Variation in the Molecular Structure and Radiocarbon Abundance of Mineral-Associated Organic Matter across a Lithosequence of Forest Soils


Abstract
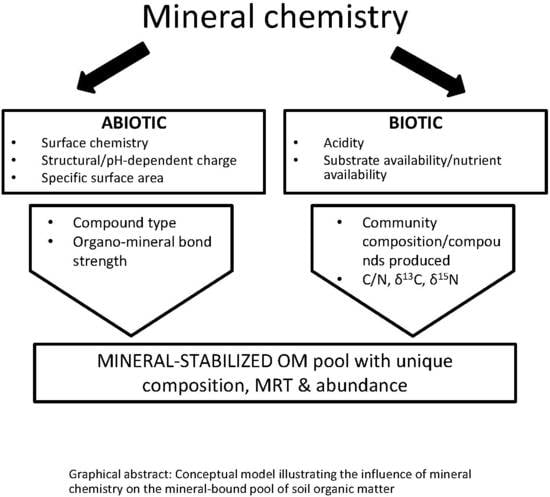
:
1. Introduction
- Does the composition and stability of mineral-bound organic matter vary among soils of differing mineral assemblage?
- If so, is variation in composition and/or radiocarbon abundance associated with specific soil physicochemical properties?
- Can any connection be made between composition of mineral-associated organics and microbial community characteristics?
2. Materials and Methods
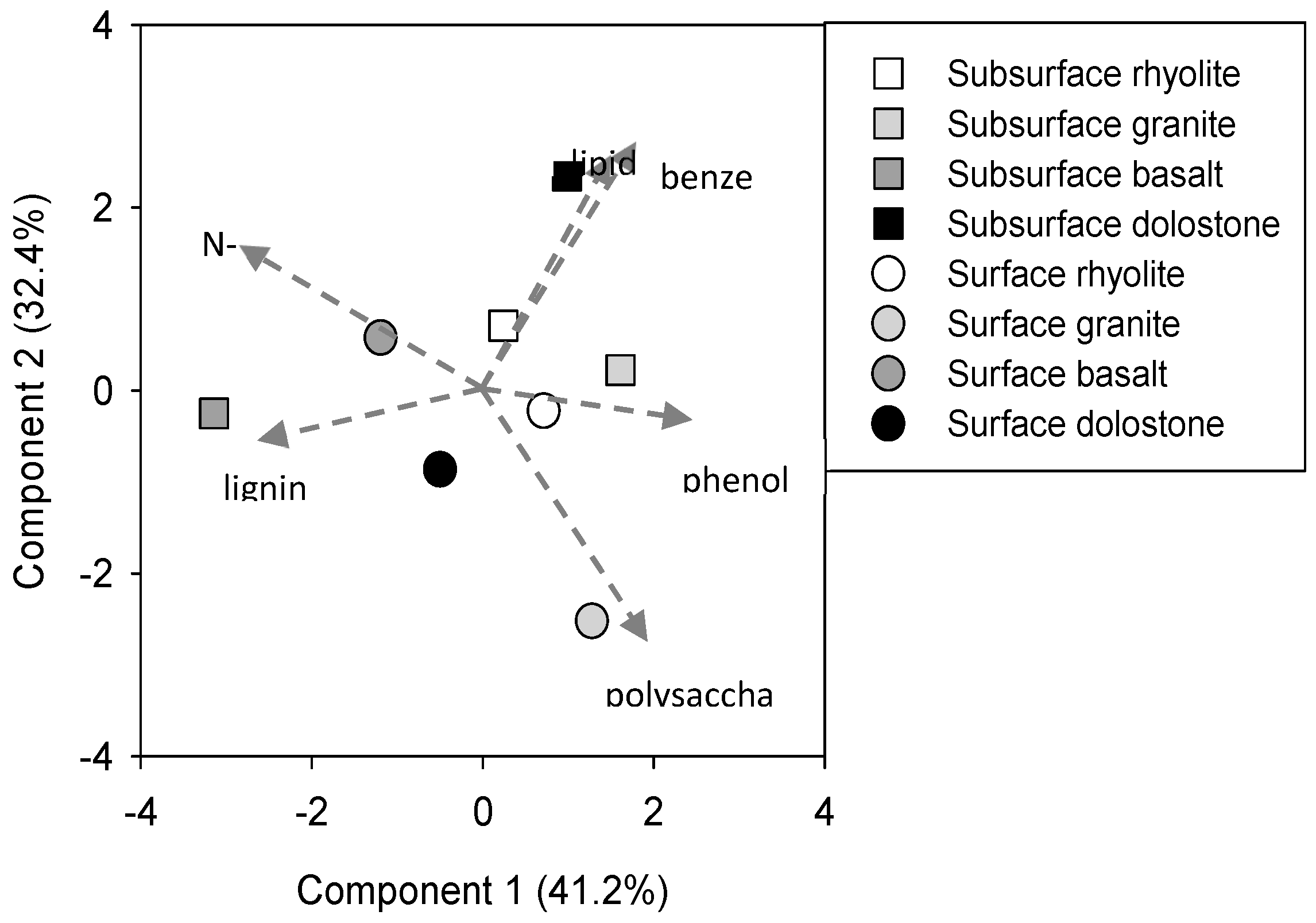
3. Results
4. Discussion
4.1. Competitive Sorption and Selective Preservation
4.2. Feedbacks between Soil Physicochemical Properties and Microbial Communities
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Namjesnik-Dejanovic, K.; Maurice, P.A.; Aiken, G.R.; Cabaniss, S.; Chin, Y.-P.; Pullin, M.J. Adsorption and fractionation of muck fulvic acid on kaolinite and goethite at pH 3.7, 6, and 8. Soil Sci. 2000, 165, 545–559. [Google Scholar] [CrossRef]
- Zhou, Q.H.; Maurice, P.A.; Cabaniss, S.E. Size fractionation upon adsorption of fulvic acid on goethite: Equilibrium and kinetic studies. Geochim. Cosmochim. Acta 2001, 65, 803–812. [Google Scholar] [CrossRef]
- Guo, M.X.; Chorover, J. Transport and fractionation of dissolved organic matter in soil columns. Soil Sci. 2003, 168, 108–118. [Google Scholar] [CrossRef]
- Heckman, K.; Vazquez-Ortega, A.; Gao, X.D.; Chorover, J.; Rasmussen, C. Changes in water extractable organic matter during incubation of forest floor material in the presence of quartz, goethite and gibbsite surfaces. Geochim. Cosmochim. Acta 2011, 75, 4295–4309. [Google Scholar] [CrossRef]
- Lichtfouse, E.; Chenu, C.; Baudin, F.; Leblond, C.; Da Silva, M.; Béhar, F.; Derenne, S.; Largeau, C.; Wehrung, P.; Albrecht, P. A novel pathway of soil organic matter formation by selective preservation of resistant straight-chain biopolymers: chemical and isotope evidence. Org. Geochem. 1998, 28, 411–415. [Google Scholar] [CrossRef] [Green Version]
- Lützow, M.V.; Kögel-Knabner, I.; Ekschmitt, K.; Matzner, E.; Guggenberger, G.; Marschner, B.; Flessa, H. Stabilization of organic matter in temperate soils: mechanisms and their relevance under different soil conditions—A review. Eur. J. Soil Sci. 2006, 57, 426–445. [Google Scholar]
- Gleeson, D.B.; Clipson, N.; Melville, K.; Gadd, G.M.; McDermott, F.P. Characterization of fungal community structure on a weathered pegmatitic granite. Microb. Ecol. 2005, 50. [Google Scholar] [CrossRef] [PubMed]
- Carson, J.K.; Campbell, L.; Rooney, D.; Clipson, N.; Gleeson, D.B. Minerals in soil select distinct bacterial communities in their microhabitats. FEMS Microb. Ecol. 2009, 67, 381–388. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heckman, K.; Welty-Bernard, A.; Rasmussen, C.; Schwartz, E. Geologic controls of soil carbon cycling and microbial dynamics in temperate conifer forests. Chem. Geol. 2009, 267, 12–23. [Google Scholar] [CrossRef]
