Methanol Oxidation on Graphenic-Supported Platinum Catalysts

 ,
, 

Abstract
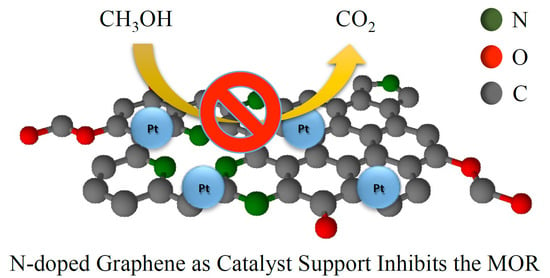
:
1. Introduction
2. Materials and Methods
2.1. Synthesis of Graphene Oxide
2.2. Synthesis of Reduced Graphene Oxide Materials
2.3. Synthesis of Pt/C, Pt/N-rGO, and Pt/rGO Catalysts
2.4. Physicochemical Characterization
2.5. Electrochemical Characterization
3. Results and Discussion
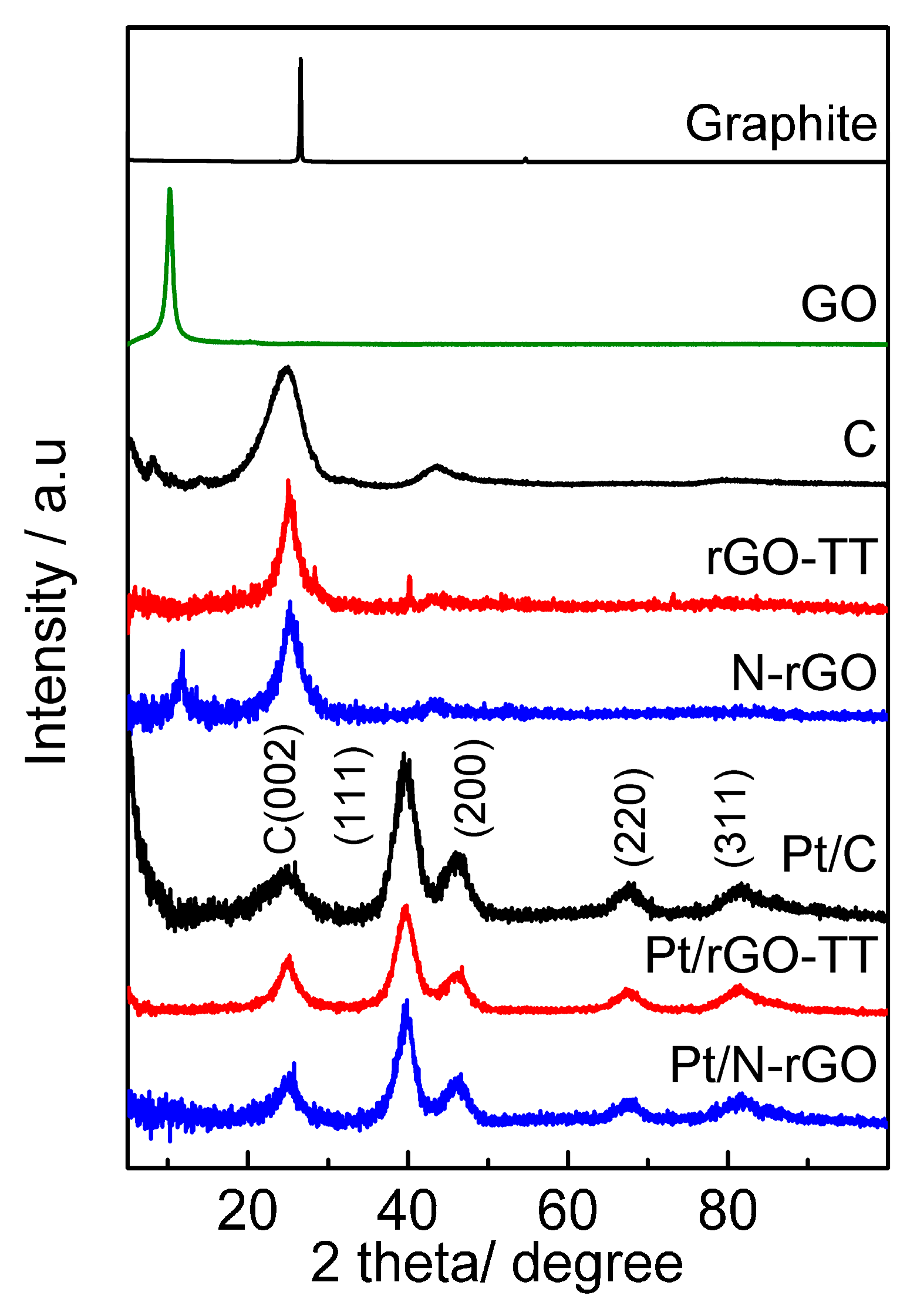
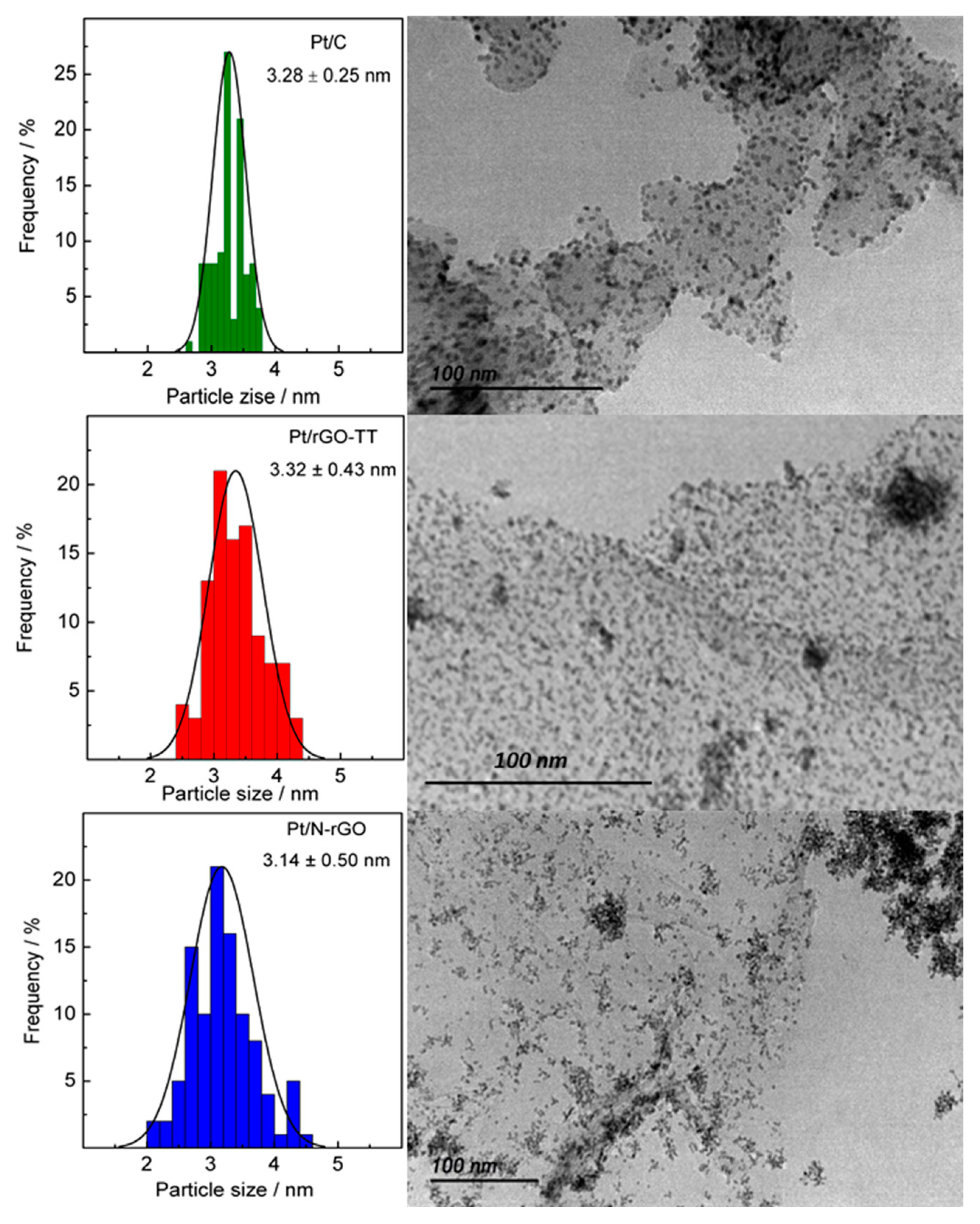
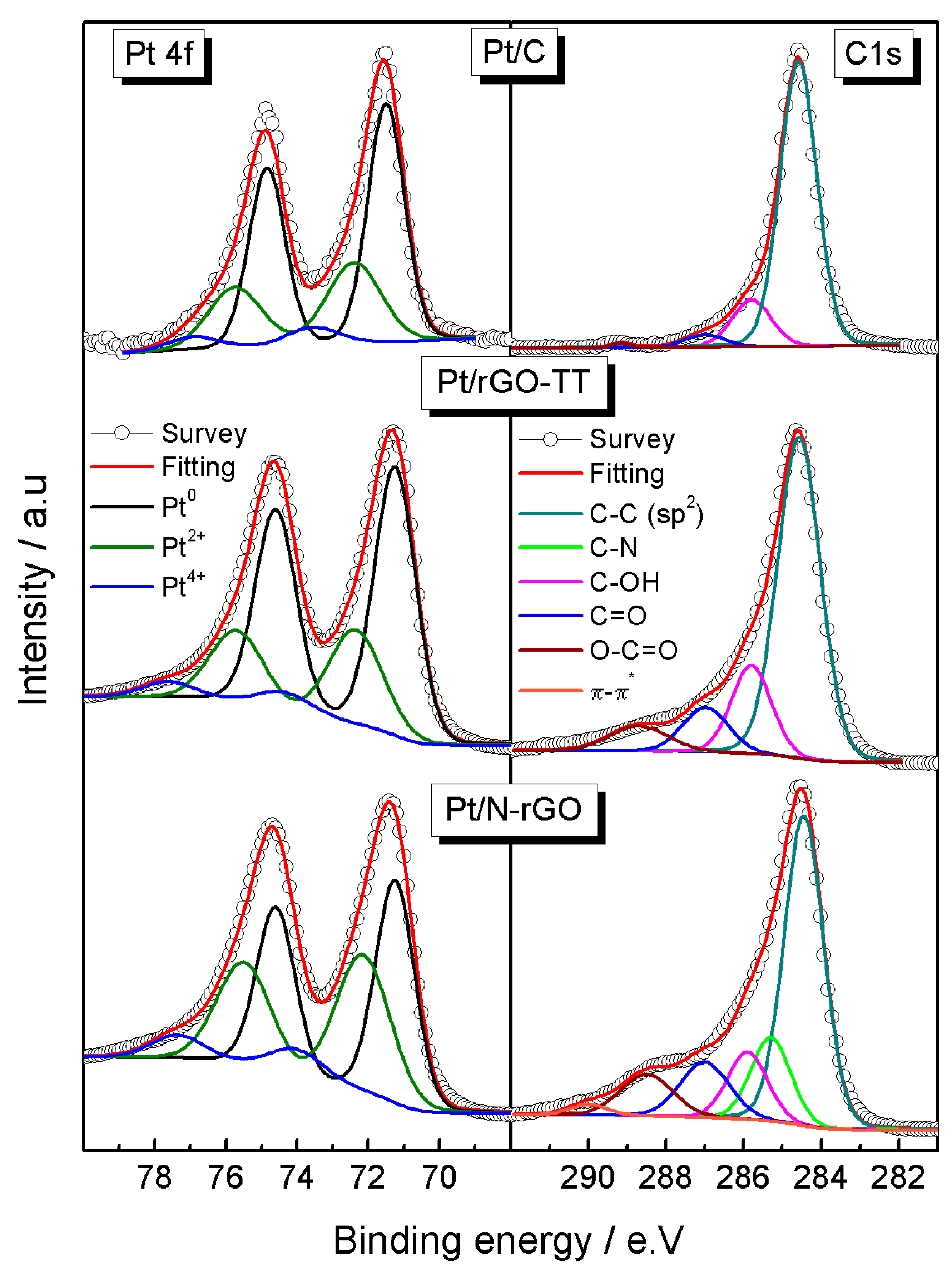
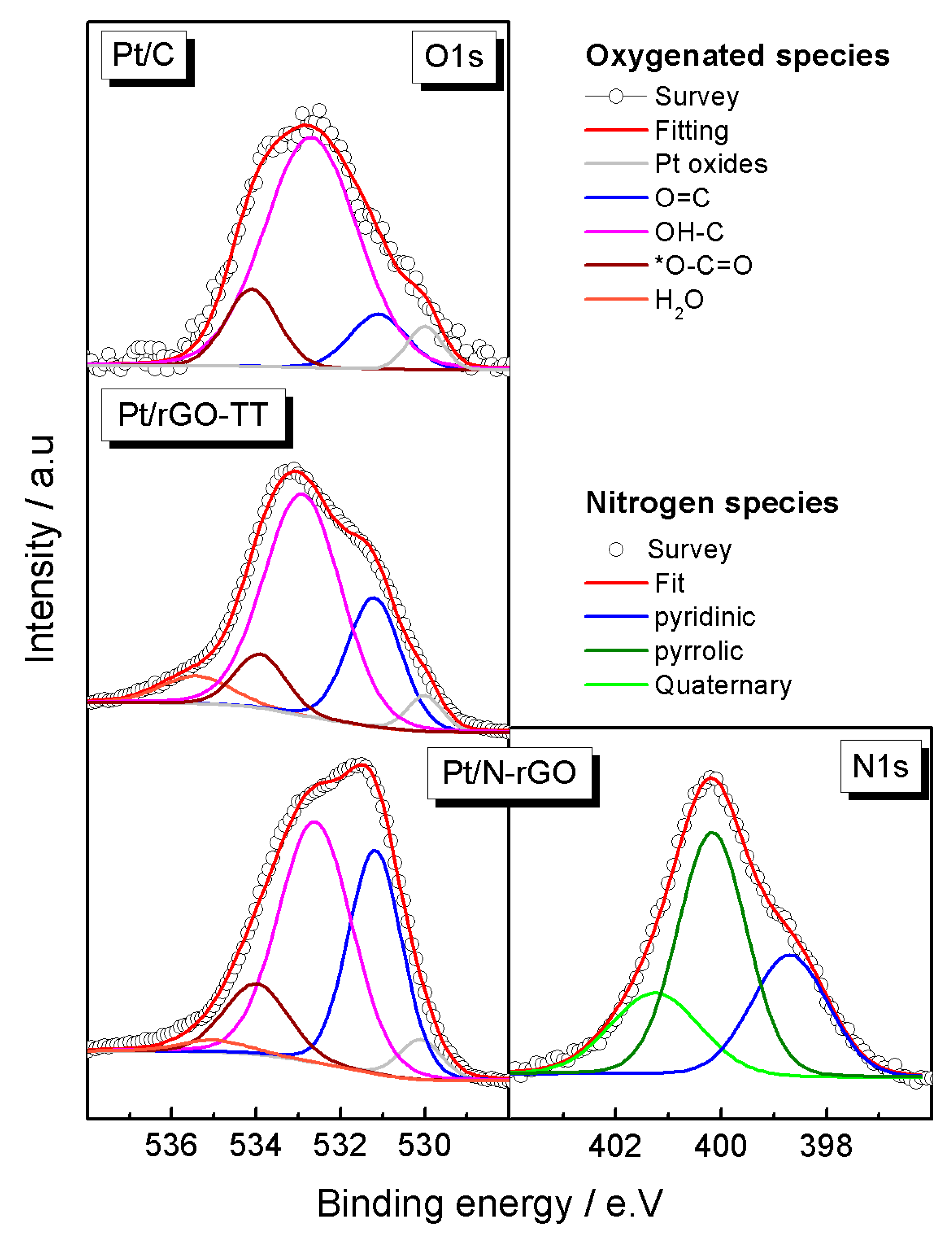
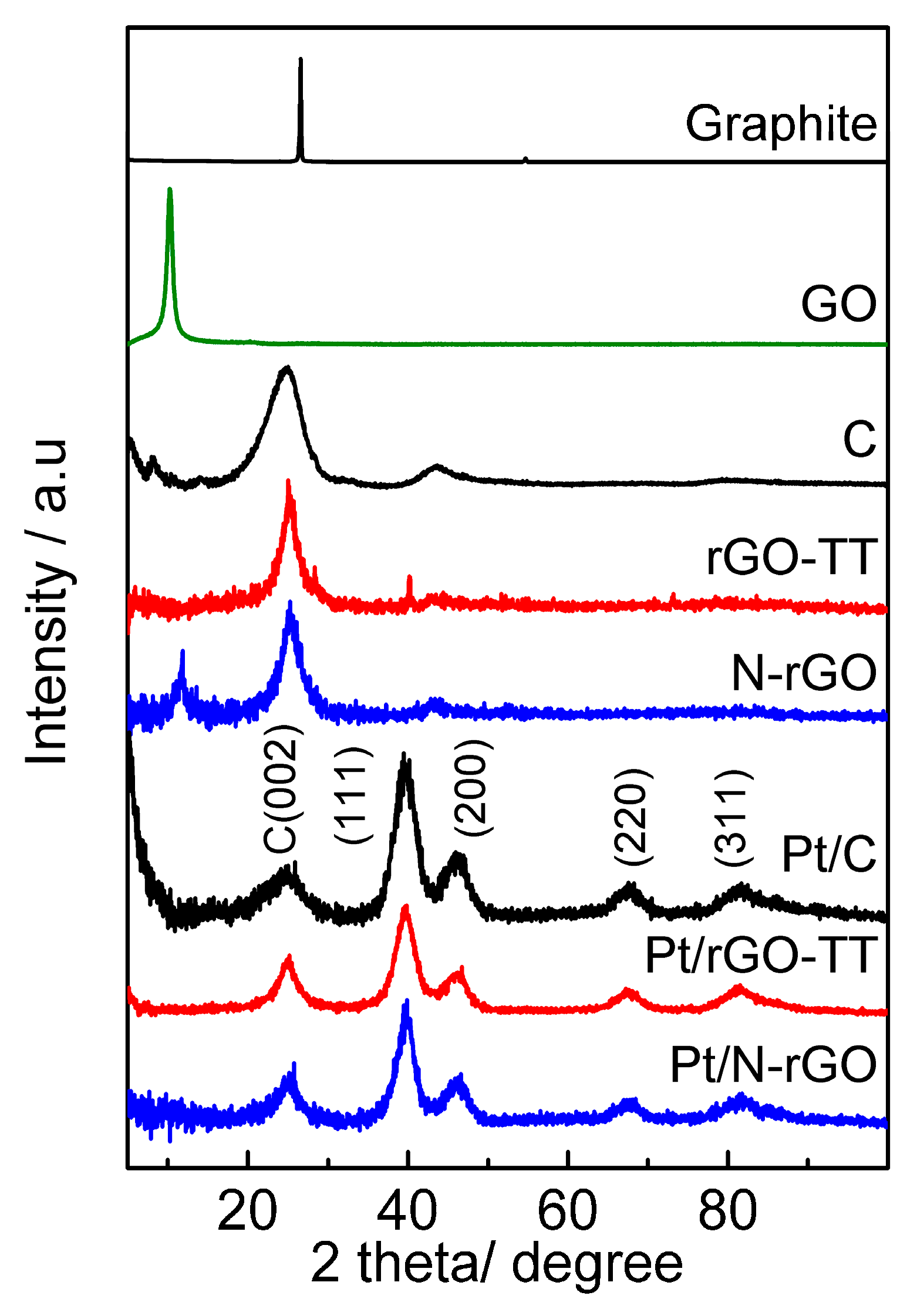
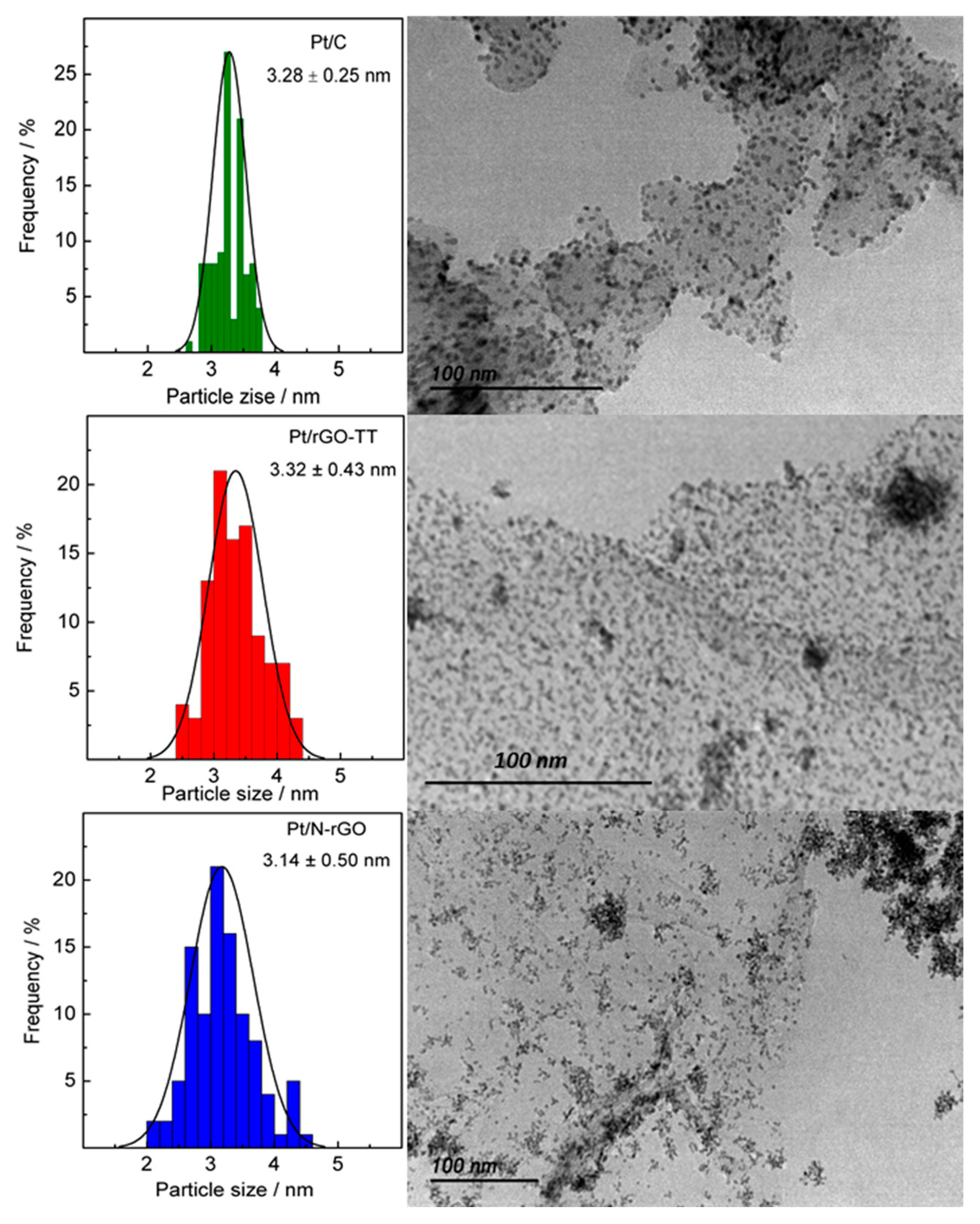
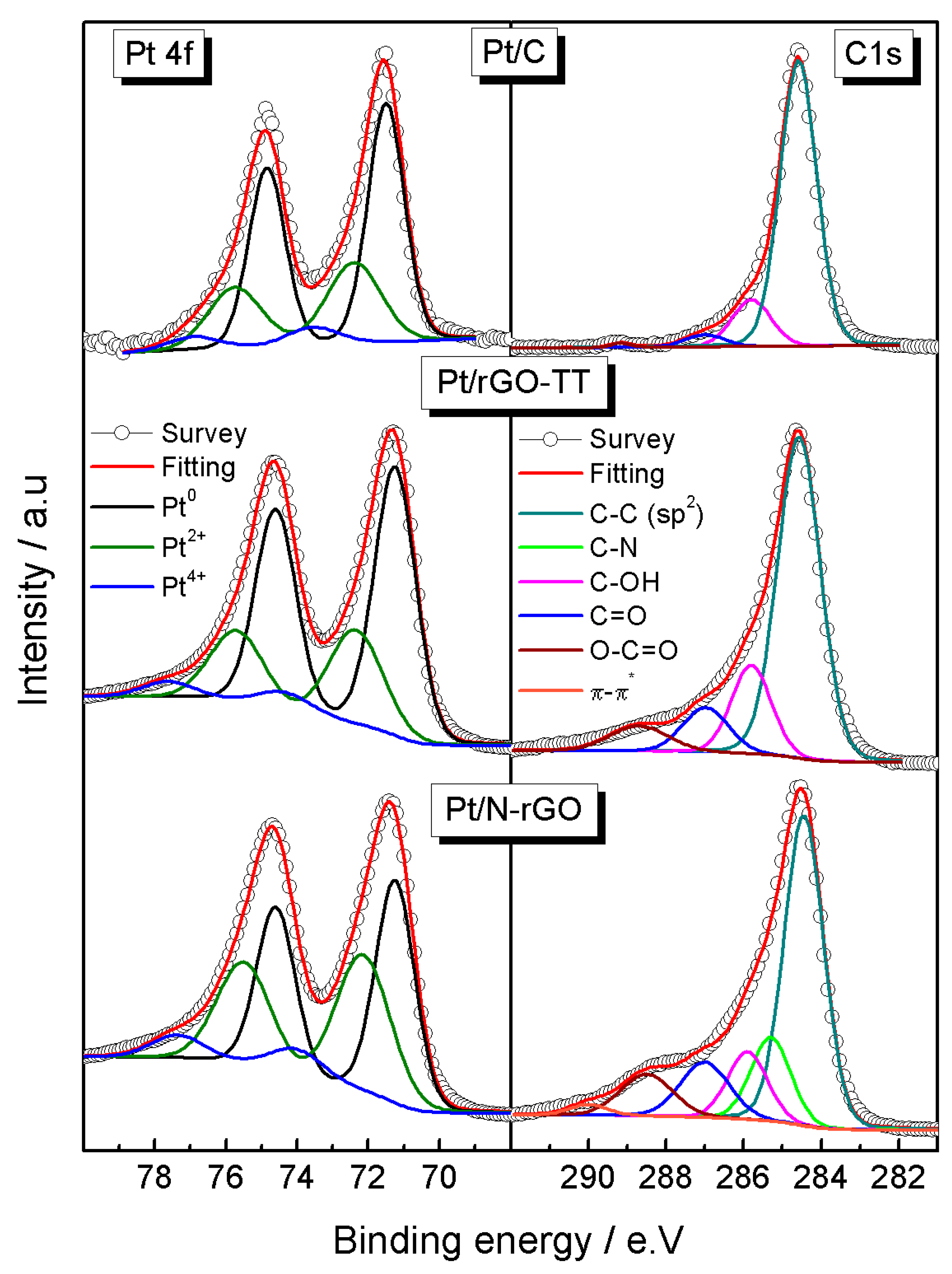
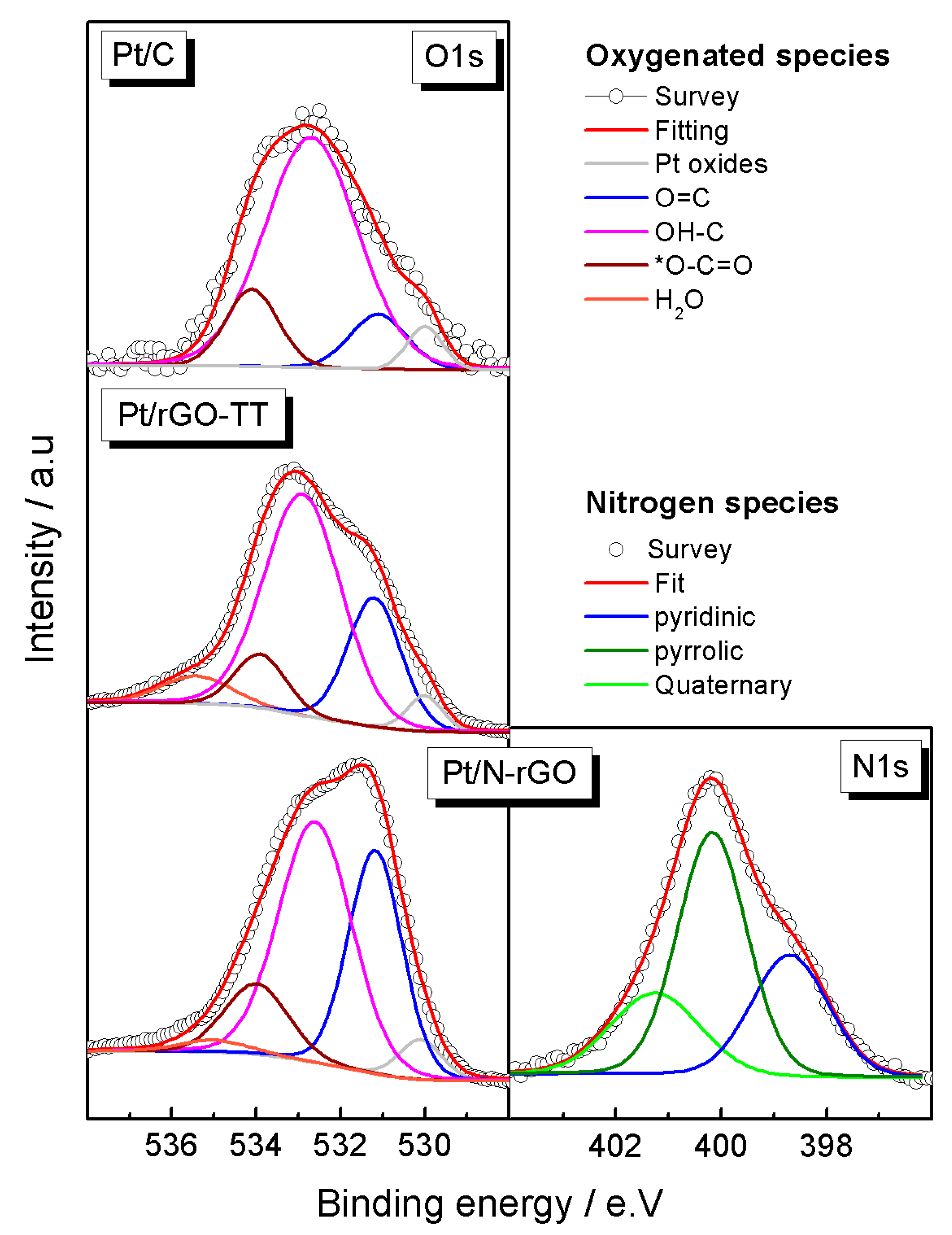
3.1. Physicochemical Characterization
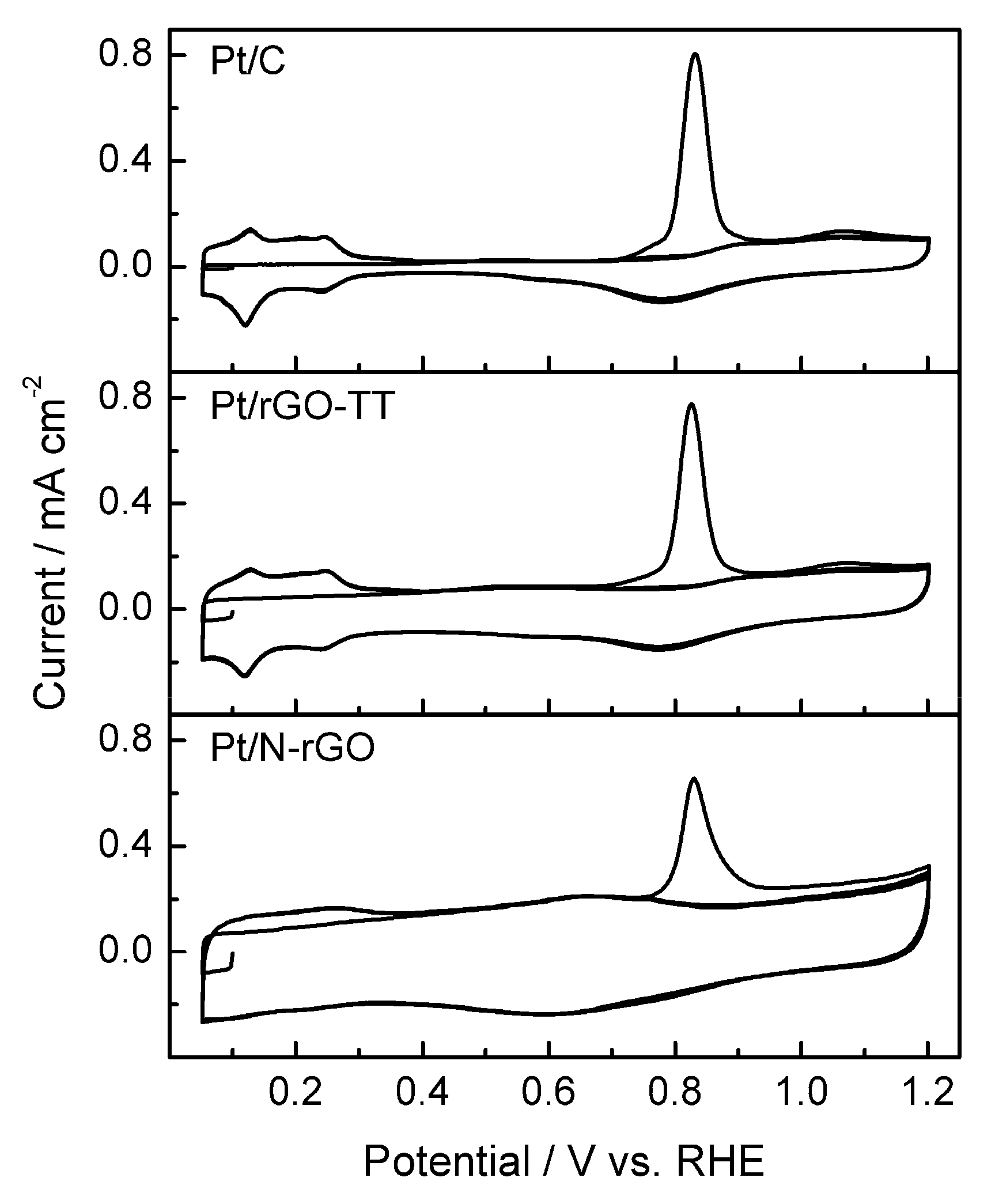
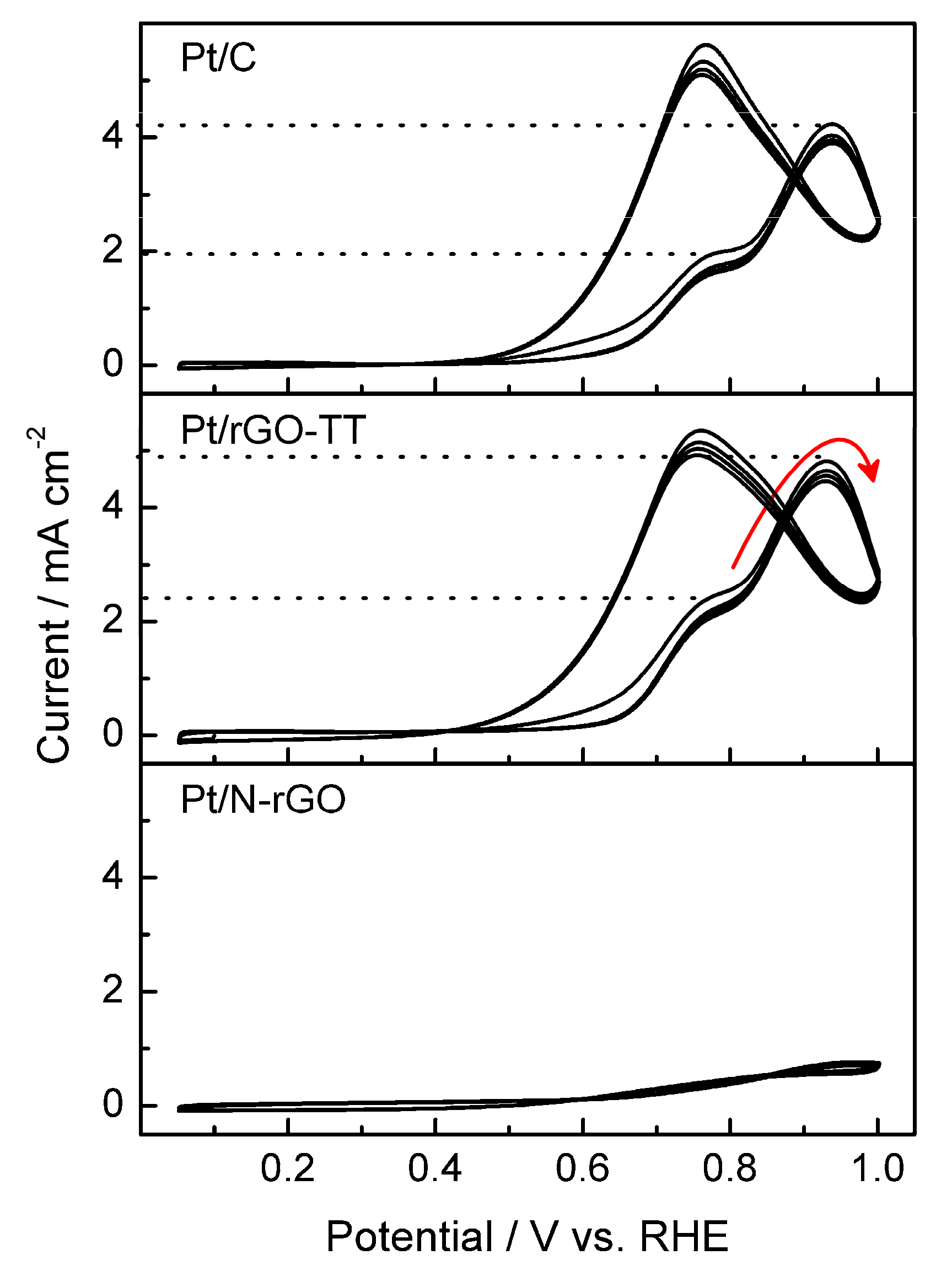
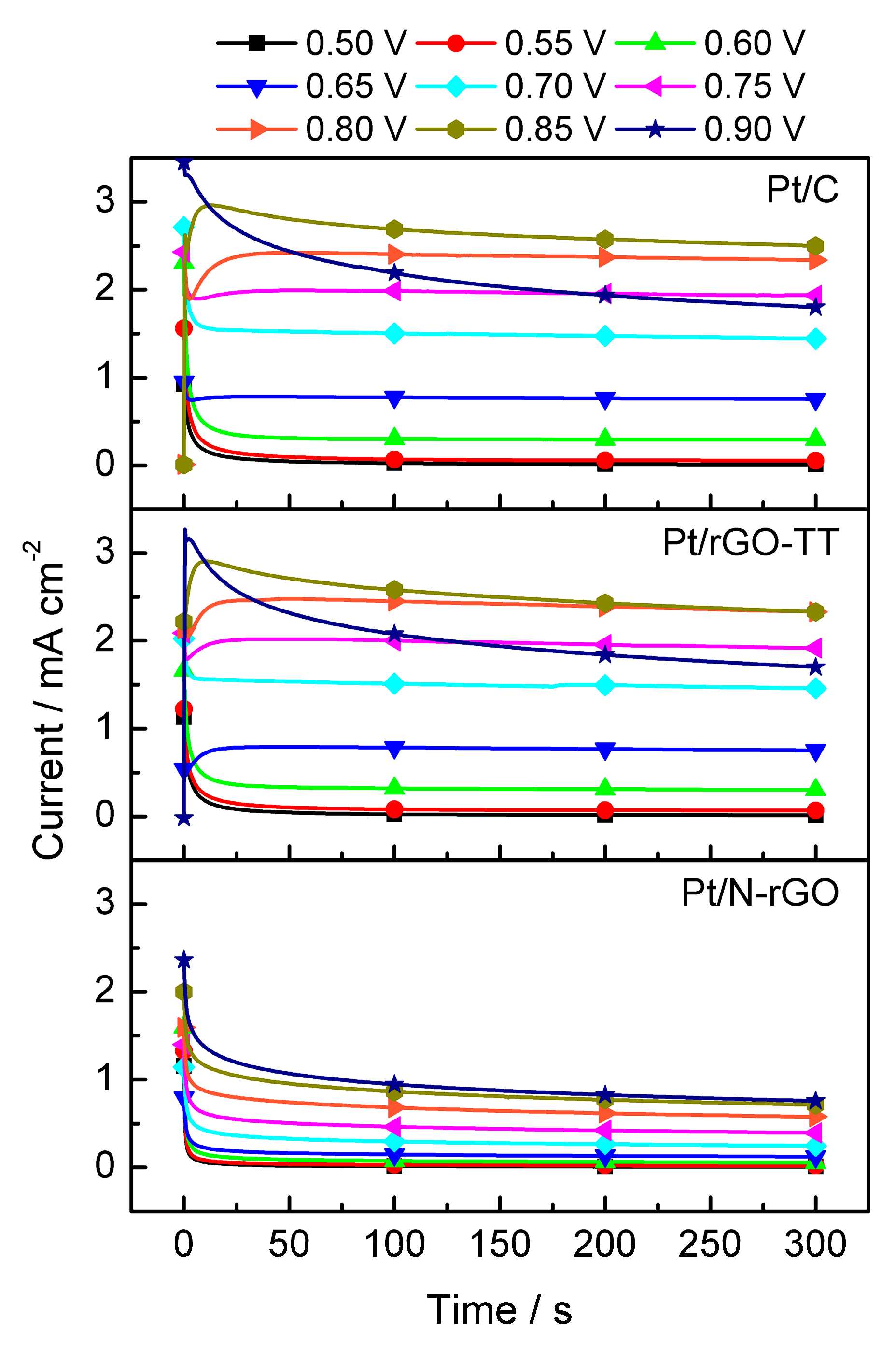
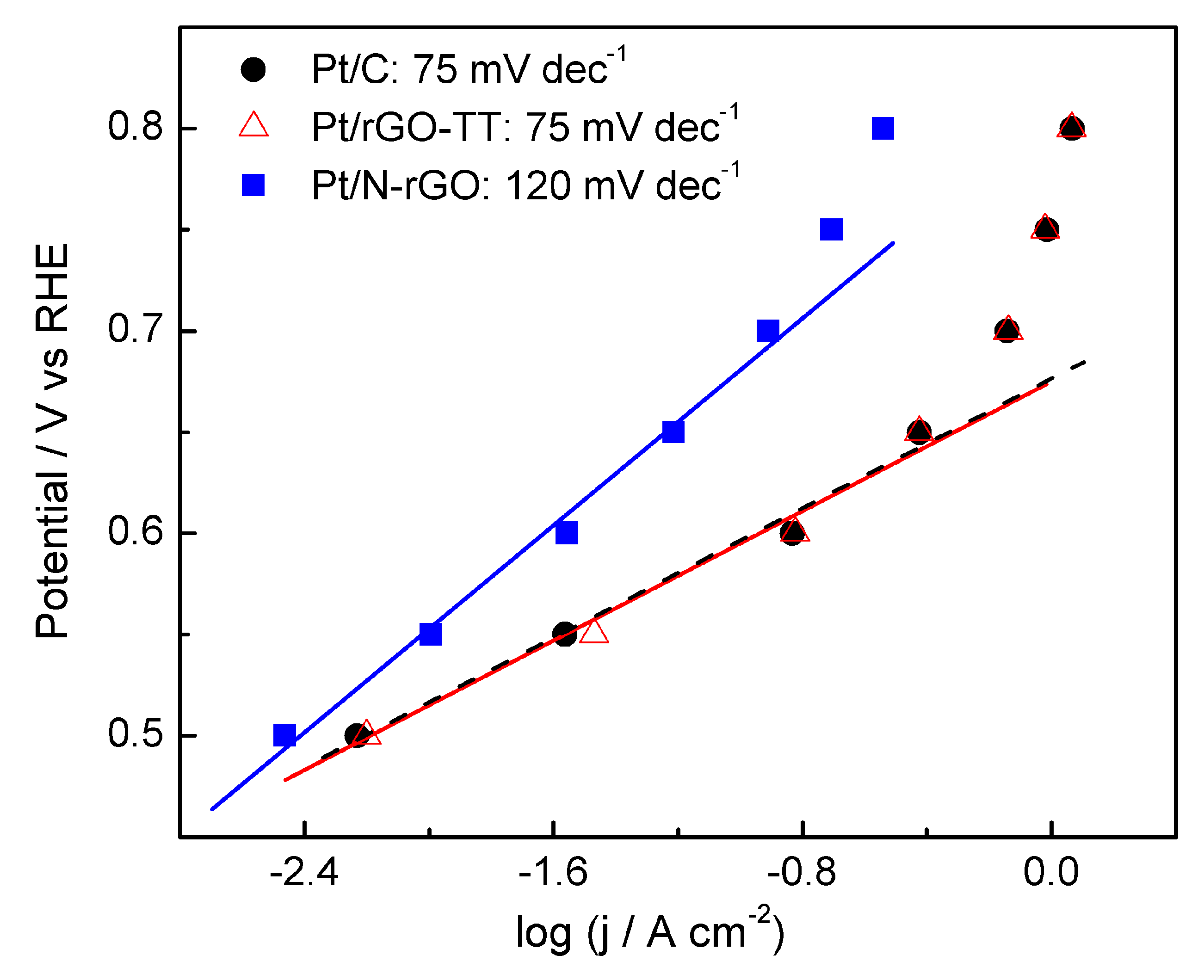
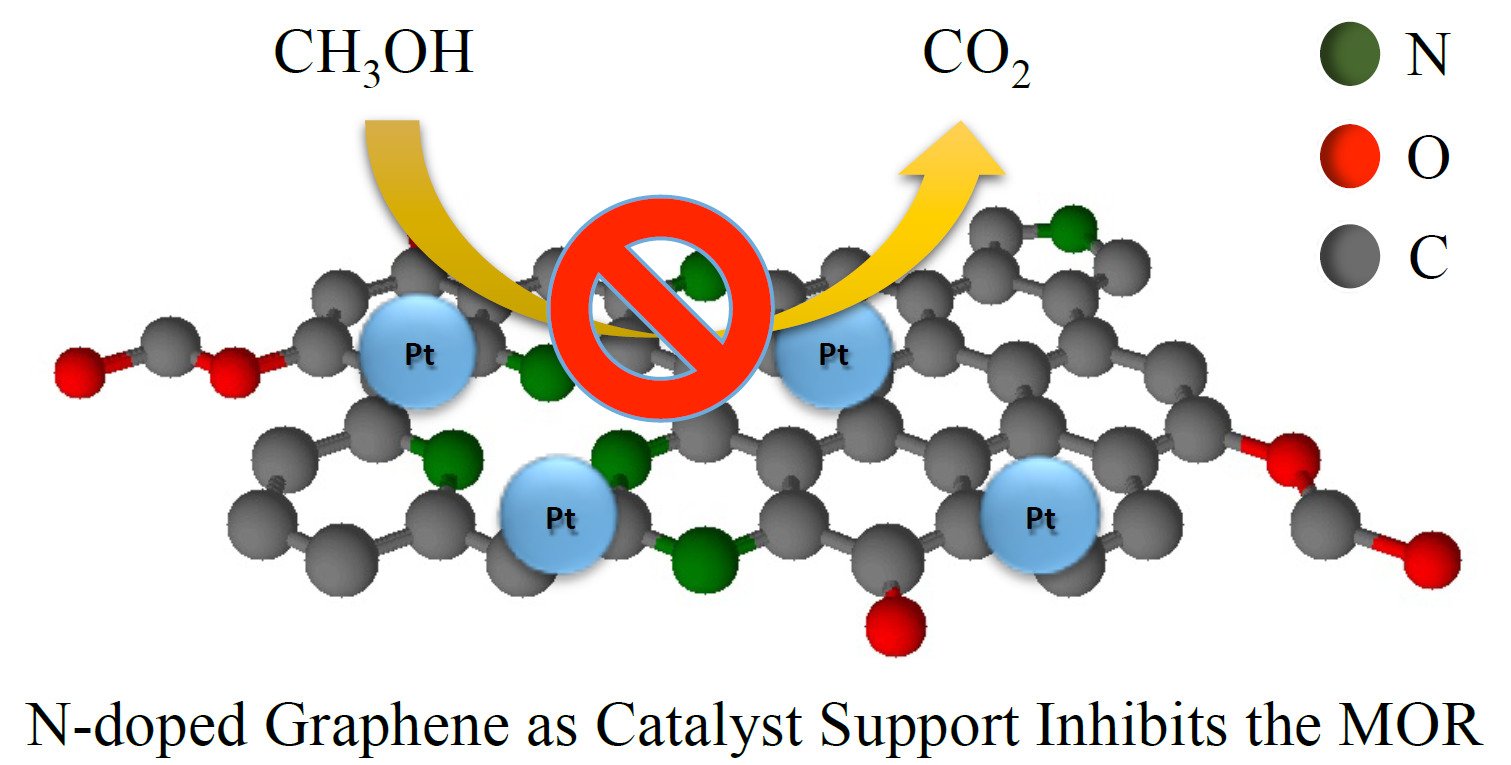
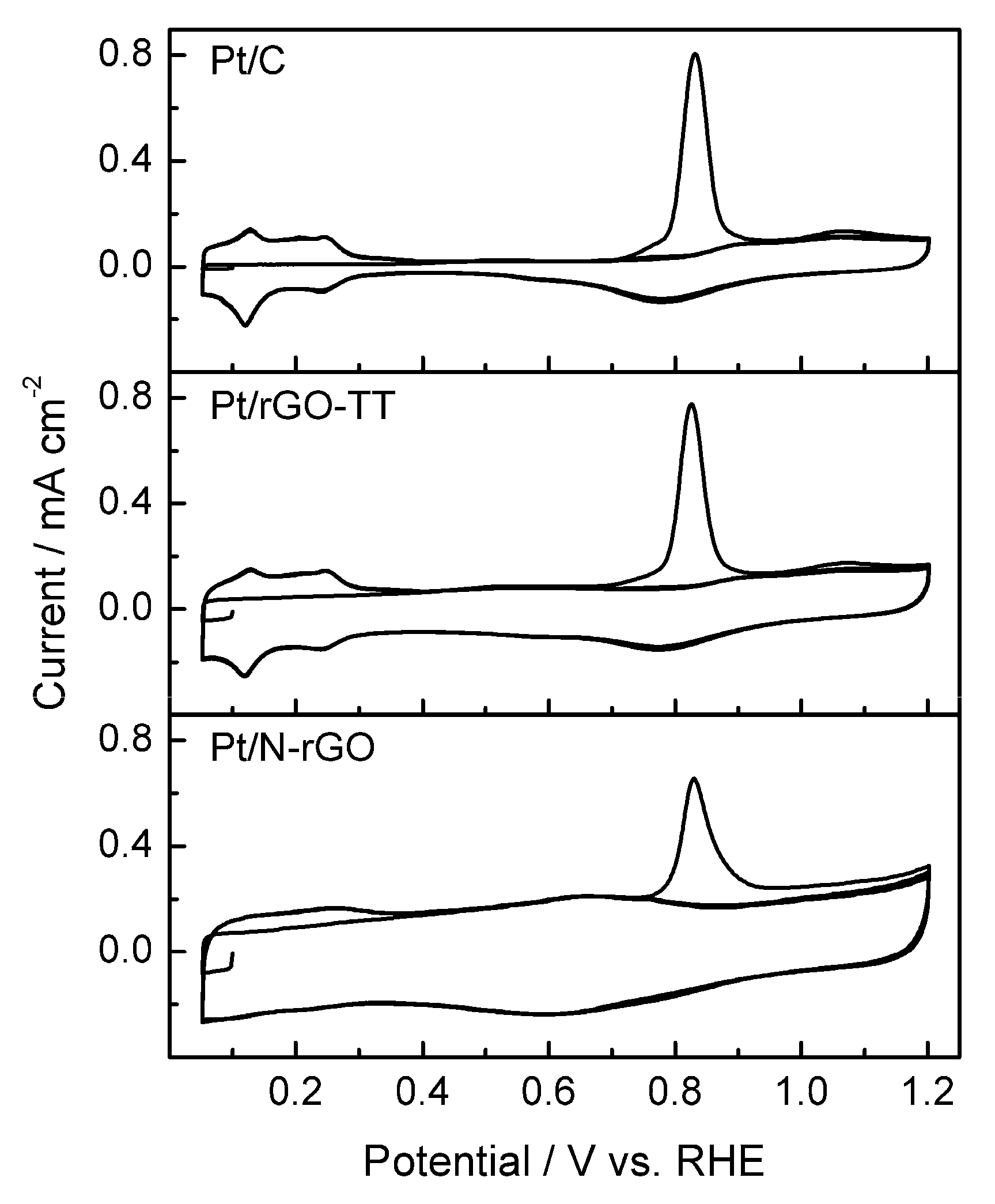
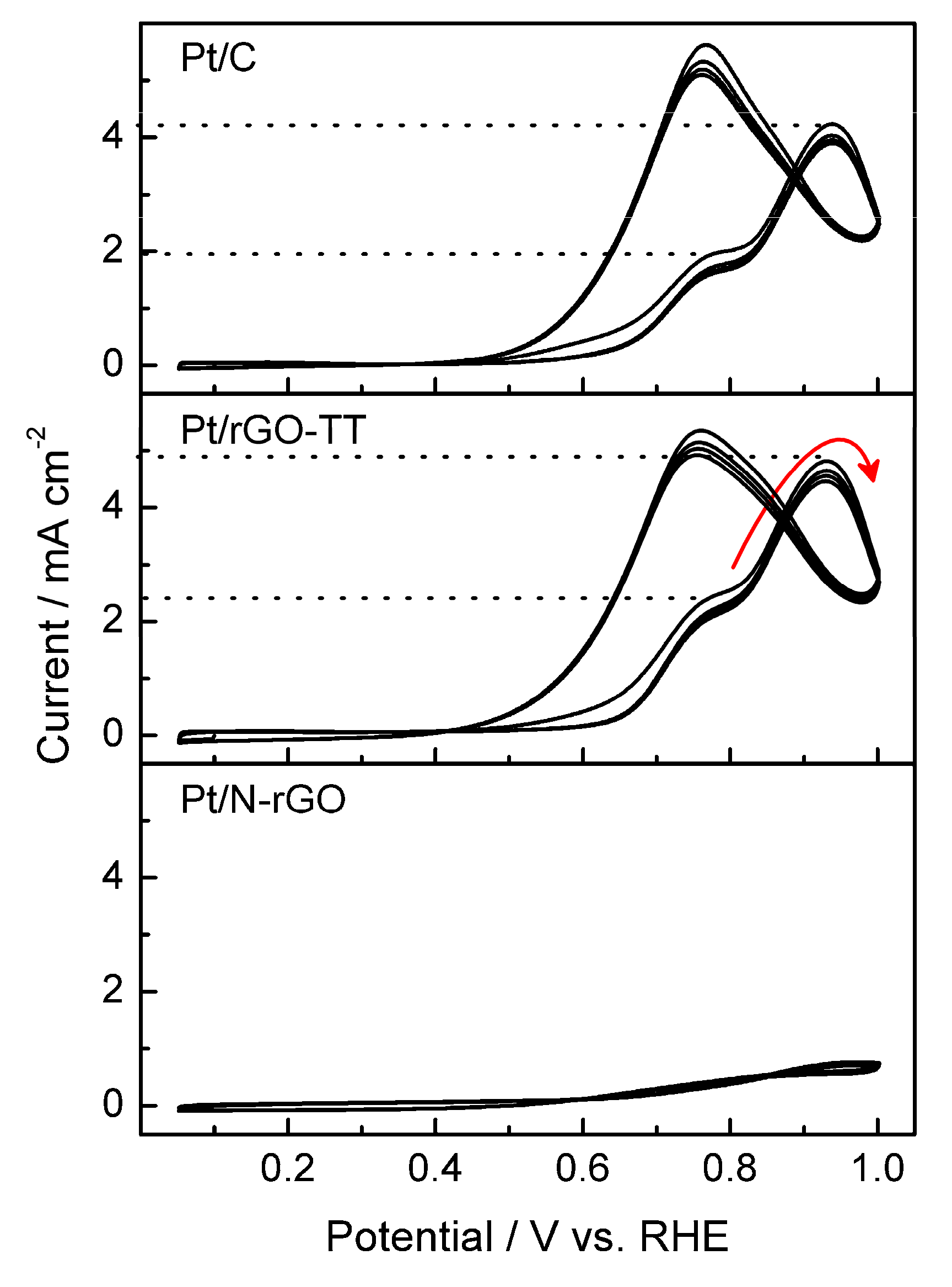
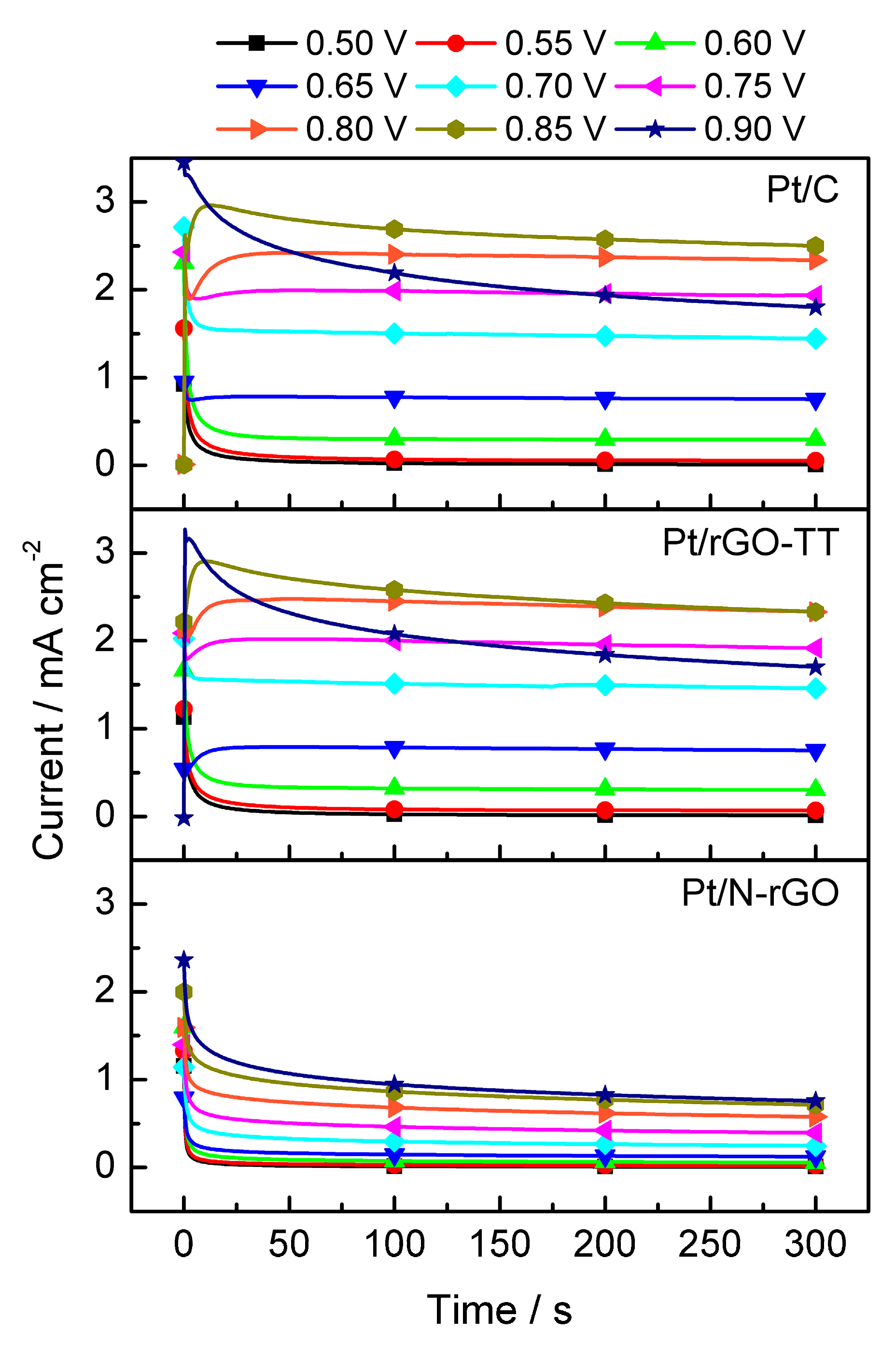
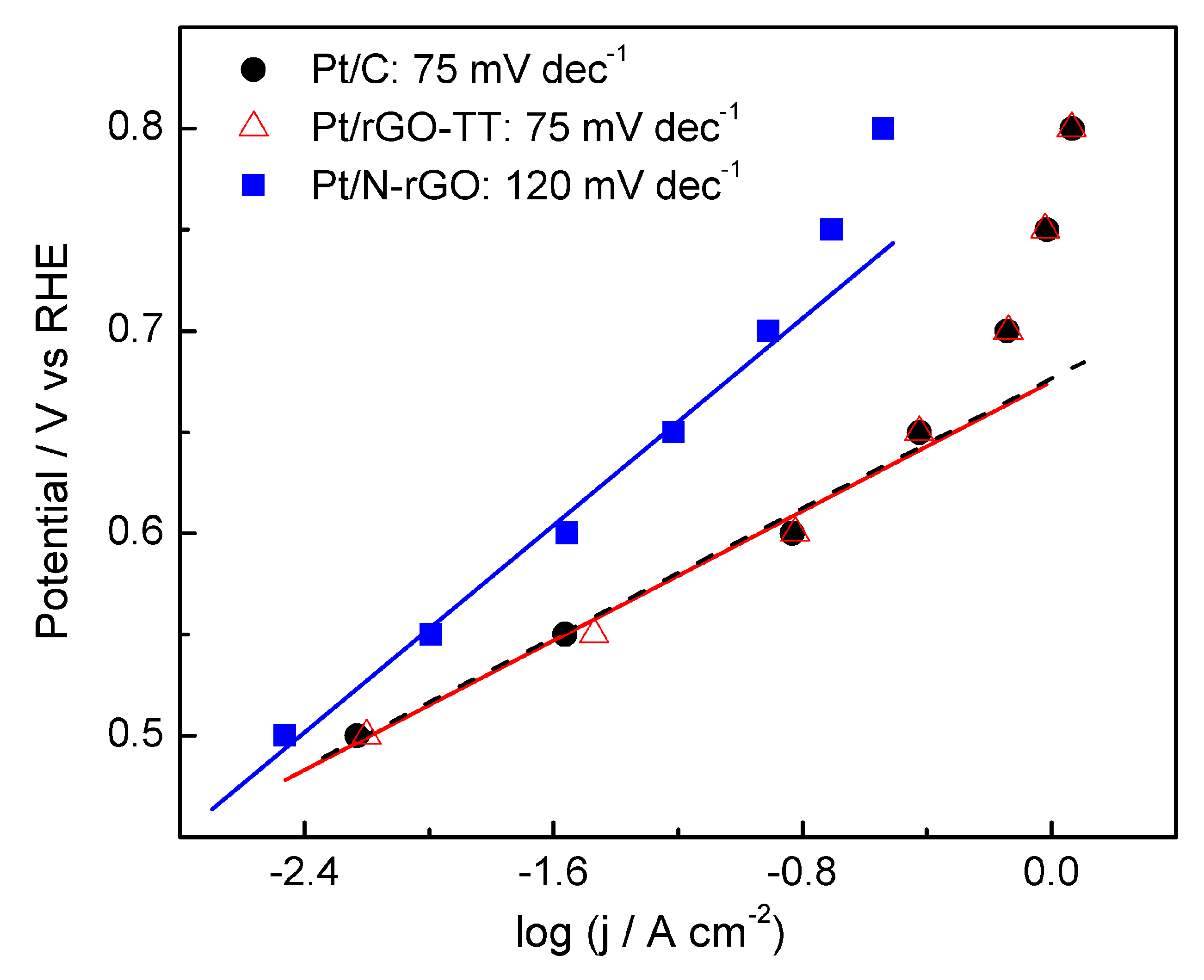
3.2. Electrochemical Characterization
4. Conclusions
Supplementary Materials
Author Contributions
Acknowledgments
Conflicts of Interest
References
- Guillén-Villafuerte, O.; Guil-López, R.; Nieto, E.; García, G.; Rodríguez, J.L.; Pastor, E.; Fierro, J.L.G. Electrocatalytic Performance of Different Mo-Phases Obtained during the Preparation of Innovative Pt-MoC Catalysts for DMFC Anode. Int. J. Hydrog. Energy 2012, 37, 7171–7179. [Google Scholar] [CrossRef]
