1. Introduction
Soils are among the most heterogeneous ecosystems on Earth, and in spite of unfavorable conditions in terms of nutrient availability and predation, thousands of microbial species find their primary habitat in them. Estimates indicate that one gram of soil may harbor between 2 × 10
3 and 8.3 × 10
5 different species [
1,
2]. Currently, surveys and counts rely on high throughput metagenomic approaches [
3,
4,
5,
6,
7,
8].
Bacteria, archaea and fungi adapted to live in soil can regulate plant growth and health through their direct role in processes, such as organic matter decomposition, nutrient cycling and pathogen control. The structure of soil microbial communities thus entails a special importance for its primary productivity. A better understanding of the mechanisms regulating soil life and functionality in response to external inputs in agriculture is fundamental to improve soil management, and in particular nitrogen (N) requirement and fate in agriculture, and to reduce the threats to the environment, and in turn to human health.
N inputs to soil can have important impacts on the environment, and many studies have focused on their effects on plant growth, N and carbon cycles, and emissions of greenhouse gases to the atmosphere [
9,
10,
11,
12]. Less well understood are the impacts of fertilizer inputs on soil bacterial communities, even though bacteria represent a major portion of living biomass in terrestrial ecosystems [
13] and their activities are intimately tied to belowground processes. Some studies have reported shifts of bacterial community composition in response to N amendments or particular fertilization practices [
14,
15,
16,
17,
18,
19].
The aim of the present work was to detect specific responses of various bacterial groups in the presence of a series of innovative fertilizers, and to compare their effects on soil bacterial communities over a two-month period. The innovative aspect in the products used consists in the fact that these fertilizers are novel mixtures recently set up by the ILSA S.p.A., Arzignano VI, Italy, and recycle different agricultural or industrial byproducts, whose properties on plants are under investigation.
The working hypothesis of the present investigation was that, notwithstanding the equal level of N delivered, different microbial groups would be affected depending on the fertilizer composition. This rests on the assumption that, although N is a major limiting nutrient for soil life, microbial communities would respond differently, depending on the form in which it is supplied. For this reason, the N dosage was set equal in all fertilizers. To frame this rationale, it needs to be considered that organic carbon, along with N, can be a strong determinant of microbial population changes. In our fertilizers, we have chosen a series in which organic carbon is the variable and an equal amount of N is bound to it in different ways (the amino group within peptides in the organic fraction or bound to carbonyl in urea or as the heterocyclic ring with N in Oxy-Amino-Triazine (OATr)). The microbial proficiency in extracting nitrogen from the different Carbon-based substrates and exploiting it structurally for their detectable fluctuation dynamics is, in this respect, the phenomenon under study.
A deeper understanding of these aspects, along with the corresponding plant productivity responses, will allow to plan improved fertilization strategies to exploit the plant-soil-microbe continuum in its ideal balance conditions for plant growth promotion.
The fertilizers included products of biological origin as Thermal-Hydrolyzed-Leather (THL), Castor Cake (CC), Neem (Nm), and Yeast-based fertilizer (Y), their combinations with urea consisting in: Urea-THL-Yeast (UTY) and Urea-THL-Neem (UTN), as well as the synthetic fertilizer OATr. The fertilizers were tested on an agricultural low-nutrient soil, in a greenhouse study using the creeping perennial grass
Cynodon dactylon (
C. dactylon) as the model plant species. Bacterial diversity was analyzed via deep 454 sequencing of 16S rDNA amplicons obtained from extracted DNA from soil samples taken 9 days and 58 days after fertilizer application. Soil sampling time points were selected based on our previous study, which made use of the same set up. In that study, the abundance of genes involved in the N cycle was measured by quantitative polymerase chain reaction (qPCR) and indications were obtained that, 9 days after application, compositional shifts are clearly detectable, whereas 58 days after application, a stabilization and partial reversion to the original conditions is observed [
20].
2. Material and Methods
2.1. Greenhouse Fertilization Trial
A greenhouse trial was set up in Summer 2012 with the aim to compare the effects of different fertilizers on plant growth and soil bacterial communities. The fertilization experiment included 8 treatments (one unfertilized control and seven fertilizers). Each treatment was replicated 4 times in a randomized design, resulting in overall 32 pots. The names and the descriptions of the fertilizers, which were all provided by a commercial supplier, (ILSA S.p.A., Arzignano, Vicenza, Italy) are given in
Table 1. In order to minimize nutrient background and to put in evidence the effects of the different organic fertilizers, the soil used was a mixture of sand (50%) and a low fertility soil which had been purposely impoverished in N through several years of repeated monoculture cropping with cereals, without any fertilization. The soil pH value was 7.6 (CaCl
2), 8.1 (H
2O) and its C/N ratio was 13.7. Further detailed chemical and physical properties of the soil used for the test have been previously reported [
20].
