Abstract
Soil mineral assemblage influences the abundance and mean residence time of soil organic matter both directly, through sorption reactions, and indirectly, through influences on microbial communities. Though organo-mineral interactions are at the heart of soil organic matter cycling, current models mostly lack parameters describing specific mineral assemblages or phases, and treat the mineral-bound pool as a single homogenous entity with a uniform response to changes in climatic conditions. We used pyrolysis GC/MS in combination with stable isotopes and radiocarbon abundance to examine mineral-bound soil organic matter fractions from a lithosequence of forest soils. Results suggest that different mineral assemblages tend to be associated with soil organics of specific molecular composition, and that these unique suites of organo-mineral complexes differ in mean residence time. We propose that mineralogy influences the composition of the mineral-bound soil organic matter pool through the direct influence of mineral surface chemistry on organo-mineral bond type and strength in combination with the indirect influence of soil acidity on microbial community composition. The composition of the mineral-bound pool of soil organic matter is therefore partially dictated by a combination of compound availability and sorption affinity, with compound availability controlled in part by microbial community composition. Furthermore, results are suggestive of a preferential sorption of N-containing moieties in Fe-rich soils. These bonds appear to be highly stable and confer extended mean residence times.
1. Introduction
The influence of soil mineral chemistry on the composition and stability of organics bound to mineral surfaces has become of interest to both experimentalists and modelers in the context of disturbances such as climate change and land use change. Benchtop experiments have suggested that mineral surface chemistry may influence the soil C cycle through processes such as competitive sorption [1,2,3,4], selective preservation [5,6], and feedbacks with microbial communities [7,8,9,10]. Here we investigate these interactions in naturally formed forest soils from a lithosequence under ponderosa pine.
Controlled laboratory experiments using reactive flow-through vessels or batch sorption experiments suggest strong competition among organic moieties for sorption to mineral phases which varies both with the surface chemistry of the solid phase and with the pH and ionic strength of the dissolved phase [11], and references therein. These competitive sorption, or sorptive fractionation, effects can result in predictable variation in the concentration, composition, and stability of mineral-bound organics in laboratory experiments which utilize a handful of model compounds. More recently, benchtop experiments examining organo-mineral interactions and sorptive fractionation have increased in complexity and in the utilization of emerging analytical capabilities such as nanoSIMS (nanoscale secondary ion mass spectrometry) and FTICR-MS (Fourier-transform ion cyclotron resonance). Not all of these studies find evidence of sorptive fractionation, for example, Fluery et al. [12] found selective sorption on Al oxide, but not on kaolinite. In an incubation experiment comparing protective capacity of various phyllosilicates, ferrihydrite and boehmite, no significant treatment effect was found [13]. These results are in contrast to recent findings utilizing dissolved organic matter on natural soils [14] and Fe oxides of varying crystallinity [15]. A portion of the discrepancy in conclusions can be attributed to different approaches to taking into account the influence of pH and the diversity of organic compounds available. Examination of naturally formed organo-mineral complexes may offer several advantages in comparison to utilizing modeled systems in that they have developed over very long time scales in a biotic environment, allowing for the complex interactions among microbes, substrate, and secondary mineral development to come into equilibrium. Additionally, these samples include the secondary influences of mineral chemistry on qualities of the soil matrix such as acidity and dissolved ion concentrations, which in turn affect organo-mineral qualities. However, the influence of competitive sorption on the composition and/or abundance of soil organic matter (SOM) in naturally formed soils has proven difficult to illustrate [16,17], with the strongest linkages having been found between SOM abundances and short-range-order mineral phases in the unique physicochemical environment of Andisols [18,19], and more recently the association of highly stabilized aromatic and carboxylate moieties on Al and Fe oxides [20]. A significant gap remains in regards to our understanding of the sorptive fractionation mechanisms dictating the formation of naturally occurring organo-mineral complexes.
Selective preservation, or the persistence of certain organics throughout the decomposition process, was long believed to be the product of the heterogeneous inherent structural characteristics of so-called humics [21]. More recently, molecular-scale investigations of the composition of SOM have revealed that the majority of SOM is composed of recognizable compounds or their constituent building blocks [22]. Therefore, with the exception of pyrogenic compounds [23], selective preservation is now hypothesized to depend on the strength of organo-mineral bonds formed [24], not solely the molecular structure of the organic component [25]. Proximity to the mineral surface and microbial conditioning of mineral surfaces are also thought to play a part in determining persistence of organo-mineral complexes [26], as in the zonal self-assembly model of Kleber et al. [27]. As is the case with sorptive fractionation, evidence of selective preservation (with the exception of pyrogenic materials) has been difficult to illustrate in naturally formed soils.
Indirect connections have been made between soil mineralogy and soil microbial characteristics, such as the dependence of fungal-to-bacterial ratios and bacterial diversity on pH [28,29]. Recent work has suggested that this connection between microbial community characteristics and pH may actually be attributed to the influence of toxic trivalent Al, the concentration of which is highly dependent on pH [30]. Trivalent Al and H concentrations are both a product of mineral assemblage and weathering, but are also influenced by the acidity of plant inputs, e.g., [31]. The influence of soil mineral chemistry may additionally shape microbial communities through factors such as elemental abundance [7], especially the abundance of macro- and micro-nutrients such as P, K, and Ca [32]. Differences in the character of microbial residues may then translate directly into differences in SOM characteristics [26].
With the objective of investigating the influence of mineral surface chemistry on characteristics of the mineral-bound SOM pool in naturally formed soils, we examined the molecular composition and radiocarbon abundances of the mineral-bound pool of organic matter across a lithosequence of four soils under ponderosa pine with the following questions in mind:
- Does the composition and stability of mineral-bound organic matter vary among soils of differing mineral assemblage?
- If so, is variation in composition and/or radiocarbon abundance associated with specific soil physicochemical properties?
- Can any connection be made between composition of mineral-associated organics and microbial community characteristics?