- Marsh, T.L.; Long, D.; Voice, T. Bacterial transformations of metals in soils. In Handbook of Soil Sciences: Resource Management and Environmental Impacts, 2nd ed.; Huang, P.M., Li, Y., Sumner, M.E., Eds.; CRC Press: Boca Raton, FL, USA, 2012; pp. 16–22. [Google Scholar]
- Kleber, M.; Eusterhues, K.; Keiluweit, M.; Mikutta, C.; Mikutta, R.; Nico, P.S. Chapter one-mineral–organic associations: formation, properties, and relevance in soil environments. Adv. Agron. 2015, 130, 1–140. [Google Scholar] [CrossRef]
- Fleury, G.; Del Nero, M.; Barillon, R. Effect of mineral surface properties (alumina, kaolinite) on the sorptive fractionation mechanisms of soil fulvic acids: Molecular-scale ESI-MS studies. Geochim. Cosmochim. Acta 2017, 196, 1–17. [Google Scholar] [CrossRef]
- Pronk, G.J.; Heister, K.; Kögel-Knabner, I. Is turnover and development of organic matter controlled by mineral composition? Soil Biol. Biochem. 2013, 67, 235–344. [Google Scholar] [CrossRef]
- Avneri-Katz, S.; Young, R.B.; McKenna, A.M.; Chen, H.; Corilo, Y.E.; Polubesova, T.; Borch, T.; Chefetz, B. Adsorptive fractionation of dissolved organic matter (DOM) by mineral soil: Macroscale approach and molecular insight. Org. Geochem. 2017, 103, 113–124. [Google Scholar] [CrossRef]
- Coward, E.K.; Ohno, T.; Plante, A.F. Adsorption and molecular fractionation of dissolved organic matter on iron-bearing mineral matrices of varying crystallinity. Environ. Sci. Technol. 2018, 52, 1036–1044. [Google Scholar] [CrossRef] [PubMed]
- Sollins, P.; Swanston, C.; Kleber, M.; Filley, T.; Kramer, M.; Crow, S.; Calwell, B.A.; Lajtha; Bowden, K.R. Organic C and N stabilization in a forest soil: Evidence from sequential density fractionation. Soil Biol. Biochem. 2006, 38, 3313–3324. [Google Scholar] [CrossRef] [Green Version]
- Sollins, P.; Kramer, M.G.; Swanston, C.; Lajtha, K.; Filley, T.; Aufdenkampe, A.K.; Wagai, R.; Bowden, R.D. Sequential density fractionation across soils of contrasting mineralogy: Evidence for both microbial-and mineral-controlled soil organic matter stabilization. Biogeochemistry 2009, 96, 209–231. [Google Scholar] [CrossRef]
- Garrido, E.; Matus, F. Are organo-mineral complexes and allophane content determinant factors for the carbon level in Chilean volcanic soils? Catena 2012, 92, 106–112. [Google Scholar] [CrossRef]
- Giardina, C.P.; Litton, C.M.; Crow, S.E.; Asner, G.P. Warming-related increases in soil CO2 efflux are explained by increased below-ground carbon flux. Nat. Clim. Chang. 2014, 4, 822–827. [Google Scholar] [CrossRef] [Green Version]
- Yeasmin, S.; Singh, B.; Johnson, C.T.; Sparks, D.L. Organic carbon characteristics in density fractions of soils with contrasting mineralogies. Geochim. Cosmochim. Acta. 2017, 218, 215–236. [Google Scholar] [CrossRef]
