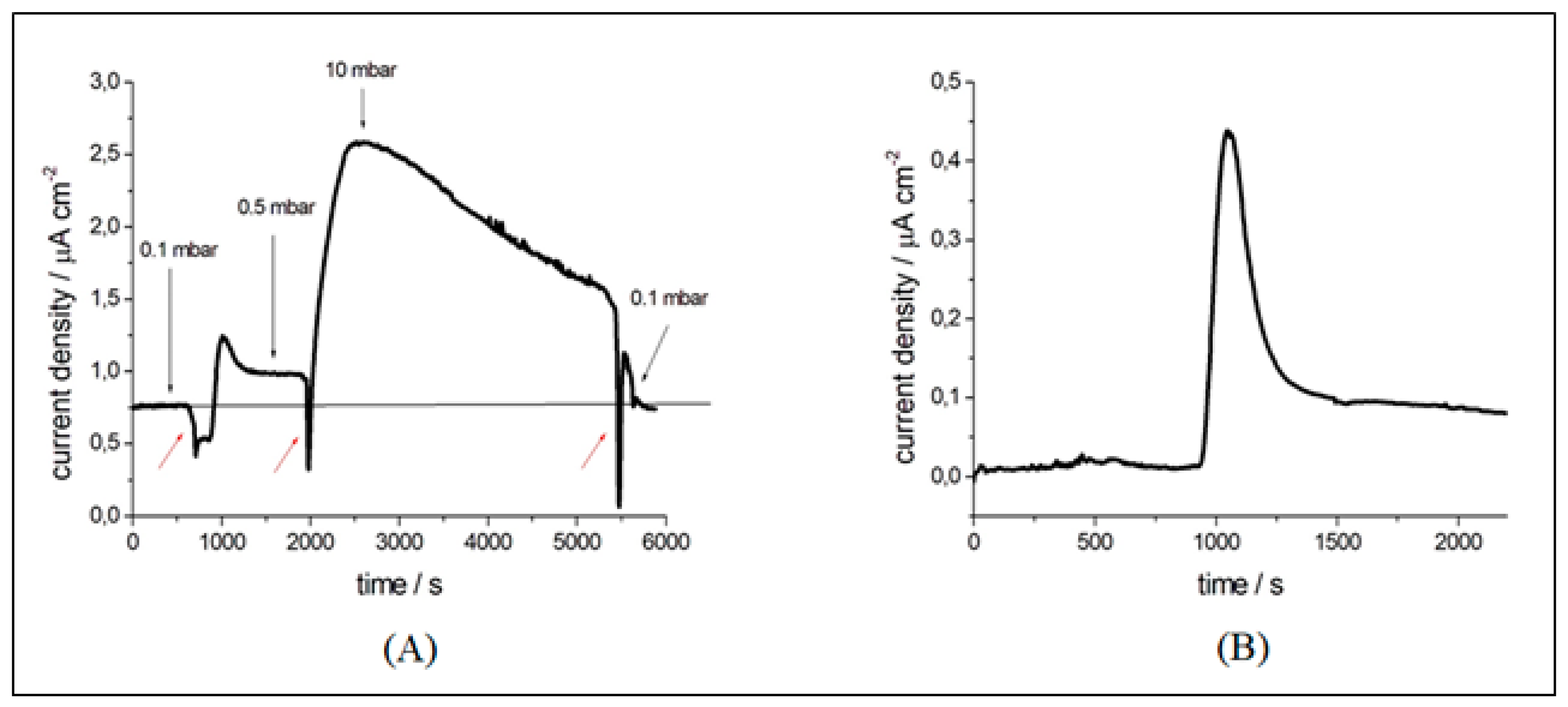
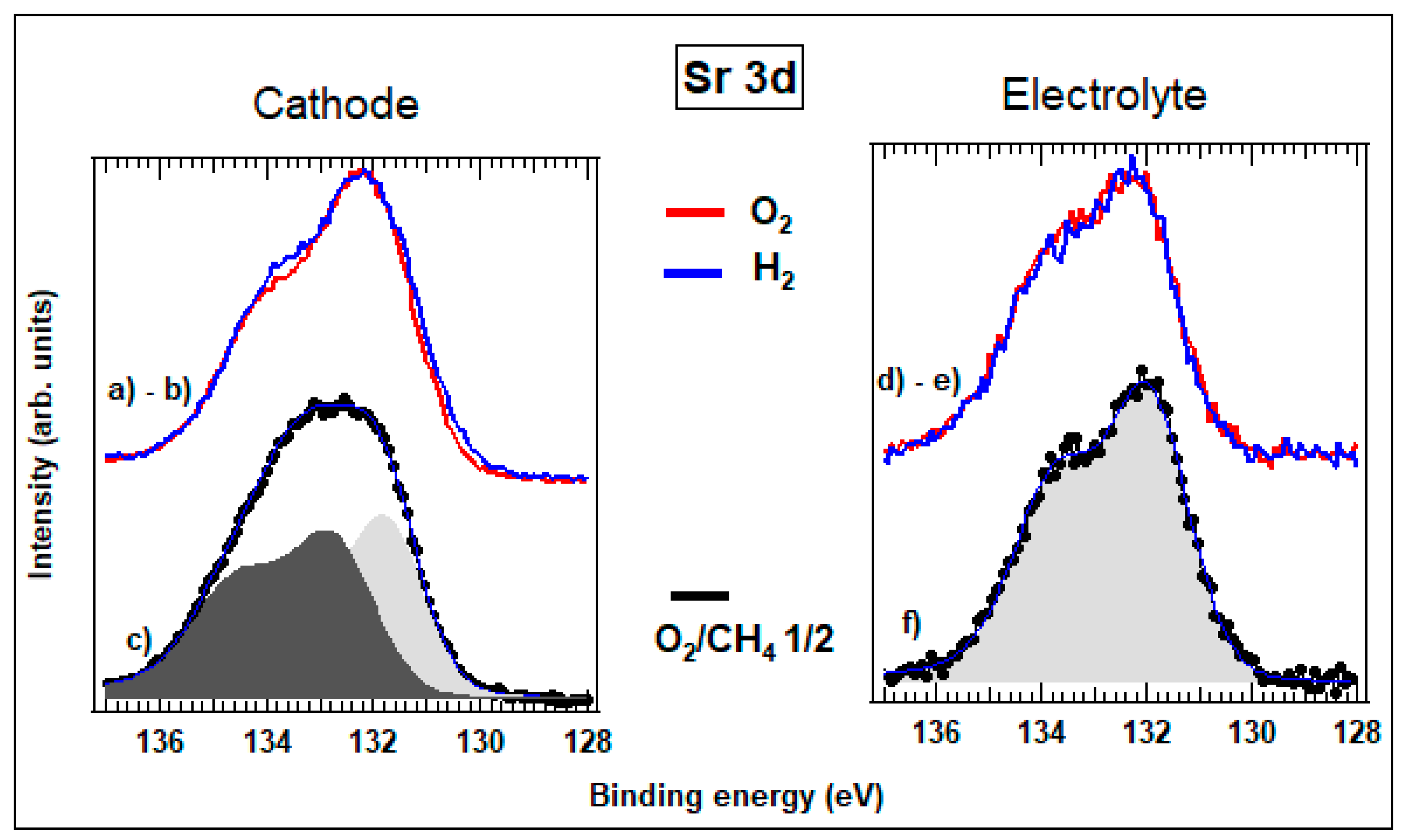
3.3. Operando Characterization of a Single-chamber Solid Oxide Fuel Cell at Near-ambient Pressure
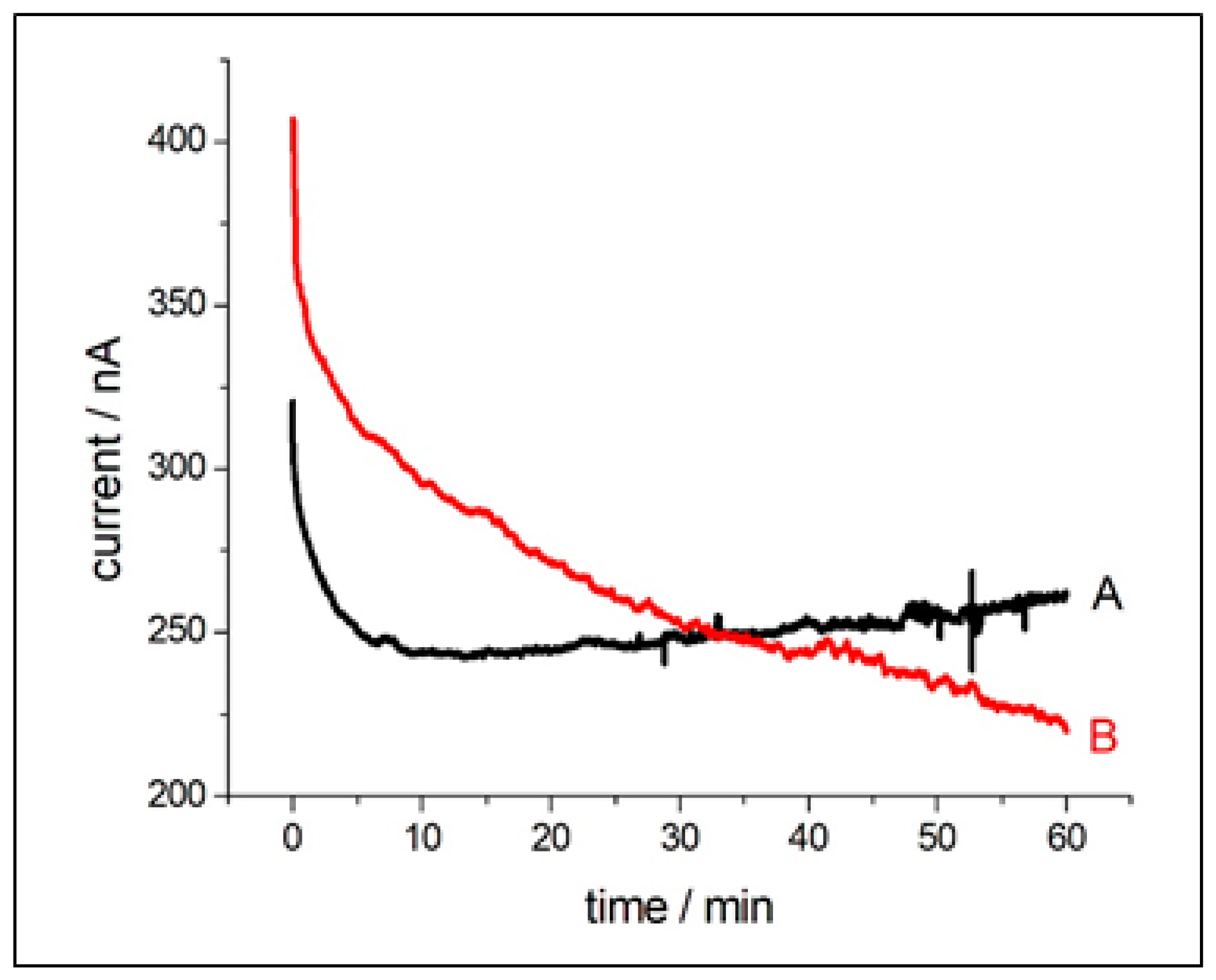
3.3.1. Electrochemical Measurements
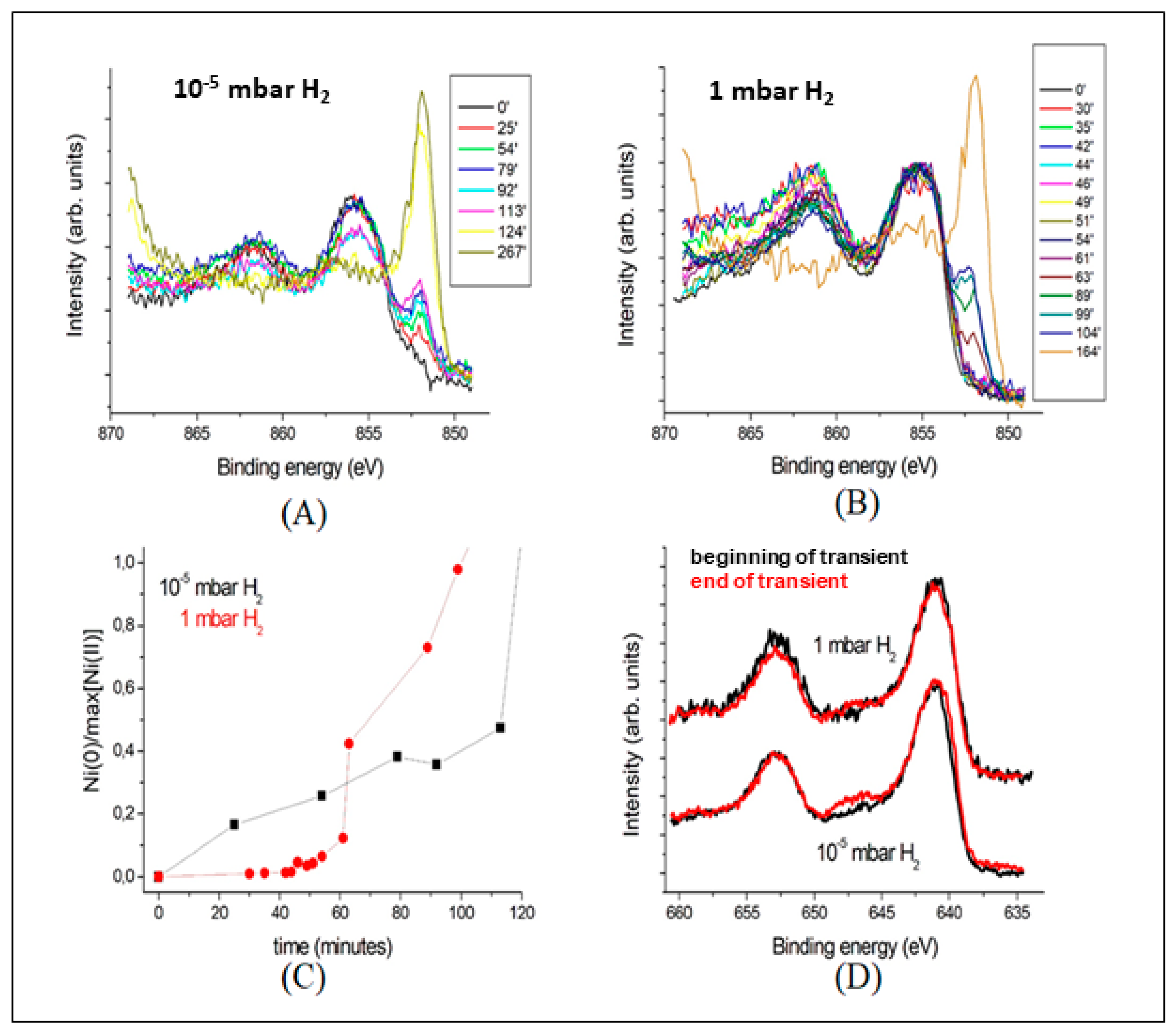
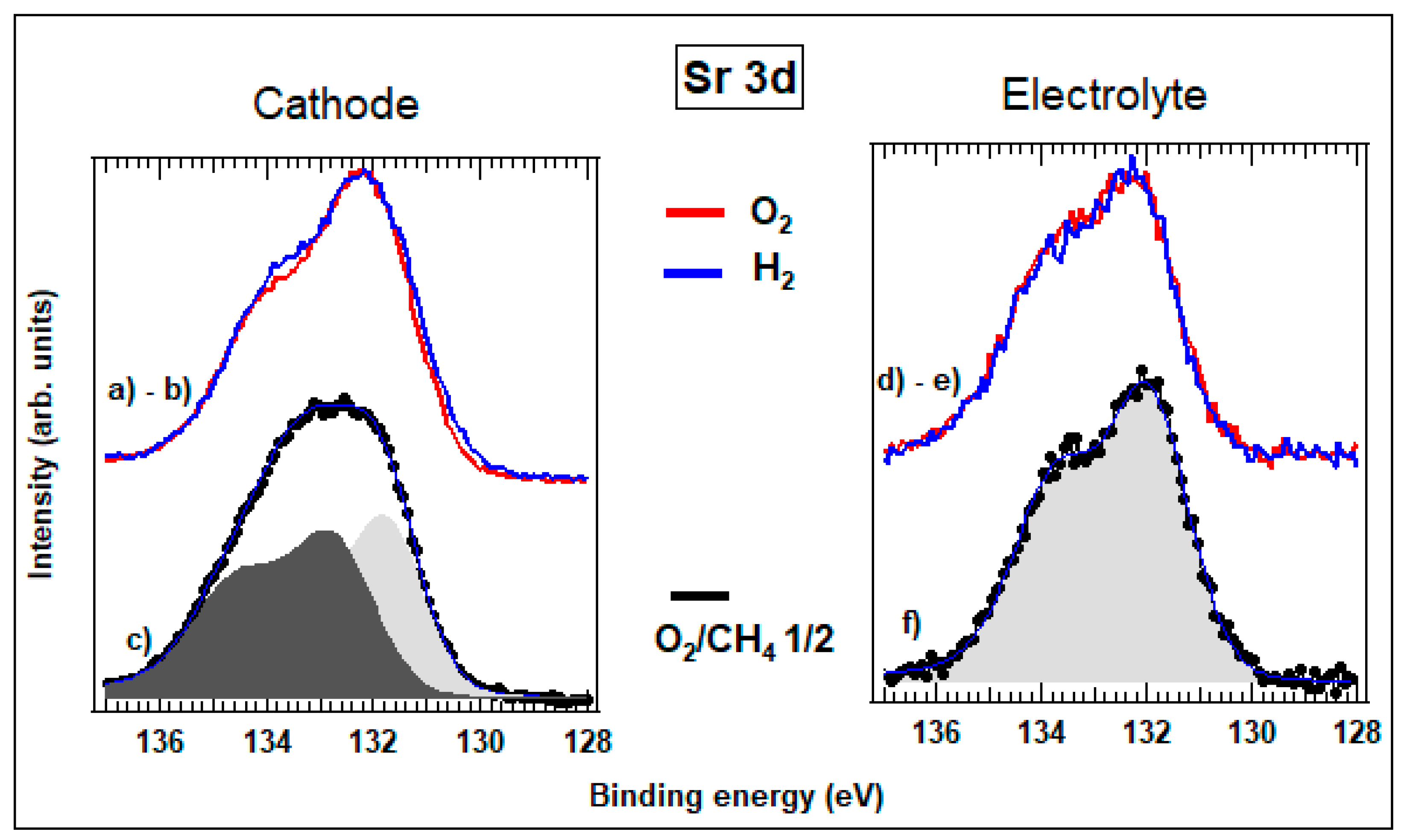
3.3.2. Spatially Resolved Photoemission Measurements at Near Ambient Pressure
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Nasirpouri, F. An Overview to Electrochemistry. In Electrodeposition of Nanostructured Materials; Springer Series in Surface Science 62; Springer: Berlin, Germany, 2017; Volume 62, pp. 43–73. [Google Scholar]
- Buckley, D.N.; O’Dwyer, C.; Quill, N.; Lynch, R.P. Electrochemical energy storage. In Energy Storage Options and Their Environmental Impact; Hester, R.E., Harrison, R.M., Eds.; The Royal Society of Chemistry: London, UK, 2019; pp. 115–149. [Google Scholar]
- Lin, F.; Liu, Y.; Yu, X.; Cheng, L.; Singer, A.; Shpyrko, O.G.; Xin, H.L.; Tamura, N.; Tian, C.; Weng, T.-C.; et al. Synchrotron X-ray Analytical Techniques for Studying Materials Electrochemistry in Rechargeable Batteries. Chem. Rev. 2017, 117, 13123–13186. [Google Scholar]
- Margaritondo, G.; Cerrina, F. Overview of soft-X-ray photoemission spectromicroscopy. Nuclear Instrum. Methods A 1990, 191, 26–35. [Google Scholar] [CrossRef]
- Ade, H.; Kirz, J.; Hulbert, S.; Johnson, E.; Anderson, E.; Kern, D. Scanning photoemission microscopy with synchrotron radiation. Vac. Sci. Technol. 1991, A9, 1902. [Google Scholar]
- Günther, S.; Kaulich, B.; Gregoratti, L.; Kiskinova, M. Photoelectron microscopy and applications in surface and materials science. Prog. Surf. Sci. 2002, 70, 187–260. [Google Scholar] [CrossRef]