Previous work across this lithosequence, utilizing the same sample set as this manuscript, indicated strong linkages among mineralogy, microbial communities, and bulk soil C abundance. Variation in bulk C abundance and bacterial community composition were associated with variation in Al and Fe phases of differing crystallinity. Data suggested a gradient in the dominant mechanism of SOM stabilization across sites, with chemical recalcitrance and metal–humus complexation the dominant control in soils of the acidic rhyolite and granite sites, and mineral adsorption the dominant factor in the basic dolostone and basalt sites [9]. Accompanying microbial community characterization and manipulative laboratory studies highlighted the role of Al toxicity in shaping microbial community composition across the acidity gradient offered by this lithosequence [30]. Examination of the factors associated with the abundance of free particulate organic matter and organics preserved through occlusion within aggregates highlighted the importance of fire and aggregate formation in determining the abundance and stability of these pools of organic matter [33]. In the current manuscript, we seek to identify and understand parameters associated with variation in the molecular composition and stability of the mineral-bound pool of organic matter.
2. Materials and Methods
Data presented in this manuscript includes compilation and reexamination of a large body of previously published work in combination with newly presented data. Consequently, only a brief explanation of laboratory methods is given along with appropriate references to published works. All previously published work was conducted on the same soil samples, but the number of replicates varied according to the expense and labor associated with different instrumental methods.
Soils were sampled by genetic horizon from a lithosequence of four soils under Pinus ponderosa (ponderosa pine) in Arizona (Table 1). Parent materials and associated soils included dolostone (Loamy, mixed, superactive, mesic Lithic Argiustoll), basalt (Clayey-skeletal, mixed superactive, mesic Typic Paleustoll), granite (Loamy-skeletal, mixed superactive, mesic Typic Ustorthent), and rhyolite (Loamy, mixed, superactive, mesic Typic Haplustept). Three pedons were sampled from each site. Soils varied from slightly acidic to neutral in pH, and from sand to clay in texture. C inputs and C quality are assumed to be highly similar among sites due to the small variation in landscape position, aspect, elevation, precipitation, and vegetation. All soils were sampled from moderately flat (5–20% slope) ENE- or WNW-facing slopes in Pinus ponderosa-dominated forests. Mean annual precipitation and temperature ranged from 685 to 815 mm and 9–12 °C, respectively. Sample site coordinates and detailed site descriptions are given in Heckman et al. [9]. Only the mineral-associated fraction of organics (here termed the heavy fraction) was examined in this manuscript since this investigation is focused on the mechanisms determining the composition and stability of mineral-bound organics specifically.

Table 1.
Bulk properties of selected surface and subsurface horizons.
Soil particle size distribution was determined by the pipette method [34]. Soil pH was measured 1:1 (wt/wt) in H2O, and 1:1 in 1 M KCl [35]. Qualitative mineralogical analysis by X-ray diffraction was conducted on the clay (<2 μm), silt (2–53 μm), and very fine sand (53–105 μm) fractions for all genetic horizons at each site [36]. Iron oxide, short-range-order iron- and aluminum-oxyhydroxide, and metal–humus content of bulk soil were measured using standard selective dissolution techniques using sodium dithionite (FeD, AlD), acid ammonium oxalate (FeO, AlO), and sodium pyrophosphate (FePY, ALPY) [35]. Specific surface area (SSA) was measured on bulk soils after organic matter removal using a Beckman Coulter SA 3100 Surface Area and Pore Size Analyzer (Fullerton, CA, USA). Samples were analyzed for total organic C, total N, δ13C, and δ15N by dry combustion at 1000 °C with an elemental analyzer (Costech Analytical Technologies, Valencia, CA, USA) coupled to a continuous-flow mass spectrometer (Finnigan Delta PlusXL, San Jose, CA, USA) at the University of Arizona Stable Isotope Laboratory. Surface area was measured under N2 and modeled using the BET equation [37]. Soils were density separated into free/light (F/L), occluded (OCC) and heavy fractions (HF) using a sodium polytungstate (SPT) solution adjusted to a density of 1.8 g cm−3 [33,38]. In brief, soils were mixed with SPT and centrifuged to separate low density organics (F/L) from the rest of the soil. The F/L was aspirated and rinsed. Sonication at a rate of 1500 J/gsoil was applied to disrupt aggregates and release low-density organics (OCC). Following centrifugation, the OCC was aspirated and rinsed. The remaining pellet was assumed to contain only mineral material and the organics intimately associated with mineral surfaces, termed the heavy fraction (HF). Density fractions of bulk soils were characterized by pyrolysis GC/MS [33,39]. In brief, ground composited subsamples were pyrolyzed in pyrofoils or platinum cups. Samples analyzed by pyrolysis GC/MS were composited from three pedons per site. Pyrolysis products were identified through comparison with published and stored NIST and Wiley library data. Pyrolysis products were assigned to one of seven chemical classes: benzene, lignin, lipid, phenol, polysaccharide, and nitrogenous. Chemical class assignments were made on the basis of comparison to published works. Soils additionally underwent incubation for estimates of biomass abundances and C use efficiencies [9]. Radiocarbon abundance of bulk soils and density separates were measured by accelerator mass spectrometry at the NSF—Arizona AMS Laboratory at the University of Arizona (Tucson, AZ, USA) [40], the Centro Nacional de Aceleradores (Seville, Spain) [41], and the Center for AMS at Lawrence Livermore National Laboratory (Livermore, CA, USA) [42,43]. For each horizon, density/aggregate fractions and bulk soils from each site were composited for AMS analysis. Radiocarbon abundance measurements were converted to steady-state mean residence time (MRT) values following Trumbore [44] and Torn et al. [45]. Estimated MRT values are not meant to represent absolute years of residence in the soil, but are used to allow for ease of comparison among samples (i.e., to allow for quick comparison of decadal versus millennial cycling rates).