- Li, M.; Adzic, R.R. Low-Platinum-Content Electrocatalysts for Methanol and Ethanol Electrooxidation. In Lecture Notes in Energy; Shao, M., Ed.; Springer: London, UK, 2013; Volume 9, pp. 1–25. [Google Scholar]
- Pérez-Rodríguez, S.; Corengia, M.; García, G.; Zinola, C.F.; Lázaro, M.J.; Pastor, E. Gas Diffusion Electrodes for Methanol Electrooxidation Studied by a New DEMS Configuration: Influence of the Diffusion Layer. Int. J. Hydrog. Energy 2012, 37, 7141–7151. [Google Scholar] [CrossRef]
- Huang, L.; Zhang, X.; Wang, Q.; Han, Y.; Fang, Y.; Dong, S. Shape-Control of Pt–Ru Nanocrystals: Tuning Surface Structure for Enhanced Electrocatalytic Methanol Oxidation. J. Am. Chem. Soc. 2018, 140, 1142–1147. [Google Scholar] [CrossRef] [PubMed]
- Roca-Ayats, M.; García, G.; Peña, M.A.; Martínez-Huerta, M.V. Titanium Carbide and Carbonitride Electrocatalyst Supports: Modifying Pt-Ti Interface Properties by Electrochemical Potential Cycling. J. Mater. Chem. A 2014, 2, 18786–18790. [Google Scholar] [CrossRef]
- Calderón, J.C.; García, G.; Querejeta, A.; Alcaide, F.; Calvillo, L.; Lázaro, M.J.; Rodríguez, J.L.; Pastor, E. Carbon Monoxide and Methanol Oxidations on Carbon Nanofibers Supported Pt-Ru Electrodes at Different Temperatures. Electrochim. Acta 2015, 186, 359–368. [Google Scholar] [CrossRef]
- Flórez-Montaño, J.; García, G.; Rodríguez, J.L.; Pastor, E.; Cappellari, P.; Planes, G.A. On the Design of Pt Based Catalysts. Combining Porous Architecture with Surface Modification by Sn for Electrocatalytic Activity Enhancement. J. Power Sources 2015, 282, 34–44. [Google Scholar] [CrossRef]
- Rizo, R.; García, G.; Pastor, E. Methanol Oxidation on Bimetallic Electrode Surfaces. In Encyclopedia of Interfacial Chemistry: Surface Science and Electrochemistry; Wandelt, K., Ed.; Elsevier: Amsterdam, The Netherlands, 2018; Volume 5, pp. 719–729. [Google Scholar]