- Amati, M.; Barinov, A.; Feyer, V.; Gregoratti, L.; Al-Hada, M.; Locatelli, A.; Mentes, T.O.; Sezen, H.; Schneider, C.M.; Kiskinova, M. Photoelectron microscopy at Elettra: Recent advances and perspectives. J. Electron. Spectrosc. Relat. Phenom. 2018, 224, 59–67. [Google Scholar] [CrossRef]
- Horiba, K.; Nakamura, Y.; Nagamura, N.; Toyoda, S.; Kumigashira, H.; Oshima, M.; Amemiya, K.; Senba, Y.; Ohashi, H. Scanning photoelectron microscope for nanoscale three-dimensional spatial-resolved electron spectroscopy for chemical analysis. Rev. Sci. Instrum. 2011, 82, 113701-1. [Google Scholar] [CrossRef]
- Bozzini, B.; Amati, M.; Gregoratti, L.; Kazemian, M.; Prasciolu, M.; Tondo, E.; Trygub, A.L.; Kiskinova, M. In Situ Electrochemical X-ray Spectromicroscopy Investigation of the Reduction/Reoxidation Dynamics of N-Cu Solid Oxide Fuel Cell Anodic Material in Contact with a Cr Interconnect in 2 × 10−6 mbar O2. J. Phys. Chem. C 2012, 116, 7243–7248. [Google Scholar] [CrossRef]
- Huber, A.; Falk, M.; Rohnke, M.; Luerssen, B.; Gregoratti, L.; Amati, M.; Janek, J. In situ study of electrochemical activation and surface segregation of the SOFC electrode material La0.75Sr0.25Cr0.5Mn0.5O3+/-δ. Phys. Chem. Chem. Phys. 2012, 14, 751–758. [Google Scholar] [CrossRef]
- Backhaus-Ricoult, M.; Adib, K.; Work, K.; Badding, M.; Ketcham, T.; Amati, M.; Gregoratti, L. In-situ scanning photoelectron microscopy study of operating (La,Sr)FeO3-based NOx-sensing surfaces. Solid State Ion. 2012, 225, 716–726. [Google Scholar] [CrossRef]
- Jirgensen, A.; Esser, N.; Hergenrçder, R. Near ambient pressure XPS with a conventional X-ray source. Surf. Interface Anal. 2012, 44, 1100–1103. [Google Scholar] [CrossRef]
