

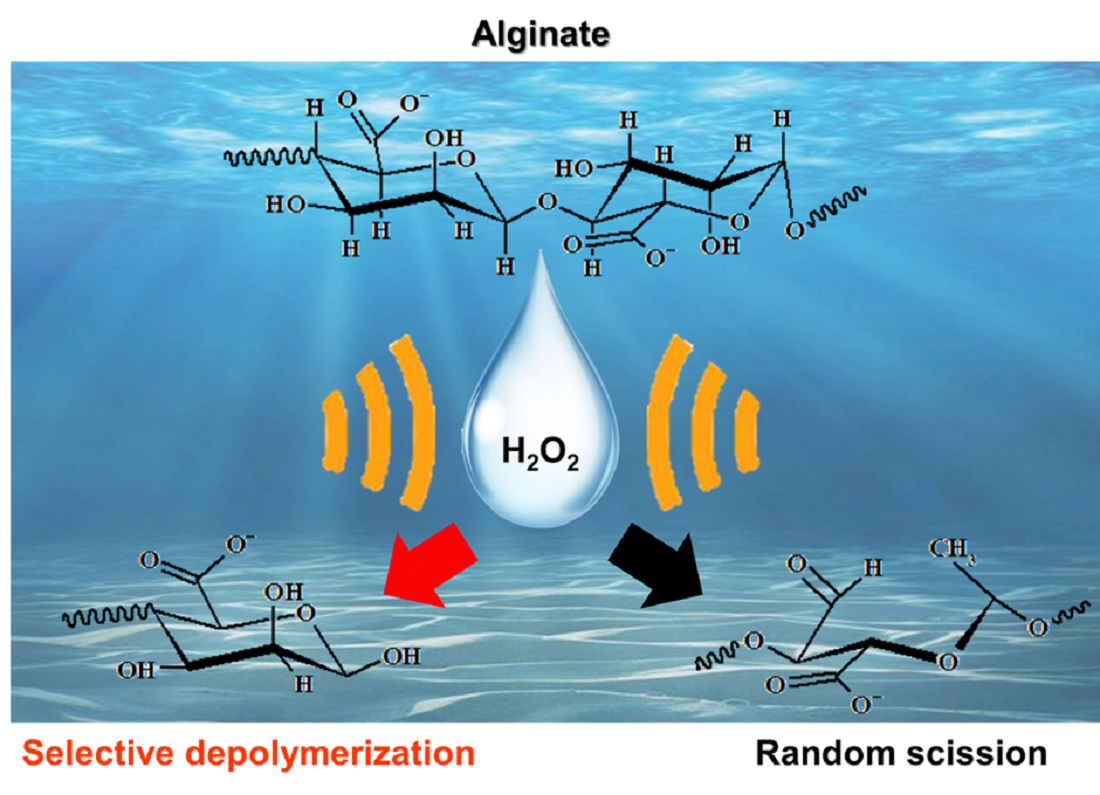
Eco-Friendly Depolymerization of Alginates by H2O2 and High-Frequency Ultrasonication

Abstract
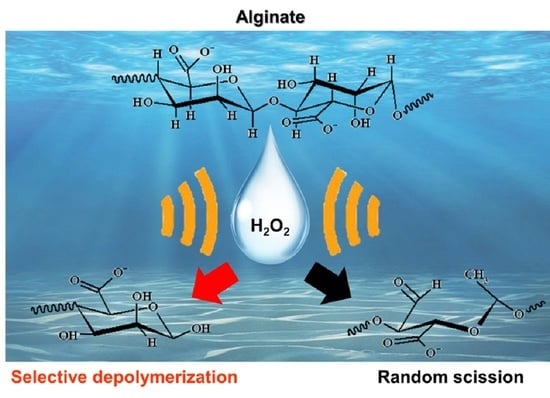
:
1. Introduction
2. Materials and Methods
2.1. Chemical Materials
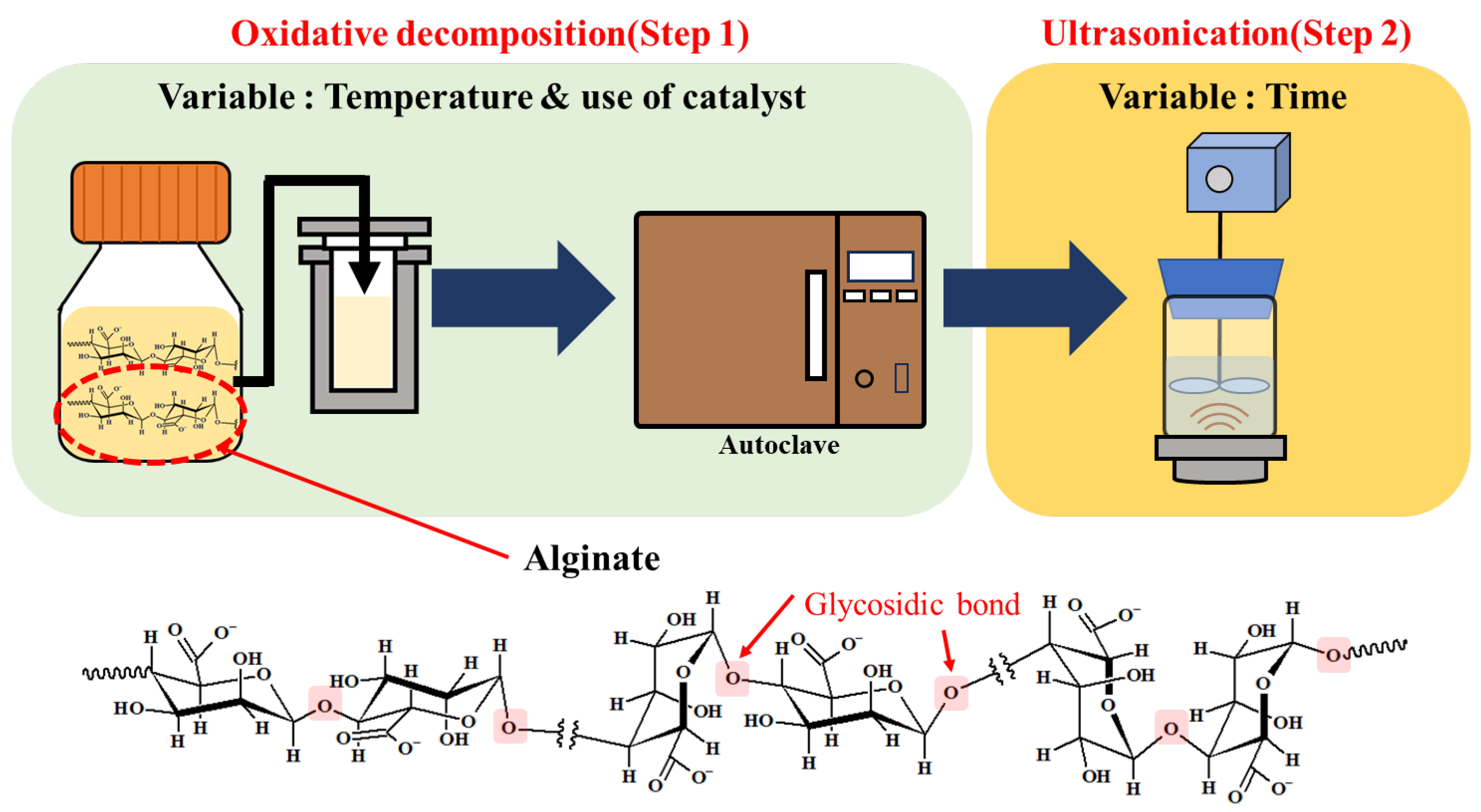
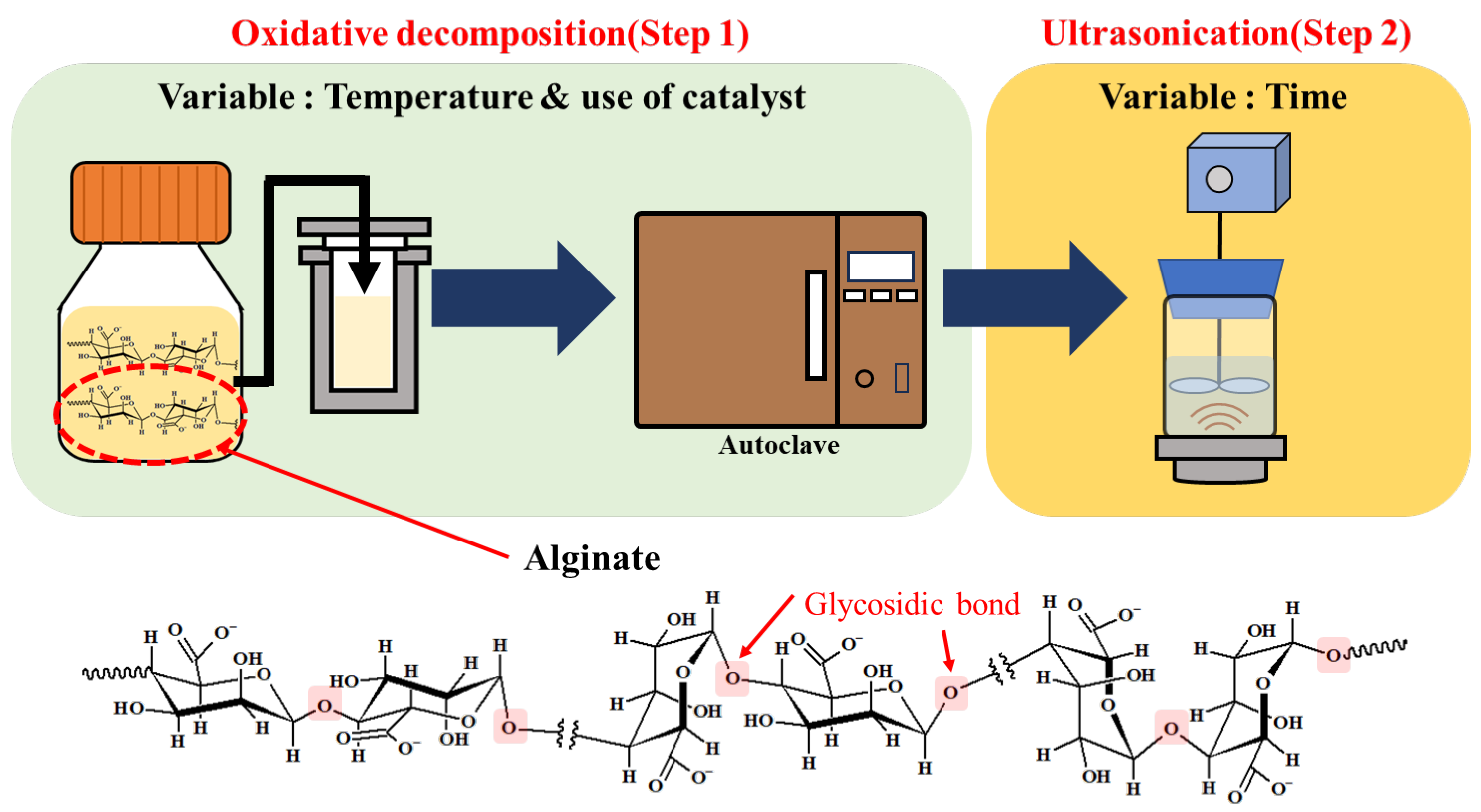
2.2. Oxidative Decomposition by H2O2 (Step 1)
2.3. High-Frequency Ultrasonication (Step 2)
2.4. Product Characterizations
3. Results and Discussion
3.1. Oxidative Decomposition (Step 1)
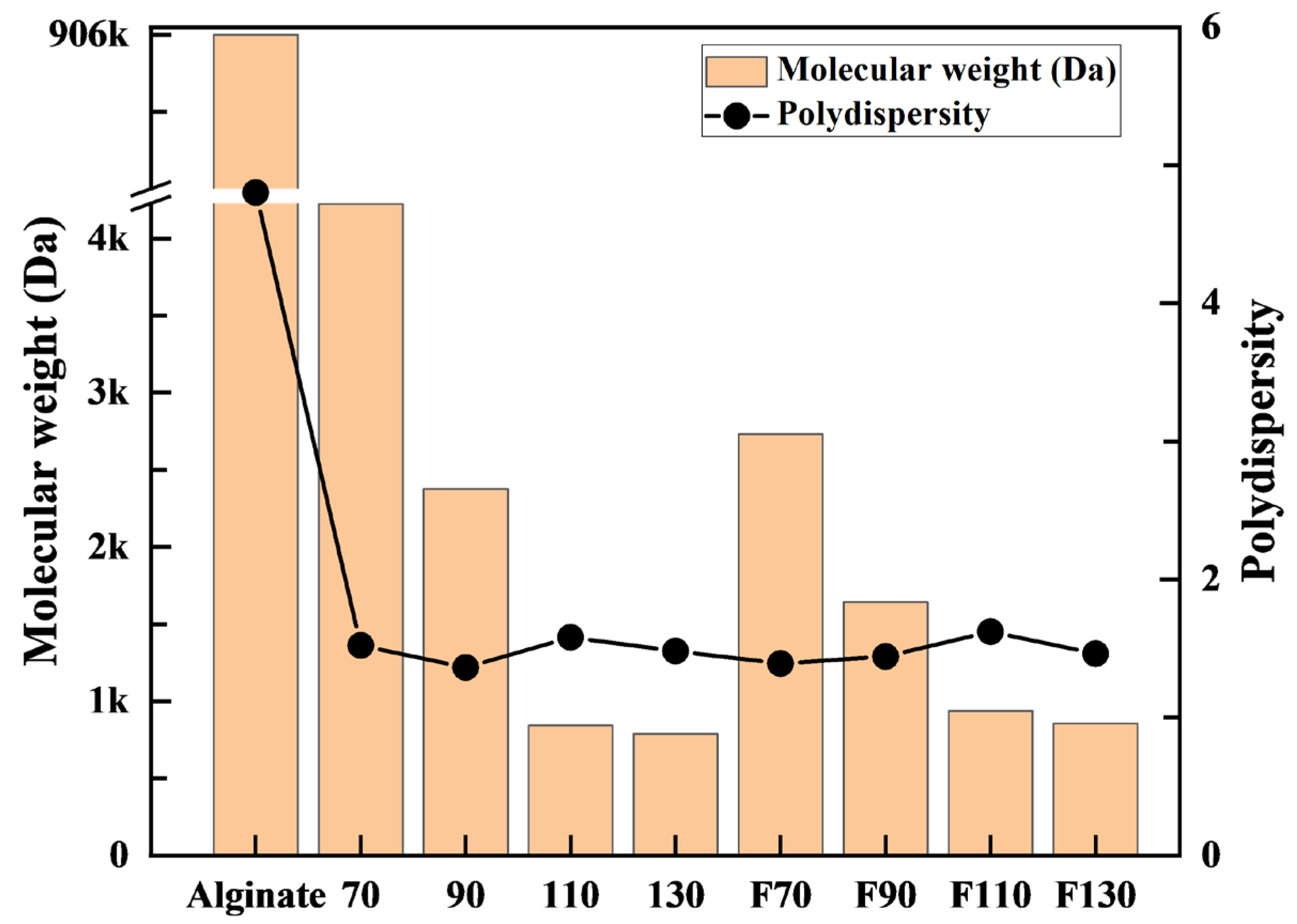
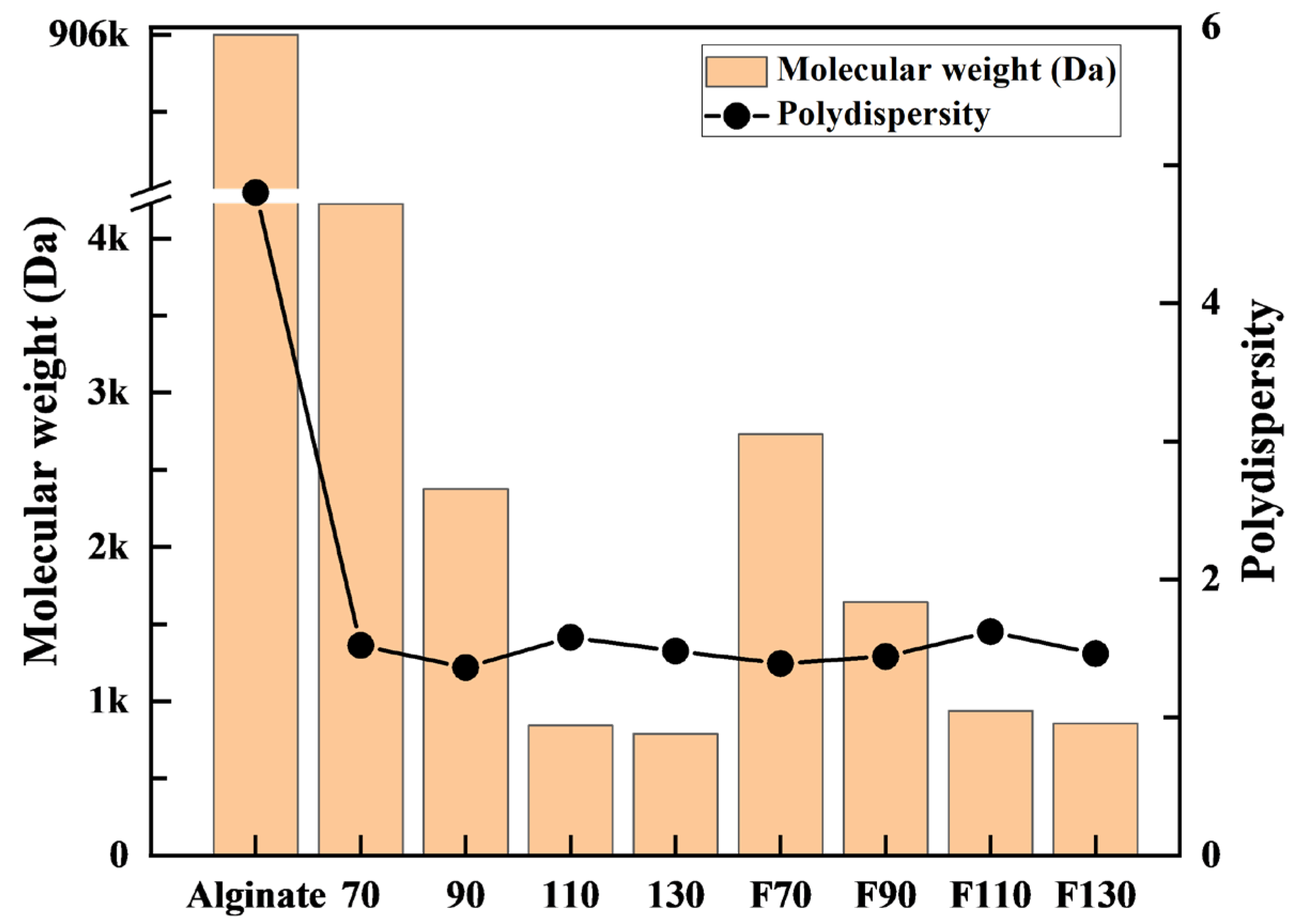
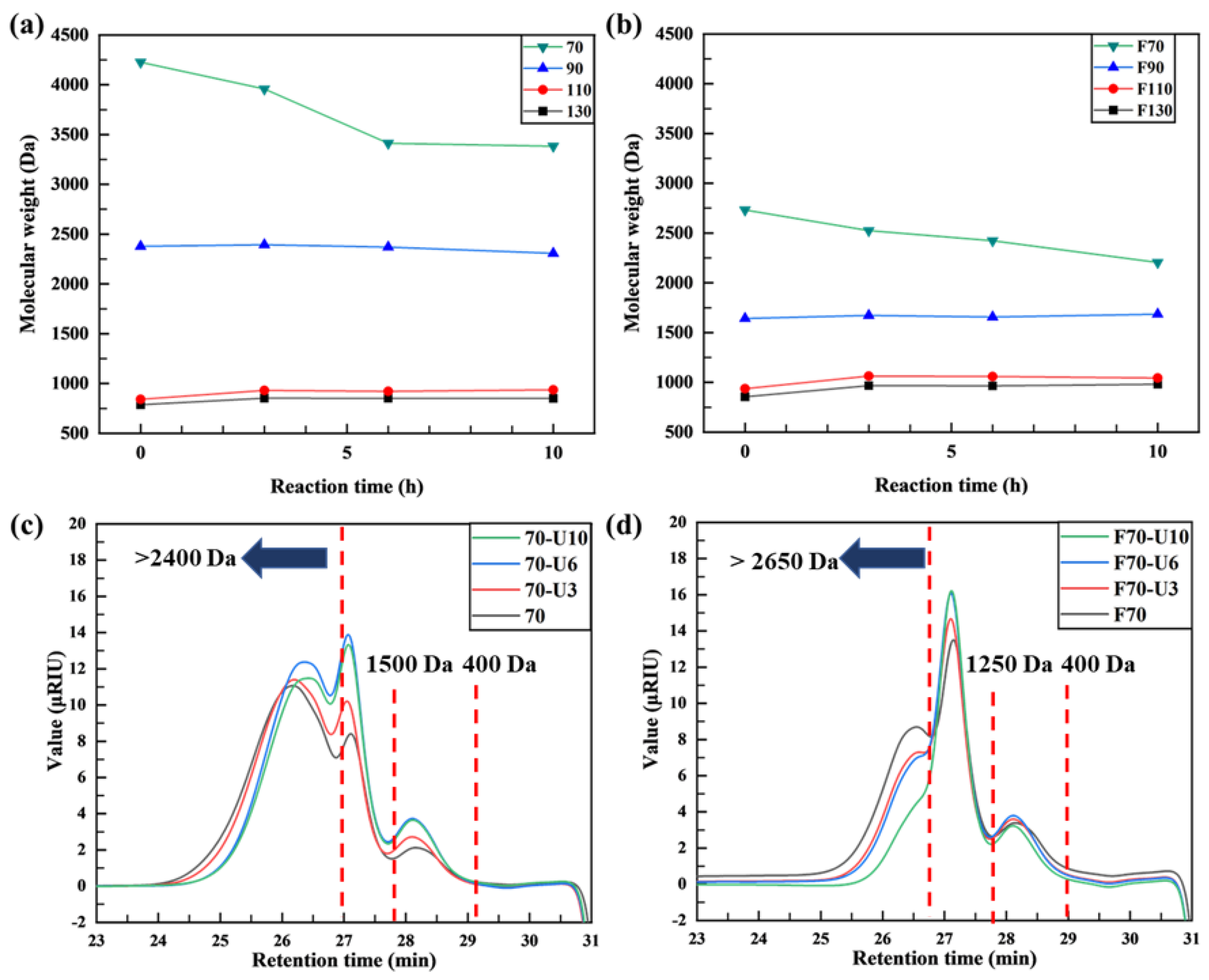
3.1.1. GPC Analysis
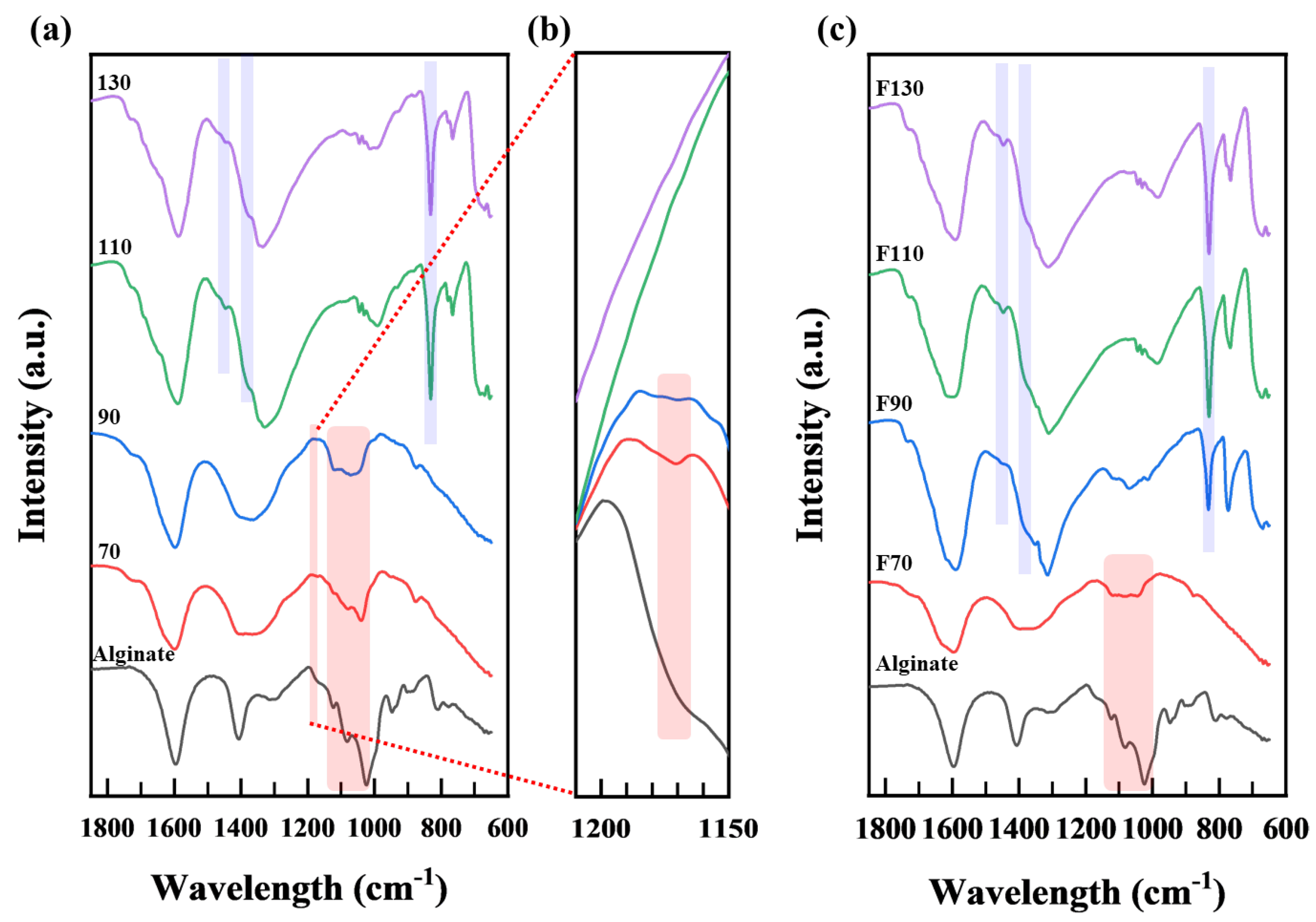
3.1.2. FT−IR Analysis
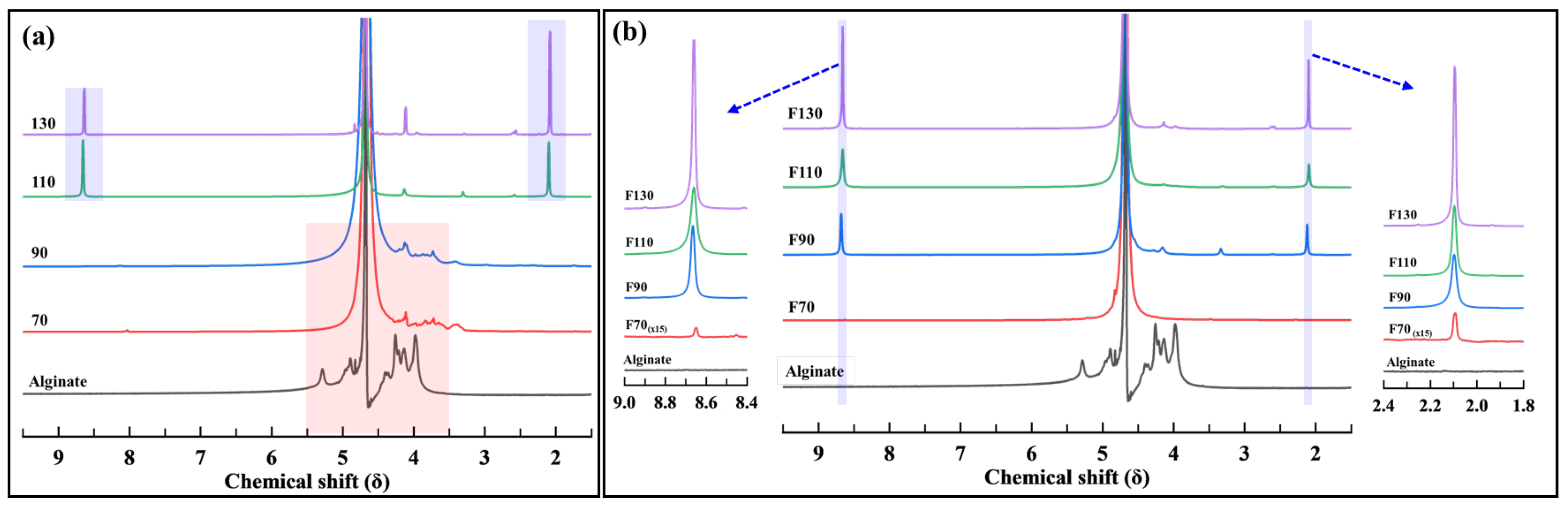
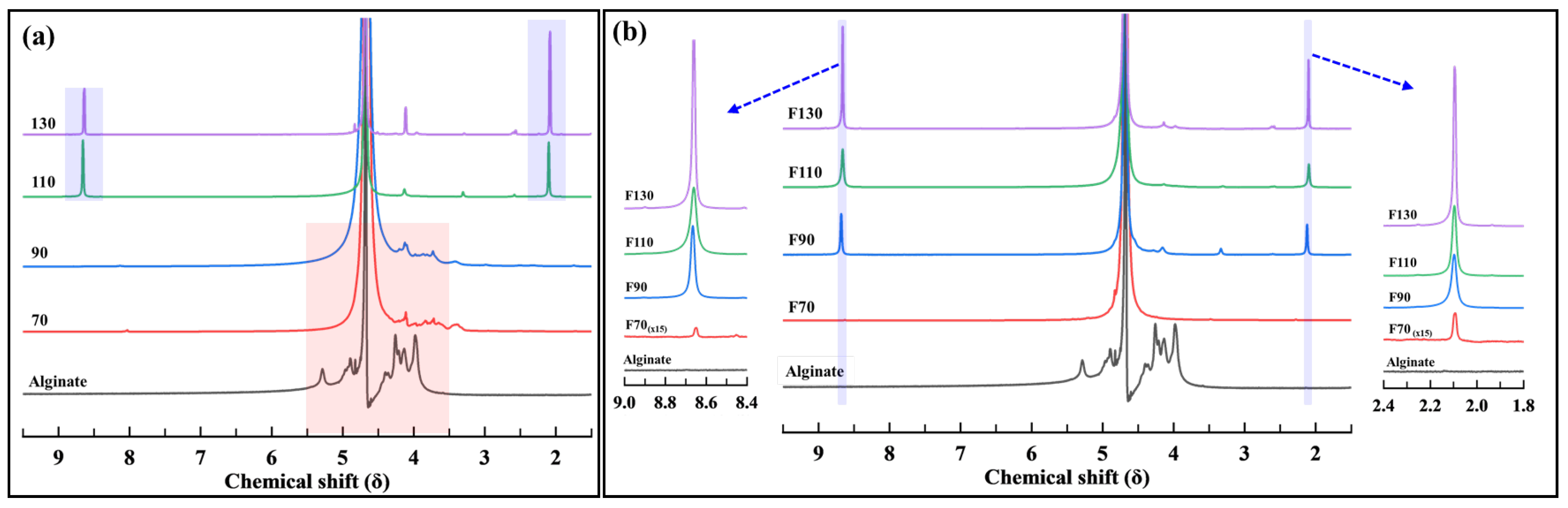
3.1.3. NMR Analysis
3.2. High-Frequency Ultrasonication (Step 2)
3.2.1. GPC Analysis
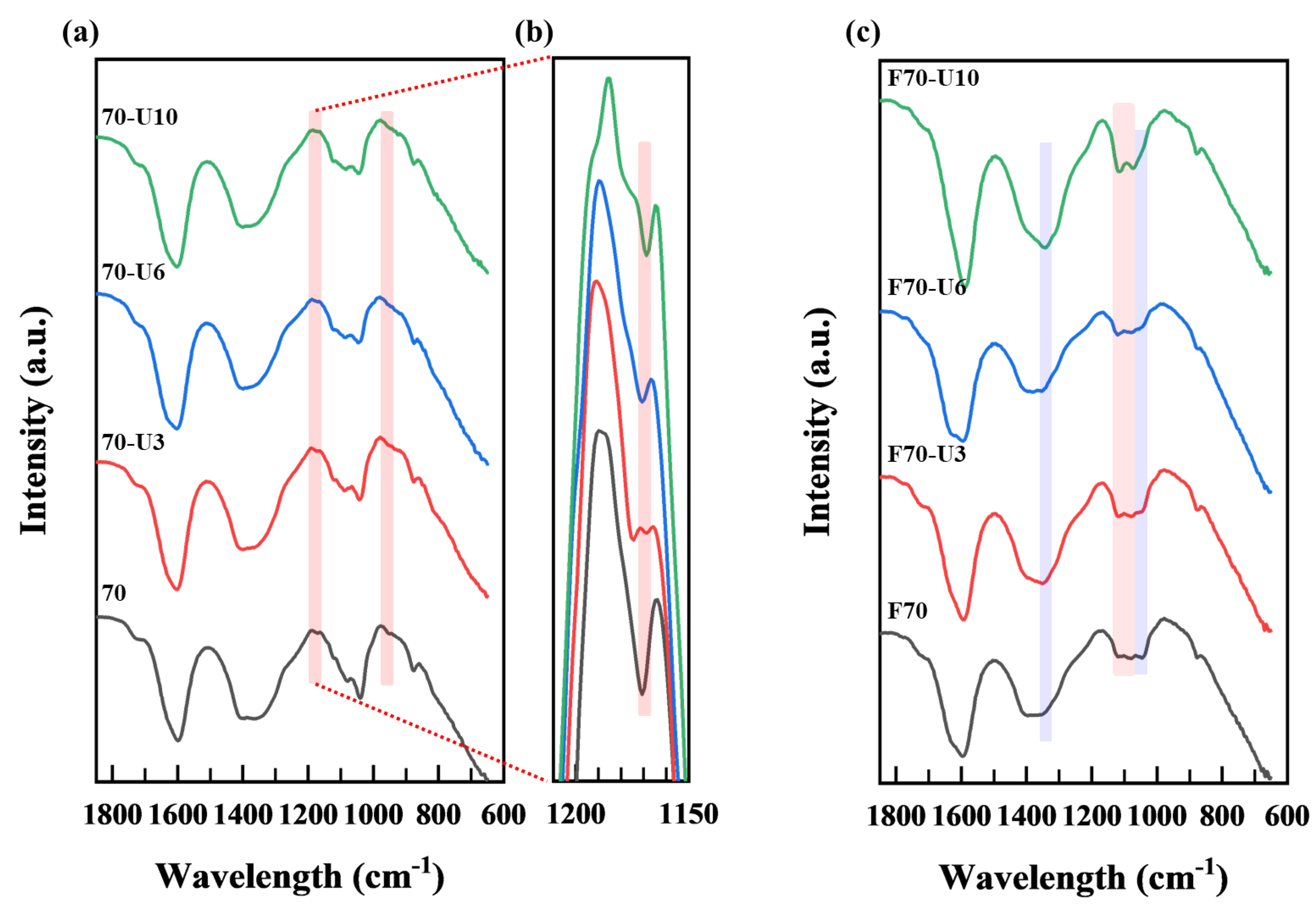
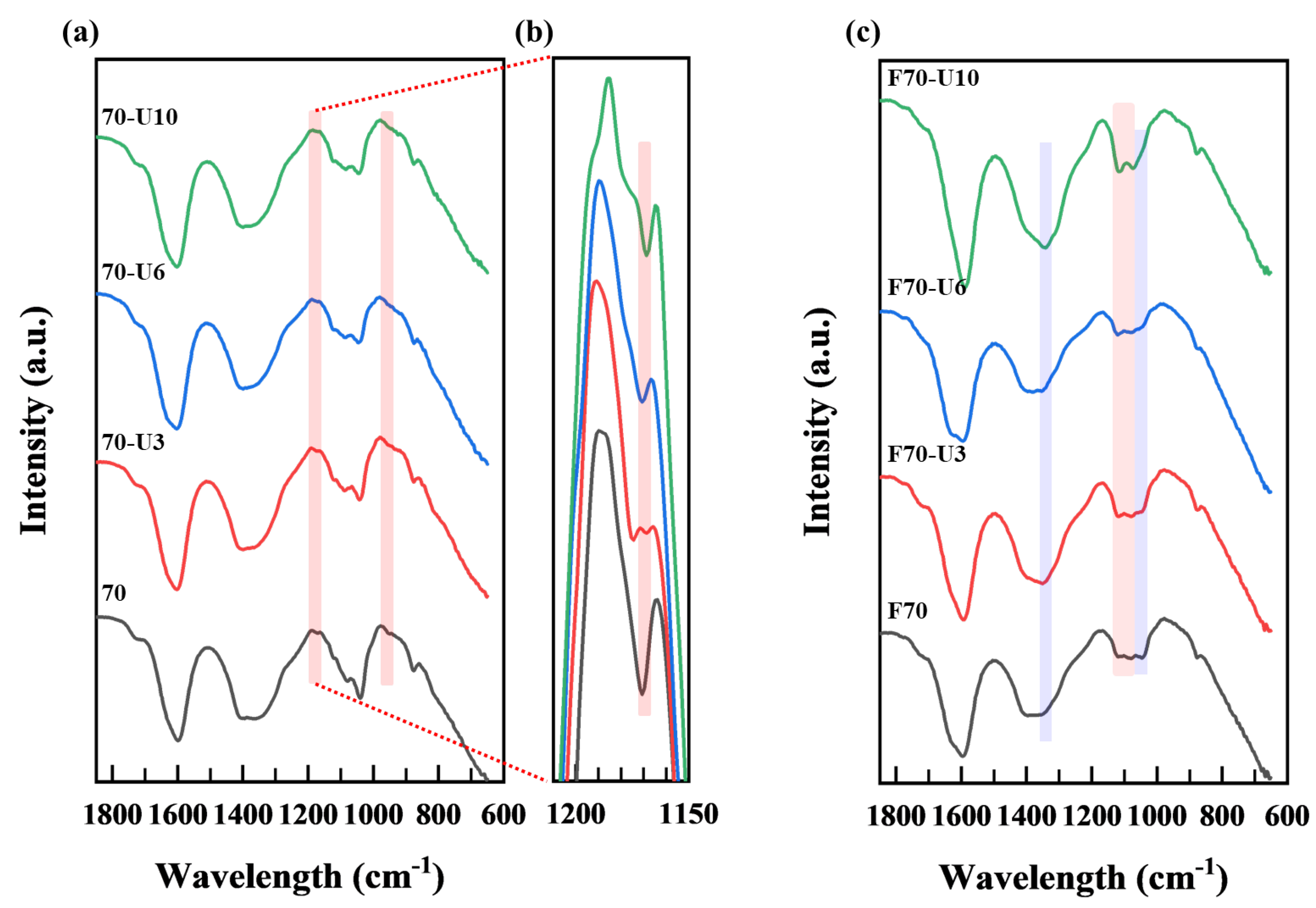
3.2.2. FT−IR Analysis
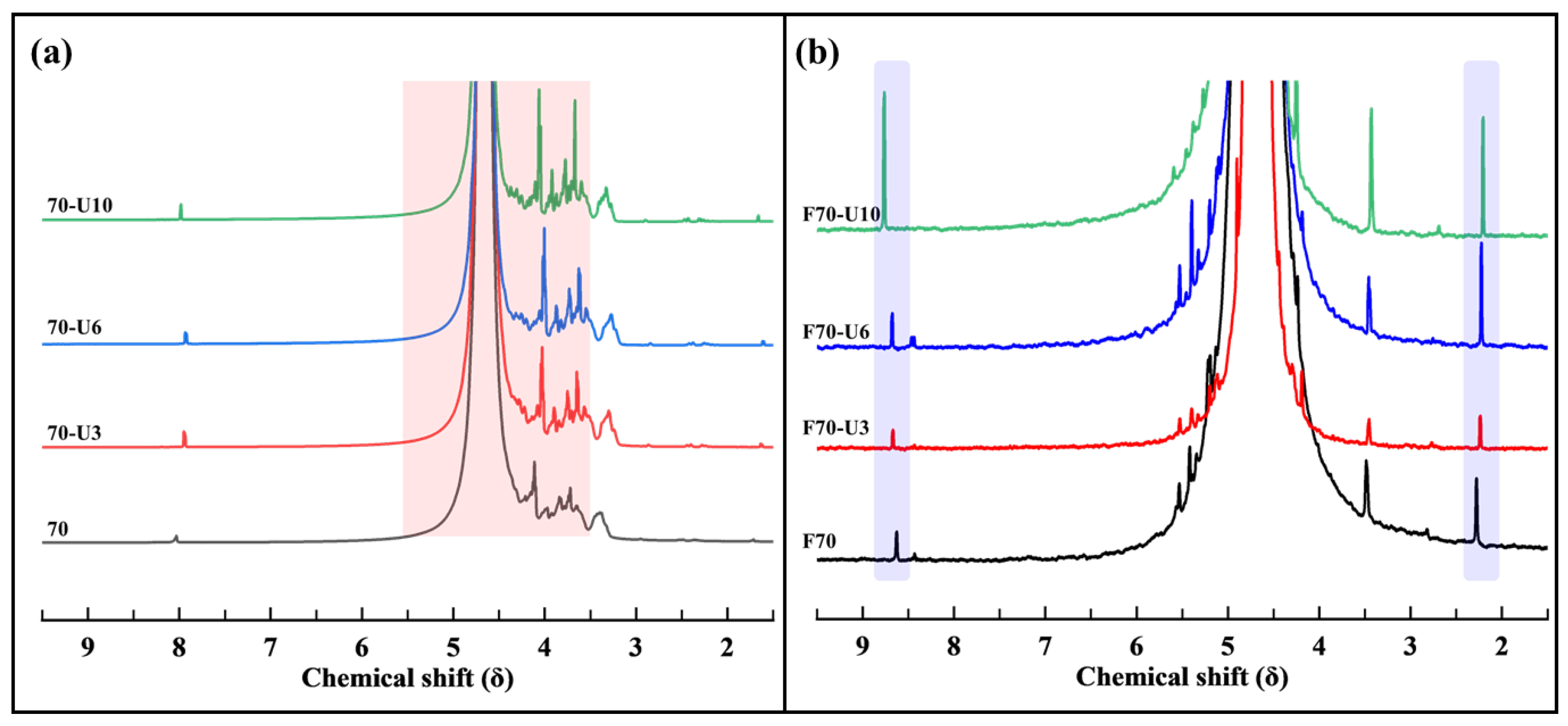
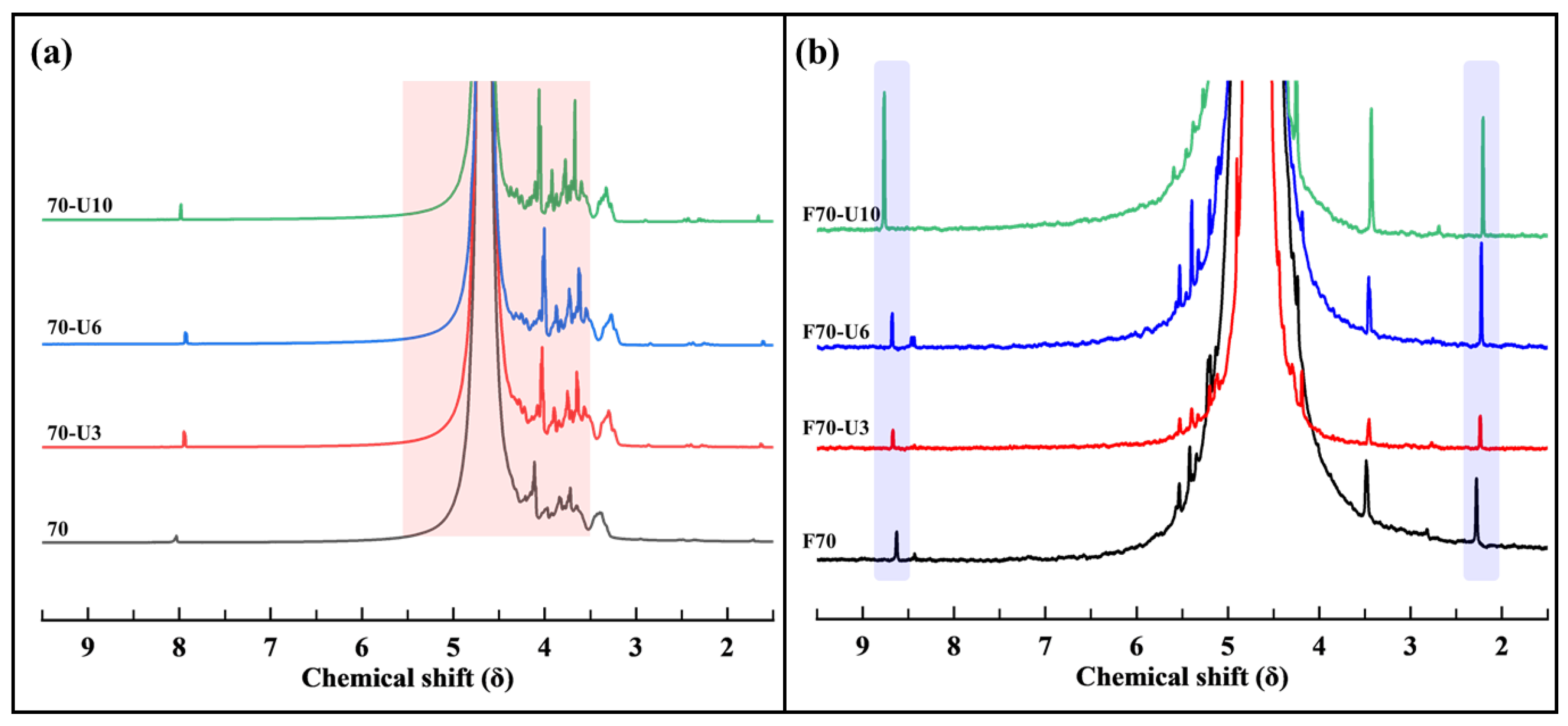
3.2.3. NMR Analysis
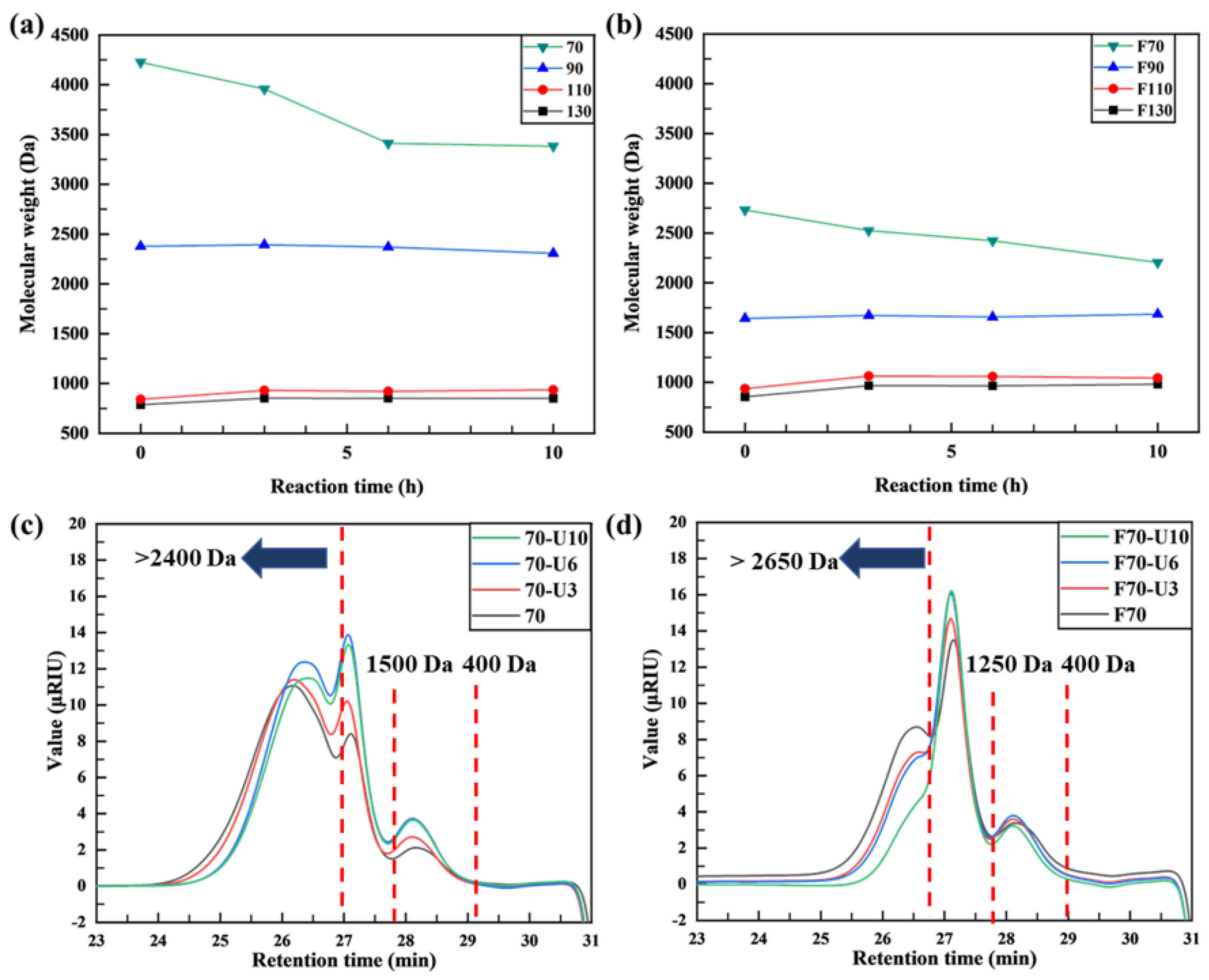
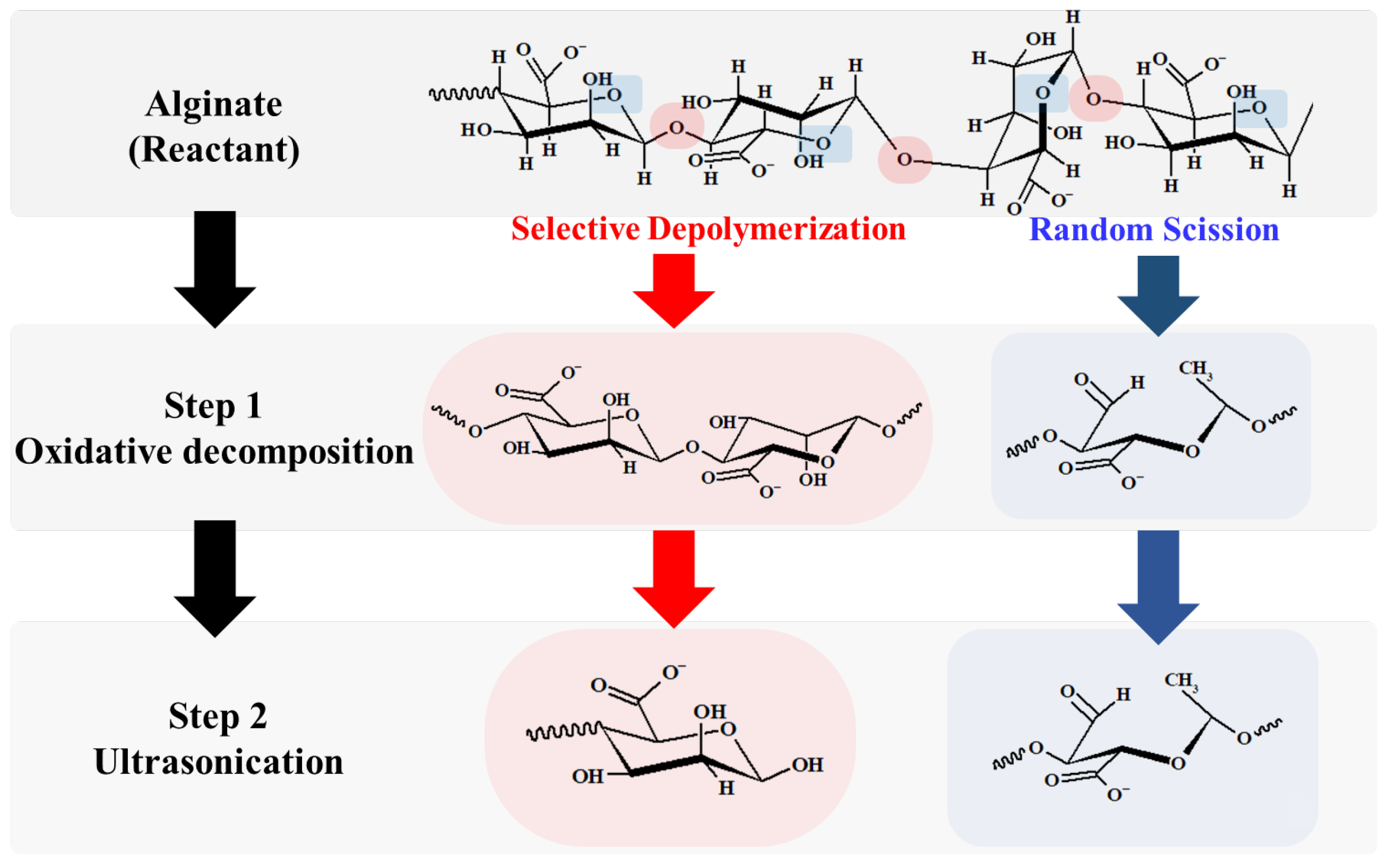
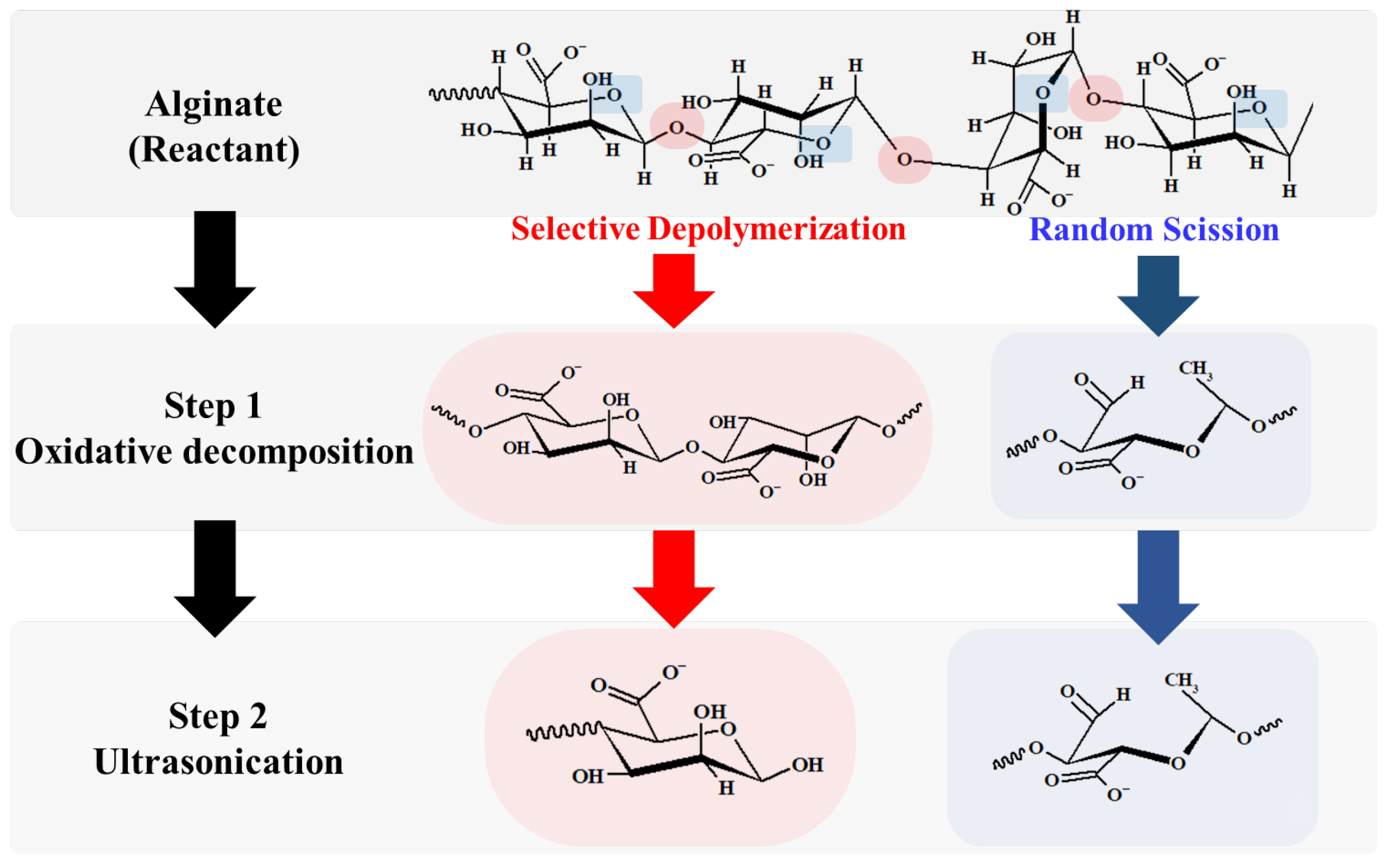
- For sample 70, ultrasonication led to the selective cleavage of glycosidic bonds within the alginate structure with increasing reaction time. This process generated low-molecular-weight polymers, particularly dimers to heptamers, from high-molecular-weight polymers (>2400 Da) through a selective depolymerization process.
