Deciphering and Predicting Microscale Controls on Radon Production in Soils, Sediments and Rock


Abstract
:
1. Introduction
2. Materials and Methods
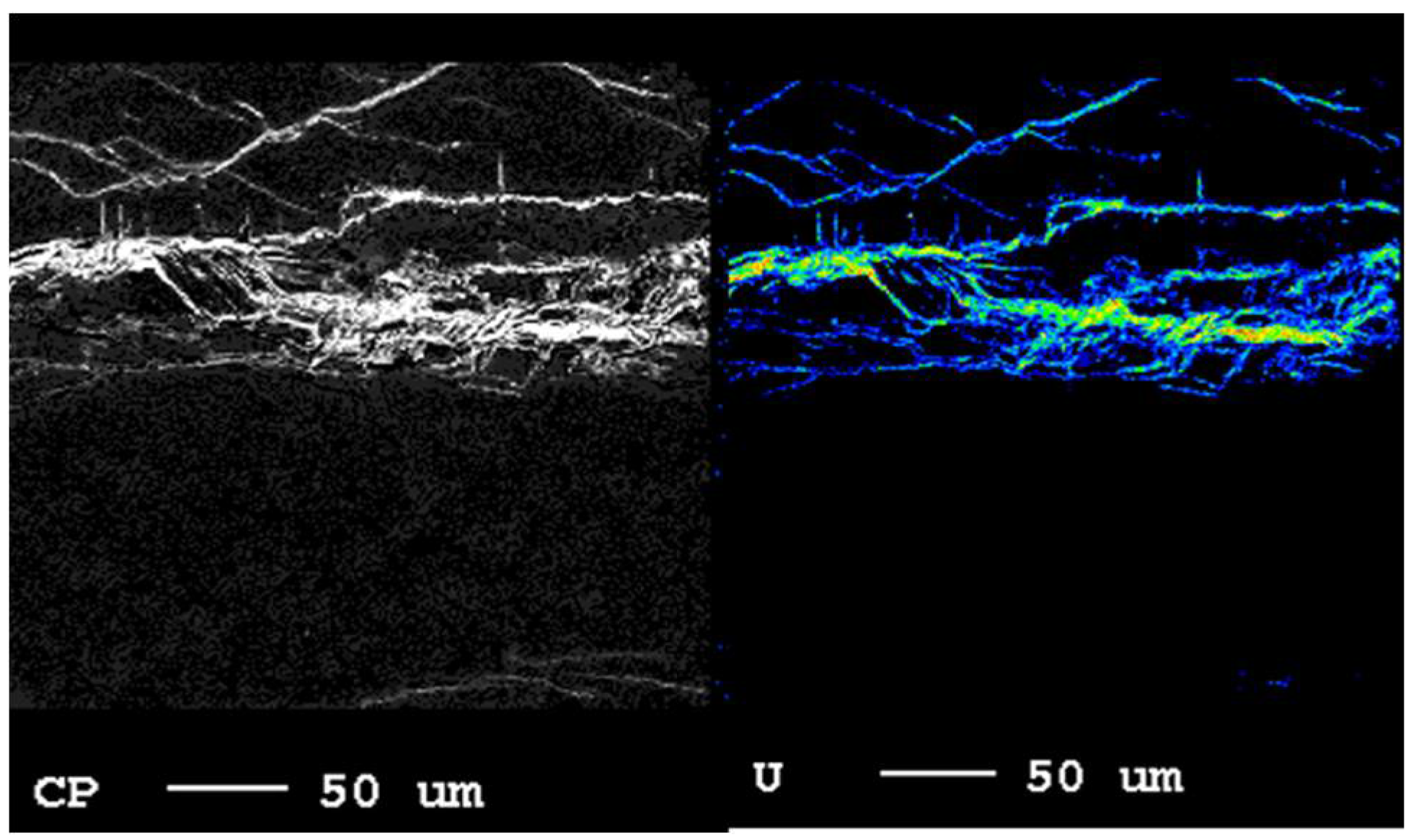