- Bluhm, H.; Havecker, M.; Knop-Gericke, A.; Kiskinova, M.; Schlögl, R.; Salmeron, M. In Situ X-ray Photoelectron Spectroscopy Studies of Gas-Solid Interfaces at Near-Ambient Conditions. MRS Bull. 2007, 32, 1022–1030. [Google Scholar] [CrossRef]
- Crumlin, E.J.; Bluhm, H.; Liu, Z. In situ investigation of electrochemical devices using ambient pressure photoelectron spectroscopy. J. Electron. Spectrosc. Relat. Phenom. 2013, 190, 84–92. [Google Scholar] [CrossRef]
- Starr, D.E.; Liu, Z.; Hävecker, M.; Knop-Gericke, A.; Bluhm, H. Investigation of solid/vapor interfaces using ambient pressure X-ray photoelectron spectroscopy. Chem. Soc. Rev. 2013, 42, 5833–5857. [Google Scholar] [CrossRef]
- Crumlin, E.J.; Liu, Z.; Bluhm, H.; Yang, W.L.; Guo, J.H.; Hussain, Z. X-Ray Spectroscopy of Energy Materials under in Situ/Operando Conditions. J. Electron. Spectrosc. Relat. Phenom. 2015, 200, 264–273. [Google Scholar] [CrossRef]
- Takagi, Y.; Uruga, T.; Tada, M.; Iwasawa, Y.; Yokoyama, T. Pressure Hard X-ray Photoelectron Spectroscopy for Functional Material Systems as Fuel Cells under Working Conditions. Acc. Chem. Res. 2018, 51, 719–727. [Google Scholar] [CrossRef]
- Favaro, M.; Valero-Vidal, C.; Eichhorn, J.; Toma, F.M.; Ross, P.N.; Yano, J.; Liu, Z.; Crumlin, E.J. Elucidating the alkaline oxygen evolution reaction mechanism on platinum. J. Mater. Chem. A 2017, 5, 11634–11643. [Google Scholar] [CrossRef]
- Lichterman, M.F.; Richter, M.H.; Hu, S.; Crumlin, E.J.; Axnanda, S.; Favaro, M.; Drisdell, W.; Hussain, Z.; Brunschwig, B.S.; Lewis, N.S.; et al. An electrochemical, microtopographical and ambient pressure X-ray photoelectron spectroscopic investigation of Si/TiO2/Ni/Electrolyte Interfaces. J. Electrochem. Soc. 2016, 163, H139–H146. [Google Scholar] [CrossRef]