- Piccolo, A. The supramolecular structure of humic substances. Soil Sci. 2001, 166, 810–832. [Google Scholar] [CrossRef]
- Sutton, R.; Sposito, G. Molecular structure in soil humic substances: The new view. Environ. Sci. Technol. 2005, 39, 9009–9015. [Google Scholar] [CrossRef] [PubMed]
- Santín, C.; Doerr, S.H.; Kane, E.S.; Masiello, C.A.; Ohlson, M.; Rosa, J.M.; Preston, C.M.; Dittmar, T. Towards a global assessment of pyrogenic carbon from vegetation fires. Global Chang. Biol. 2016, 22, 76–91. [Google Scholar] [CrossRef] [PubMed]
- Newcomb, C.J.; Qafoku, N.P.; Grate, J.W.; Bailey, V.L.; De Yoreo, J.J. Developing a molecular picture of soil organic matter–mineral interactions by quantifying organo–mineral binding. Nature Commun. 2017, 8, 396. [Google Scholar] [CrossRef] [PubMed]
- Kleber, M.; Nico, P.S.; Plante, A.; Filley, T.; Kramer, M.; Swanston, C.; Sollins, P. Old and stable soil organic matter is not necessarily chemically recalcitrant: Implications for modeling concepts and temperature sensitivity. Glob. Chang. Biol. 2011, 17, 1097–1107. [Google Scholar] [CrossRef]
- Kallenbach, C.M.; Frey, S.D.; Grandy, A.S. Direct evidence for microbial-derived soil organic matter formation and its ecophysiological controls. Nat. Commun. 2016, 7, 13630. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kleber, M.; Sollins, P.; Sutton, R.A. conceptual model of organo-mineral interactions in soils: Self-assembly of organic molecular fragments into zonal structures on mineral surfaces. Biogeochemistry 2007, 85, 9–24. [Google Scholar] [CrossRef]
- Bååth, E.; Anderson, T.H. Comparison of soil fungal/bacterial ratios in a pH gradient using physiological and PLFA-based techniques. Soil Biol. Biochem. 2003, 35, 955–963. [Google Scholar] [CrossRef] [Green Version]
- Fierer, N.; Jackson, R.B. The diversity and biogeography of soil bacterial communities. PNAS 2006, 103, 626–631. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Welty-Bernard, A.T. Al, Fe, and Ph Effects on Soil Microbial Communities. Ph.D. Thesis, Northern Arizona University, Flagstaff, AZ, USA, May 2014. [Google Scholar]
- Essington, M.E. Soil and Water Chemistry: An Integrative Approach; CRC Press: Boca Raton, FL, USA, 2004. [Google Scholar]
- Carson, J.K.; Rooney, D.; Gleeson, D.B.; Clipson, N. Altering the mineral composition of soil causes a shift in microbial community structure. FEMS Microbiol. Ecol. 2007, 61, 414–423. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jackson, M.L. Soil Chemical Analysis: Advanced Course, revised 2nd ed.; Paralled Press University of Wisconsin-Madison Libraries: Madison, WI, USA, 1985. [Google Scholar]
- Soil Survey Staff, National Soil Survey Laboratory. Methods Manual: Soil Survey Investigations Report No. 42, Version 4.0; United States Department of Agriculture, Natural Resources Conservation Service, National Soil Survey Center: Lincoln, NE, USA, 2004.