- Conversely, when sample F70 was subjected to ultrasonication, the alginate was randomly depolymerized by the opening of the pyranose rings within the alginate structure or the cleavage of glycosidic bonds in the monomer chains. Consequently, low-molecular-weight polymers in the range of 1250–2650 Da were generated because of this random depolymerization process.
- In the case of alginate sources other than the aforementioned samples, ultrasonication did not result in further depolymerization. This suggests that the hydroxyl radicals generated by ultrasound primarily target glycosidic bonds that connect the monomer units of the polymer, leading to selective depolymerization and generation of low-molecular-weight species, such as dimers or oligomers.
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Song, M.; Pham, H.D.; Seon, J.; Woo, H.C. Marine brown algae: A conundrum answer for sustainable biofuels production. Renew. Sustain. Energy Rev. 2015, 50, 782–792. [Google Scholar] [CrossRef]
- Luo, L.; van der Voet, E.; Huppes, G. Biorefining of lignocellulosic feedstock–Technical, economic and environmental considerations. Bioresour. Technol. 2010, 101, 5023–5032. [Google Scholar] [CrossRef] [PubMed]
- Draget, K.I.; Skjåk-Bræk, G.; Smidsrød, O. Alginate based new materials. Int. J. Biol. Macromol. 1997, 21, 47–55. [Google Scholar] [CrossRef] [PubMed]
- Zia, K.M.; Zia, F.; Zuber, M.; Rehman, S.; Ahmad, M.N. Alginate based polyurethanes: A review of recent advances and perspective. Int. J. Biol. Macromol. 2015, 79, 377–387. [Google Scholar] [CrossRef] [PubMed]