- Anitha, V.C.; Zazpe, R.; Krbal, M.; Yoo, J.; Sopha, H.; Prikryl, J.; Cha, G.; Slang, S.; Schmuki, P.; Macak, J.M. Anodic TiO 2 Nanotubes Decorated by Pt Nanoparticles Using ALD: An Efficient Electrocatalyst for Methanol Oxidation. J. Catal. 2018, 365, 86–93. [Google Scholar] [CrossRef]
- Wasmus, S.; Küwer, A. Methanol Oxidation and Direct Methanol Fuel Cells: A Selective Review. J. Electroanal. Chem. 1999, 461, 14–31. [Google Scholar] [CrossRef]
- Planes, G. a.; García, G.; Pastor, E. High Performance Mesoporous Pt Electrode for Methanol Electrooxidation. A DEMS Study. Electrochem. Commun. 2007, 9, 839–844. [Google Scholar] [CrossRef]
- Martínez Huerta, M.V.; García, G. Fabrication of Electro-Catalytic Nano-Particles and Applications to Proton Exchange Membrane Fuel Cells. In Micro & Nano-Engineering of Fuel Cells; Leung, D.Y.C., Xuan, J., Eds.; Sustainable Energy Developments; CRC Press: London, UK, 2015; pp. 95–129. [Google Scholar]
- Rizo, R.; Sebastián, D.; Rodríguez, J.L.; Lázaro, M.J.; Pastor, E. Influence of the Nature of the Carbon Support on the Activity of Pt/C Catalysts for Ethanol and Carbon Monoxide Oxidation. J. Catal. 2017, 348, 22–28. [Google Scholar] [CrossRef]
- Rizo, R.; Arán-Ais, R.M.; Padgett, E.; Muller, D.A.; Lázaro, M.J.; Solla-Gullón, J.; Feliu, J.M.; Pastor, E.; Abruña, H.D. Pt-Richcore/Sn-Richsubsurface/Ptskin Nanocubes As Highly Active and Stable Electrocatalysts for the Ethanol Oxidation Reaction. J. Am. Chem. Soc. 2018, 140, 3791–3797. [Google Scholar] [CrossRef] [PubMed]