- Heckman, K.; Rasmussen, C. Lithologic controls on regolith weathering and mass flux in forested ecosystems of the southwestern USA. Geoderma 2011, 164, 3, 99–111. [Google Scholar] [CrossRef]
- Brunauer, S.; Emmett, P.H.; Teller, E. Adsorption of gases in multimolecular layers. J. Am. Chem. Soc. 1938, 60, 309–319. [Google Scholar] [CrossRef]
- Swanston, C.W.; Torn, M.S.; Hanson, P.J.; Southon, J.R.; Garten, C.T.; Hanlon, E.M.; Ganio, L. Initial characterization of processes of soil carbon stabilization using forest stand-level radiocarbon enrichment. Geoderma 2005, 128, 52–62. [Google Scholar] [CrossRef]
- Heckman, K.; Throckmorton, H.; Clingensmith, C.; Vila, F.J.G.; Horwath, W.R.; Knicker, H.; Rasmussen, C. Factors affecting the molecular structure and mean residence time of occluded organics in a lithosequence of soils under ponderosa pine. Soil Biol. Biochem. 2014, 77, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Throckmorton, H.M. Diverse Microbial Carbon as a Source of Soil Organic Matter. Ph.D. Thesis, University of California, Davis, Davis, CA, USA, 2012. [Google Scholar]
- Donahue, D.J.; Jull, A.J.T.; Toolin, L.J. Radiocarbon measurements at the University of Arizona AMS facility. Nucl. Instr. Meth. Phys. Res. B 1990, 52, 224–228. [Google Scholar] [CrossRef]
- Arèvalo, F.J.S.; Martínez, I.G.; Leon, M.G. Radiocarbon measurement program at the Centro Nacional de Aceleradores (CAN), Spain. Radiocarbon 2009, 51, 883–889. [Google Scholar] [CrossRef]
- Vogel, J.S.; Southon, J.R.; Nelson, D.E. Catalyst and binder effects in the use of filamentous graphite for AMS. Nucl. Instrum. Meth. B 1987, 29, 50–56. [Google Scholar] [CrossRef]
- Davis, J.C.; Proctor, I.D.; Southon, J.R.; Caffee, M.W.; Heikkinen, D.W.; Roberts, M.L.; Moore, T.L.; Turteltaub, K.W.; Nelson, D.E.; Loyd, D.H.; et al. LLNL/US AMS facility and research program. Nucl. Instr. Meth. Phys. Res. B 1990, 52, 269–272. [Google Scholar] [CrossRef]
- Trumbore, S.E. Comparison of carbon dynamics in tropical and temperate soils using radiocarbon measurements. Glob. Biogeochem. Cycles 1993, 7, 275–290. [Google Scholar] [CrossRef]
- Torn, M.S.; Lapenis, A.G.; Timofeev, A.; Fischer, M.L.; Babikov, B.V.; Harden, J.W. Organic carbon and carbon isotopes in modern and 100-year-old-soil archives of the Russian steppe. Glob. Chang. Biol. 2002, 8, 941–953. [Google Scholar] [CrossRef]