- Jeon, W.; Ban, C.; Park, G.; Yu, T.-K.; Suh, J.-Y.; Woo, H.C.; Kim, D.H. Catalytic hydrothermal conversion of macroalgae-derived alginate: Effect of pH on production of furfural and valuable organic acids under subcritical water conditions. J. Mol. Catal. A Chem. 2015, 399, 106–113. [Google Scholar] [CrossRef]
- Jeon, W.; Ban, C.; Park, G.; Kim, J.E.; Woo, H.C.; Kim, D.H. Catalytic conversion of macroalgae-derived alginate to useful chemicals. Catal. Surv. Asia 2016, 20, 195–209. [Google Scholar] [CrossRef]
- Koyanagi, S.; Tanigawa, N.; Nakagawa, H.; Soeda, S.; Shimeno, H. Oversulfation of fucoidan enhances its anti-angiogenic and antitumor activities. Biochem. Pharmacol. 2003, 65, 173–179. [Google Scholar] [CrossRef] [PubMed]
- Schaeffer, D.J.; Krylov, V.S. Anti-HIV activity of extracts and compounds from algae and cyanobacteria. Ecotoxicol. Environ. Saf. 2000, 45, 208–227. [Google Scholar] [CrossRef]
- Zhang, C.; Li, M.; Rauf, A.; Khalil, A.A.; Shan, Z.; Chen, C.; Rengasamy, K.R.; Wan, C. Process and applications of alginate oligosaccharides with emphasis on health beneficial perspectives. Crit. Rev. Food Sci. Nutr. 2023, 63, 303–329. [Google Scholar] [CrossRef]
- Wang, M.; Chen, L.; Zhang, Z. Potential applications of alginate oligosaccharides for biomedicine–A mini review. Carbohydr. Polym. 2021, 271, 118408. [Google Scholar] [CrossRef]
- Choi, S.K.; Choi, Y.S. Depolymerization of alginates by hydrogen peroxide/ultrasonic irradiation. Polymers 2011, 35, 444–450. [Google Scholar]
- Tecson, M.G.; Abad, L.V.; Ebajo, V.D., Jr.; Camacho, D.H. Ultrasound-assisted depolymerization of kappa-carrageenan and characterization of degradation product. Ultrason. Sonochem. 2021, 73, 105540. [Google Scholar] [CrossRef] [PubMed]