The pots had a diameter of 20 cm, and contained 5.5 kg of soil. The fertilizers were mixed in the upper 5 cm of the substrate, and subsequently a sod with pre-grown C. dactylon was placed on the top.
Fertilizers were added to obtain a final N input corresponding to 150 kg/ha. Moreover, P2O5 and K2O5 were added to deliver 50 kg/ha and 100 kg/ha, respectively. The fertilization trial lasted for 2 months, during which plant growth measurements and soil sampling were done.
Watering was carried out by capillary infiltration from the bottom of the pots by keeping a constant level of water in plastic planter saucers placed under each pot. Refilled water dosage corresponded to the daily restitution of evapotranspiration, which spanned between 3 mm and 5 mm. The mean temperature in the greenhouse across the trial period was 26.7 °C. The light regime was the natural daylight span occurring at coordinates 45_34007.9800 N; 11_37014.6300 E from mid-June to mid-August (15 h 40’ and 14 h 5’).
Plant growth in terms of height, fresh weight and dry weight (after cutting at the ground level) of C. dactylon were measured in all treatments three times during the two months period, at days 20, 35 and 56 after fertilization.
2.2. Soil Sampling and DNA Extraction
Soil was sampled with a 1.5-cm-diameter corer at a 5-cm depth in the center of the pots under the C. dactylon sod, 9 and 58 days after the addition of the fertilizers. Sixty-four samples were taken for amplicon pyrosequencing, representing 4 biological replicates of 8 treatments at two sampling times. After sampling root portions were discarded if present and 2 g of soil were immediately frozen in liquid nitrogen, and then stored at −80 °C until the extraction of nucleic acids.
Nucleic acids were extracted from 0.45 g of thawed and air-dried soil. Soil was added to a Lysing Matrix E tube (MP Biomedicals, Santa Ana, CA, USA), with 750 µL of 120 mM NaPO4 buffer (pH 8) and 250 µL of TNS (500 mM Tris-HCl pH 8.0, 100 mM NaCl, 10% SDS w/v). To perform cell lysis, the tube was shaken 30 s at 5.5 m per second in a bead beater (Precellys Bead Beater, Bertin Technologies, Montigny-le-Bretonneux, France). Soil and cell debris were pelleted by centrifugation at 13,200× g at 4 °C for 20 min, and the supernatant was transferred to a 2-mL fresh RNase-free tube. Nucleic acids were extracted and purified with phenol/chloroform/isoamylalcohol (25:24:1, pH 8, v/v) and subsequently with chloroform/isoamylalcohol (24:1, v/v). The obtained aqueous phase was mixed with 2 volumes of PEG solution (30% w/v polyethylene glycol 6000 in 1.6 M NaCl), incubated at 4 °C for 2 h and centrifuged at 13,200× g at 4 °C for 15 min. The nucleic acid pellet was washed with 70% ethanol, air-dried and resuspended in 50 µL (DEPC)-water. All steps were performed on ice. DNA was quantified spectrophotometrically on a Nanodrop (Thermo Scientific, Tewksbury, MA, USA). DNA samples were stored at −80 °C.
2.3. 16S rRNA Gene Barcoded Amplicon Sequencing
Amplicon pyrosequencing was performed on a 454 GS FLX Titanium system (Roche, Penzberg, Germany) under the following operating conditions. Barcoded amplicons for multiplexing were prepared using the primers Ba27f (5′-AGA GTT TGA TCM TGG CTC AG-3′) and Ba519r (5′-TAT TAC CGC GGC KGC tg-3′) [
21] extended with the respective A or B adapters, key sequence and multiplex identifiers (MID) as recommended by Roche. As advised by Berry and co-workers [
22], a two-step PCR approach was selected, with the addition of the MID-primers only in the second PCR amplification, to avoid biases and increase reproducibility. Each amplification was done in triplicate. The first PCR was performed with the following cycling conditions: initial denaturation (94 °C, 10 min), followed by 22 cycles of denaturation (94 °C, 30 s), annealing (52 °C, 30 s) and extension (72 °C, 60 s) and by a final extension step at 72 °C for 10 min. Each 25-µL PCR reaction contained 1x PCR buffer (containing MgCl
2), 0.2 mM dNTPs, 1.25 U Fast Start HiFi Polymerase (Roche, Penzberg, Germany), 0.24 mg/mL BSA, 0.25 mM of each primer and 1 µL of template DNA (at a concentration of 20 ng/µL). The second PCR was performed with the same cycling conditions as the first, but with only 6 cycles of denaturation, annealing and extension. Each 25-µL PCR reaction contained 1x PCR buffer (including MgCl
2), 0.2 mM dNTPs, 1.25 U Fast Start HiFi Polymerase (Roche), 0.24 mg/mL BSA, 0.25 mM of each MID-primer (Biomers Pte Ltd., Ulm, Germany) and 5 µL of the previous PCR product. Amplicons were purified using Agencourt AMPure XP beads (Beckman Coulter, Brea, CA, USA), and quantified with the Quant-iT PicoGreen dsDNA quantification kit (Invitrogen, Thermo Fisher Sciebntific Inc. Monza, Italy). The purification was checked using an Agilent Bioanalyzer 2100 (Agilent Technologies, Santa Clara, CA, USA). Amplicons were diluted to 10
9 molecules/µL and equimolarly pooled in 4 pools of 16 samples.
