1. Introduction
Taurine is abundant in mammalian tissue, especially in excitable tissues, such as the heart. Taurine functions as a compatible organic osmolyte, thereby assisting in the regulation of intracellular osmotic balance as well as betaine (also called glycine betaine), glycerophosphocholine (GPC), sorbitol, free amino acids, etc. [
1]. Taurine also possesses various cellular actions, such as modulation of ion movement and calcium handling [
2]. Maintenance of certain species, such as cats, on a taurine-deficient diet leads to development of dilated cardiomyopathy [
3]. We previously demonstrated that knocking out the taurine transporter (TauT; Slc6a6) of mice (TauTKO mice) also caused a taurine-deficient cardiomyopathy characterized by ventricular wall thinning and induction of heart failure marker genes [
4]. Despite the severe depletion of taurine (less than 1% of wild-type mice), cardiac output is normal in the young animal although it declines with age. Moreover, the decrease in longevity is not severe (median lifespan is about 18 months) [
4,
5,
6]. The mild condition of the TauTKO phenotype may relate to homeostatic mechanisms that compensate for detrimental effects caused by taurine depletion.
Warskulat et al. reported that some amino acids, including alanine, glutamine, and glutamate, accumulate in the heart of TauTKO mice [
7], which may be caused by a compensatory event associated with taurine loss. We previously performed LC-MS-based metabolome analysis in TauTKO mice to comprehensively analyze the changes in metabolites [
8] and observed increases in organic osmolytes, such as betaine, GPC, and amino acids. These observations imply the importance of metabolic control of organic osmolytes in providing protection to the stressed heart.
In the present study, we investigated the genetic mechanisms involved in taurine-depletion-induced metabolome changes of the TauTKO mouse that could contribute to compensation for disturbances of cellular osmoregulation.
4. Discussion
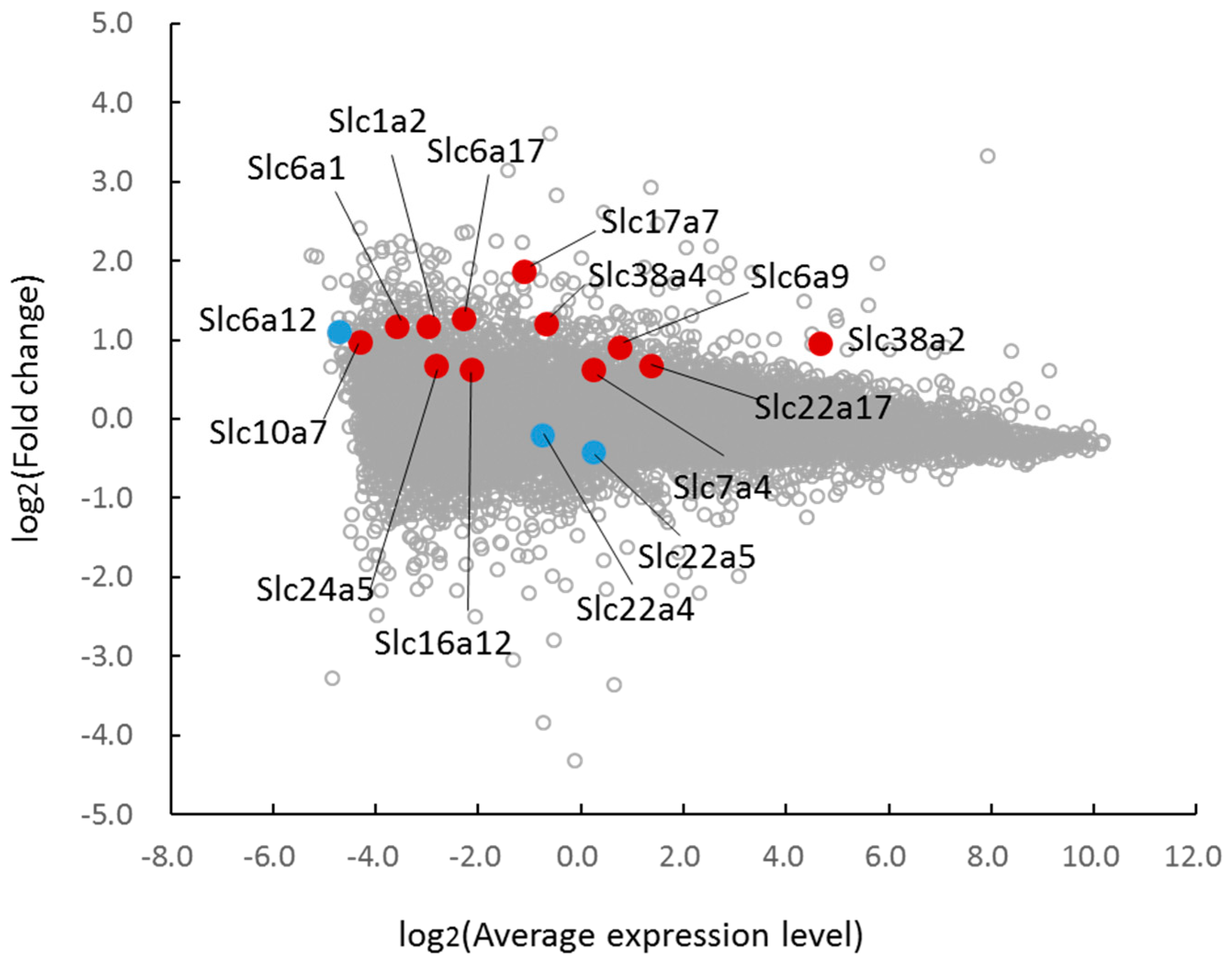
One of the most important functions of taurine in the cell is the regulation of intracellular osmolality. When cells are exposed to the hyperosmolar milieu, the content of organic osmolytes, including those of taurine, betaine, GPC, and amino acids, are increased, which contributes to the establishment of an ionic balance and minimizes changes in cell volume [