- Sezen, H.; Alemán, B.; Amati, M.; Dalmiglio, M.; Gregoratti, L. Spatially Resolved Chemical Characterization with Scanning Photoemission Spectromicroscopy: Towards Near-Ambient-Pressure Experiments. ChemCatChem 2015, 7, 3665–3673. [Google Scholar] [CrossRef]
- Amati, M.; Abyaneh, K.M.; Gregoratti, L. Dynamic High Pressure: A novel approach toward near ambient pressure photoemission spectroscopy and spectromicroscopy. J. Instrum. 2013, 8, T05001. [Google Scholar] [CrossRef]
- Gregoratti, L.; Barinov, A.; Benfatto, E.; Cautero, G.; Fava, C.; Lacovig, P.; Lonza, D.; Kiskinova, M.; Tommasini, R.; Mahl, S. 48-Channel electron detector for photoemission spectroscopy and microscopy. Rev. Sci. Instrum. 2004, 75, 64–68. [Google Scholar] [CrossRef]
- Miller, D.R. Atomic and Molecular Beam Methods, 1st ed.; Oxford University Press: Oxford, UK, 1988. [Google Scholar]
- Soukhanovskii, V.A.; Kugel, H.W.; Kaita, R.; Majeski, R.; Roquemore, A.L. Supersonic gas injector for fueling and diagnostic applications on the national spherical torus experiment. Rev. Sci. Instrum. 2004, 75, 4320–4323. [Google Scholar] [CrossRef]
- Piseri, P.; Podestà, A.; Barborini, E.; Milani, P. Production and characterization of highly intense and collimated cluster beams by inertial focusing in supersonic expansions. Rev. Sci. Instrum. 2001, 72, 2261–2267. [Google Scholar] [CrossRef]