Emulsion PCR, emulsion breaking and sequencing were performed applying the GS FLX Titanium chemistry following the supplier’s protocol.
2.4. Data Analysis and Statistics
Raw sequence analyses were conducted using the QIIME pipeline [
23]. About 450,000 raw sequences were generated by the run; the subsequent procedure of quality filtering and barcode assignment involved separate denoising with a pre-processing step by using split_libraries.py at recommended QIIME settings (-w 50 -g combination).
After such filtering steps and chimera removal, about 246,000 good-quality sequences were used for further analyses, with a mean of about 3800 sequences per sample. Phylotypes were selected at a 97% sequence similarity level and the taxonomic identity was determined using the Ribosomal Database Project (RDP) database scheme. Sequence data have been deposited at the European Biotechnology Institute (EBI) database (
http://www.ebi.ac.uk/) and are available under the code PRJEB21947.
The QIIME software (version 1,
http://qiime.org/) was also used to calculate and draw rarefaction curves based on the number of observed species and the Shannon index.
The relative abundance data of each taxon were used for further analysis, to compare different soil samples and to deduce individual effects caused by different fertilizers on soil bacterial communities.
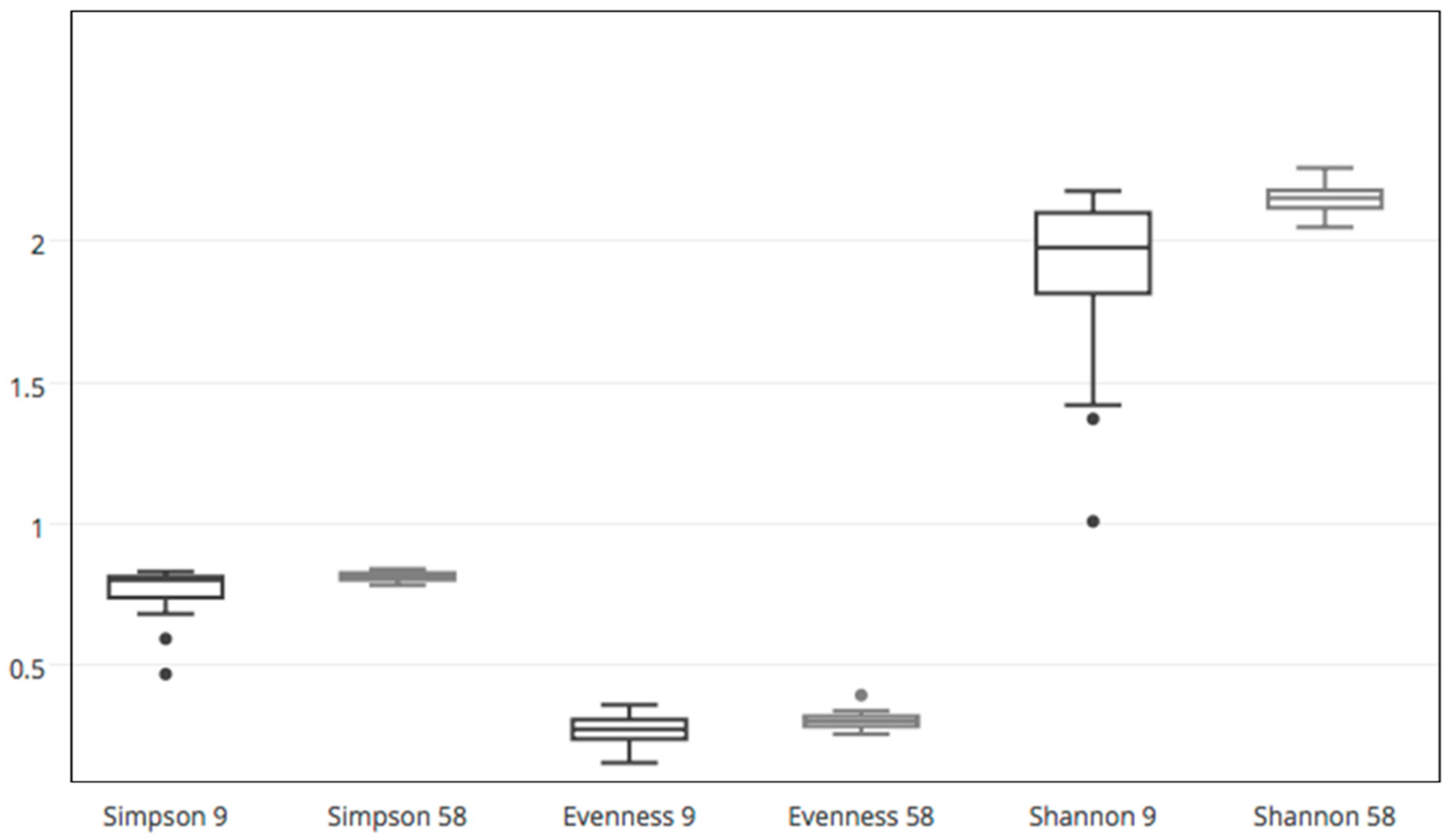
Diversity indices (Simpson index 1-D and Evenness e^H/S) were calculated using the Past Software (Paleontological Statistics Software Package, version 2.12); after normality distribution evaluation, statistical differences were analyzed with T tests for paired samples (normally distributed data) or the non-parametric Wilcoxon test (non-normally distributed data).
4. Discussion
The analysis revealed a considerable extent of diversity within the chosen soil, in spite of its sandy texture and low nutrient content. Rarefaction analysis showed that a higher number of sequences would be required to approximate the number of OTUs in the considered soils, and this kind of situation was also reported in other works [
18]. Schloss and Handelsman [
1] suggested that as many as 18,000 sequences per sample should be required to fully describe soil community diversity. However, this was not the main goal of this study, as the focus here was on the detection of the major relative changes in soil microbial communities induced by the presence of a certain fertilizer.
As regards the expected scenarios in this kind of analysis, extensive studies focusing on the impact of manure or chemical fertilizers in arable soils report that the application of those could significantly change the soil bacterial community [
8]. The phenomenon can be neither general nor permanent, as some authors demonstrated that specific soil taxa as
Pseudomonas could be temporarily affected, depending on the type of organic fertilizer [
5].
The major phyla detected in our soil—Proteobacteria, Acidobacteria, Actinobacteria, Firmicutes, Bacteroidetes, Chloroflexi, Gemmatimonadetes—were found to be predominating in general in soils of different countries [
14,
15,
17,
18,
19]. The relatively high abundance of Firmicutes found in our study is instead controversially discussed in literature. Whereas Roesch et al. [