Principal components analysis and multiple regression were used to explore relationships among organic, physicochemical, and microbial variables. The following is a listing of all parameters considered when constructing simple multiple regression models, with the total number of degrees of freedom (n) given in parentheses: HF %C (44), HF %N (44), HF δ13C (44), HF δ15N (4), HF Δ14C (15), FePY (44), AlPY (44), FeO (44), AlO (44), FeD (44), horizon depth (44), SSA (14), Py GC/MS compound class composition (8), microbial respiration (24), microbial biomass N abundance (24), microbial biomass C abundance (24), pH H2O (44), pH KCl (44).
The primarily featured data in this manuscript is the pyrolysis GC/MS data. Due to the time associated with data processing, only eight individual heavy fractions were measured, one surface and one subsurface horizon from each of the four sites. We attempted to include horizons from each site that were at similar depths across sites. The uppermost A horizon was not considered a desirable horizon due to the fact that the influence of mineralogy would be more difficult to detect in horizons with an abundance of organic matter. However, since the dolostone soil had only two horizons total, the surface A horizon was used.
3. Results
Bulk soil properties for the horizons selected for pyrolysis GC/MS measurement are given in Table 1. This lithosequence of soils was selected in order to represent a spectrum of pH and textural values. Basalt soils had the highest abundance of clay and pedogenic Fe (as estimated from chemical extraction with dithionite) as well as the highest SSA. Rhyolite, granite, and dolostone soils were depleted in these qualities in comparison to the basalt soils. Soils followed a pH gradient from slightly acidic to neutral in the following order: rhyolite < granite < basalt < dolostone. Clay mineralogy varied considerably across the soils. All soils contained the nonexpansible phase, kaolinite. The highly expansible phase, smectite, was present in rhyolite and basalt soils. Granite and dolostone soils contained partially expansible phases, vermiculite, illite, and hydroxyl-interlayered vermiculite.
Properties of the heavy fractions are given in Table 2. Because the pyrolysis GC/MS data is not replicated, principal components analysis and simple linear regression were used to assess relationships among soil parent material type, organic matter characteristics, and heavy fraction physicochemical characteristics. There was considerable variation in the composition of the mineral-bound organic matter pool (Figure 1). Polysaccharide-to-nitrogenous compound ratios were used to illustrate the relative enrichment in nitrogen-bearing compounds in the Fe-rich basalt soils (Figure 2). Mineral-bound organics from the basalt soils also had the longest MRTs in comparison to the other soils. The relative abundance of nitrogenous compounds in the mineral-bound organics was positively related to the enrichment of 13C and the specific surface area of the heavy fractions (Figure 3a,b). Total N content of the heavy fractions was moderately correlated with soil microbial biomass N abundance (Figure 3c).
Table 2.
Heavy fraction SOM characteristics for selected surface and subsurface horizons.
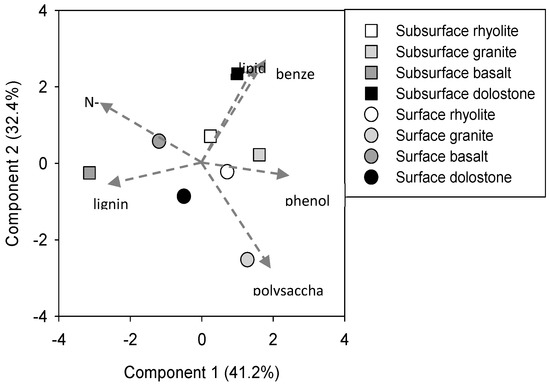
Figure 1.
Principle components analysis of heavy fraction molecular composition as measured by pyrolysis GC/MS.
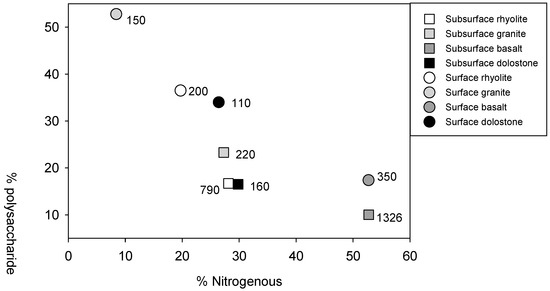
Figure 2.
Heavy fraction polysaccharide vs. nitrogen-bearing compound abundance as determined by pyrolysis GC/MS. Parent material is indicated by shade and depth is indicated by symbol. Numbers accompanying symbols reflect the estimated steady-state mean residence time of the sample.
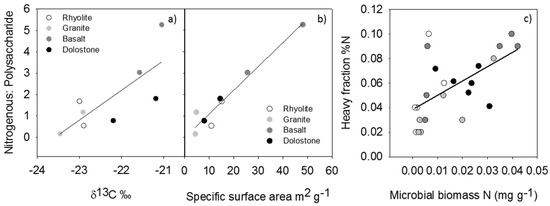
Figure 3.
(a) Heavy fraction δ13C abundance vs. nitrogenous: polysaccharide, r2 = 0.60, p = 0.0241; (b) heavy fraction specific surface area vs. nitrogenous: polysaccharide, r2 = 0.95, p < 0.0001; (c) heavy fraction %N vs. microbial biomass N, r2 = 0.39, p = 0.0015.
Stepwise regression was used to identify explanatory variables for the C and N abundance and MRTs of the heavy fraction (Table 3 and Table S1). N abundance showed a relationship with %C, δ15N, FePY, and biomass N. C abundance was related to depth and AlPY+FePY. The Δ14C values of the heavy fraction (used in the estimation of MRTs) were strongly related to both FeD and SSA.
Table 3.
Explanatory variables for characteristics of the heavy fractions.