1]. In certain types of cells, such as kidney cells, the mRNA of the TauT (Slc6a6), the BGT-1 (Slc6a12), and the amino acid transporter system A (Slc38a2) are increased by hyperosmotic stress, which in turn stimulates the uptake of their respective substrates. These cellular responses against a change in osmolality are regulated by the transcription factor TonEBP (tonicity-response element binding protein; also called NFAT5). In the case of the heart, we observed that taurine depletion (TauT knockout) also alters organic osmolyte content, suggesting that disturbances in cellular osmoregulation caused by the loss of taurine may be compensated for by alterations in other osmolytes. However, the expression of BGT-1 is not significantly different between TauTKO and wild-type mice, implying the involvement of an unidentified mechanism in the change in betaine content. Transcriptome analysis also revealed the induction of other transporters, including Slc38a2, Slc38a4, Slc6a9 (glycine transporter-1), and Slc7a4 (a member of the cationic amino acid transporter y+ system), in the TauTKO heart. In addition to BGT-1, other betaine transport systems exist: Slc6A20 (betaine/proline transporter) and Slc7a6 (another member of a member of the cationic amino acid transporter y+ system, y+ LAT2), which are expressed in mouse oocytes [
17,
18]. The Slc38 protein family is also a potential transport system for betaine [
17]. While these transporters are candidates for betaine transport in the heart, further study is necessary to identify the transporter which contributes to osmoregulation of betaine in the heart.
Carnitine also functions as an organic osmolyte [
19]. In the transcriptome profile, levels of the carnitine transporter genes Slc22a4 and Slc22a5 are not altered in TauTKO mice. By contrast, the content of another member of the organic cation transporter family, Slc22a17, is increased in the TauTKO heart; however, carnitine does not function as a substrate of that transporter [
12]. Activation of carnitine transport in TauTKO mice may be related to post-translational modifications, such as phosphorylation.
