Iron and Manganese Biogeochemistry in Forested Coal Mine Spoil


Abstract
:
1. Introduction
2. Materials and Methods
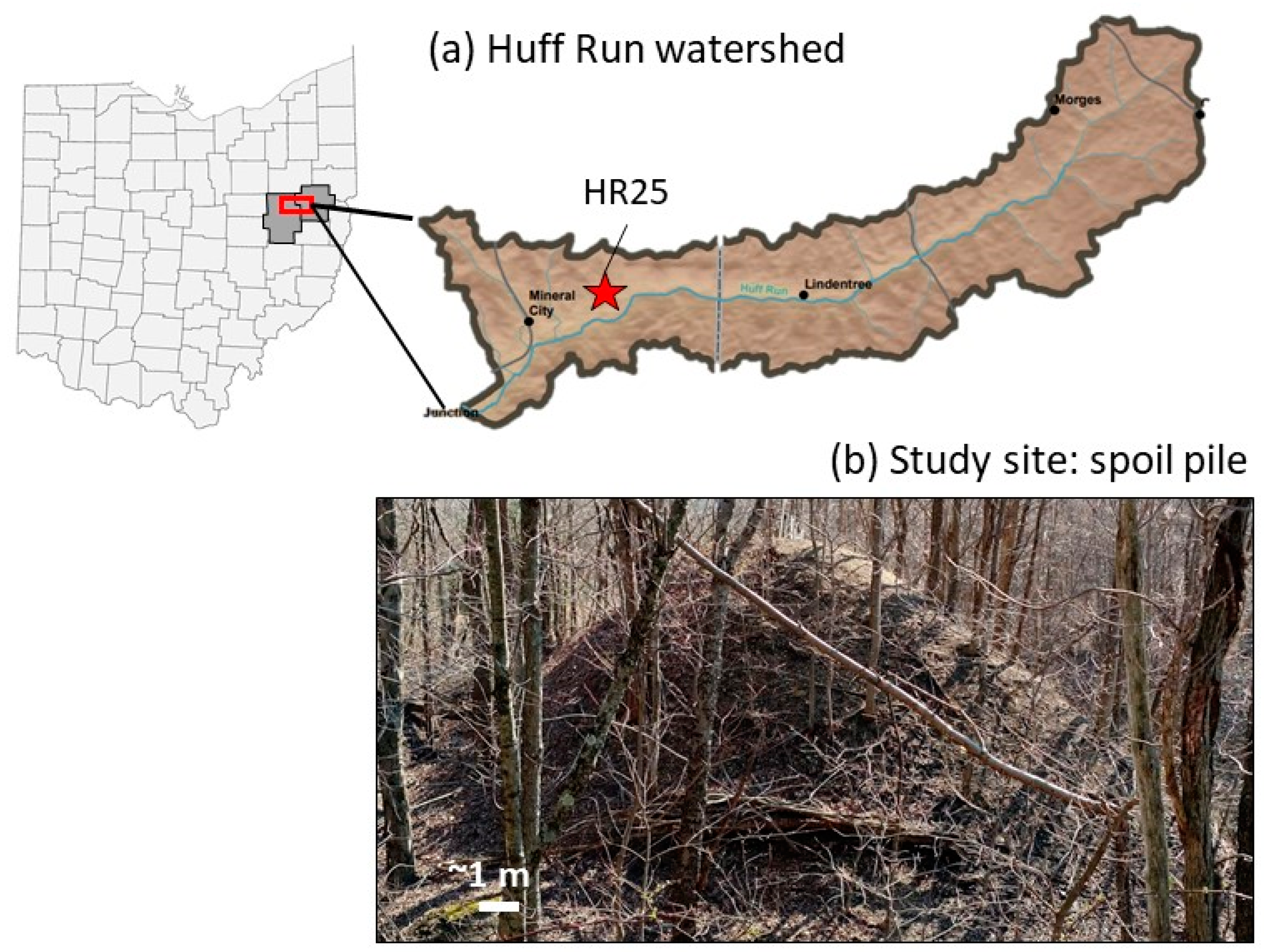
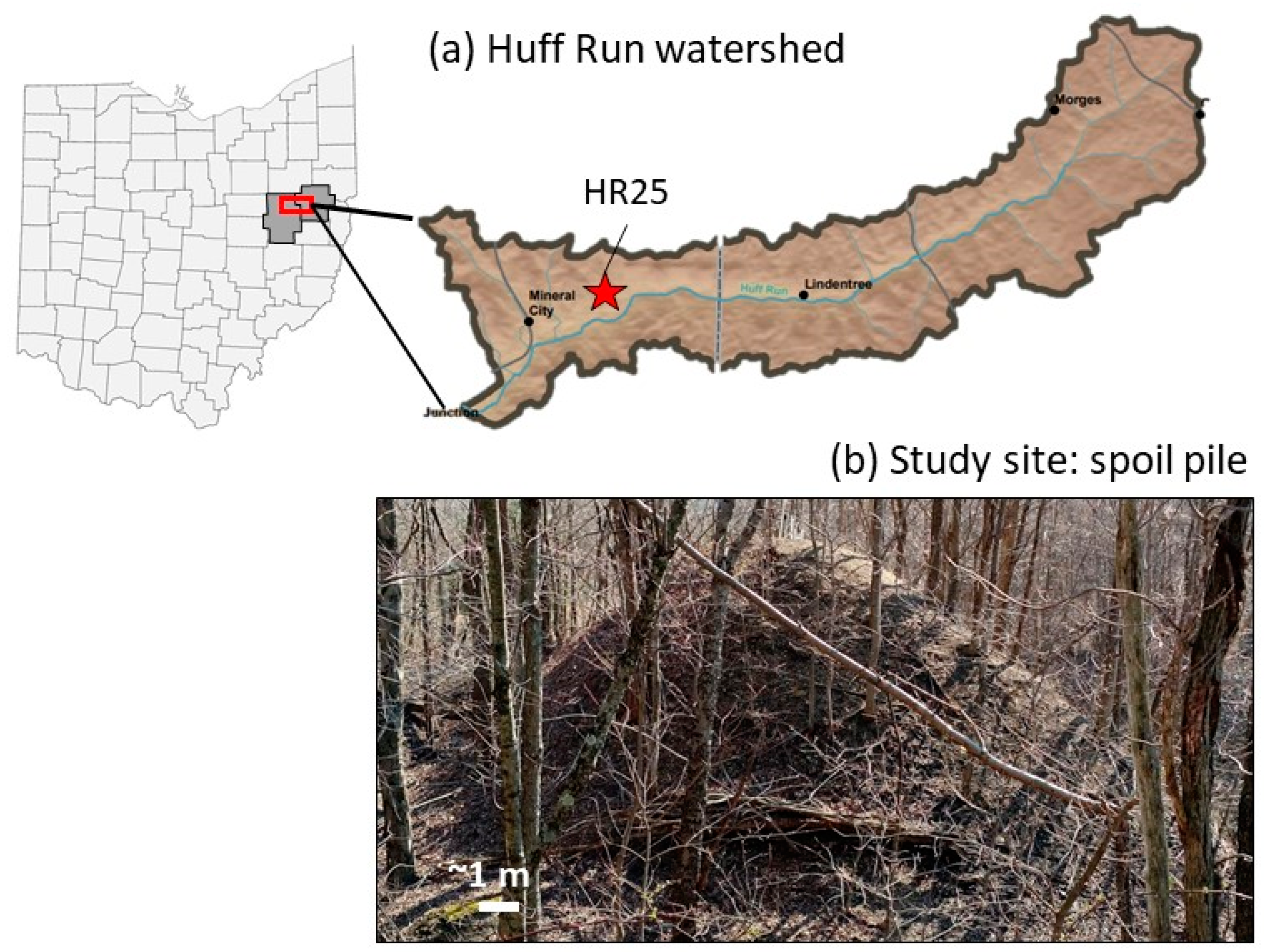
2.1. Site Description
2.2. Weather Data and Evapotranspiration Calculations