- Rivera, L.M.; Fajardo, S.; Arévalo, M.D.C.; García, G.; Pastor, E. S- and N-Doped Graphene Nanomaterials for the Oxygen Reduction Reaction. Catalysts 2017, 7, 278. [Google Scholar] [CrossRef]
- Zhu, Y.; Chen, G.; Zhong, Y.; Zhou, W.; Shao, Z. Rationally Designed Hierarchically Structured Tungsten Nitride and Nitrogen-Rich Graphene-Like Carbon Nanocomposite as Efficient Hydrogen Evolution Electrocatalyst. Adv. Sci. 2018, 5, 1700603. [Google Scholar] [CrossRef] [PubMed]
- Liang, H.; Li, C.; Chen, T.; Cui, L.; Han, J.; Peng, Z.; Liu, J. Facile Preparation of Three-Dimensional Co 1-x S/Sulfur and Nitrogen-Codoped Graphene/Carbon Foam for Highly Efficient Oxygen Reduction Reaction. J. Power Sources 2018, 378, 699–706. [Google Scholar] [CrossRef]
- Rivera, L.M.; García, G.; Pastor, E. Novel Graphene Materials for the Oxygen Reduction Reaction. Curr. Opin. Electrochem. 2018, 9, 233–239. [Google Scholar] [CrossRef]
- Sun, M.; Liu, H.; Liu, Y.; Qu, J.; Li, J. Graphene-based transition metal oxide nanocomposites for the oxygen reduction reaction. Nanoscale 2015, 7, 1250–1269. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, D.; Feng, H.; Li, J. Graphene Oxide: Preparation, Functionalization, and Electrochemical Applications. Chem. Rev. 2012, 112, 6027–6053. [Google Scholar] [CrossRef] [PubMed]
- Chen, D.; Tang, L.; Li, J. Graphene-based materials in electrochemistry. Chem. Soc. Rev. 2010, 39, 3157–3180. [Google Scholar] [CrossRef]
- Chetty, R.; Kundu, S.; Xia, W.; Bron, M.; Schuhmann, W.; Chirila, V.; Brandl, W.; Reinecke, T.; Muhler, M. PtRu Nanoparticles Supported on Nitrogen-Doped Multiwalled Carbon Nanotubes as Catalyst for Methanol Electrooxidation. Electrochim. Acta 2009, 54, 4208–4215. [Google Scholar] [CrossRef]