4. Discussion
There is much debate over the relative importance of different factors in determining the abundance and character of the mineral-bound pool of SOM. The community has moved from the long-held paradigm that the character of organic inputs is the largest determinant of SOM character to a more holistic paradigm where the influence of a myriad of ecosystem properties are considered [46]. Recent work highlighting the microbial community as drivers of SOM production and character is creating a new understanding of how C is transformed and stabilized in soils [26,47]. Understanding of the relative importance of physicochemical stabilization mechanisms across ecosystem types is also improving [48]. However, the role of mineral chemistry in C stabilization is still not well defined in natural soils, despite decades of lab work with model systems showing sorptive fractionation and selective preservation effects. One reason for this may be that mineral influences are difficult to detect in bulk soils, and difficult to identify among the myriad of other influential factors. The use of a lithosequence allows for clearer examination of the role of mineral assemblage by limiting variation in other soil-forming factors. The use of density separation additionally isolates the heavy fraction, and therefore isolates the mineral-bound pool of organics from particulate organics. Below, we use a lithosequence of heavy fractions from ponderosa pine forests as a case study to illustrate how mineral chemistry influences the character and persistence of mineral-bound organics in naturally formed soils.
Across this lithosequence of forest soils, C, N, and 14C abundances of the heavy fractions were specifically linked to mineralogical parameters, namely Fe and Al abundances in the form of oxyhydroxide or organo-metal phases. Associated data on the molecular composition of heavy fraction organics suggests the preferential binding of nitrogenous compounds on Fe-rich mineral surfaces. Increasing abundance of nitrogenous compounds on Fe-rich surfaces was accompanied by depletion in 14C, suggesting that organics on Fe-rich surfaces are more tightly bound relative to the other mineral assemblages examined here.
4.1. Competitive Sorption and Selective Preservation
If we assume that the free/light fraction is the primary source of microbial growth substrate [49,50], we can use differences between the composition of the free/light fraction organics and the heavy fraction organics to look for evidence of competitive sorption and/or selective preservation. In comparison to the composition of the free/light fraction, mineral-bound organics were enriched in benzene-based compounds and nitrogenous compounds, while depleted in lignin, phenol, and lipids (38; Figure S1).
The abundance of benzene-based compounds in the heavy fraction was not related to any physicochemical or microbial property available in the current dataset, but may be attributable to structural recalcitrance, as previous work on these soils suggests that these compounds may be pyrogenic in origin. Fire is a natural part of Pinus ponderosa systems, and previous measurements on these soils utilizing 13C solid-state nuclear magnetic resonance indicated that 5–10% of bulk SOM was pyrogenic [33]. Strong pi-pi bonding between these benzene compounds and mineral surfaces may additionally account for their selective preservation [51].
Nitrogenous compound abundance was higher in general in heavy fractions in comparison to free/light fractions (Figure S1), but was especially concentrated in the Fe-rich basalt soils (Figure 2). The preferential binding of N-bearing moieties on Fe-rich mineral surfaces at circum-neutral pH has been illustrated in both laboratory settings and natural soils [52,53,54,55,56]. Recently, atomic force microscopy was used to illustrate the strong binding of N to goethite, where ammonia bonds were on average 3 times stronger than carboxylate, phosphate, or methyl bonds [24]. Keiluweit et al. [52] hypothesized that this preferential bonding of N-bearing compounds to Fe-rich surfaces was due to competitive sorption of phosphorylated proteins with hydroxylated Fe oxide surfaces [57]. The relationship of heavy fraction %N with δ15N and FePY (Table 3 and Table S1) adds additional support to the argument of competitive sorption of microbially-derived nitrogenous compounds with Fe, as δ15N increases with degree of microbial processing, and FePY is representative of the pool of Fe most available for reaction with organic constituents in the soil.
In addition to illustrating competitive sorption behavior, bonding to Fe surfaces seems to confer greater stability to the bound organics. In previous work exploring the possibility of an influence of mineral chemistry on organic matter composition and stability, no clear patterns were found aside from a consistent increase in MRT associated with increasing concentration of Fe-rich mineral phases [17]. In the dataset examined here, heavy fraction MRT was explained almost equally by FeD and SSA (Table 3 and Table S1). The production of surface area (SSA) and pedogenic Fe go hand-in-hand as weathering progresses, and both may act as a proxy for the general developmental stage of the soil [58,59] (absolute abundance of clay and pedogenic Fe also varying with parent material composition). However, mounting evidence suggests that Fe-rich phases play a unique and highly influential role in the stabilization and preservation of SOM. Iron- and Al-oxyhydroxide phases, especially short-range-order phases, are hypothesized to play a prominent role in metal- and mineral-organic matter precipitation and bonding reactions due to their ubiquity in soils [60] and their abundance of reactive surface area [61]. Al-oxyhydroxide phases are often recognized for their unique role in SOM cycling in acidic soils and Andisols, whereas Fe-oxyhydroxide phases may exert more influence in soils with circum-neutral pH values, such as those examined in the current lithosequence. A large body of work has accumulated that examines the conditions influencing coprecipitation and surface sorption to oxyhydroxide phases of differing crystallinity, as well as the relative stability of these organo-mineral/metal compounds e.g., [62,63,64]. Across a diversity of naturally-formed soils, Mayes et al. [65] identified a prominent role of Fe-oxyhydroxide content for predicting sorptive capacity across a diversity of bulk soils. The long-term persistence of organo-mineral/metal compounds in natural soils has yet to be quantified, though recent radiocarbon analysis has suggested a significant dependence of heavy fraction MRT on the abundance of Al- and Fe-oxyhydroxide phases [66].
4.2. Feedbacks between Soil Physicochemical Properties and Microbial Communities
Mineral surface chemistry influences the character of mineral-bound organics directly through the mostly abiotic processes outlined above, but also has an influence on SOM composition through its impacts on microbial communities. Because soil microbes live on mineral surfaces, mineral surface characteristics are likely to affect them. Microbial community structure has been linked to the presence/absence of particular elements in the mineral surface that may have toxic effects (e.g., Al) or are nutrients essential for life (e.g., Ca, K, P) [7]. Mineral composition also influences the abundance of Fe, which can be employed as a terminal electron acceptor by certain soil microbes, as well as determining the abundance of specific surface area and buffering capacity. Furthermore, a significant portion of bulk SOM is composed of microbial metabolites and necromass [26], and much of this microbially-derived SOM is associated with mineral surfaces as indicated by heavy fraction organic characteristics such as low C:N ratios, enrichment in heavy isotopes, and strong indications of microbial alteration including degree of oxidation and thermal lability [25,67].