2.1. Rock Type and Characterization
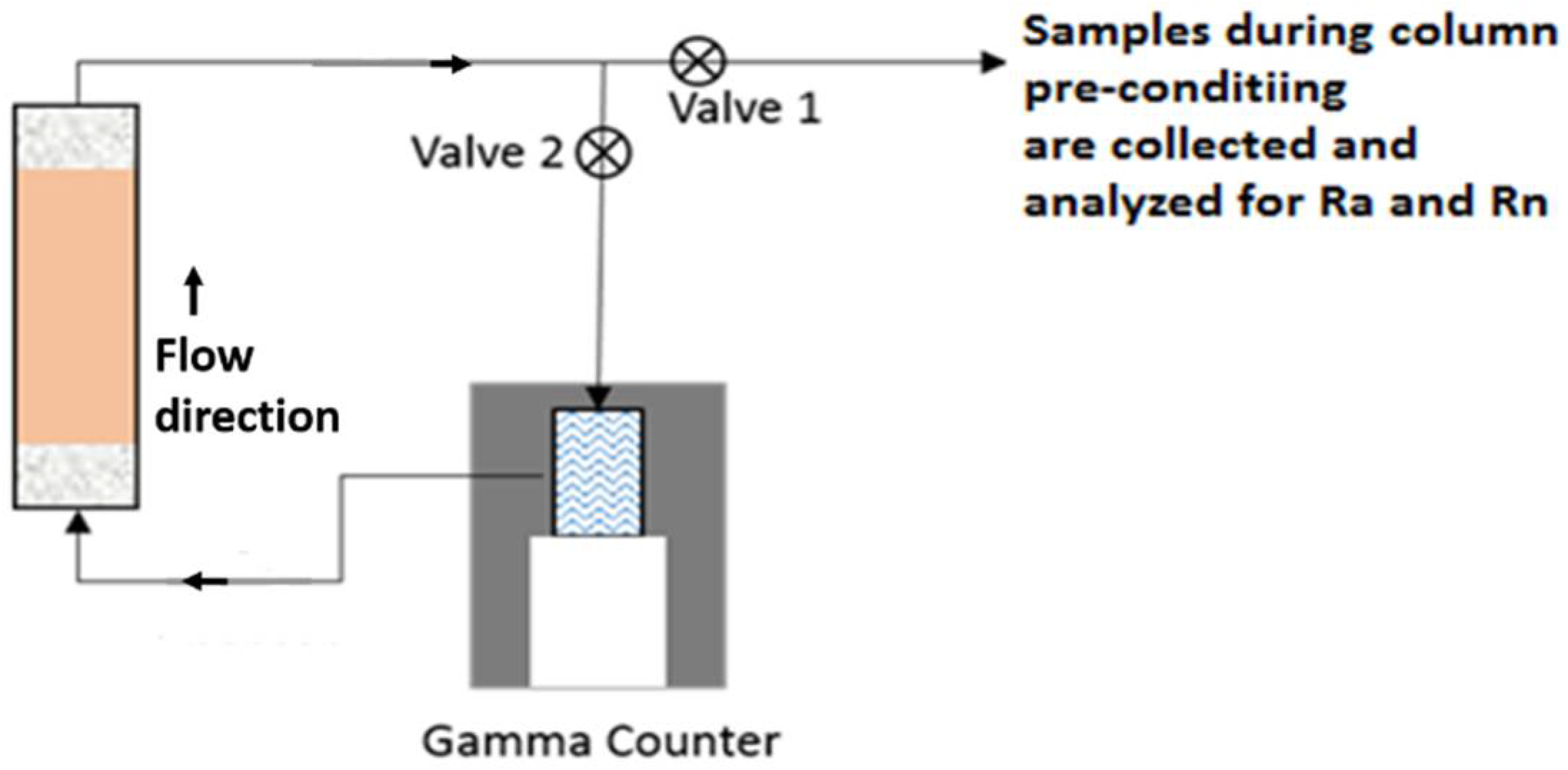