- Maiyalagan, T. Synthesis and Electro-Catalytic Activity of Methanol Oxidation on Nitrogen Containing Carbon Nanotubes Supported Pt Electrodes. Appl. Catal. B Environ. 2008, 80, 286–295. [Google Scholar] [CrossRef]
- Sebastián, D.; Nieto-Monge, M.J.; Pérez-Rodríguez, S.; Pastor, E.; Lázaro, M.J. Nitrogen Doped Ordered Mesoporous Carbon as Support of PtRu Nanoparticles for Methanol Electro-Oxidation. Energies 2018, 11, 831. [Google Scholar] [CrossRef]
- Antonietti, M.; Oschatz, M. The Concept of “Noble, Heteroatom-Doped Carbons,” Their Directed Synthesis by Electronic Band Control of Carbonization, and Applications in Catalysis and Energy Materials. Adv. Mater. 2018, 30, 1706836. [Google Scholar] [CrossRef] [PubMed]
- Ambrosi, A.; Chua, C.K.; Latiff, N.M.; Loo, A.H.; Wong, C.H.A.; Eng, A.Y.S.; Bonanni, A.; Pumera, M. Graphene and Its Electrochemistry—An Update. Chem. Soc. Rev. 2016, 45, 2458–2493. [Google Scholar] [CrossRef] [PubMed]
- Zickler, G.A.; Smarsly, B.; Gierlinger, N.; Peterlik, H.; Paris, O. A Reconsideration of the Relationship between the Crystallite Size La of Carbons Determined by X-Ray Diffraction and Raman Spectroscopy. Carbon N. Y. 2006, 44, 3239–3246. [Google Scholar] [CrossRef]
- Cançado, L.G.; Takai, K.; Enoki, T.; Endo, M.; Kim, Y.A.; Mizusaki, H.; Jorio, A.; Coelho, L.N.; Magalhães-Paniago, R.; Pimenta, M.A. General Equation for the Determination of the Crystallite Size La of Nanographite by Raman Spectroscopy. Appl. Phys. Lett. 2006, 88, 163106. [Google Scholar] [CrossRef]
- Wagner, C.; Riggs, W.M.; Davis, L.E.; Moulder, J.F.; Muilenberg, G.E. Handbook of X-ray Photoelectron Spectroscopy; Muilenberg, G.E., Ed.; Perkin-Elmer Corporation: Eden Prairie, MN, USA, 1979. [Google Scholar]