2.3. Soil Collection and Analysis
2.3.1. Bulk Characterization
2.3.2. X-ray Absorption Spectroscopy
2.4. Water Collection and Analysis
2.5. Vegetation Collection and Analysis
2.6. Mass Balance Model
3. Results
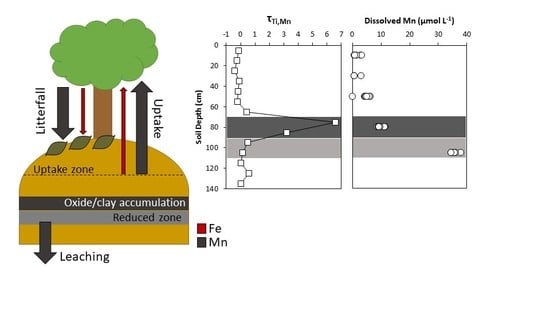
3.1. Soil Geochemistry
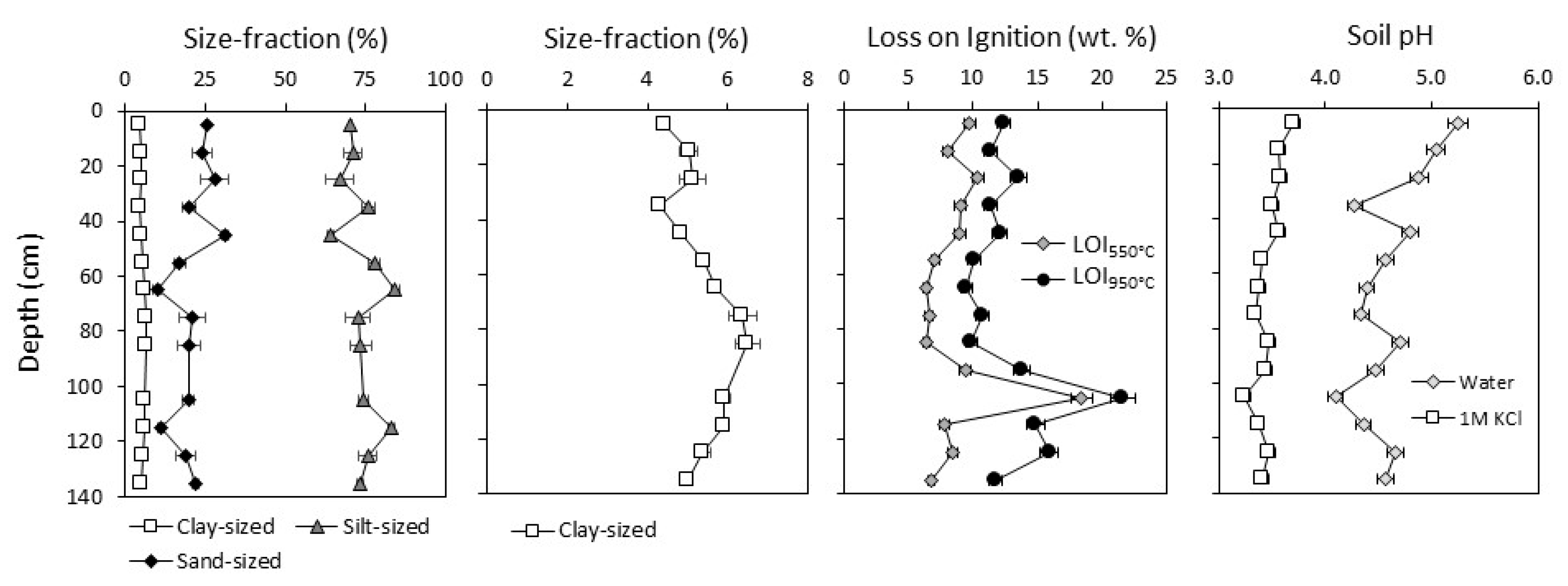
3.1.1. Soil Properties
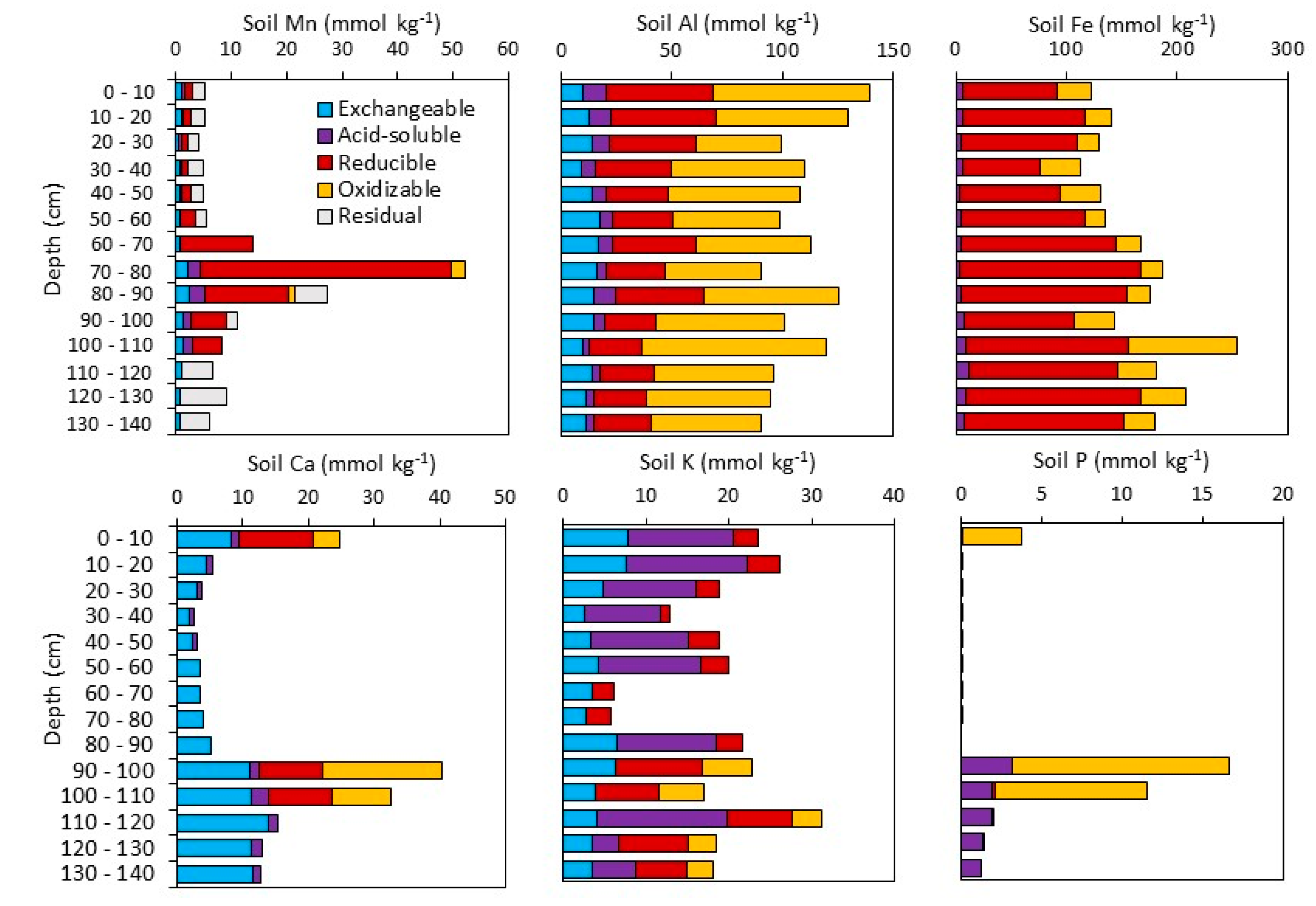
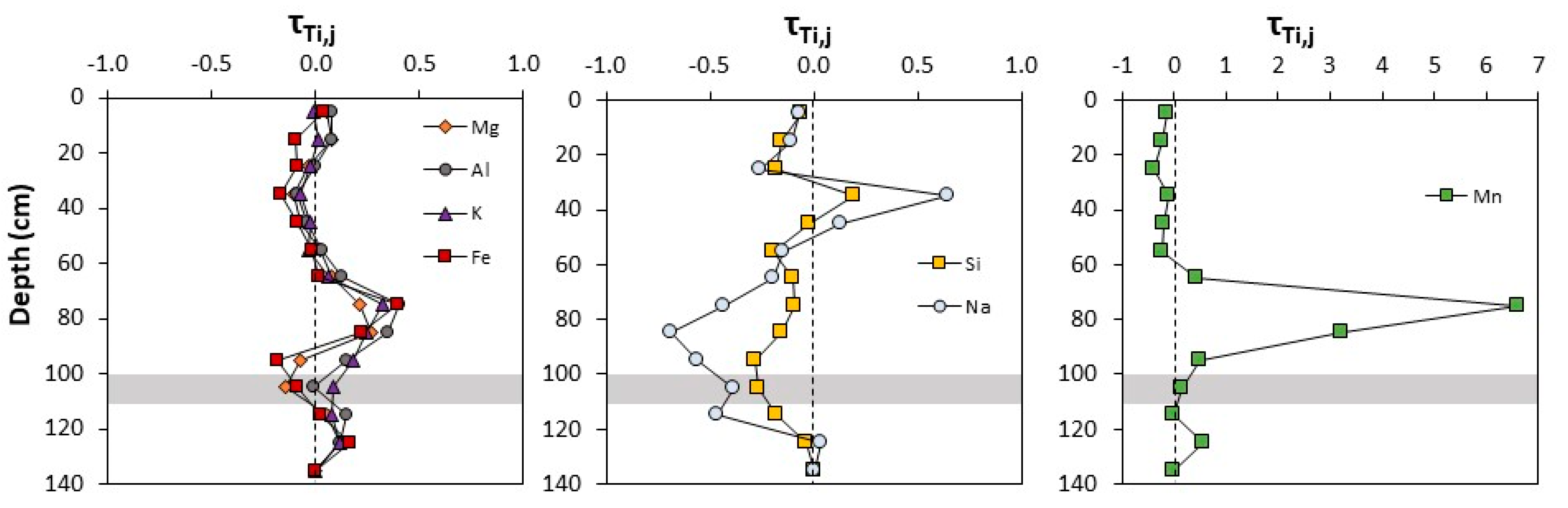
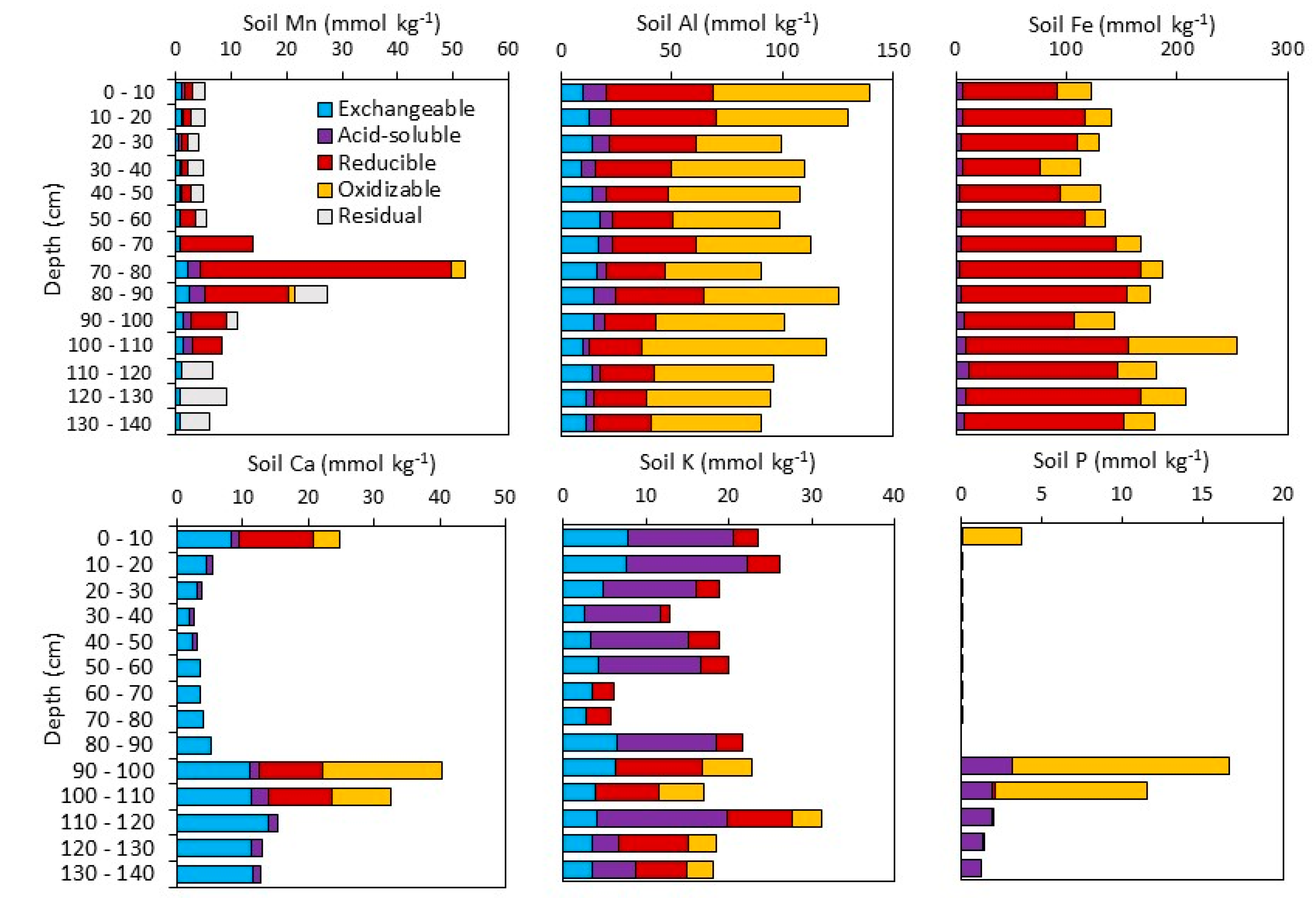
3.1.2. Sequential Extractions
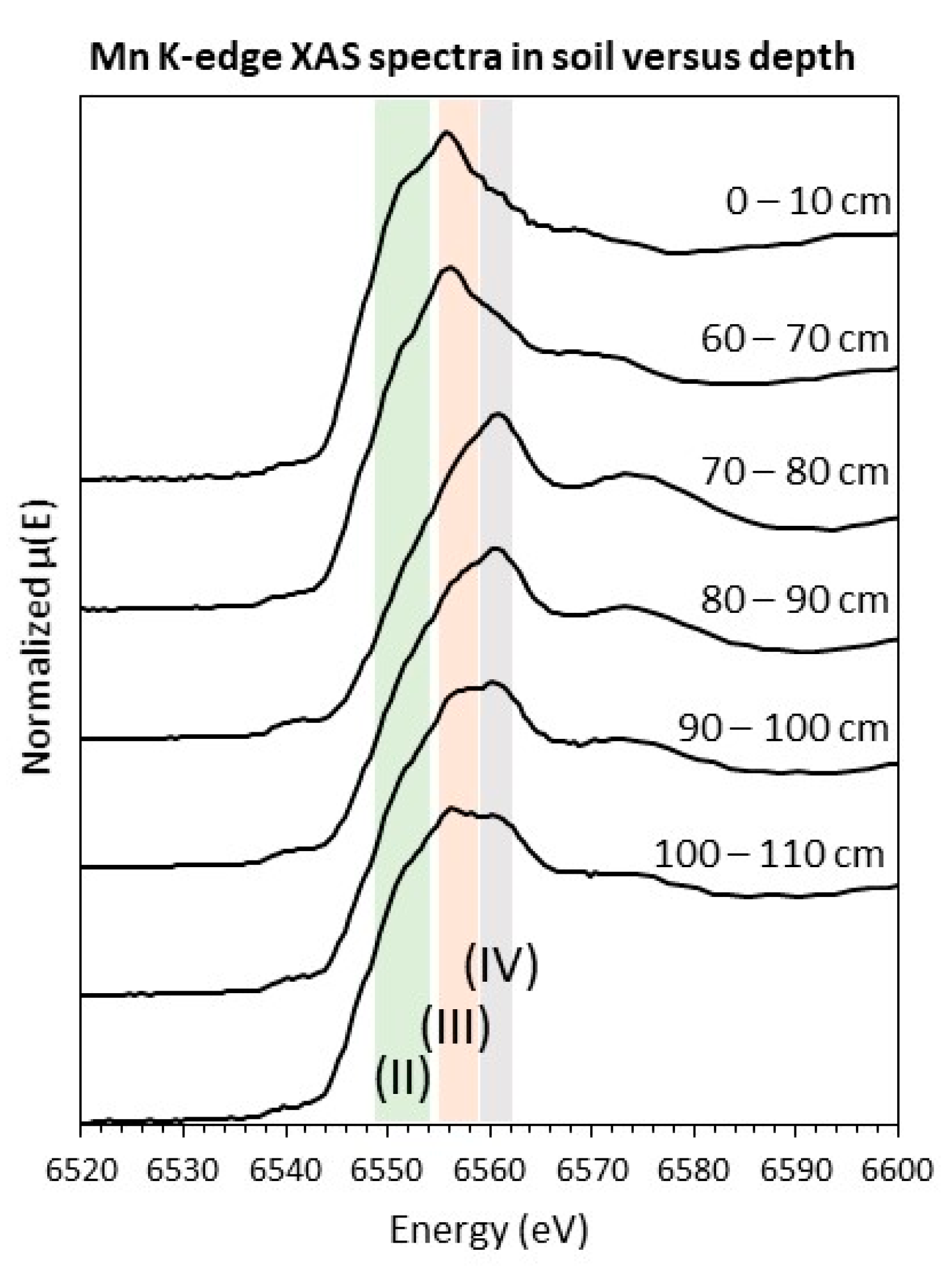