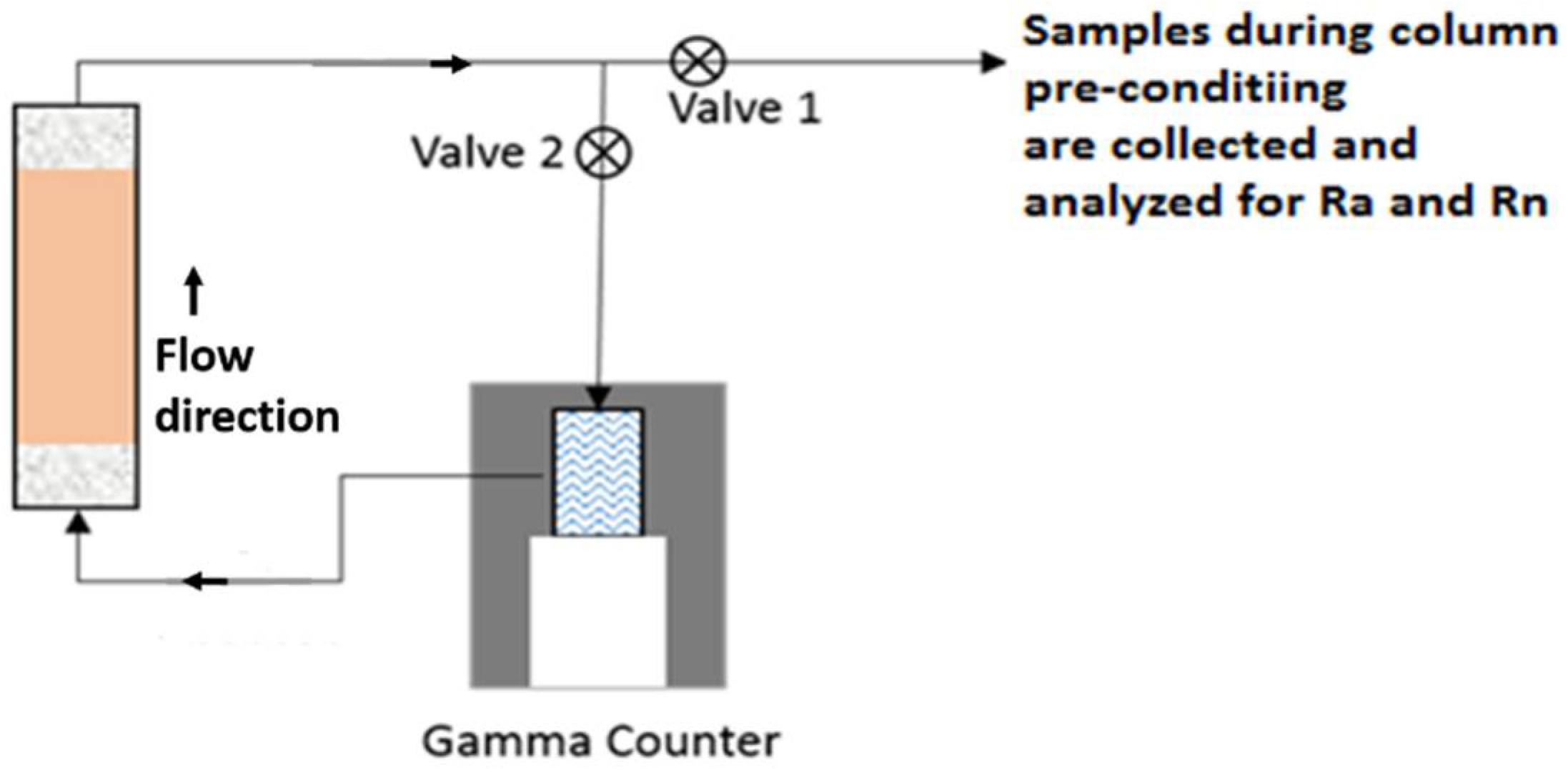
2.2. Column Setup
2.3. Radon Emanation Coefficient
2.4. Estimating