- Ganguly, A.; Sharma, S.; Papakonstantinou, P.; Hamilton, J. Probing the Thermal Deoxygenation of Graphene Oxide Using High-Resolution In Situ X-Ray-Based Spectroscopies. J. Phys. Chem. C 2011, 115, 17009–17019. [Google Scholar] [CrossRef]
- Liu, D.; Li, L.; You, T. Superior catalytic performances of platinum nanoparticles loaded nitrogen-doped graphene toward methanol oxidation and hydrogen evolution reaction. J. Colloid Interface Sci. 2017, 487, 330–335. [Google Scholar] [CrossRef]
- Xiong, B.; Zhou, Y.; Zhao, Y.; Wang, J.; Chen, X.; O’Hayre, R.; Shao, Z. The use of nitrogen-doped graphene supporting Pt nanoparticles as a catalyst for methanol electrocatalytic oxidation. Carbon N. Y. 2013, 52, 181–192. [Google Scholar] [CrossRef]
- Chung, D.Y.; Lee, K.J.; Sung, Y.E. Methanol Electro-Oxidation on the Pt Surface: Revisiting the Cyclic Voltammetry Interpretation. J. Phys. Chem. C 2016, 120, 9028–9035. [Google Scholar] [CrossRef]
- García, G.; Koper, M.T.M. Carbon Monoxide Oxidation on Pt Single Crystal Electrodes: Understanding the Catalysis for Low Temperature Fuel Cells. ChemPhysChem 2011, 12, 2064–2072. [Google Scholar] [CrossRef]
- García, G. Correlation between CO Oxidation and H Adsorption/Desorption on Pt Surfaces in a Wide PH Range: The Role of Alkali Cations. ChemElectroChem 2017, 4, 459–462. [Google Scholar] [CrossRef]
- Iwasita, T. Electrocatalysis of Methanol Oxidation. Electrochim. Acta 2002, 47, 3663–3674. [Google Scholar] [CrossRef]
- Guillén-Villafuerte, O.; García, G.; Guil-López, R.; Nieto, E.; Rodríguez, J.L.; Fierro, J.L.G.; Pastor, E. Carbon monoxide and methanol oxidations on Pt/X@MoO3/C (X = Mo2C, MoO2, Mo0) electrodes at different temperatures. J. Power Sources 2013, 231, 163–172. [Google Scholar] [CrossRef]