It has been shown that mineralogy directly influences microbial community composition [8,30,32], with composition differing even among mineral grains of differing phases within the same bulk material [7]. Fungal:bacterial ratios are also strongly influenced by soil pH, with ratios increasing with decreasing pH [28]. The C:N ratios of bacteria are lower than that of fungi, which has led to C:N being employed to roughly approximate shifts in fungal:bacterial ratios, e.g., [68], but also leads to a difference in C:N ratio of microbial residues/necromass and therefore differences in the abundance and availability of N-bearing compounds.
A detailed examination of microbial communities from this lithosequence of soils additionally highlights the influence of soil mineralogy on microbial communities. Variation in bacterial communities, as measured by terminal restriction fragment length polymorphism, was clearly divided by soil parent material type, with variation primarily explained by pH, trivalent Al, and amorphous Fe-oxyhydroxide content [9].
Our work is consistent with the literature, with the correlation among microbial biomass N abundance and heavy fraction N abundance in our lithosequence (Figure 3c) likely arising from a combination of two major processes: (1) variation in microbial community composition resulting from the influences of soil pH and mineral assemblage; and (2) the selective sorption and preservation of N-bearing compounds on the Fe-rich basalt surfaces. Enrichment in δ13C is associated with increasing degree of microbial processing, since the heavier C isotope is preferentially incorporated into biomass during respiration and growth. In soils, decreasing C:N ratios are also associated with advancing degree of biodegradation, because as substrate is processed into microbial biomass, it begins to be more N-enriched. Therefore, we suggest that increased δ13C enrichment associated with increased nitrogenous compound: polysaccharide (Figure 3a) also points to an increase in microbial processing associated with an increase in mineral-bound nitrogenous compound abundance.
5. Conclusions
There is strong evidence in the literature showing that mineral assemblage can influence the character and stability of the mineral-bound pool of organics, with mineral Fe content playing a large role in competitive sorption and selective preservation mechanisms, leading to an enrichment of N-containing compounds associated with Fe-rich surfaces. We suggest that the composition and stability of the mineral-bound pool of organics, often isolated through density fractionation and termed the heavy fraction, reflects dynamic feedbacks among biotic and abiotic soil components and processes. As mineral assemblage shapes the physicochemical environment of the soil through its influence on soil acidity, nutrient availability, and specific mineral surface characteristics such as Fe abundance, microbial communities influence the character of organics made available for interaction with mineral surfaces. Our work is consistent with previous findings of a unique role of Fe-N bonding in the retention and long-term preservation of mineral-bound organics.
Supplementary Materials
The following are available online at http://www.mdpi.com/2571-8789/2/2/36/s1, Figure S1: Pyrolysis GC/MS compound class abundance, Table S1: Full regression models.
Author Contributions
Conceptualization, K.H., C.R., W.R.H., and C.W.S.; Formal Analysis, K.H. and C.R.; Investigation, K.H. and H.T.; Resources, K.H. and H.T.; Writing-Original Draft Preparation, K.H., C.R., C.W.S.; Writing-Review & Editing, K.H., H.T., W.R.H., C.W.S., C.R.; Visualization, K.H., C.R. and C.W.S.; Supervision, C.R. and W.R.H.; Project Administration, C.R. and W.R.H.; Funding Acquisition, C.R.
Funding
This research was funded by National Science Foundation grant number [0543130].
Acknowledgments
We acknowledge the efforts and input of Assistant Editor, Janey Zhao, and three anonymous reviewers who helped to improve the quality of this manuscript.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Namjesnik-Dejanovic, K.; Maurice, P.A.; Aiken, G.R.; Cabaniss, S.; Chin, Y.-P.; Pullin, M.J. Adsorption and fractionation of muck fulvic acid on kaolinite and goethite at pH 3.7, 6, and 8. Soil Sci. 2000, 165, 545–559. [Google Scholar] [CrossRef]
- Zhou, Q.H.; Maurice, P.A.; Cabaniss, S.E. Size fractionation upon adsorption of fulvic acid on goethite: Equilibrium and kinetic studies. Geochim. Cosmochim. Acta 2001, 65, 803–812. [Google Scholar] [CrossRef]
- Guo, M.X.; Chorover, J. Transport and fractionation of dissolved organic matter in soil columns. Soil Sci. 2003, 168, 108–118. [Google Scholar] [CrossRef]
- Heckman, K.; Vazquez-Ortega, A.; Gao, X.D.; Chorover, J.; Rasmussen, C. Changes in water extractable organic matter during incubation of forest floor material in the presence of quartz, goethite and gibbsite surfaces. Geochim. Cosmochim. Acta 2011, 75, 4295–4309. [Google Scholar] [CrossRef]
- Lichtfouse, E.; Chenu, C.; Baudin, F.; Leblond, C.; Da Silva, M.; Béhar, F.; Derenne, S.; Largeau, C.; Wehrung, P.; Albrecht, P. A novel pathway of soil organic matter formation by selective preservation of resistant straight-chain biopolymers: chemical and isotope evidence. Org. Geochem. 1998, 28, 411–415. [Google Scholar] [CrossRef]
- Lützow, M.V.; Kögel-Knabner, I.; Ekschmitt, K.; Matzner, E.; Guggenberger, G.; Marschner, B.; Flessa, H. Stabilization of organic matter in temperate soils: mechanisms and their relevance under different soil conditions—A review. Eur. J. Soil Sci. 2006, 57, 426–445. [Google Scholar]
- Gleeson, D.B.; Clipson, N.; Melville, K.; Gadd, G.M.; McDermott, F.P. Characterization of fungal community structure on a weathered pegmatitic granite. Microb. Ecol. 2005, 50. [Google Scholar] [CrossRef] [PubMed]