- Schmidt, M.W.; Torn, M.S.; Abiven, S.; Dittmar, T.; Guggenberger, G.; Janssens, I.A.; Kleber, M.; Kögel-Knabner, I.; Lehmann, J.; Manning, D.A.; et al. Persistence of soil organic matter as an ecosystem property. Nature 2011, 478, 49–56. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paul, E.A. The nature and dynamics of soil organic matter: Plant inputs, microbial transformations, and organic matter stabilization. Soil Biol. Biochem. 2016, 98, 109–126. [Google Scholar] [CrossRef] [Green Version]
- Rasmussen, C.; Heckman, K.; Wieder, W.R.; Keiluweit, M.; Lawrence, C.R.; Berhe, A.A.; Blankinship, J.C.; Crow, S.E.; Druhan, J.L.; Pries, C.E.H.; et al. Beyond clay: Towards an improved set of variables for predicting soil organic matter content. Biogeochemistry 2018, 137, 297–306. [Google Scholar] [CrossRef]
- Alvarez, R.; Alvarez, C.R. Soil organic matter pools and their associations with carbon mineralization kinetics. Soil Sci. Soc. Am. J. 2000, 64, 184–189. [Google Scholar] [CrossRef]
- McLauchlan, K.K.; Hobbie, S.E. Comparison of labile soil organic matter fractionation techniques. Soil Sci. Soc. Am. J. 2004, 68, 1616–1625. [Google Scholar] [CrossRef]
- Keiluweit, M.; Kleber, M. Molecular-level interactions in soils and sediments: The role of aromatic π-systems. Environ. Sci. Technol. 2009, 43, 3421–3429. [Google Scholar] [CrossRef] [PubMed]
- Keiluweit, M.; Bougoure, J.J.; Zeglin, L.H.; Myrold, D.D.; Weber, P.K.; Pett-Ridge, J.; Kleber, M.; Nico, P.S. Nano-scale investigation of the association of microbial nitrogen residues with iron (hydr) oxides in a forest soil O-horizon. Geochim. Cosmochim. Acta 2012, 95, 213–226. [Google Scholar] [CrossRef]
- Chassé, A.W.; Ohno, T.; Higgins, S.R.; Amirbahman, A.; Yildirim, N.; Parr, T.B. Chemical force spectroscopy evidence supporting the layer-by-layer model of organic matter binding to iron (oxy)hydroxide mineral surfaces. Environ. Sci. Technol. 2015, 49, 9733–9741. [Google Scholar] [CrossRef] [PubMed]
- Swensen, T.L.; Bowen, B.P.; Nico, P.S.; Northen, T.R. Competitive sorption of microbial metabolites on an iron oxide mineral. Soil Biol. Biochem. 2015, 90, 34–41. [Google Scholar] [CrossRef] [Green Version]