- Bouanati, T.; Colson, E.; Moins, S.; Cabrera, J.-C.; Eeckhaut, I.; Raquez, J.-M.; Gerbaux, P. Microwave-assisted depolymerization of carrageenans from Kappaphycus alvarezii and Eucheuma spinosum: Controlled and green production of oligosaccharides from the algae biomass. Algal Res. 2020, 51, 102054. [Google Scholar] [CrossRef]
- Holme, H.K.; Lindmo, K.; Kristiansen, A.; Smidsrød, O. Thermal depolymerization of alginate in the solid state. Carbohydr. Polym. 2003, 54, 431–438. [Google Scholar] [CrossRef]
- Haouache, S.; Karam, A.; Chave, T.; Clarhaut, J.; Amaniampong, P.N.; Fernandez, J.M.G.; Vigier, K.D.O.; Capron, I.; Jérôme, F. Selective radical depolymerization of cellulose to glucose induced by high frequency ultrasound. Chem. Sci. 2020, 11, 2664–2669. [Google Scholar] [CrossRef]
- Wong, T.Y.; Preston, L.A.; Schiller, N.L. Alginate lyase: Review of major sources and enzyme characteristics, structure-function analysis, biological roles, and applications. Annu. Rev. Microbiol. 2000, 54, 289–340. [Google Scholar] [CrossRef]
- Yamasaki, M.; Ogura, K.; Hashimoto, W.; Mikami, B.; Murata, K. A structural basis for depolymerization of alginate by polysaccharide lyase family-7. J. Mol. Biol. 2005, 352, 11–21. [Google Scholar] [CrossRef] [PubMed]
- Burana-Osot, J.; Hosoyama, S.; Nagamoto, Y.; Suzuki, S.; Linhardt, R.J.; Toida, T. Photolytic depolymerization of alginate. Carbohydr. Res. 2009, 344, 2023–2027. [Google Scholar] [CrossRef]
- Soukaina, B.; Zainab, E.A.-T.; Guillaume, P.; Halima, R.; Philippe, M.; Cherkaoui, E.M.; Cédric, D. Radical depolymerization of alginate extracted from Moroccan brown seaweed Bifurcaria bifurcata. Appl. Sci. 2020, 10, 4166. [Google Scholar] [CrossRef]