- Carson, J.K.; Campbell, L.; Rooney, D.; Clipson, N.; Gleeson, D.B. Minerals in soil select distinct bacterial communities in their microhabitats. FEMS Microb. Ecol. 2009, 67, 381–388. [Google Scholar] [CrossRef] [PubMed]
- Heckman, K.; Welty-Bernard, A.; Rasmussen, C.; Schwartz, E. Geologic controls of soil carbon cycling and microbial dynamics in temperate conifer forests. Chem. Geol. 2009, 267, 12–23. [Google Scholar] [CrossRef]
- Marsh, T.L.; Long, D.; Voice, T. Bacterial transformations of metals in soils. In Handbook of Soil Sciences: Resource Management and Environmental Impacts, 2nd ed.; Huang, P.M., Li, Y., Sumner, M.E., Eds.; CRC Press: Boca Raton, FL, USA, 2012; pp. 16–22. [Google Scholar]
- Kleber, M.; Eusterhues, K.; Keiluweit, M.; Mikutta, C.; Mikutta, R.; Nico, P.S. Chapter one-mineral–organic associations: formation, properties, and relevance in soil environments. Adv. Agron. 2015, 130, 1–140. [Google Scholar] [CrossRef]
- Fleury, G.; Del Nero, M.; Barillon, R. Effect of mineral surface properties (alumina, kaolinite) on the sorptive fractionation mechanisms of soil fulvic acids: Molecular-scale ESI-MS studies. Geochim. Cosmochim. Acta 2017, 196, 1–17. [Google Scholar] [CrossRef]
- Pronk, G.J.; Heister, K.; Kögel-Knabner, I. Is turnover and development of organic matter controlled by mineral composition? Soil Biol. Biochem. 2013, 67, 235–344. [Google Scholar] [CrossRef]
- Avneri-Katz, S.; Young, R.B.; McKenna, A.M.; Chen, H.; Corilo, Y.E.; Polubesova, T.; Borch, T.; Chefetz, B. Adsorptive fractionation of dissolved organic matter (DOM) by mineral soil: Macroscale approach and molecular insight. Org. Geochem. 2017, 103, 113–124. [Google Scholar] [CrossRef]
- Coward, E.K.; Ohno, T.; Plante, A.F. Adsorption and molecular fractionation of dissolved organic matter on iron-bearing mineral matrices of varying crystallinity. Environ. Sci. Technol. 2018, 52, 1036–1044. [Google Scholar] [CrossRef] [PubMed]
- Sollins, P.; Swanston, C.; Kleber, M.; Filley, T.; Kramer, M.; Crow, S.; Calwell, B.A.; Lajtha; Bowden, K.R. Organic C and N stabilization in a forest soil: Evidence from sequential density fractionation. Soil Biol. Biochem. 2006, 38, 3313–3324. [Google Scholar] [CrossRef]
- Sollins, P.; Kramer, M.G.; Swanston, C.; Lajtha, K.; Filley, T.; Aufdenkampe, A.K.; Wagai, R.; Bowden, R.D. Sequential density fractionation across soils of contrasting mineralogy: Evidence for both microbial-and mineral-controlled soil organic matter stabilization. Biogeochemistry 2009, 96, 209–231. [Google Scholar] [CrossRef]
- Garrido, E.; Matus, F. Are organo-mineral complexes and allophane content determinant factors for the carbon level in Chilean volcanic soils? Catena 2012, 92, 106–112. [Google Scholar] [CrossRef]
- Giardina, C.P.; Litton, C.M.; Crow, S.E.; Asner, G.P. Warming-related increases in soil CO2 efflux are explained by increased below-ground carbon flux. Nat. Clim. Chang. 2014, 4, 822–827. [Google Scholar] [CrossRef]
- Yeasmin, S.; Singh, B.; Johnson, C.T.; Sparks, D.L. Organic carbon characteristics in density fractions of soils with contrasting mineralogies. Geochim. Cosmochim. Acta. 2017, 218, 215–236. [Google Scholar] [CrossRef]
- Piccolo, A. The supramolecular structure of humic substances. Soil Sci. 2001, 166, 810–832. [Google Scholar] [CrossRef]
- Sutton, R.; Sposito, G. Molecular structure in soil humic substances: The new view. Environ. Sci. Technol. 2005, 39, 9009–9015. [Google Scholar] [CrossRef] [PubMed]
- Santín, C.; Doerr, S.H.; Kane, E.S.; Masiello, C.A.; Ohlson, M.; Rosa, J.M.; Preston, C.M.; Dittmar, T. Towards a global assessment of pyrogenic carbon from vegetation fires. Global Chang. Biol. 2016, 22, 76–91. [Google Scholar] [CrossRef] [PubMed]
- Newcomb, C.J.; Qafoku, N.P.; Grate, J.W.; Bailey, V.L.; De Yoreo, J.J. Developing a molecular picture of soil organic matter–mineral interactions by quantifying organo–mineral binding. Nature Commun. 2017, 8, 396. [Google Scholar] [CrossRef] [PubMed]
- Kleber, M.; Nico, P.S.; Plante, A.; Filley, T.; Kramer, M.; Swanston, C.; Sollins, P. Old and stable soil organic matter is not necessarily chemically recalcitrant: Implications for modeling concepts and temperature sensitivity. Glob. Chang. Biol. 2011, 17, 1097–1107. [Google Scholar] [CrossRef]
- Kallenbach, C.M.; Frey, S.D.; Grandy, A.S. Direct evidence for microbial-derived soil organic matter formation and its ecophysiological controls. Nat. Commun. 2016, 7, 13630. [Google Scholar] [CrossRef] [PubMed]
- Kleber, M.; Sollins, P.; Sutton, R.A. conceptual model of organo-mineral interactions in soils: Self-assembly of organic molecular fragments into zonal structures on mineral surfaces. Biogeochemistry 2007, 85, 9–24. [Google Scholar] [CrossRef]
- Bååth, E.; Anderson, T.H. Comparison of soil fungal/bacterial ratios in a pH gradient using physiological and PLFA-based techniques. Soil Biol. Biochem. 2003, 35, 955–963. [Google Scholar] [CrossRef]
- Fierer, N.; Jackson, R.B. The diversity and biogeography of soil bacterial communities. PNAS 2006, 103, 626–631. [Google Scholar] [CrossRef] [PubMed]
- Welty-Bernard, A.T. Al, Fe, and Ph Effects on Soil Microbial Communities. Ph.D. Thesis, Northern Arizona University, Flagstaff, AZ, USA, May 2014. [Google Scholar]
- Essington, M.E. Soil and Water Chemistry: An Integrative Approach; CRC Press: Boca Raton, FL, USA, 2004. [Google Scholar]
- Carson, J.K.; Rooney, D.; Gleeson, D.B.; Clipson, N. Altering the mineral composition of soil causes a shift in microbial community structure. FEMS Microbiol. Ecol. 2007, 61, 414–423. [Google Scholar] [CrossRef] [PubMed]
- Jackson, M.L. Soil Chemical Analysis: Advanced Course, revised 2nd ed.; Paralled Press University of Wisconsin-Madison Libraries: Madison, WI, USA, 1985. [Google Scholar]
- Soil Survey Staff, National Soil Survey Laboratory. Methods Manual: Soil Survey Investigations Report No. 42, Version 4.0; United States Department of Agriculture, Natural Resources Conservation Service, National Soil Survey Center: Lincoln, NE, USA, 2004.