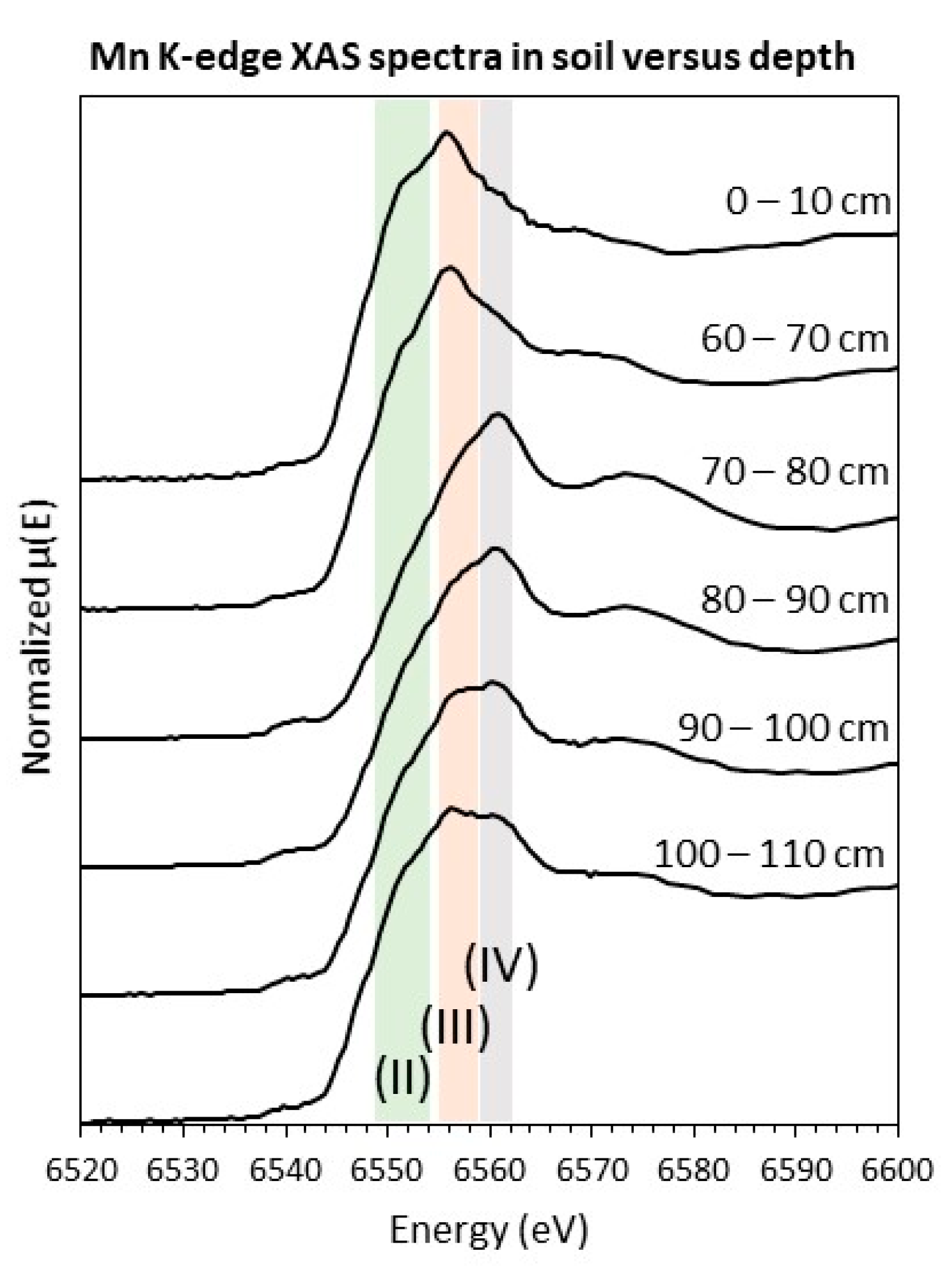
3.1.3. Spectroscopy
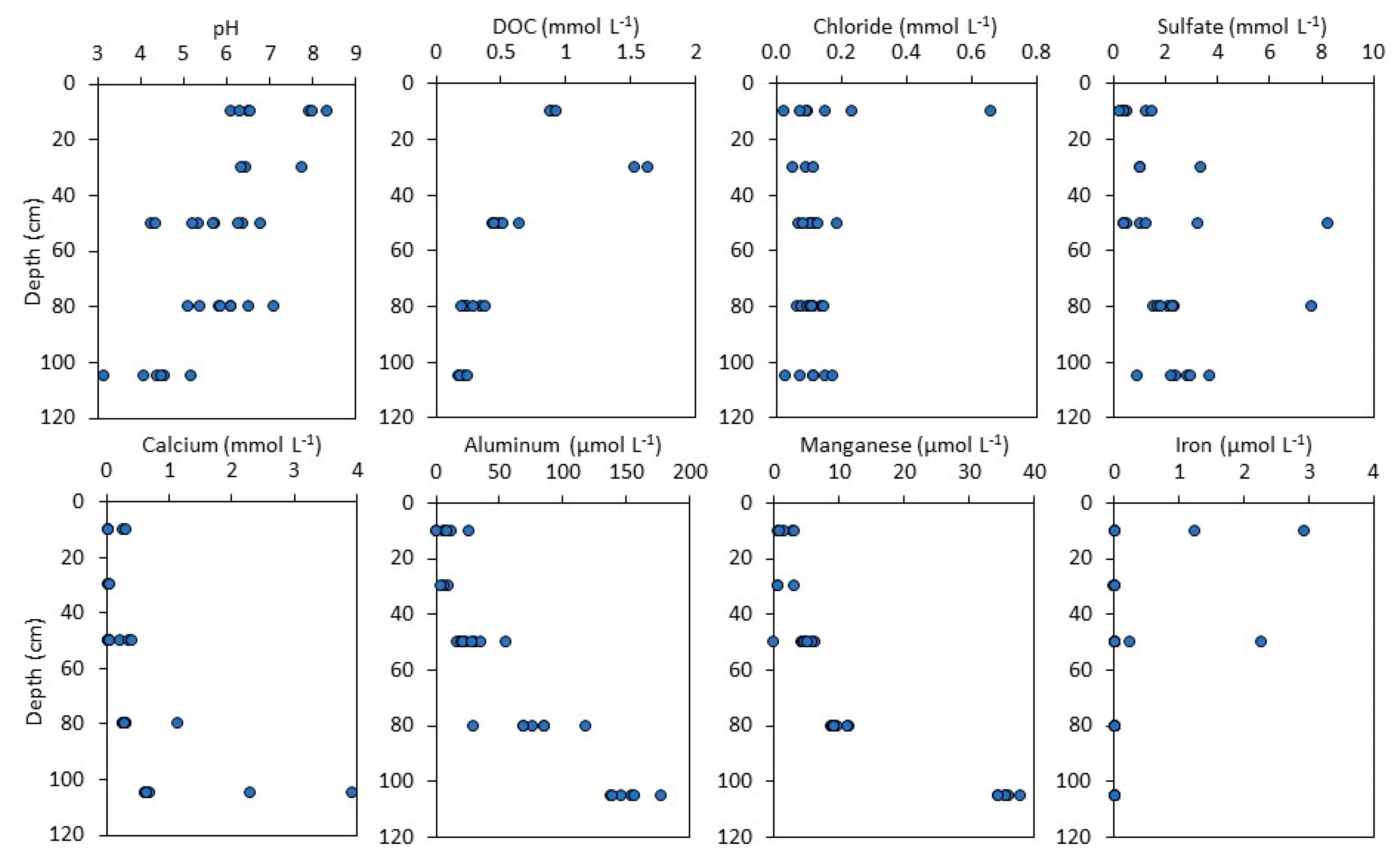
3.2. Soil Water Chemistry
3.3. Vegetation Chemistry
4. Discussion
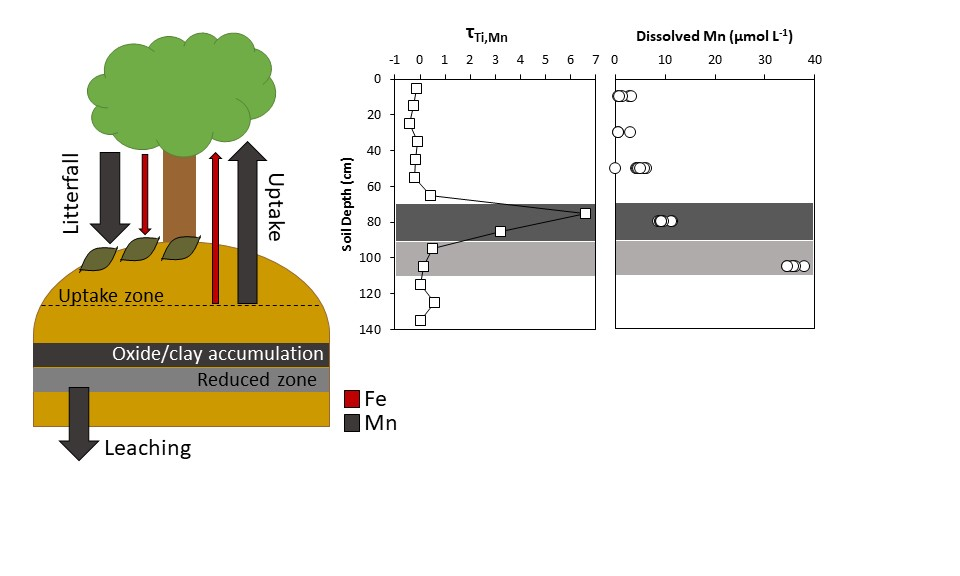
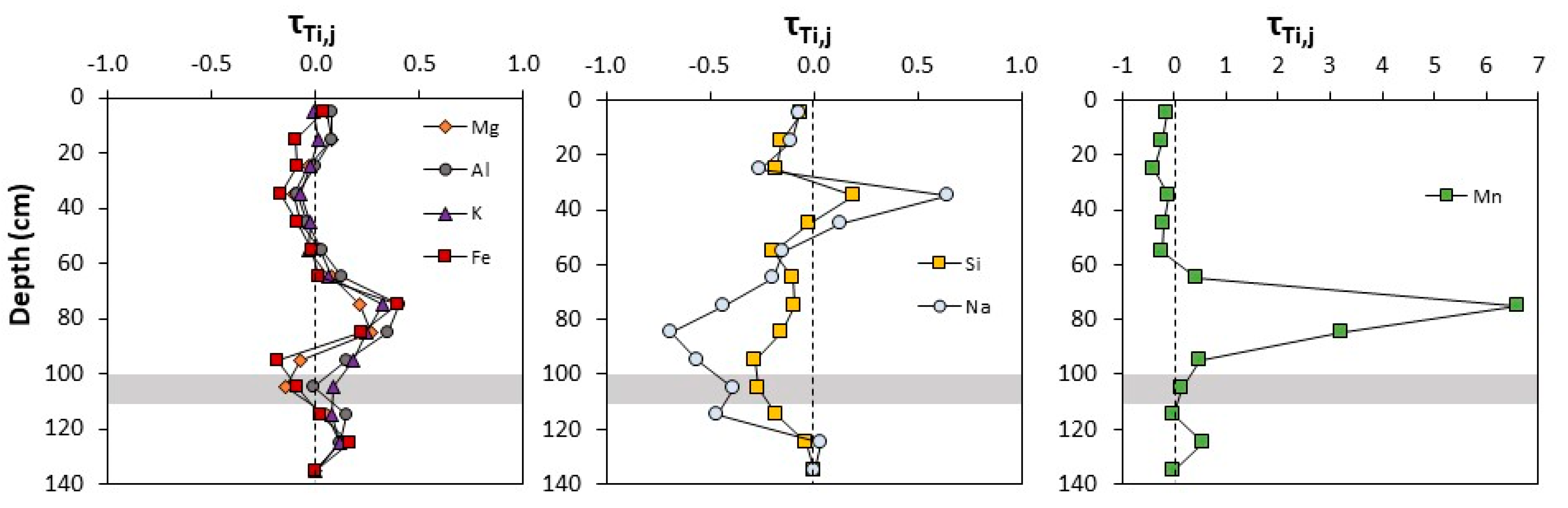
4.1. Soil and Pore Water Geochemistry
4.2. Biogeochemical Cycling between Plants and Mine Spoil
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- U.S. Department of the Interior. Office of Surface Mining Reclamation and Enforcement. Available online: https://www.osmre.gov/ (accessed on 10 November 2018).