- Haber, F.; Weiss, J. The catalytic decomposition of hydrogen peroxide by iron salts. Proc. R. Soc. London Ser. A Math. Phys. Sci. 1934, 147, 332–351. [Google Scholar]
- Jérôme, F.; Chatel, G.; Vigier, K.D.O. Depolymerization of cellulose to processable glucans by non-thermal technologies. Green Chem. 2016, 18, 3903–3913. [Google Scholar] [CrossRef]
- McKenzie, T.G.; Karimi, F.; Ashokkumar, M.; Qiao, G.G. Ultrasound and sonochemistry for radical polymerization: Sound synthesis. Chem. Eur. J. 2019, 25, 5372–5388. [Google Scholar] [CrossRef] [PubMed]
- Roy, K.; Moholkar, V.S. p–nitrophenol degradation by hybrid advanced oxidation process of heterogeneous Fenton assisted hydrodynamic cavitation: Discernment of synergistic interactions and chemical mechanism. Chemosphere 2020, 283, 131114. [Google Scholar] [CrossRef] [PubMed]
- Ahn, H.J.; Kang, K.S.; Song, Y.H.; Lee, D.H.; Kim, M.H.; Lee, J.; Woo, H.C. Effect of cardanol content on the antibacterial films derived from alginate-PVA blended matrix. Clean Technol. 2022, 28, 24–31. [Google Scholar]
- Huang, R.; Pal, R.; Moon, G. Characteristics of sodium alginate membranes for the pervaporation dehydration of ethanol–water and isopropanol–water mixtures. J. Membr. Sci. 1999, 160, 101–113. [Google Scholar] [CrossRef]
- Zain, N.A.M.; Suhaimi, M.S.; Idris, A. Development and modification of PVA–alginate as a suitable immobilization matrix. Process Biochem. 2011, 46, 2122–2129. [Google Scholar] [CrossRef]