- Heckman, K.; Rasmussen, C. Lithologic controls on regolith weathering and mass flux in forested ecosystems of the southwestern USA. Geoderma 2011, 164, 3, 99–111. [Google Scholar] [CrossRef]
- Brunauer, S.; Emmett, P.H.; Teller, E. Adsorption of gases in multimolecular layers. J. Am. Chem. Soc. 1938, 60, 309–319. [Google Scholar] [CrossRef]
- Swanston, C.W.; Torn, M.S.; Hanson, P.J.; Southon, J.R.; Garten, C.T.; Hanlon, E.M.; Ganio, L. Initial characterization of processes of soil carbon stabilization using forest stand-level radiocarbon enrichment. Geoderma 2005, 128, 52–62. [Google Scholar] [CrossRef]
- Heckman, K.; Throckmorton, H.; Clingensmith, C.; Vila, F.J.G.; Horwath, W.R.; Knicker, H.; Rasmussen, C. Factors affecting the molecular structure and mean residence time of occluded organics in a lithosequence of soils under ponderosa pine. Soil Biol. Biochem. 2014, 77, 1–11. [Google Scholar] [CrossRef]
- Throckmorton, H.M. Diverse Microbial Carbon as a Source of Soil Organic Matter. Ph.D. Thesis, University of California, Davis, Davis, CA, USA, 2012. [Google Scholar]
- Donahue, D.J.; Jull, A.J.T.; Toolin, L.J. Radiocarbon measurements at the University of Arizona AMS facility. Nucl. Instr. Meth. Phys. Res. B 1990, 52, 224–228. [Google Scholar] [CrossRef]
- Arèvalo, F.J.S.; Martínez, I.G.; Leon, M.G. Radiocarbon measurement program at the Centro Nacional de Aceleradores (CAN), Spain. Radiocarbon 2009, 51, 883–889. [Google Scholar] [CrossRef]
- Vogel, J.S.; Southon, J.R.; Nelson, D.E. Catalyst and binder effects in the use of filamentous graphite for AMS. Nucl. Instrum. Meth. B 1987, 29, 50–56. [Google Scholar] [CrossRef]
- Davis, J.C.; Proctor, I.D.; Southon, J.R.; Caffee, M.W.; Heikkinen, D.W.; Roberts, M.L.; Moore, T.L.; Turteltaub, K.W.; Nelson, D.E.; Loyd, D.H.; et al. LLNL/US AMS facility and research program. Nucl. Instr. Meth. Phys. Res. B 1990, 52, 269–272. [Google Scholar] [CrossRef]
- Trumbore, S.E. Comparison of carbon dynamics in tropical and temperate soils using radiocarbon measurements. Glob. Biogeochem. Cycles 1993, 7, 275–290. [Google Scholar] [CrossRef]
- Torn, M.S.; Lapenis, A.G.; Timofeev, A.; Fischer, M.L.; Babikov, B.V.; Harden, J.W. Organic carbon and carbon isotopes in modern and 100-year-old-soil archives of the Russian steppe. Glob. Chang. Biol. 2002, 8, 941–953. [Google Scholar] [CrossRef]
- Schmidt, M.W.; Torn, M.S.; Abiven, S.; Dittmar, T.; Guggenberger, G.; Janssens, I.A.; Kleber, M.; Kögel-Knabner, I.; Lehmann, J.; Manning, D.A.; et al. Persistence of soil organic matter as an ecosystem property. Nature 2011, 478, 49–56. [Google Scholar] [CrossRef] [PubMed]
- Paul, E.A. The nature and dynamics of soil organic matter: Plant inputs, microbial transformations, and organic matter stabilization. Soil Biol. Biochem. 2016, 98, 109–126. [Google Scholar] [CrossRef]
- Rasmussen, C.; Heckman, K.; Wieder, W.R.; Keiluweit, M.; Lawrence, C.R.; Berhe, A.A.; Blankinship, J.C.; Crow, S.E.; Druhan, J.L.; Pries, C.E.H.; et al. Beyond clay: Towards an improved set of variables for predicting soil organic matter content. Biogeochemistry 2018, 137, 297–306. [Google Scholar] [CrossRef]
- Alvarez, R.; Alvarez, C.R. Soil organic matter pools and their associations with carbon mineralization kinetics. Soil Sci. Soc. Am. J. 2000, 64, 184–189. [Google Scholar] [CrossRef]
- McLauchlan, K.K.; Hobbie, S.E. Comparison of labile soil organic matter fractionation techniques. Soil Sci. Soc. Am. J. 2004, 68, 1616–1625. [Google Scholar] [CrossRef]
- Keiluweit, M.; Kleber, M. Molecular-level interactions in soils and sediments: The role of aromatic π-systems. Environ. Sci. Technol. 2009, 43, 3421–3429. [Google Scholar] [CrossRef] [PubMed]
- Keiluweit, M.; Bougoure, J.J.; Zeglin, L.H.; Myrold, D.D.; Weber, P.K.; Pett-Ridge, J.; Kleber, M.; Nico, P.S. Nano-scale investigation of the association of microbial nitrogen residues with iron (hydr) oxides in a forest soil O-horizon. Geochim. Cosmochim. Acta 2012, 95, 213–226. [Google Scholar] [CrossRef]