- Chassé, A.W.; Ohno, T. Higher molecular mass organic matter molecules compete with orthophosphate for adsorption to iron (oxy)hydroxide. Environ. Sci. Technol. 2016, 50, 7461–7469. [Google Scholar] [CrossRef] [PubMed]
- Ohno, T.; Heckman, K.A.; Plante, A.F.; Fernandez, I.J.; Parr, T.B. 14C Mean Residence Time and Its Relationship with Thermal Stability and Molecular Composition of Soil Organic Matter: A Case Study of Deciduous and Coniferous Forest Types. Geoderma 2017, 308, 1–8. [Google Scholar] [CrossRef]
- Omoike, A.; Chorover, J.; Kwon, K.; Kubicki, J. Adhesion of bacterial exopolymers to alpha-FeOOH: inner-sphere complexation of phosphodiester groups. Langmuir 2004, 20, 11108–11114. [Google Scholar] [CrossRef] [PubMed]
- Torn, M.S.; Trumbore, S.E.; Chadwick, O.A.; Vitousek, P.M.; Hendricks, D.M. Mineral control of soil organic carbon storage and turnover. Nature 1997, 389, 170–173. [Google Scholar] [CrossRef]
- Bailey, V.L.; Bond-Lamberty, B.; DeAngelis, K.; Grandy, A.S.; Hawkes, C.V.; Heckman, K.; Lajtha, K.; Phillips, R.P.; Sulman, B.N.; Todd-Brown, K.E.O.; et al. Soil carbon cycling proxies: Understanding their critical role in predicting climate change feedbacks. Global Chang. Biol. 2017, 24, 895–905. [Google Scholar] [CrossRef] [PubMed]
- Kampf, N.; Scheinost, A.L.; Schulze, D.G. Oxide minerals. In Handbook of Soil Science; Sumner, M.E., Ed.; CRC Press: Boca Raton, FL, USA, 2000; pp. 125–168. [Google Scholar]
- Dixon, J.B.; Schulze, D.G. Soil Mineralogy with Environmental Applications; Soil Science Society of America, Inc.: Madison, WI, USA, 2002. [Google Scholar]
- Wagai, R.; Mayer, L.M. Sorptive stabilization of organic matter in soils by hydrous iron oxides. Geochim. Cosmochim. Acta 2006, 71, 25–35. [Google Scholar] [CrossRef]
- Eusterhues, K.; Neidhardt, J.; Hädrich, A.; Küsel, K.; Totsche, K.U. Biodegradation of ferrihydrite-associated organic matter. Biogeochemistry 2014, 119, 45–50. [Google Scholar] [CrossRef]
- Mikutta, R.; Lorenz, D.; Guggenberger, G.; Haumaier, L.; Freund, A. Properties and reactivity of Fe-organic matter associations formed by coprecipitation versus adsorption: Clues from arsenate batch adsorption. Geochim. Cosmochim. Acta 2014, 144, 258–276. [Google Scholar] [CrossRef]