- Fertah, M.; Belfkira, A.; Taourirte, M.; Brouillette, F. Extraction and characterization of sodium alginate from Moroccan Laminaria digitata brown seaweed. Arab. J. Chem. 2017, 10, S3707–S3714. [Google Scholar] [CrossRef]
- González-López, N.; Moure, A.; Domínguez, H. Hydrothermal fractionation of Sargassum muticum biomass. J. Appl. Phycol. 2012, 24, 1569–1578. [Google Scholar] [CrossRef]
- Margenot, A.J.; Calderón, F.J.; Parikh, S.J. Limitations and potential of spectral subtractions in Fourier-transform infrared spectroscopy of soil samples. Soil Sci. Soc. Am. J. 2016, 80, 10–26. [Google Scholar] [CrossRef]
- Anjali, T. Modification of carboxymethyl cellulose through oxidation. Carbohydr. Polym. 2012, 87, 457–460. [Google Scholar] [CrossRef]
- Vijayakumar, S.; Malaikozhundan, B.; Parthasarathy, A.; Saravanakumar, K.; Wang, M.-H.; Vaseeharan, B. Nano biomedical potential of biopolymer chitosan-capped silver nanoparticles with special reference to antibacterial, antibiofilm, anticoagulant and wound dressing material. J. Clust. Sci. 2020, 31, 355–366. [Google Scholar] [CrossRef]
- Singh, K.; Devi, S.; Bajaj, H.; Ingole, P.; Choudhari, J.; Bhrambhatt, H. Optical resolution of racemic mixtures of amino acids through nanofiltration membrane process. Sep. Sci. Technol. 2014, 49, 2630–2641. [Google Scholar] [CrossRef]
- Caykara, T.; Demirci, S. Preparation and characterization of blend films of poly (vinyl alcohol) and sodium alginate. J. Macromol. Sci. Part A 2006, 43, 1113–1121. [Google Scholar] [CrossRef]
- El Nemr, A.; Eleryan, A.; Mashaly, M.; Khaled, A. Rapid synthesis of cellulose propionate and its conversion to cellulose nitrate propionate. Polym. Bull. 2021, 78, 4149–4182. [Google Scholar] [CrossRef]