- U.S. Congress. Surface mining control and reclamation act of 1977. Public Law 1977, 95, 30. [Google Scholar]
- Dixon, E.L.; Bilbrey, K. Abandoned Mine Land Program: A Policy Analysis for Central Appalachia and the Nation; AML Policy Priorities Group; Appalachian Citizen’s Law Center, The Alliance for Appalachia: Whitesburg, KY, USA, 2015. [Google Scholar]
- U.S. Environmental Protection Agency. The Effects of Mountaintop Mines and Valley Fills on Aquatic Ecosystems of the Central Appalachian Coalfields; EPA/600/R-09/138F; U.S. Environmental Protection Agency: Washington, DC, USA, 2011.
- Cravotta, C.A. Dissolved metals and associated constituents in abandoned coal-mine discharges, Pennsylvania, USA. Part 1: Constituent quantities and correlations. Appl. Geochem. 2008, 23, 166–202. [Google Scholar] [CrossRef]
- Blowes, D.W.; Jambor, J.L. The Pore-Water Geochemistry and The Mineralogy of The Vadose Zone of Sulfide Tailings, Waite-Amulet, Quebec, Canada. Appl. Geochem. 1990, 5, 327–346. [Google Scholar] [CrossRef]
- Nordstrom, D.K. Hydrogeochemical processes governing the origin, transport and fate of major and trace elements from mine wastes and mineralized rock to surface waters. Appl. Geochem. 2011, 26, 1777–1791. [Google Scholar] [CrossRef]
- Akcil, A.; Koldas, S. Acid Mine Drainage (AMD): Causes, treatment and case studies. J. Clean. Prod. 2006, 14, 1139–1145. [Google Scholar] [CrossRef]
- Gray, N.F. Environmental impact and remediation of acid mine drainage: A management problem. Environ. Geol. 1997, 30, 62–71. [Google Scholar] [CrossRef]
- Johnson, D.B.; Hallberg, K.B. Acid mine drainage remediation options: A review. Sci. Total Environ. 2005, 338, 3–14. [Google Scholar] [CrossRef] [PubMed]
- Singer, P.C.; Stumm, W. Acidic mine drainage: The rate-determining step. Science 1970, 167, 1121–1123. [Google Scholar] [CrossRef] [PubMed]
- Moses, C.O.; Nordstrom, D.K.; Herman, J.S.; Mills, A.L. Aqueous pyrite oxidation by dissolved oxygen and by ferric iron. Geochim. Cosmochim. Acta 1987, 51, 1561–1571. [Google Scholar] [CrossRef]
- Evangelou, V.P. Pyrite Oxidation and Its Control; CRC Press, Taylor Francis Group: Boca Raton, FL, USA, 1995. [Google Scholar]
- Dang, Z.; Liu, C.; Haigh, M.J. Mobility of heavy metals associated with the natural weathering of coal mine spoils. Environ. Pollut. 2002, 118, 419–426. [Google Scholar] [CrossRef]
- Cravotta, C.A. Dissolved metals and associated constituents in abandoned coal-mine discharges, Pennsylvania, USA. Part 2: Geochemical controls on constituent concentrations. Appl. Geochem. 2008, 23, 203–226. [Google Scholar] [CrossRef]
- Hallberg, K.B.; Johnson, D.B. Biological manganese removal from acid mine drainage in constructed wetlands and prototype bioreactors. Sci. Total Environ. 2005, 338, 115–124. [Google Scholar] [CrossRef] [PubMed]
- Jamieson, H.E.; Walker, S.R.; Parsons, M.B. Mineralogical characterization of mine waste. Appl. Geochem. 2015, 57, 85–105. [Google Scholar] [CrossRef]
- Diem, D.; Stumm, W. Is dissolved Mn2+ being oxidized by O2 in absence of Mn-bacteria or surface catalysts? Geochim. Cosmochim. Acta 1984, 48, 1571–1573. [Google Scholar] [CrossRef]
- Vogel, W.G. A Guide for Revegetating Coal Minesoils in the Eastern United States; No. PB-81-245011, GTR-NE-68; Forest Service Northeastern Forest Experiment Station: Broomall, PA, USA, 1981.
- Sheoran, V.; Sheoran, A.S.; Poonia, P. Soil Reclamation of Abandoned Mine Land by Revegetation: A Review. Int. J. Soil Sediment Water 2010, 3, 1–21. [Google Scholar]
- Berner, E.; Berner, R.; Moulton, K. Plants and mineral weathering: Present and past. Treatise Geochem. 2003, 5, 169–188. [Google Scholar]
- Bormann, B.T.; Wang, D.; Snyder, M.C.; Bormann, F.H.; Benoit, G.; April, R. Rapid, plant-induced weathering in an aggrading experimental ecosystem. Biogeochemistry 1998, 43, 129–155. [Google Scholar] [CrossRef]
- Drever, J.I. The effect of land plants on weathering rates of silicate minerals. Geochim. Cosmochim. Acta 1994, 58, 2325–2332. [Google Scholar] [CrossRef]
- Taylor, L.L.; Leake, J.R.; Quirk, J.; Hardy, K.; Banwart, S.A.; Beerling, D.J. Biological weathering and the long-term carbon cycle: Integrating mycorrhizal evolution and function into the current paradigm. Geobiology 2009, 7, 171–191. [Google Scholar] [CrossRef] [PubMed]
- Akafia, M.A.; Harrington, J.M.; Bargar, J.R.; Duckworth, O.W. Metal oxyhydroxide dissolution as promoted by structurally diverse siderophores and oxalate. Geochim. Cosmochim. Acta 2014, 141, 258–269. [Google Scholar] [CrossRef]
- Duckworth, O.W.; Bargar, J.R.; Sposito, G. Coupled biogeochemical cycling of iron and manganese as mediated by microbial siderophores. BioMetals 2009, 22, 605–613. [Google Scholar] [CrossRef] [PubMed]
- Hausrath, E.M.; Neaman, A.; Brantley, S.L. Elemental release rates from dissolving basalt and granite with and without organic ligands. Am. J. Sci. 2009, 309, 633–660. [Google Scholar] [CrossRef]
- Balogh-Brunstad, Z.; Keller, C.K.; Bormann, B.T.; O’Brien, R.; Wang, D.; Hawley, G. Chemical weathering and chemical denudation dynamics through ecosystem development and disturbance. Glob. Biogeochem. Cycles 2008, 22. [Google Scholar] [CrossRef]
- Herndon, E.M.; Jin, L.; Andrews, D.M.; Eissenstat, D.M.; Brantley, S.L. Importance of vegetation for manganese cycling in temperate forested watersheds. Glob. Biogeochem. Cycles 2015, 29, 160–174. [Google Scholar] [CrossRef]
- Guo, L.; Cutright, T. Remediation of Acid Mine Drainage (AMD)-Contaminated Soil by Phragmites Australis And Rhizosphere Bacteria. Environ. Sci. Pollut. Res. 2014, 21, 7350–7360. [Google Scholar] [CrossRef] [PubMed]
- Johnson, N.M.; Likens, G.E.; Bormann, F.H.; Fisher, D.W.; Pierce, R.S. A working model for the variation in stream water chemistry at the Hubbard Brook Experimental Forest, New Hampshire. Water Resour. Res. 1969, 5, 1353–1363. [Google Scholar] [CrossRef]
- Lamborn, R.E. Geology of Tuscarawas County; DNR Division of Geological Survey Bulletin: Columbus, OH, USA, 1956; Volume 55.