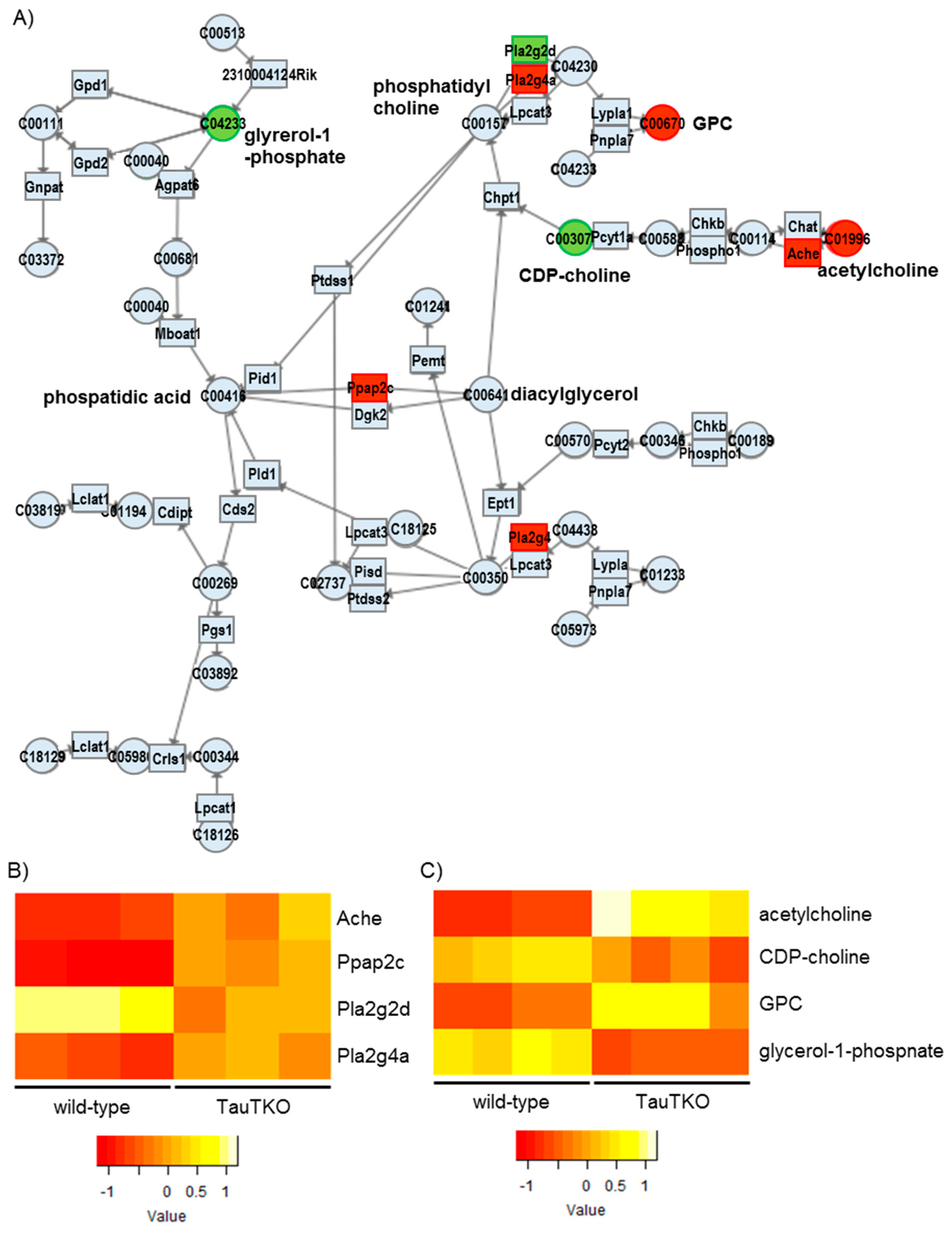
GPC is converted from phosphatidylcholine to lysophosphatidylcholine by Phospholipase A2 and Glycerophosphocholine phosphodiesterase. Alternatively, Phospholipase B (PLB) catalyzes the direct conversion of phosphatidylcholine to GPC [
1]. It has been reported that cellular exposure to a hyperosmolar condition increases the content of PLB mRNA, which should mediate an increase in GPC in the kidney cell. In the present study, we observed in the TauTKO heart induction of one of the phospholipase genes, Pla2g4, but not that of the PLB gene (Fold change = 1.243,
p = 0.237), indicating that a different osmotic-related pathway may function in the heart to control GPC synthesis. In addition, Acetylcholinesterase (Ache) is induced in the TauTKO mouse while acetylcholine level increases and CDP-choline content falls. This pathway may provide the choline portion of GPC’s structure. Moreover, phosphatidic acid phosphatase-2c (Ppap2c), which catalyzes the conversion of phosphatidic acid to diacylglycerol, a precursor of phosphatidylcholine, is also induced in the TauTKO mouse. These coordinated activations of the metabolic pathway likely contribute to the effectiveness of GPC as an osmoregulator in the heart.