- Chassé, A.W.; Ohno, T.; Higgins, S.R.; Amirbahman, A.; Yildirim, N.; Parr, T.B. Chemical force spectroscopy evidence supporting the layer-by-layer model of organic matter binding to iron (oxy)hydroxide mineral surfaces. Environ. Sci. Technol. 2015, 49, 9733–9741. [Google Scholar] [CrossRef] [PubMed]
- Swensen, T.L.; Bowen, B.P.; Nico, P.S.; Northen, T.R. Competitive sorption of microbial metabolites on an iron oxide mineral. Soil Biol. Biochem. 2015, 90, 34–41. [Google Scholar] [CrossRef]
- Chassé, A.W.; Ohno, T. Higher molecular mass organic matter molecules compete with orthophosphate for adsorption to iron (oxy)hydroxide. Environ. Sci. Technol. 2016, 50, 7461–7469. [Google Scholar] [CrossRef] [PubMed]
- Ohno, T.; Heckman, K.A.; Plante, A.F.; Fernandez, I.J.; Parr, T.B. 14C Mean Residence Time and Its Relationship with Thermal Stability and Molecular Composition of Soil Organic Matter: A Case Study of Deciduous and Coniferous Forest Types. Geoderma 2017, 308, 1–8. [Google Scholar] [CrossRef]
- Omoike, A.; Chorover, J.; Kwon, K.; Kubicki, J. Adhesion of bacterial exopolymers to alpha-FeOOH: inner-sphere complexation of phosphodiester groups. Langmuir 2004, 20, 11108–11114. [Google Scholar] [CrossRef] [PubMed]
- Torn, M.S.; Trumbore, S.E.; Chadwick, O.A.; Vitousek, P.M.; Hendricks, D.M. Mineral control of soil organic carbon storage and turnover. Nature 1997, 389, 170–173. [Google Scholar] [CrossRef]
- Bailey, V.L.; Bond-Lamberty, B.; DeAngelis, K.; Grandy, A.S.; Hawkes, C.V.; Heckman, K.; Lajtha, K.; Phillips, R.P.; Sulman, B.N.; Todd-Brown, K.E.O.; et al. Soil carbon cycling proxies: Understanding their critical role in predicting climate change feedbacks. Global Chang. Biol. 2017, 24, 895–905. [Google Scholar] [CrossRef] [PubMed]
- Kampf, N.; Scheinost, A.L.; Schulze, D.G. Oxide minerals. In Handbook of Soil Science; Sumner, M.E., Ed.; CRC Press: Boca Raton, FL, USA, 2000; pp. 125–168. [Google Scholar]
- Dixon, J.B.; Schulze, D.G. Soil Mineralogy with Environmental Applications; Soil Science Society of America, Inc.: Madison, WI, USA, 2002. [Google Scholar]
- Wagai, R.; Mayer, L.M. Sorptive stabilization of organic matter in soils by hydrous iron oxides. Geochim. Cosmochim. Acta 2006, 71, 25–35. [Google Scholar] [CrossRef]
- Eusterhues, K.; Neidhardt, J.; Hädrich, A.; Küsel, K.; Totsche, K.U. Biodegradation of ferrihydrite-associated organic matter. Biogeochemistry 2014, 119, 45–50. [Google Scholar] [CrossRef]
- Mikutta, R.; Lorenz, D.; Guggenberger, G.; Haumaier, L.; Freund, A. Properties and reactivity of Fe-organic matter associations formed by coprecipitation versus adsorption: Clues from arsenate batch adsorption. Geochim. Cosmochim. Acta 2014, 144, 258–276. [Google Scholar] [CrossRef]
- Mayes, M.A.; Heal, K.R.; Brandt, C.C.; Phillips, J.R.; Jardine, P.M. Relation between soil order and sorption of dissolved organic carbon in temperate subsoils. Soil Sci. Soc. Am. J. 2012, 76, 1027–1037. [Google Scholar] [CrossRef]
- Heckman, K.; Lawrence, C.R.; Harden, J.W. A sequential selective dissolution method to quantify storage and stability of organic carbon associated with Al and Fe hydroxide phases. Geoderma 2018, 312, 24–35. [Google Scholar] [CrossRef]
- Rasmussen, C.; Torn, M.S.; Southard, R.J. Mineral assemblage and aggregates control carbon dynamics in a California conifer forest. Soil Sci. Soc. Am. J. 2005, 69, 1711–1721. [Google Scholar] [CrossRef]
- De Ruiter, P.C.; Moore, J.C.; Zwart, K.B.; Bouwman, L.A.; Hassink, J.; Bloem, J.; de Vos, J.A.; Marinissen, J.C.Y.; Didden, W.A.M.; Lebbink, G.; et al. Simulation of nitrogen mineralization in the below-ground food webs of two winter wheat fields. J. Appl. Ecol. 1993, 30, 95–106. [Google Scholar] [CrossRef]
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).