- Mayes, M.A.; Heal, K.R.; Brandt, C.C.; Phillips, J.R.; Jardine, P.M. Relation between soil order and sorption of dissolved organic carbon in temperate subsoils. Soil Sci. Soc. Am. J. 2012, 76, 1027–1037. [Google Scholar] [CrossRef]
- Heckman, K.; Lawrence, C.R.; Harden, J.W. A sequential selective dissolution method to quantify storage and stability of organic carbon associated with Al and Fe hydroxide phases. Geoderma 2018, 312, 24–35. [Google Scholar] [CrossRef]
- Rasmussen, C.; Torn, M.S.; Southard, R.J. Mineral assemblage and aggregates control carbon dynamics in a California conifer forest. Soil Sci. Soc. Am. J. 2005, 69, 1711–1721. [Google Scholar] [CrossRef]
- De Ruiter, P.C.; Moore, J.C.; Zwart, K.B.; Bouwman, L.A.; Hassink, J.; Bloem, J.; de Vos, J.A.; Marinissen, J.C.Y.; Didden, W.A.M.; Lebbink, G.; et al. Simulation of nitrogen mineralization in the below-ground food webs of two winter wheat fields. J. Appl. Ecol. 1993, 30, 95–106. [Google Scholar] [CrossRef]
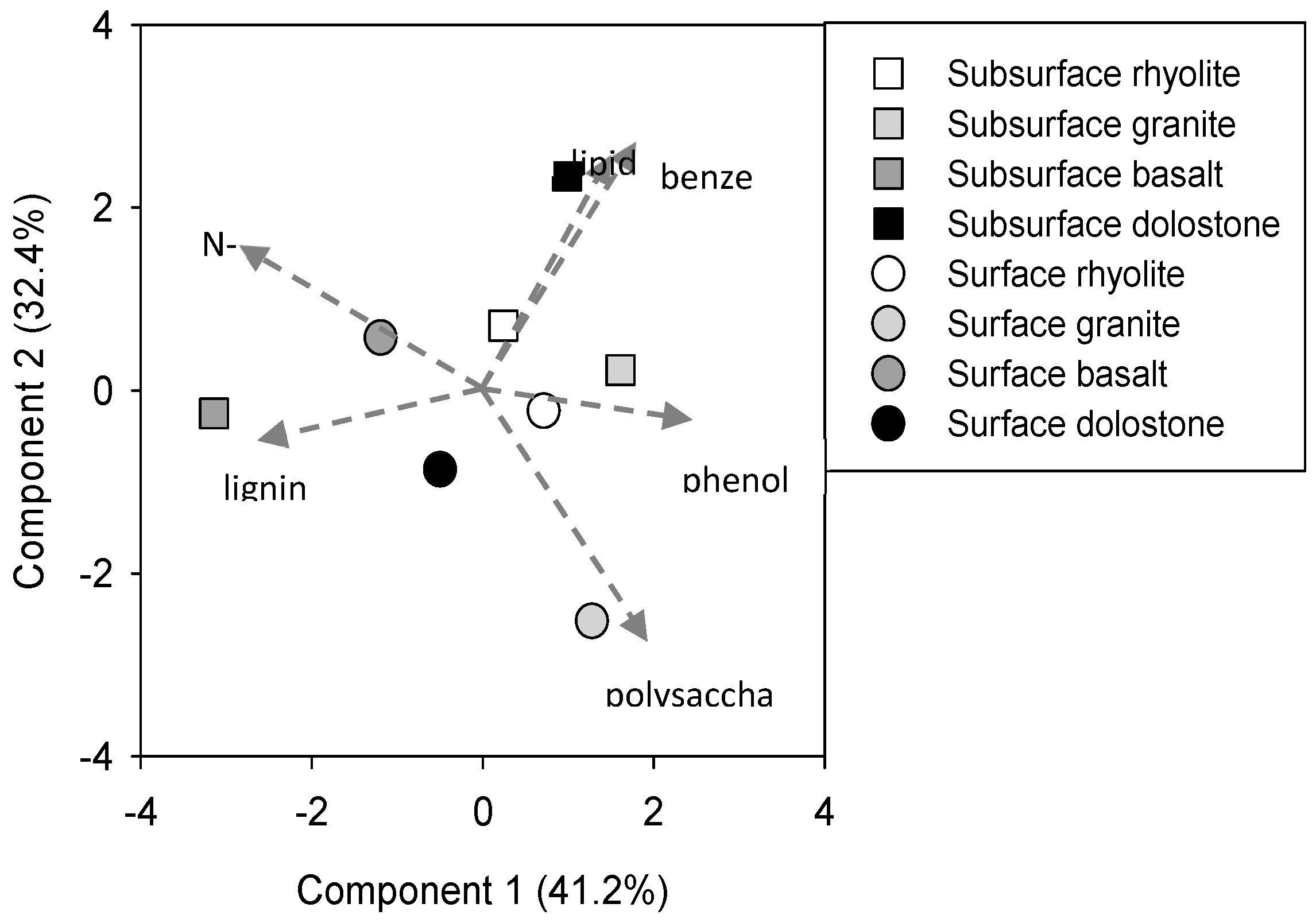
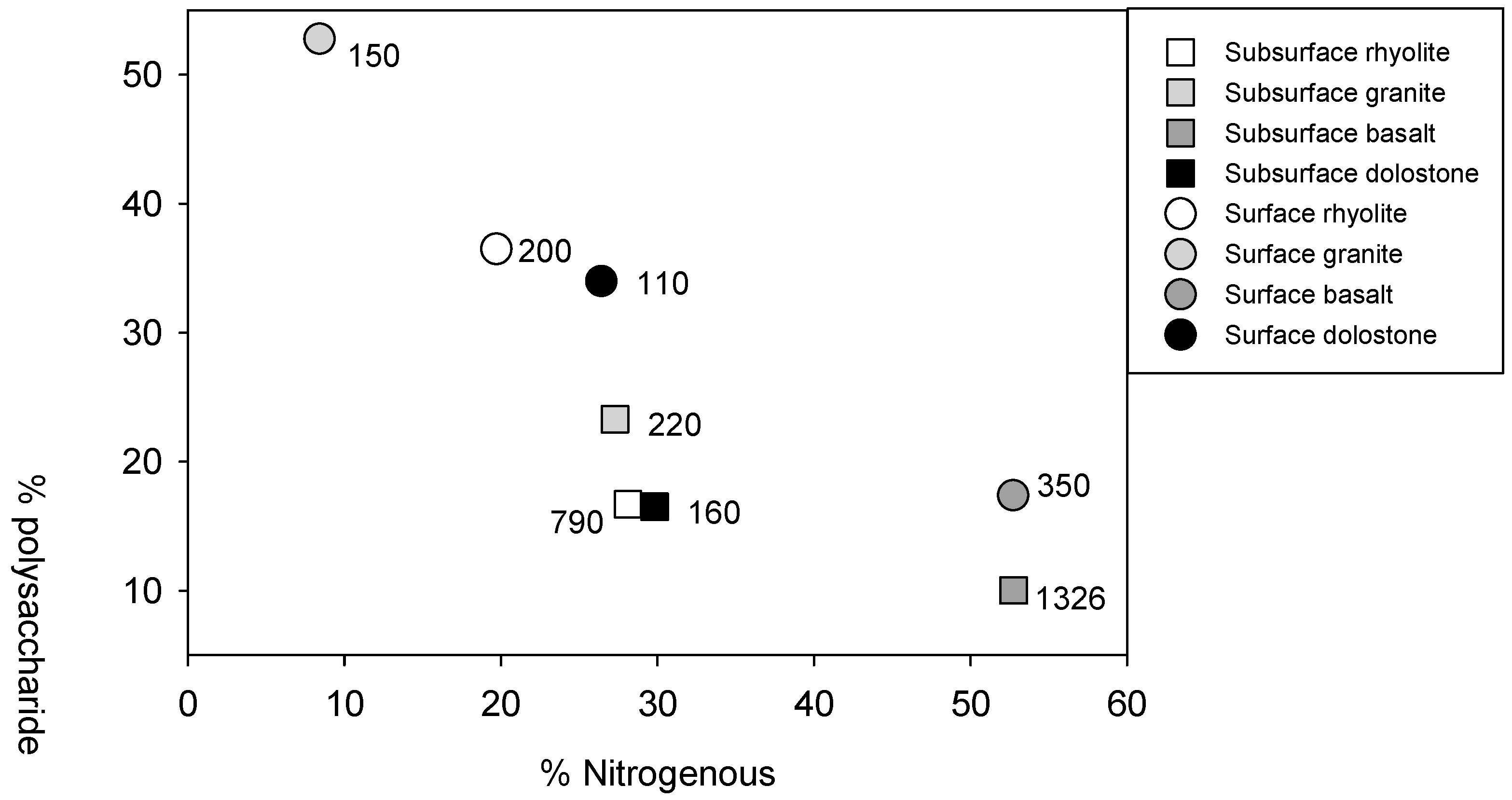
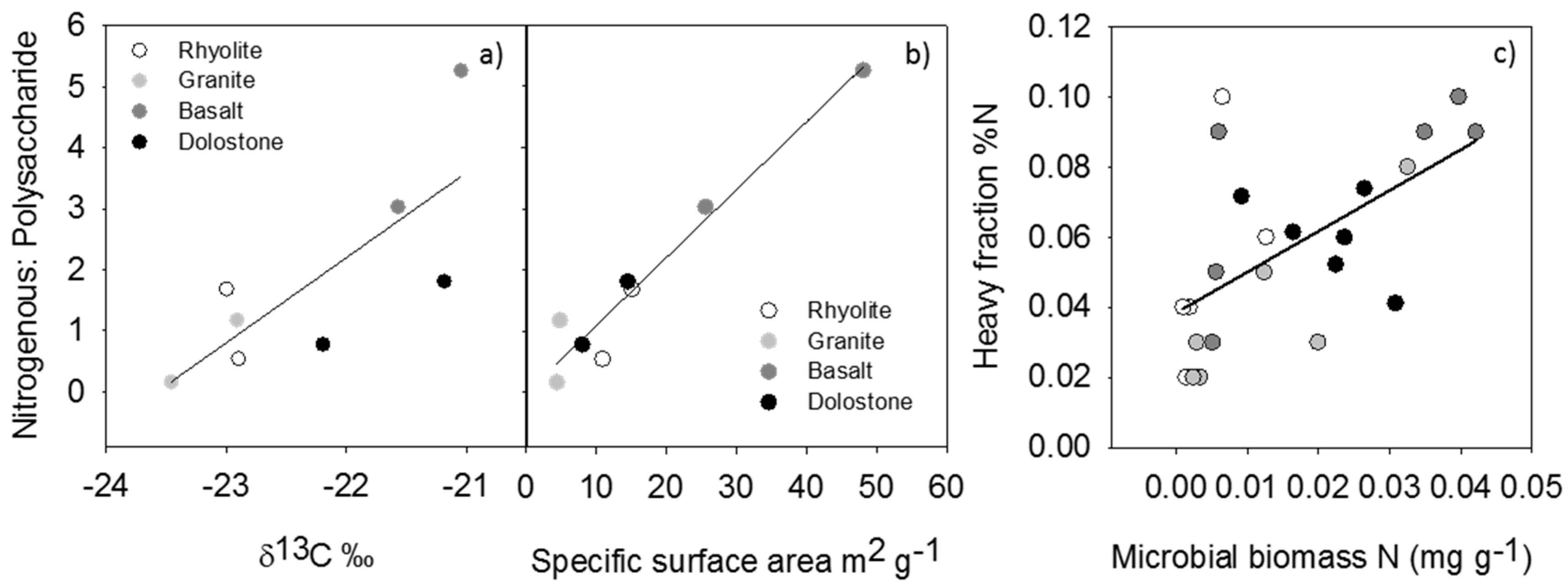

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Horizon | Parent | Horizon | pH | Clay | FePY + AlPY | FeD | SSA * | Secondary | |
|---|---|---|---|---|---|---|---|---|---|
| Material | Midpoint (cm) | 1:1 H2O | (%) | (g kg−1) | (m2 g−1) | Phyllosilicates | |||
| Surface | A2 | Rhyolite | 9.5 | 5.7 (0.3) | 10 (1) | 4.4 (0.2) | 6.4 (0.6) | 10.9 | Ka, Vm, Sm |
| A2 | Granite | 16 | 5.8 (0.2) | 6 (0) | 2.3 (0.3) | 8.1 (0.5) | 4.4 | Ka, HV, Il | |
| Bt1 | Basalt | 16.5 | 6.2 (0.1) | 28 (6) | 2.1 (0.7) | 26.3 (0.3) | 25.6 | Ka, Sm, Vm | |
| A | Dolostone | 2 | 6.6 (0.1) | 9 (2) | 1.8 (0.4) | 6.6 (0.6) | 8.0 | Ka, Il, HV | |
| Subsurface | Bw1 | Rhyolite | 35.5 | 5.3 (0.2) | 10 (1) | 1.9 (0.6) | 6.3 (0.7) | 15.1 | Ka, Vm, Sm |
| AC | Granite | 29.5 | 5.6 (0.2) | 6 (0) | 2.3 (0.5) | 7.9 (0.2) | 4.8 | Ka, HV, Il | |
| Bt2 | Basalt | 34.5 | 6.2 | 39 (3) | 1.9 (0.5) | 28.3 (2.3) | 48.1 | Ka, Sm, Vm | |
| Bt1 | Dolostone | 12 | 7.1 (0.2) | 16 (3) | 1.7 (0.2) | 8.8 (2.0) | 14.5 | Ka, Il, HV | |
| Horizon | Parent | Benzene | Lignin | Lipid | Phenol | Polysaccharide | Nitrogenous | C/N | δ13C | δ15N | Δ14C (‰) | MRT | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Material | % Abundance | (‰) | (Years) | ||||||||||