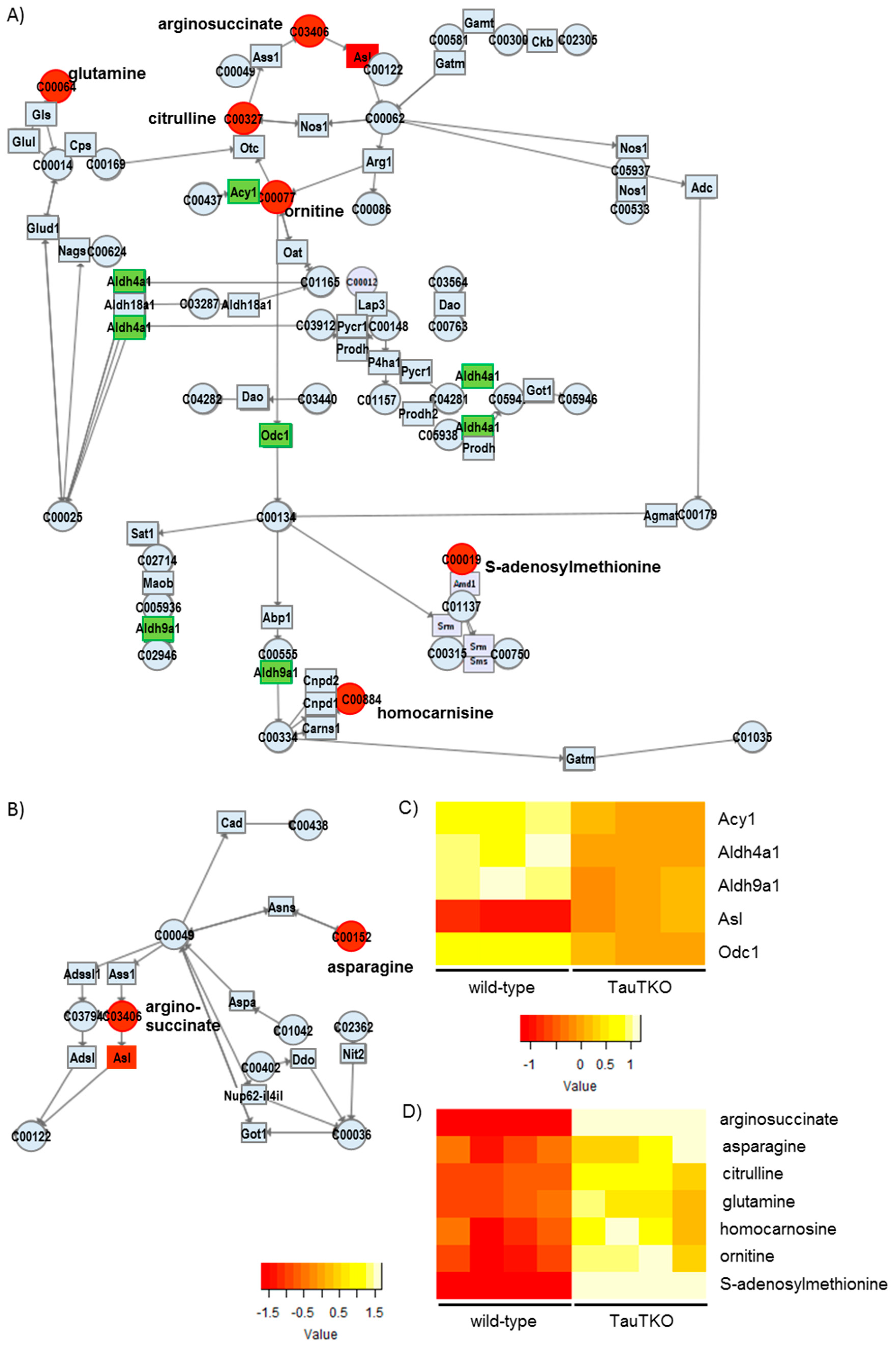
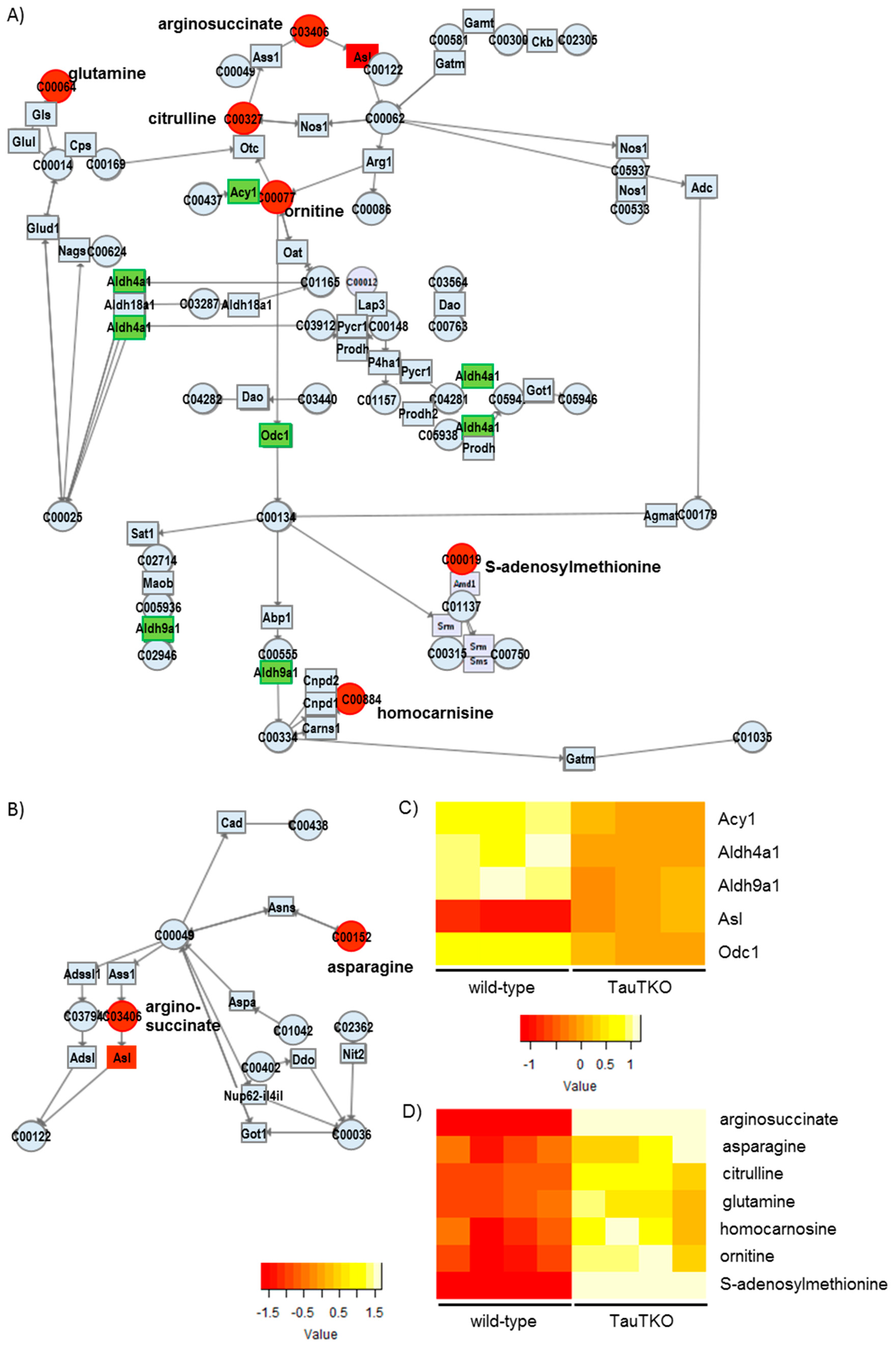
Besides examining the modulation of various osmolytes in the TauTKO heart, we observed an induction of arginosuccinate lyase, concomitant with an increase in the levels of arginosuccinate, ornithine, and citrulline, in TauTKO mice. Citrulline and arginosuccinate are important intermediates in the production of NO in most tissues, the exception being the liver [
13]. The knockout of Asl in mice causes a decrease in protein nitrosylation and nitrite in the heart, evidence of an attenuation of NO synthesis [
14]. Therefore, the activation of this pathway in the TauTKO heart may increase NO production.