- Bozzini, B.; Amati, M.; Dobrovolska, T.; Gregoratti, L.; Krastev, I.; Sgura, I.; Taurino, A.; Kiskinova, M. Depth-dependent scanning photoelectron microspectroscopy unravels the origin of dynamic pattern formation in alloy electrodeposition. J. Phys. Chem. C 2018, 112, 15996–16007. [Google Scholar] [CrossRef]
- Dobrovolska, T.; Jovic, V.D.; Jovic, B.M.; Krastev, I. Phase Identification in Electrodeposited Ag-In Alloys by ALSV technique. J. Electroanal. Chem. 2007, 611, 232–240. [Google Scholar] [CrossRef]
- Dobrovolska, T.; Beck, G.; Krastev, I.; Zielonka, A. Phase Composition of Electrodeposited Silver-Indium Alloys. J. Solid State Electrochem. 2008, 12, 1461–1467. [Google Scholar] [CrossRef]
- Briggs, D.; Seah, M.P. Practical Surface Analysis, Auger and X-ray Photoelectron Spectroscopy, 2nd ed.; Wiley: Chichester, UK, 1990; Volume 1. [Google Scholar]
- Bozzini, B. Effect of Sputter-Induced and other Roughness on Auger Electron Intensities. Il Vuoto 1997, 26, 18–29. [Google Scholar]
- Bozzini, B.; Amati, M.; Gregoratti, L.; Kiskinova, M. In-situ Photoelectron Microspectroscopy and Imaging of Spontaneous Electrochemical Processes at the Electrodes of a Self-Driven Cell. Sci. Rep. 2013, 3, 2848–2852. [Google Scholar] [CrossRef]
- Bozzini, B.; Kuscer, D.; Drnovšek, S.; Al-Hada, M.; Amati, M.; Sezen, H.; Gregoratti, L. Spatially resolved photoemission and electrochemical characterization of a single-chamber solid oxide fuel cell. Top. Catal. 2018, 61, 2185–2194. [Google Scholar] [CrossRef]
- Yano, M.; Tomita, A.; Sano, M.; Hibino, T. Recent advances in single-chamber solid oxide fuel cells: A review. Solid State Ion. 2007, 77, 3351–3359. [Google Scholar] [CrossRef]
- Kuhn, M.; Napporn, T.W. Single-Chamber Solid Oxide Fuel Cell Technology—From Its Origins to Today’s State of the Art. Energies 2010, 3, 57–134. [Google Scholar] [CrossRef]