Recoil Range in Solids
3. Results
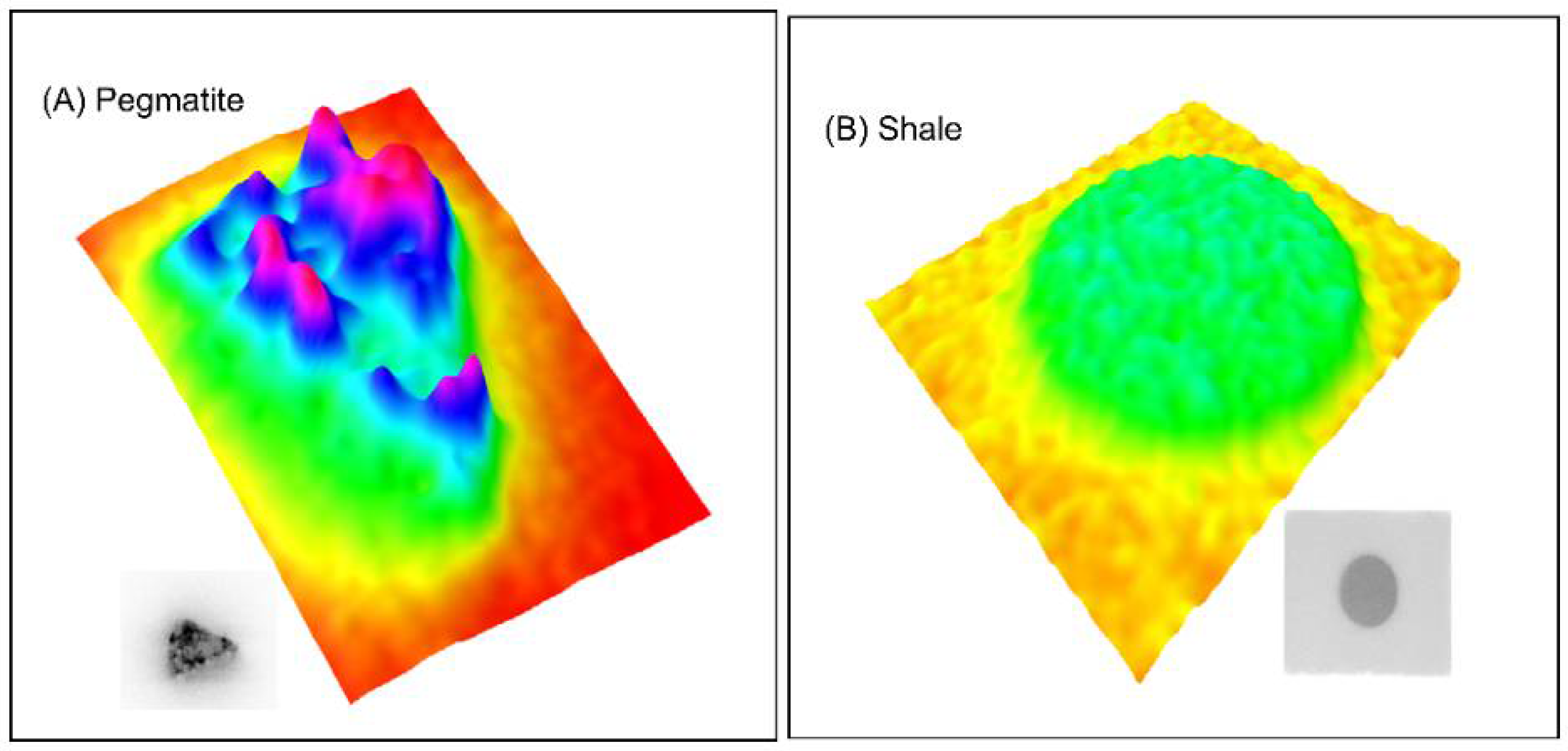