| Surface | A2 | Rhyolite | 27 | 8 | 1 | 3 | 37 | 20 | 10.5 (0.8) | −22.90 (0.18) | 3.9 (1.0) | 32.8 (4.2) | 203 |
| A2 | Granite | 16 | 8 | <1 | 11 | 53 | 8 | 10.4 (0.7) | −23.46 (0.16) | 1.3 (2.5) | 55.7 (4.3) | 148 | |
| Bt1 | Basalt | 25 | 5 | <1 | <1 | 17 | 53 | 9.6 (1.5) | −21.57 (0.32) | 5.6 (1.0) | −5.4 (4.1) | 349 | |
| A | Dolostone | 23 | 14 | <1 | 2 | 34 | 26 | 14.2 (1.7) | −22.20 (0.39) | 1.7 (0.8) | 76.4 (4.3) | 113 | |
| Subsurface | Bw1 | Rhyolite | 33 | 12 | 1 | 10 | 17 | 28 | 21.1 (10.5) | −23.00 (0.25) | −0.7 (1.1) | −72.0 (6.1) | 785 |
| AC | Granite | 28 | 3 | 1 | 18 | 23 | 27 | 14.5 (1.1) | −22.91 (0.16) | −4.5 (5.5) | 27.5 (4.4) | 219 | |
| Bt2 | Basalt | 13 | 22 | <1 | 2 | 10 | 53 | 10.7 (0.6) | −21.05 (0.13) | 2.6 (2.3) | −131.4 (5.6) | 1326 | |
| Bt1 | Dolostone | 36 | 9 | 4 | 6 | 17 | 30 | 11.7 (0.2) | −21.19 (0.13) | 4.5 (0.7) | 51.5 (4.5) | 157 | |
| Parameter | Explanatory Variables | R2 | p-Value | n |
|---|---|---|---|---|
| Heavy fraction %N | %C, Heavy fraction δ15N, FePY | 0.73 | <0.0001 | 44 |
| Heavy fraction %N | Biomass N | 0.35 | 0.0028 | 24 |
| Heavy fraction %C | Depth, AlPY + FePY | 0.58 | <0.0001 | 44 |
| Heavy fraction Δ14C | Depth, FeD | 0.84 | <0.0001 | 15 |
| Heavy fraction Δ14C | Depth, SSA | 0.91 | <0.0001 | 14 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Heckman, K.; Throckmorton, H.; Horwath, W.R.; Swanston, C.W.; Rasmussen, C. Variation in the Molecular Structure and Radiocarbon Abundance of Mineral-Associated Organic Matter across a Lithosequence of Forest Soils. Soil Syst. 2018, 2, 36. https://doi.org/10.3390/soilsystems2020036
Heckman K, Throckmorton H, Horwath WR, Swanston CW, Rasmussen C. Variation in the Molecular Structure and Radiocarbon Abundance of Mineral-Associated Organic Matter across a Lithosequence of Forest Soils. Soil Systems. 2018; 2(2):36. https://doi.org/10.3390/soilsystems2020036
Chicago/Turabian StyleHeckman, Katherine, Heather Throckmorton, William R. Horwath, Christopher W. Swanston, and Craig Rasmussen. 2018. "Variation in the Molecular Structure and Radiocarbon Abundance of Mineral-Associated Organic Matter across a Lithosequence of Forest Soils" Soil Systems 2, no. 2: 36. https://doi.org/10.3390/soilsystems2020036