- Hibino, T. Single Chamber Solid Oxide Fuel Cell. In Encyclopedia of Applied Electrochemistry; Kreysa, G., Ota, K., Savinell, R.F., Eds.; Springer: New York, NY, USA, 2014. [Google Scholar]
- Bozzini, B.; Amati, M.; Gregoratti, L.; Lacitignola, D.; Sgura, I.; Krastev, I.; Dobrovolska, T. Intermetallics as key to Spiral Formation in In-Co Electrodeposition. A Study based on Photoelectron Microspectroscopy, Mathematical Modelling and Numerical Approximations. J. Phys. D 2015, 48, 395502. [Google Scholar] [CrossRef]
- Lacitignola, D.; Bozzini, B.; Peipmann, R.; Sgura, I. Cross-diffusion effects on a morphochemical model for electrodeposition. Appl. Math. Model. 2018, 57, 492–513. [Google Scholar] [CrossRef]
- Zhang, C.; Zheng, Y.; Ran, R.; Shao, Z.; Jin, W.; Xu, N.; Ahn, J. Initialization of a methane-fueled single-chamber solid-oxide fuel cell with NiO+ SDC anode and BSCF+ SDC cathode. J. Power Sources 2008, 179, 640–648. [Google Scholar] [CrossRef]
- Lacitignola, D.; Bozzini, B.; Sgura, I. Spatio-temporal organization in a morphochemical electrodeposition model: Analysis and numerical simulation of spiral waves. Acta Appl. Math. 2014, 132, 377–389. [Google Scholar] [CrossRef]
- Lacitignola, D.; Bozzini, B.; Frittelli, M.; Sgura, I. Turing Pattern Formation on the Sphere for a Morphochemical Reaction-Diffusion Model for Electrodeposition. Commun. Nonlinear Sci. Numer. Simul. 2017, 48, 484–508. [Google Scholar] [CrossRef]
- Zhang, C.; Lin, Y.; Ran, R.; Shao, Z. Activation of a single-chamber solid oxide fuel cell by a simple catalyst-assisted in-situ process. Electrochem. Commun. 2009, 11, 1563–1566. [Google Scholar] [CrossRef]