14] listed Firmicutes in the prominent phyla present in soils, which is in agreement with our study, other authors analyzing tundra soil [
15,
18], grassland sites and other agricultural soils [
17,
18,
19] did not describe this phylum as an highly abundant part of soil microbiomes.
Our main focus was on the effect of N, which is an acknowledged key variable in microbial ecology. Cederlund and coworkers [
6] demonstrated that N fertilization was the most important driver for changes in the relative abundances of soil bacteria and that no specific taxa were indicative of organic amendment in general.
Several authors reported, by comparing N fertilized soils with controls, shifts in the abundance of determined bacterial groups, such as Bacteroidetes, some members of Proteobacteria and Acidobacteria [
17,
18,
19], Gemmatimonadetes and Verrucomicrobia [
15]. Interestingly, in our study, Firmicutes were highly responding to the applied fertilizers as their apparent percentage increase was favored in the presence of fertilizers, such as Y, THL, CC or N. Urea-based fertilizers (UTY and UTN) did not show this positive effect on this phylum. As a possible explanation of this result, the presence of N in complex macromolecules, such as in organic fertilizers, could have enriched particular groups of organisms endowed with prompt adaptation to metabolize those (in this case, Firmicutes representing a competent heterotrophic group). Conversely, the presence of urea, which is an easy source of N for both plants and microorganisms, could have played a more neutral role, not favoring a specific phylum over others. For bacteria of the phylum Bacteroidetes, a higher relative abundance in the presence of Y was observed, while bacteria of this phylum decreased in relative abundance when the fertilizer UTN was used. Bacteroidetes are considered copiotrophic (r-selected), and their increase in abundance in the presence of easily accessible N fertilization has already been reported [
15,
17,
18,
19]. Armatimonadetes and Nitrospirae, in contrast, appeared negatively affected by the Y, which was expected as easily available N induces an out-competition mainly of the autotrophic microbes belonging to Nitrospirae [
17]. In general, the presence of the Y induced a decrease in abundance of many bacterial groups, favoring only a few abundant ones. In contrast, the presence of OATr fertilizer in soils brought about the relative increase of many low-abundant phyla. These proportional changes at the community level did not reflect significant differences in the calculated diversity indices. Results obtained by Fierer et al. [
19] are in line with these observations. In fact, they detected no significant effects of N fertilization on bacterial diversity, but significant shifts in community composition. In contrast, other authors [
18] found a decrease in bacterial diversity with N additions. These two contrasting visions suggest that the effects of N amendments on bacterial diversity levels within soil are variable and dependent on the site as well as the fertilizer quality, or even other effects such as changes in soil pH or micronutrient status.