- Sakugawa, K.; Ikeda, A.; Takemura, A.; Ono, H. Simplified method for estimation of composition of alginates by FTIR. J. Appl. Polym. Sci. 2004, 93, 1372–1377. [Google Scholar] [CrossRef]
- Margariti, C. The application of FTIR microspectroscopy in a non-invasive and non-destructive way to the study and conservation of mineralised excavated textiles. Herit. Sci. 2019, 7, 63. [Google Scholar] [CrossRef]
- Cardenas-Jiron, G.; Leal, D.; Matsuhiro, B.; Osorio-Roman, I. Vibrational spectroscopy and density functional theory calculations of poly-D-mannuronate and heteropolymeric fractions from sodium alginate. J. Raman Spectrosc. 2011, 42, 870–878. [Google Scholar] [CrossRef]
- Huamani-Palomino, R.G.; Córdova, B.M.; Pichilingue, L.E.R.; Venâncio, T.; Valderrama, A.C. Functionalization of an alginate-based material by oxidation and reductive amination. Polymers 2021, 13, 255. [Google Scholar] [CrossRef] [PubMed]
- Gomez, C.G.; Rinaudo, M.; Villar, M.A. Oxidation of sodium alginate and characterization of the oxidized derivatives. Carbohydr. Polym. 2007, 67, 296–304. [Google Scholar] [CrossRef]
- Delattre, C.; Michaud, P.; Elboutachfaiti, R.; Courtois, B.; Courtois, J. Production of oligocellouronates by biodegradation of oxidized cellulose. Cellulose 2006, 13, 63–71. [Google Scholar] [CrossRef]
- Luo, P.; Liu, L.; Xu, W.; Fan, L.; Nie, M. Preparation and characterization of aminated hyaluronic acid/oxidized hydroxyethyl cellulose hydrogel. Carbohydr. Polym. 2018, 199, 170–177. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Reaction Temperature (°C) | Use of Fe3O4 Catalyst | Product Label |