- Idris, M.A.; Bak, T.; Li, S.; Nowotny, J. Effect of Segregation on Surface and Near-Surface Chemistry of Yttria-Stabilized Zirconia. J. Phys. Chem. C. 2012, 116, 10950–10958. [Google Scholar] [CrossRef]
- Han, S.W.; Lee, J.D.; Kim, K.H.; Song, H.; Kim, W.J.; Kwon, J.; Lee, H.G.; Hwang, C.; Jeong, J.I.; Kang, J.S. Electronic structures of the CMR perovskites R1−xAxMnO3 (R = La, Pr; A = Ca, Sr, Ce) using photoelectron spectroscopy. J. Korean Phys. Soc. 2002, 40, 501–510. [Google Scholar]
- Huber, A.K.; Falk, M.; Rohnke, M.; Luerssen, B.; Amati, M.; Gregoratti, L.; Hesse, D.; Janek, J. In situ study of activation and de-activation of LSM fuel cell cathodes—Electrochemistry and surface analysis of thin-film electrodes. J. Catal. 2012, 294, 79–88. [Google Scholar] [CrossRef]
- Konysheva, E.Y.; Kuznetsov, M.V. Fluctuation of surface composition and chemical states at the hetero-interface in composites comprised of a phase with perovskite structure and a phase related to the Ruddlesden-Popper family of compounds. RSC Adv. 2013, 3, 14114–14122. [Google Scholar] [CrossRef]
- Crumlin, E.J.; Mutoro, E.; Liu, Z.; Grass, M.E.; Biegalski, M.D.; Lee, Y.L.; Morgan, D.; Christen, H.M.; Bluhm, E.; Shao-Horn, Y. Surface strontium enrichment on highly active perovskites for oxygen electrocatalysis in solid oxide fuel cells. Energy Environ. Sci. 2012, 5, 6081–6086. [Google Scholar] [CrossRef]
- Aphale, A.; Liang, C.; Hu, B.; Singh, P. Cathode degradation from airborne contaminants in solid oxide fuel cells: A Review. In Solid Oxide Fuel Cells Lifetime and Reliability, 1st ed.; Brandon, N.P., Ruiz-Trejo, E., Boldrin, P., Eds.; Academic Press: London, UK, 2017; pp. 106–108. [Google Scholar]