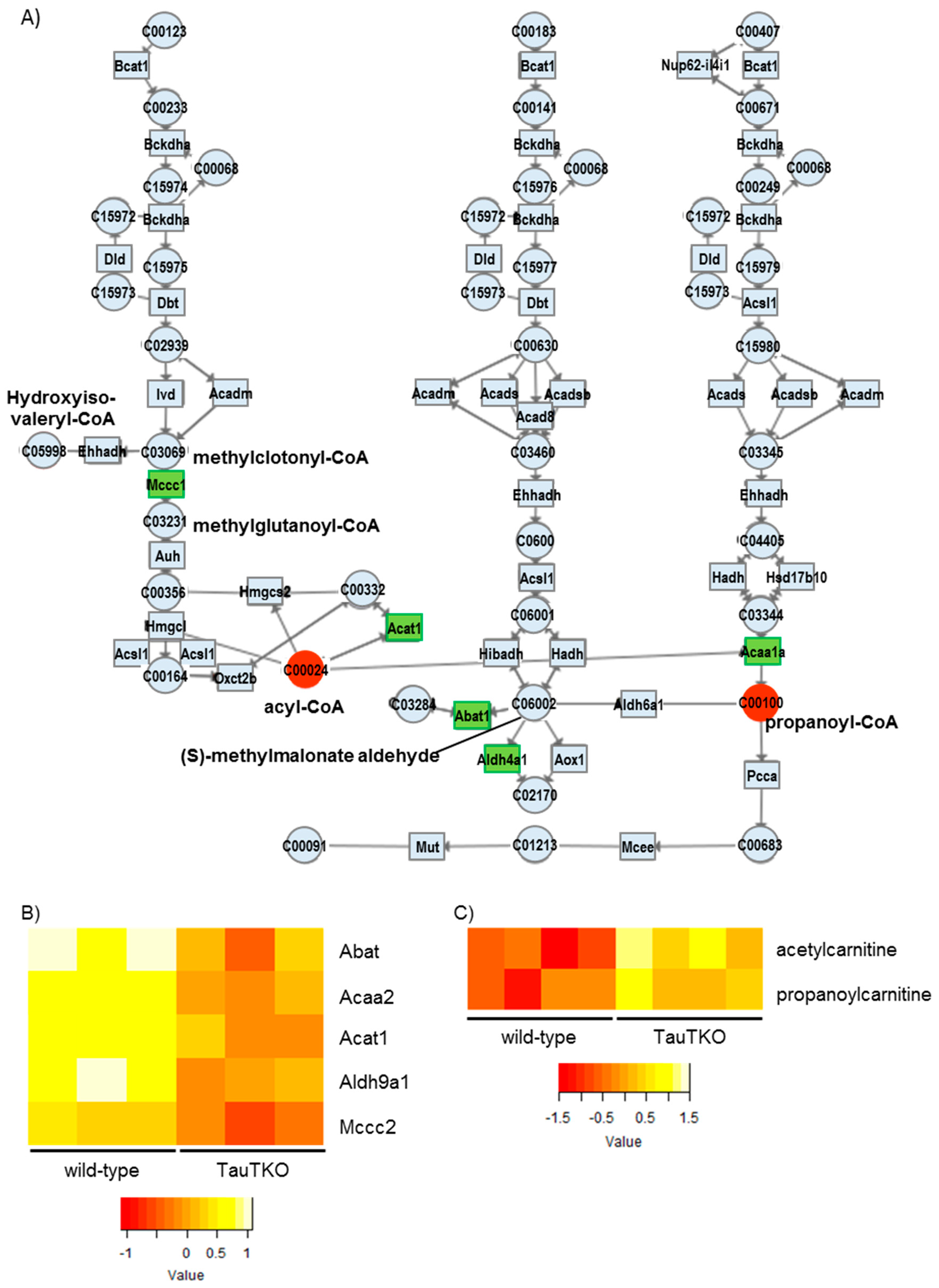
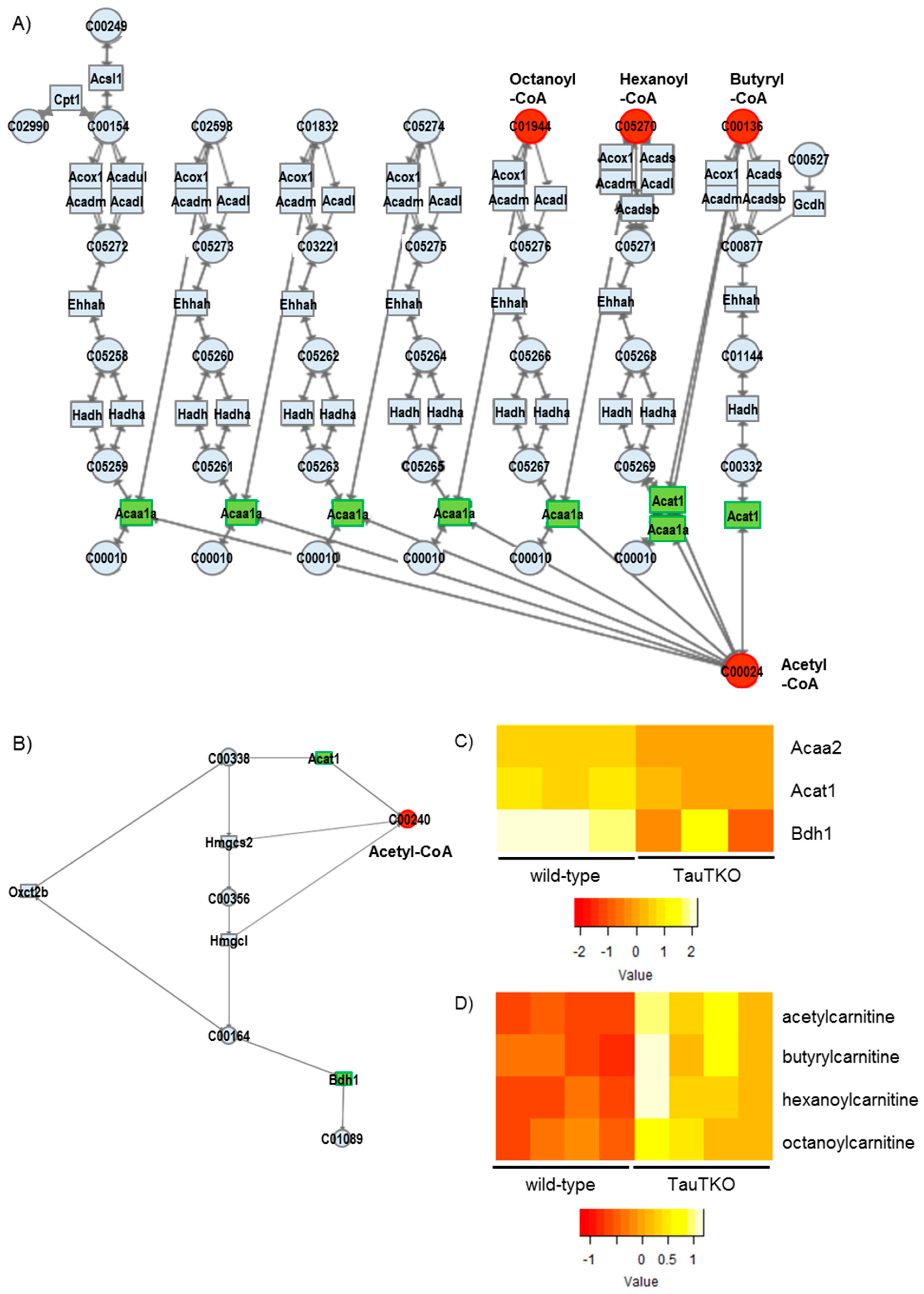
We observed reductions in some genes associated with fatty acid oxidation in the TauTKO mouse, which may cause incomplete fatty acid oxidation followed by the accumulation of the carnitine derivatives of short chain fatty acids. Furthermore, acetylcarnitine is also higher in TauTKO mice, indicating an accumulation of acetyl-CoA. Additionally, we observed that the gene expressions of Bdh1 and Acat1 that metabolize to acetyl-CoA from ketone bodies were decreased, but acetyl-CoA was increased. Acetyl-CoA not only feeds carbon into the tricarboxylic acid cycle but can also control energy metabolism by acetylation of lysine residues of key enzymes [
20]. Although the enzymes related to fatty acid oxidation, as well as the tricarboxylic acid cycle and the electron transport chain, are targets for acetylation, the effect of acetylation on the activity of fatty acid oxidation is controversial. In the case of the heart, hyperacetylation of mitochondrial proteins caused by a reduction in SIRT3, a mitochondrial NAD-dependent deacetylase, in obesity and diabetes is associated with an increase in the fatty acid oxidation rate [
21]. Lysine acetylation can also control transcription of fatty acid oxidation-related genes by modification of transcriptional regulator PGC-1alpha [
21]. These acetylation processes, which are mediated by acetyl-CoA accumulation, may slow fatty acid oxidation and ketone degradation in the heart of TauTKO mice. Moreover, the carboxylation of acetyl CoA produces malonyl CoA, which inhibits carnitine palmitoyltransferase, a key enzyme involved in fatty acid uptake and oxidation by the heart. Importantly, similar transcriptome changes related to fatty acid oxidation were observed in skeletal muscle of TauTKO mice [
22]. Therefore, this may be a result of tissue taurine depletion. Indeed, the decrease in oxidation by the taurine-deficient rat heart has been largely attributed to taurine-mediated reductions in carnitine palmitoyltransferase-1 activity [
23]. Additionally, why acetyl-CoA is increased in the heart of TauTKO mice is unclear. It is possible that the accumulated acetyl-CoA comes from glycolysis pathway activation and/or the reduction of the tricarboxylic acid (TCA) cycle. We previously observed that the glycolysis is enhanced and the TCA cycle activity is diminished in the taurine-deficient heart [
23].