Overall Bacteroidetes and Firmicutes, which were present at high relative abundances a few days after the starting of fertilization trial, showed a decrease in time in all soil samples. The majority of low-abundance phyla showed instead an increase of relative abundance over time, leading to a gain in evenness. Nevertheless, the analysis also allowed to differentiate effects for all applied fertilizers. For example, major ones were detected in response to the addition of the fertilizer Y, in particular at the beginning of the trial. Here, after 9 days, Firmicutes represented the 45% of the entire community, and decreased to 13% after two months. This situation could be explained by an initial rapid growth of this group of microorganisms in contact with Y, to the detriment of less abundant phyla that almost disappeared in this first phase. After about 60 days, the decrease in the relative abundance of Firmicutes could have caused the slight increase of the abundance of minor phyla and establishment of a more equilibrated condition. Soils treated with this organic fertilizer presented an apparent increase in the number of Proteobacteria over time, while, in other soils, this phylum remained stable.
It could be possible that the rapid changes observed soon after the addition of the treatments, apart from being due to the responses of bacteria already present in the substrate soil, could be in part also caused by a component of newly introduced microorganisms present in the fertilizers. However, as regards the influence of the bacteria carried by the fertilizers themselves, prior reports point out that their occurrence in soil does not appear to match their prevalence in the added substrates, as although
Psychrobacter was the most abundant genus in pig manure, it was not detectable in the corresponding treated soil [
7].
As a general comment, it is also worth recalling that, when analyzing these data, as inevitably the case in all metagenomics surveys, it needs to be kept in mind that they are based on percentage values and thus represent just relative abundances. Therefore, an apparent increase or decrease of a given taxon could not necessarily reflect a true active or passive dynamics, since these shifts could be the indirect effects of the behavior of other taxa.
5. Conclusions
Upon comparing the communities observed at day 9 in the unfertilized control soil with those of the soils having received the fertilizers at the same date (
Figure 1 and
Table S3), it can be observed that, in general, treatments caused a rapid change in the relative taxa abundances during the first days. The same comparisons relative to the later date (day 58) showed smaller differences between control and fertilized cases, suggesting a gradual stabilization of equilibrium with lower instances of dominance, which in turn would entail a rise of evenness. Such increase in evenness and its statistical significance was independently verified by a comparison between the two single time points by running the Wilcoxon test on the ecological indices.
The different shifts observed in soil bacteria suggest that N quality, in spite of N quantity, is the driver of community changes over time. This concurs to confirm the initial hypothesis: microbial communities responding differently according to the form in which the (constant) amount of N is supplied to them.
Particularly, evident was the effect of the fertilizer Y. This treatment caused a significant increase in plant yield as well a rapid and strong increase in the relative abundance of Firmicutes and Bacteroidetes, which in turn negatively influenced the share of less abundant phyla. Seeking for possible reasons, one could remark that Ys are complete microbial organisms and their extract is consequently rich in most life elements. While this could be beneficial for plants, as regards the effects on the growth of bacteria, it has also to be considered that those Y biomasses come in part from antibiotics-producing bioreactors, which can in turn display negative impacts on given bacteria due to such antimicrobial product residuals. The other treatment having a peculiar and significant effect on bacteria was OATr, which was the only one causing a strong relative decrease in the number of Firmicutes and an increase in abundance of the low-number phyla. The two composite fertilizers UTY and UTN often caused similar changes at the phylum level, which were different from the effects caused by single component organic treatments. Future studies will be necessary to describe the consequences of the seemingly occurring shifts in bacterial community structure for the related functional traits as well as their expression profiles.


 ,
, 




{kind=link}
{kind=link}