- Gisbert, R.; García, G.; Koper, M.T.M. Oxidation of carbon monoxide on poly-oriented and single-crystalline platinum electrodes over a wide range of pH. Electrochim. Acta 2011, 56, 2443–2449. [Google Scholar] [CrossRef]
- Gilman, S. The Mechanism of Electrochemical Oxidation of Carbon Monoxide and Methanol on Platinum. II. The “Reactant-Pair” Mechanism for Electrochemical Oxidation of Carbon Monoxide and Methanol1. J. Phys. Chem. 1964, 68, 70–80. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Catalyst | Positionhkl 2θ/° | dhkl/nm | Number of Layers | Pt Crystallite Size/nm |
|---|---|---|---|---|
| Graphite | 26.5 C(002) | 0.34 C(002) | 121 | - |
| GO | 10.7 C(002) | 0.82 C(002) | 11 | |
| Vulcan | 24.7 C(002) | 0.36 C(002) | 6 | |
| rGO-TT | 25.2 C(002) | 0.35 C(002) | 12 | |
| N-rGO | 25.3 C(002) | 0.35 C(002) | 9 | |
| Pt/C | 67.4 Pt(220) | 0.23 Pt(111) | - | 2.3 |
| Pt/rGO-TT | 67.5 Pt(220) | 0.23 Pt(111) | - | 2.4 |
| Pt/N-rGO | 67.6 Pt(220) | 0.23 Pt(111) | - | 2.0 |
| Composition (% Weight) | |||
|---|---|---|---|
| Material | C | O | N * |
| Graphite | 80.0 | 20.0 | |
| Carbon | 96.5 | 3.5 | - |
| GO | 48.0 | 52.0 | - |
| rGO-TT | 75.3 | 24.7 | - |
| N-rGO | 73.0 | 21.0 | 6.0 |
| Pt/C | 96.4 | 3.6 | - |
| Pt/rGO-TT | 86.9 | 13.1 | - |
| Pt/N-rGO | 78.3 | 15.7 | 6.0 |
| Elements | Pt4f | C1s | O1s | |||||
|---|---|---|---|---|---|---|---|---|
| Catalysts | ||||||||
| Pt/C | Pt0 | 71.5 (62) | C–C | 284.6 (81) | Pt oxides | 530 (5) | ||
| Pt2+ | 72.4 (31) | C–N | - | O=C | 531.1 (10) | |||
| Pt4+ | 73.5 (7) | C–OH | 285.8 (14) | OH–C | 532.8 (72) | |||
| C=O | 287 (4) | O–C=O | 534.1 (13) | |||||