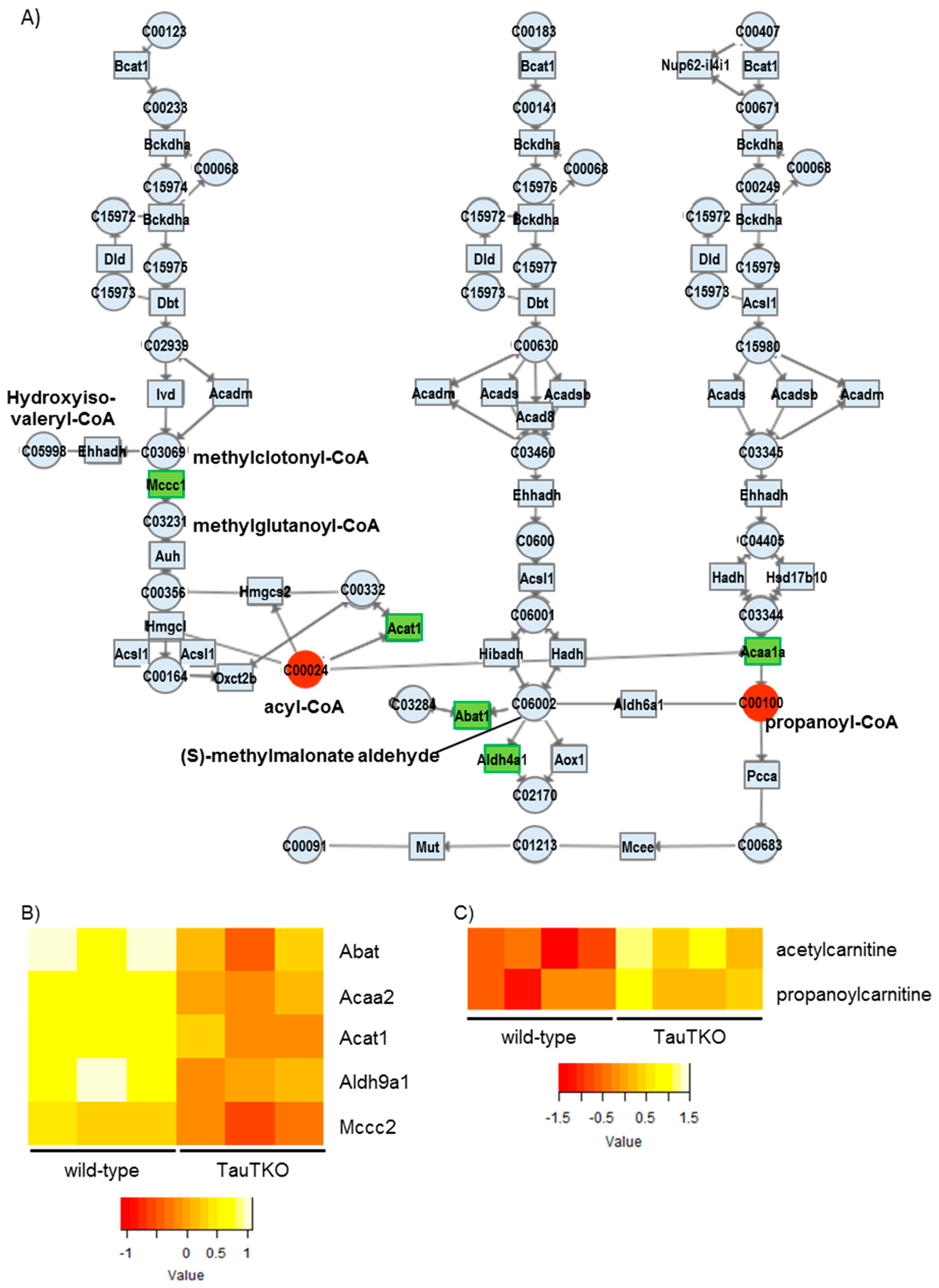
Finally, we observed a reduction in some genes associated with the branched-chain amino acid (BCAA) degradation pathways. Catabolic defects of BCAA metabolism occur in heart failure. In the case of the pressure-overload-induced failing heart of mice, most of genes of the KEGG BCAA catabolic pathway are reduced compared to those of the sham-operated heart. Moreover, the genetic changes are accompanied by an increase in the branched-chain keto acids (BCKAs), which are markers for a defect in the BCAA metabolic pathway [
24]. Importantly, it has been reported that elevations in BCKAs by knocking out PP2Cm, a BCKA dehydrogenase phosphatase, lead to impaired cardiac function. Similarly, it is attractive to suggest that suppression of the BCAA metabolic pathway in TauTKO mice may partially contribute to the aging-dependent decline in cardiac function.







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}