- Gannett Fleming, Inc. Huff Run Watershed Acid Mine Drainage and Abatement Plan; Gannett Fleming, Inc.: Camp Hill, PA, USA, 2000. [Google Scholar]
- Kinney, C. Huff Run Acid Mine Drainage Abatement and Treatment Plan Addendum; Division of Mineral Resources Managements, Ohio Department of Natural Resources: New Philadelphia, OH, USA, 2013.
- NADP. National Atmospheric Deposition Program (NRSP-3); NADP Program Office, Wisconsin State Laboratory of Hygiene: Madison, WI, USA, 2018. [Google Scholar]
- NWIS. USGS 03121850 Huff Run at Mineral City OH; U.S. Geological Survey National Water Information Systems. Available online: https://waterdata.usgs.gov/usa/nwis/ (accessed on 17 October 2016).
- Snyder, R.L.; Eching, S. Daily Reference Evapotranspiration (ETref) Calculator; Regents of the University of California: Oakland, CA, USA, 2006.
- Tessier, A.; Campbell, P.G.C.; Bisson, M. Sequential extraction procedure for the speciation of particulate trace metals. Anal. Chem. 1979, 51, 844–851. [Google Scholar] [CrossRef]
- Kraft, S.; Stümpel, J.; Becker, P.; Kuetgens, U. High resolution X-ray absorption spectroscopy with absolute energy calibration for the determination of absorption edge energies. Rev. Sci. Instrum. 1996, 67, 681–687. [Google Scholar] [CrossRef]
- Manceau, A.; Marcus, M.A.; Grangeon, S. Determination of Mn valence states in mixed-valent manganates by XANES spectroscopy. Am. Miner. 2012, 97, 816–827. [Google Scholar] [CrossRef]
- Harris, W.F.; Goldstein, R.A.; Henderson, G.S. Analysis of Forest Biomass Pools, Annual Primary Production and Turnover of Biomass for a Mixed Deciduous Forest Watershed; IUFRO Biomass Studies: Nancy, France, 1973; pp. 43–64. [Google Scholar]
- Miller, R.O. High-temperature oxidation: Dry ashing. In Handbook and Reference Methods for Plant Analysis; CRC Press: New York, NY, USA, 1998; pp. 53–56. [Google Scholar]
- McCain, D.C.; Markley, J.L. More manganese accumulates in maple sun leaves than in shade leaves. Plant Physiol. 1989, 90, 1417–1421. [Google Scholar] [CrossRef] [PubMed]
- Kirkby, E.A.; Pilbeam, D.J. Calcium as a plant nutrient. Plant Cell Environ. 1984, 7, 397–405. [Google Scholar] [CrossRef]
- Brimhall, G.H.; Dietrich, W.E. Constitutive mass balance relations between chemical composition, volume, density, porosity, and strain in metasomatic hydrochemical systems: Results on weathering and pedogenesis. Geochim. Cosmochim. Acta 1987, 51, 567–587. [Google Scholar] [CrossRef]
- Anderson, S.P.; Dietrich, W.E.; Brimhall, G.H. Weathering profiles, mass-balance analysis, and rates of solute loss: Linkages between weathering and erosion in a small, steep catchment. Bull. Geol. Soc. Am. 2002, 114, 1143–1158. [Google Scholar]
- Brantley, S.L.; Goldhaber, M.B.; Vala Ragnarsdottir, K. Crossing disciplines and scales to understand the critical zone. Elements 2007, 3, 307–314. [Google Scholar] [CrossRef]
- Herndon, E.M.; Martínez, C.E.; Brantley, S.L. Spectroscopic (XANES/XRF) characterization of contaminant manganese cycling in a temperate watershed. Biogeochemistry 2014, 121, 505–517. [Google Scholar] [CrossRef]
- Keiluweit, M.; Nico, P.; Harmon, M.E.; Mao, J.; Pett-Ridge, J.; Kleber, M. Long-term litter decomposition controlled by manganese redox cycling. Proc. Natl. Acad. Sci. USA 2015, 112, E5253–E5260. [Google Scholar] [CrossRef] [PubMed]
- Kabata-Pendias, A.; Pendias, H. Trace Elements in Soils and Plants; CRC Press Taylor Francis Group: Boca Raton, FL, USA, 1984. [Google Scholar]
- Houle, D.; Tremblay, S.; Ouimet, R. Foliar and wood chemistry of sugar maple along a gradient of soil acidity and stand health. Plant Soil 2007, 300, 173–183. [Google Scholar] [CrossRef]
- Huerta-Diaz, M.A.; Morse, J.W. Pyritization of trace metals in anoxic marine sediments. Geochim. Cosmochim. Acta 1992, 56, 2681–2702. [Google Scholar] [CrossRef]
- Morse, J.W.; Luther, G.W. Chemical influences on trace metal-sulfide interactions in anoxic sediments. Geochim. Cosmochim. Acta 1999, 63, 3373–3378. [Google Scholar] [CrossRef]
- Ussiri, D.A.N.; Lal, R. Method for Determining Coal Carbon in the Reclaimed Minesoils Contaminated with Coal. Soil Sci. Soc. Am. J. 2008, 72, 231. [Google Scholar] [CrossRef]
- Cornelissen, G.; Gustafsson, O.; Bucheli, T.D.; Jonker, M.T.; Koelmans, A.A.; van Noort, P.C. Extensive sorption of organic compounds to black carbon, coal, and kerogen in sediments and soils: Mechanisms and consequences for distribution, bioaccumulation, and biodegradation. Environ. Sci. Technol. 2005, 39, 6881–6895. [Google Scholar] [CrossRef] [PubMed]
- Jobbagy, E.G.; Jackson, R.B. The uplift of soil nutrients by plants: Biogeochemical consequences across scales. Ecology 2004, 85, 2380–2389. [Google Scholar] [CrossRef]
- Kogelmann, W.J.; Sharpe, W.E. Soil acidity and manganese in declining and nondeclining sugar maple stands in Pennsylvania. J. Environ. Qual. 2006, 35, 433–441. [Google Scholar] [CrossRef] [PubMed]
- St Clair, S.B.; Lynch, J.P. Element accumulation patterns of deciduous and evergreen tree seedlings on acid soils: Implications for sensitivity to manganese toxicity. Tree Physiol. 2005, 25, 85–92. [Google Scholar] [CrossRef] [PubMed]