| O–C=O | 289.1 (1) | H2O | - 530 (3) | |||||
| Pt/rGO-TT | Pt0 | 71.2 (63) | C–C | 284.6 (68) | Pt oxides | |||
| Pt2+ | 72.3 (30) | C–N | - | O=C | 531.2 (23) | |||
| Pt4+ | 74.3 (7) | C–OH | 285.8 (15) | OH–C | 532.9 (58) | |||
| C=O | 287 (10) | O–C=O | 533.9 (9) | |||||
| O–C=O | 288.7 (7) | H2O | 535.2 (7) | |||||
| Pt/N-rGO | Pt0 | 71.2 (49) | C–C | 284.5 (52) | Pt oxides | 530.1 (4) | ||
| Pt2+ | 72.1 (42) | C–N | 285.3 (14) | O=C | 531.2 (33) | N1s | ||
| Pt4+ | 74.0 (10) | C–OH | 285.9 (12) | OH–C | 532.7 (48) | Pyridinic | 398.7 (29) | |
| C=O | 287.1 (12) | O–C=O | 534 (12) | Pyrrolic | 400.2 (49) | |||
| O–C=O | 288.5 (10) | H2O | 535 (2) | Quaternary | 401.2 (22) | |||
| π–π | 290.0 (2) | - | - | - | - | |||
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Arteaga, G.; Rivera-Gavidia, L.M.; Martínez, S.J.; Rizo, R.; Pastor, E.; García, G. Methanol Oxidation on Graphenic-Supported Platinum Catalysts. Surfaces 2019, 2, 16-31. https://doi.org/10.3390/surfaces2010002
Arteaga G, Rivera-Gavidia LM, Martínez SJ, Rizo R, Pastor E, García G. Methanol Oxidation on Graphenic-Supported Platinum Catalysts. Surfaces. 2019; 2(1):16-31. https://doi.org/10.3390/surfaces2010002
Chicago/Turabian StyleArteaga, Gladys, Luis M. Rivera-Gavidia, Sthephanie J. Martínez, Rubén Rizo, Elena Pastor, and Gonzalo García. 2019. "Methanol Oxidation on Graphenic-Supported Platinum Catalysts" Surfaces 2, no. 1: 16-31. https://doi.org/10.3390/surfaces2010002
APA StyleArteaga, G., Rivera-Gavidia, L. M., Martínez, S. J., Rizo, R., Pastor, E., & García, G. (2019). Methanol Oxidation on Graphenic-Supported Platinum Catalysts. Surfaces, 2(1), 16-31. https://doi.org/10.3390/surfaces2010002