|---|---|---|
| 70 | No | 70 |
| 90 | No | 90 |
| 110 | No | 110 |
| 130 | No | 130 |
| 70 | Yes | F70 |
| 90 | Yes | F90 |
| 110 | Yes | F110 |
| 130 | Yes | F130 |
| Reaction Temperature (°C) | Duration of Ultrasonication (h) | Product Label |
|---|---|---|
| 70 | 3 | 70-U3 |
| 6 | 70-U6 | |
| 10 | 70-U10 | |
| 90 | 3 | 90-U3 |
| 6 | 90-U6 | |
| 10 | 90-U10 | |
| 110 | 3 | 110-U3 |
| 6 | 110-U6 | |
| 10 | 110-U10 | |
| 130 | 3 | 130-U3 |
| 6 | 130-U6 | |
| 10 | 130-U10 | |
| F70 | 3 | F70-U3 |
| 6 | F70-U6 | |
| 10 | F70-U10 | |
| F90 | 3 | F90-U3 |
| 6 | F90-U6 | |
| 10 | F90-U10 | |
| F110 | 3 | F110-U3 |
| 6 | F110-U6 | |
| 10 | F110-U10 | |
| F130 | 3 | F130-U3 |
| 6 | F130-U6 | |
| 10 | F130-U10 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Song, Y.H.; Woo, H.C.; Lee, J. Eco-Friendly Depolymerization of Alginates by H2O2 and High-Frequency Ultrasonication. Clean Technol. 2023, 5, 1402-1414. https://doi.org/10.3390/cleantechnol5040069
Song YH, Woo HC, Lee J. Eco-Friendly Depolymerization of Alginates by H2O2 and High-Frequency Ultrasonication. Clean Technologies. 2023; 5(4):1402-1414. https://doi.org/10.3390/cleantechnol5040069
Chicago/Turabian StyleSong, Yun Ha, Hee Chul Woo, and Jaekyoung Lee. 2023. "Eco-Friendly Depolymerization of Alginates by H2O2 and High-Frequency Ultrasonication" Clean Technologies 5, no. 4: 1402-1414. https://doi.org/10.3390/cleantechnol5040069
APA StyleSong, Y. H., Woo, H. C., & Lee, J. (2023). Eco-Friendly Depolymerization of Alginates by H2O2 and High-Frequency Ultrasonication. Clean Technologies, 5(4), 1402-1414. https://doi.org/10.3390/cleantechnol5040069