- Richardson, J.B.; Friedland, A.J. Influence of coniferous and deciduous vegetation on major and trace metals in forests of northern New England, USA. Plant Soil 2016, 402, 363–378. [Google Scholar] [CrossRef]
- Richardson, J.B. Manganese and Mn/Ca ratios in soil and vegetation in forests across the northeastern US: Insights on spatial Mn enrichment. Sci. Total Environ. 2017, 581, 612–620. [Google Scholar] [CrossRef] [PubMed]
- Shanley, J.B. Manganese biogeochemistry in a small Adirondack forested lake watershed. Water Resour. Res. 1986, 22, 1647–1656. [Google Scholar] [CrossRef]
- Navrátil, T.; Shanley, J.B.; Skřivan, P.; Krám, P.; Mihaljevič, M.; Drahota, P. Manganese biogeochemistry in a central Czech Republic catchment. Water. Air. Soil Pollut. 2007, 186, 149–165. [Google Scholar] [CrossRef]
- Gaines, K.P.; Stanley, J.W.; Meinzer, F.C.; McCulloh, K.A.; Woodruff, D.R.; Chen, W.; Adams, T.S.; Lin, H.; Eissenstat, D.M. Reliance on shallow soil water in a mixed-hardwood forest in central Pennsylvania. Tree Physiol. 2015, 36, 444–458. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Depth (cm) | LOI550 (%) | LOI₉₅₀ (%) | pHw | pHKCl | Na (mmol/kg) | Mg (mmol/kg) | Al (mmol/kg) | Si (mmol/kg) | K (mmol/kg) | Mn (mmol/kg) | Fe (mmol/kg) | Ti (mmol/kg) |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 0–10 | 9.75 | 12.39 | 5.3 | 3.7 | 159 | 267 | 3120 | 9871 | 685 | 5.3 | 894 | 65 |
| 10–20 | 8.07 | 11.33 | 5.0 | 3.6 | 172 | 308 | 3524 | 10034 | 785 | 5.2 | 871 | 73 |
| 20–30 | 10.39 | 13.55 | 4.9 | 3.6 | 144 | 280 | 3312 | 9820 | 771 | 4.2 | 894 | 74 |
| 30–40 | 9.00 | 11.40 | 4.3 | 3.5 | 250 | 201 | 2345 | 11130 | 567 | 4.9 | 636 | 58 |
| 40–50 | 8.99 | 12.07 | 4.8 | 3.6 | 192 | 234 | 2764 | 10286 | 668 | 4.9 | 781 | 65 |
| 50–60 | 7.06 | 10.08 | 4.6 | 3.4 | 170 | 303 | 3501 | 9889 | 779 | 5.7 | 985 | 76 |
| 60–70 | 6.37 | 9.49 | 4.4 | 3.4 | 147 | 293 | 3484 | 10136 | 782 | 9.5 | 931 | 70 |
| 70–80 | 6.59 | 10.69 | 4.3 | 3.3 | 93 | 298 | 3934 | 9214 | 881 | 45.6 | 1158 | 63 |
| 80–90 | 6.40 | 9.83 | 4.7 | 3.5 | 56 | 335 | 4081 | 9309 | 894 | 27.3 | 1099 | 68 |
| 90–100 | 9.40 | 13.75 | 4.5 | 3.4 | 89 | 279 | 3958 | 8998 | 970 | 11.1 | 834 | 77 |
| 100–110 | 18.41 | 21.63 | 4.1 | 3.2 | 118 | 241 | 3208 | 8583 | 833 | 8.0 | 878 | 73 |
| 110–120 | 7.81 | 14.85 | 4.4 | 3.4 | 100 | 292 | 3657 | 9460 | 819 | 6.8 | 963 | 71 |
| 120–130 | 8.46 | 15.91 | 4.7 | 3.5 | 166 | 268 | 3045 | 9533 | 725 | 9.2 | 943 | 61 |
| 130–140 | 6.77 | 11.75 | 4.6 | 3.4 | 167 | 248 | 2836 | 10280 | 676 | 6.1 | 843 | 64 |
| Depth (cm) | τTi,Na | τTi,Mg | τTi,Al | τTi,Si | τTi,K | τTi,Mn | τTi,Fe |
|---|---|---|---|---|---|---|---|
| 0–10 | −0.07 | 0.05 | 0.08 | −0.06 | −0.01 | −0.15 | 0.04 |
| 10–20 | −0.11 | 0.08 | 0.08 | −0.15 | 0.01 | −0.25 | −0.10 |
| 20–30 | −0.26 | −0.04 | 0.00 | −0.18 | −0.02 | −0.40 | −0.09 |
| 30–40 | 0.64 | −0.11 | −0.09 | 0.19 | −0.08 | −0.11 | −0.17 |
| 40–50 | 0.13 | −0.08 | −0.04 | −0.02 | −0.03 | −0.20 | −0.09 |
| 50–60 | −0.15 | 0.02 | 0.03 | −0.20 | −0.04 | −0.22 | −0.02 |
| 60–70 | −0.19 | 0.08 | 0.12 | −0.10 | 0.06 | 0.43 | 0.01 |
| 70–80 | −0.44 | 0.21 | 0.40 | −0.09 | 0.32 | 6.60 | 0.39 |
| 80–90 | −0.69 | 0.26 | 0.35 | −0.15 | 0.24 | 3.21 | 0.22 |
| 90–100 | −0.56 | −0.08 | 0.15 | −0.28 | 0.18 | 0.50 | −0.18 |
| 100–110 | −0.38 | −0.15 | −0.01 | −0.27 | 0.08 | 0.15 | −0.09 |
| 110–120 | −0.47 | 0.05 | 0.15 | −0.18 | 0.08 | 0.00 | 0.02 |
| 120–130 | 0.03 | 0.12 | 0.11 | −0.04 | 0.11 | 0.57 | 0.16 |
| 130–140 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 |
| Soil Depth (cm) | Average Mn | Mn(II) (%) | Mn(III) (%) | Mn(IV) (%) | Red. χ2 (× 10−4) |
|---|---|---|---|---|---|
| 0–10 | 2.5 | 42 | 58 | 0 | 6.2 |
| 60–70 | 2.8 | 25 | 70 | 5 | 4.8 |
| 70–80 | 3.7 | 16 | 1 | 82 | 3.7 |
| 80–90 | 3.4 | 19 | 21 | 60 | 1.9 |
| 90–100 | 3.1 | 26 | 34 | 40 | 3.9 |
| 100–110 | 3.0 | 32 | 41 | 27 | 4.2 |
| Fluxes (mmol m−2 y−1) | Mn | Fe | Al | Ca |
|---|---|---|---|---|
| Plant uptake | 12 ± 1 | 0.45 ± 0.04 | 1.6 ± 0.6 | 82 ± 7 |
| Litterfall | 6.6 ± 2.4 | 0.36 ± 0.10 | 0.55 ± 0.23 | 71 ± 39 |
| Atmospheric deposition | <DL | <DL | <DL | 4.3 ± 0.7 |
| Chemical weathering (upper) | 15.4 ± 5.7 | <DL | 65 ± 24 | 630 ± 234 |
| Chemical weathering (lower) | 3.6 | 0.00 | 15 | 149 |
| Element mass in the soil core (mol m−2) | 23 | 1907 | 7016 | 24 |
| Plant uptake (× 10−5 y−1) | 52 ± 4 | 0.02 ± 0.001 | 0.02 ± 0.001 | 340 ± 29 |
| Chemical weathering (× 10−5 y−1) | 67 ± 25 | <DL | 0.93 ± 0.34 | 2,570 ± 954 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Herndon, E.; Yarger, B.; Frederick, H.; Singer, D. Iron and Manganese Biogeochemistry in Forested Coal Mine Spoil. Soil Syst. 2019, 3, 13. https://doi.org/10.3390/soilsystems3010013
Herndon E, Yarger B, Frederick H, Singer D. Iron and Manganese Biogeochemistry in Forested Coal Mine Spoil. Soil Systems. 2019; 3(1):13. https://doi.org/10.3390/soilsystems3010013
Chicago/Turabian StyleHerndon, Elizabeth, Brianne Yarger, Hannah Frederick, and David Singer. 2019. "Iron and Manganese Biogeochemistry in Forested Coal Mine Spoil" Soil Systems 3, no. 1: 13. https://doi.org/10.3390/soilsystems3010013
APA StyleHerndon, E., Yarger, B., Frederick, H., & Singer, D. (2019). Iron and Manganese Biogeochemistry in Forested Coal Mine Spoil. Soil Systems, 3(1), 13. https://doi.org/10.3390/soilsystems3010013