- Chen, K.; Hyodo, J.; Al, N.; Ishihara, T.; Jiang, S.P. Boron deposition and poisoning of La0.8Sr0.2MnO3 oxygen electrodes of solid oxide electrolysis cells under accelerated operation conditions. Int. J. Hydrogen Energy 2016, 41, 1419–1431. [Google Scholar] [CrossRef]
- Hu, B.; Keane, M.; Mahapatra, M.K.; Singh, P. Stability of strontium-doped lanthanum manganite cathode in humidified air. J. Power Sources 2014, 248, 196–204. [Google Scholar] [CrossRef]
- Wang, W.; Jiang, S.P. A mechanistic study on the activation process of (La, Sr)MnO3 electrodes of solid oxide fuel cells. Solid State Ion. 2006, 177, 1361–1369. [Google Scholar] [CrossRef]





© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bozzini, B.; Kuscer, D.; Amati, M.; Gregoratti, L.; Zeller, P.; Dobrovolska, T.; Krastev, I. Spatially Resolved XPS Characterization of Electrochemical Surfaces. Surfaces 2019, 2, 295-314. https://doi.org/10.3390/surfaces2020022
Bozzini B, Kuscer D, Amati M, Gregoratti L, Zeller P, Dobrovolska T, Krastev I. Spatially Resolved XPS Characterization of Electrochemical Surfaces. Surfaces. 2019; 2(2):295-314. https://doi.org/10.3390/surfaces2020022
Chicago/Turabian StyleBozzini, Benedetto, Danjela Kuscer, Matteo Amati, Luca Gregoratti, Patrick Zeller, Tsvetina Dobrovolska, and Ivan Krastev. 2019. "Spatially Resolved XPS Characterization of Electrochemical Surfaces" Surfaces 2, no. 2: 295-314. https://doi.org/10.3390/surfaces2020022
APA StyleBozzini, B., Kuscer, D., Amati, M., Gregoratti, L., Zeller, P., Dobrovolska, T., & Krastev, I. (2019). Spatially Resolved XPS Characterization of Electrochemical Surfaces. Surfaces, 2(2), 295-314. https://doi.org/10.3390/surfaces2020022