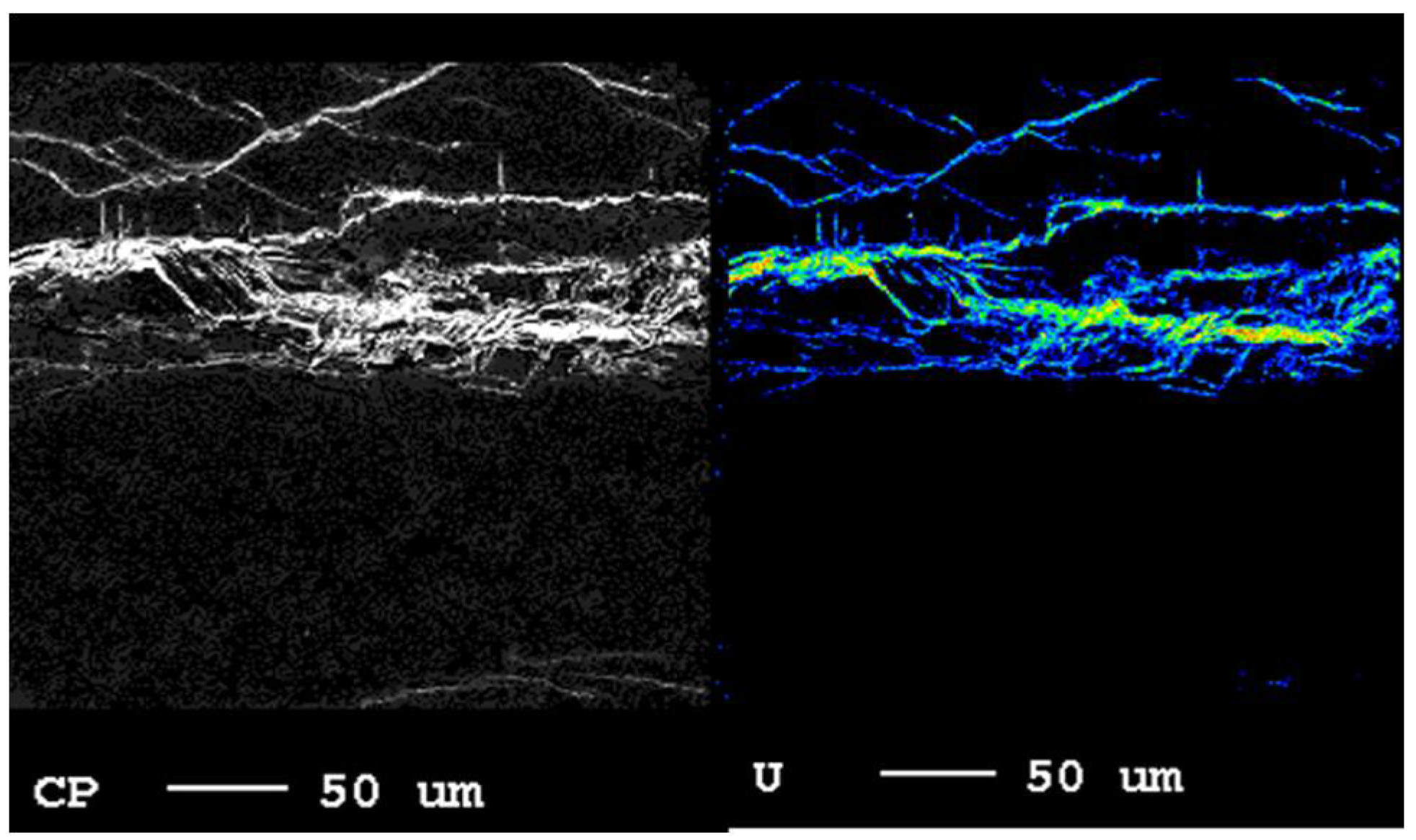
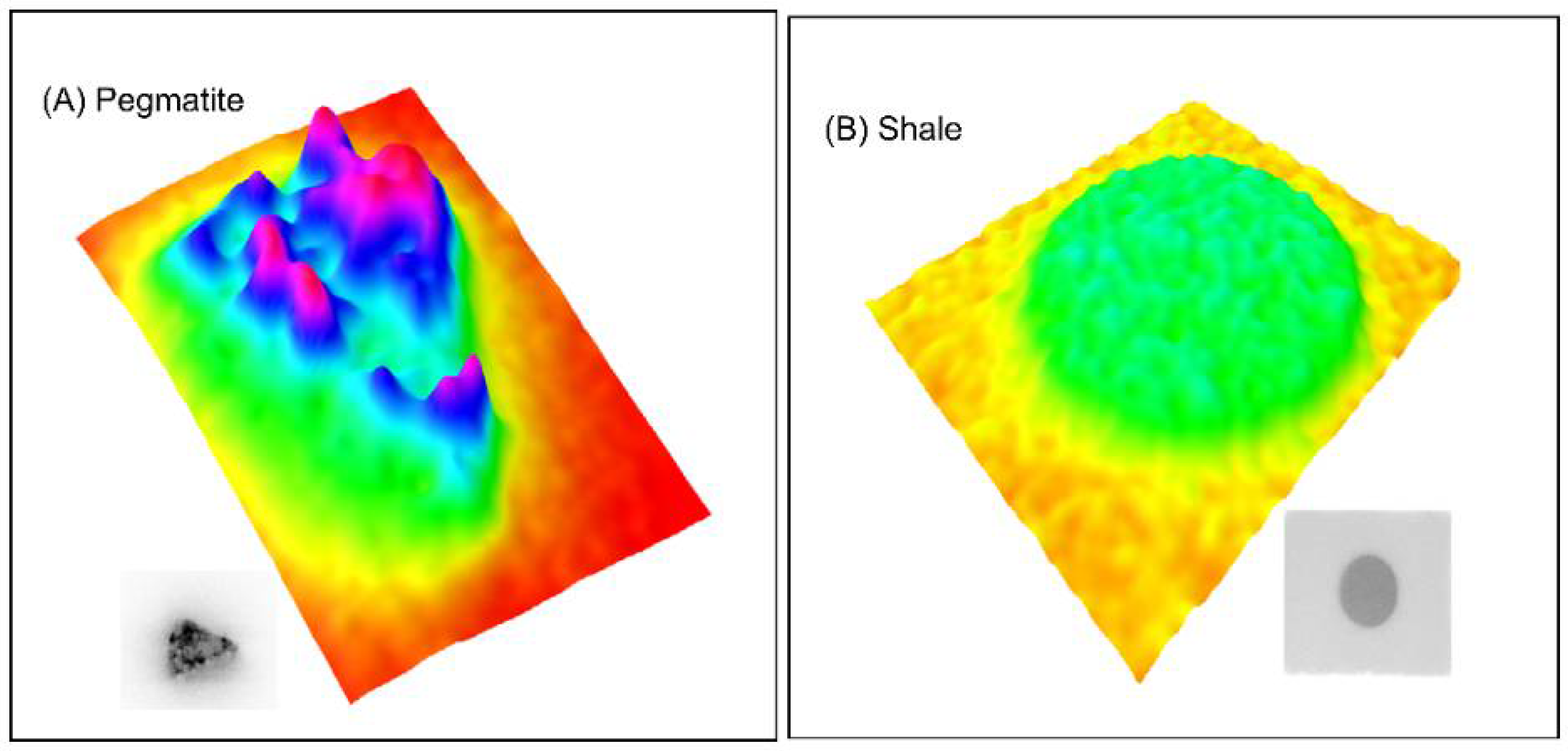
3.1. Material Characteristics
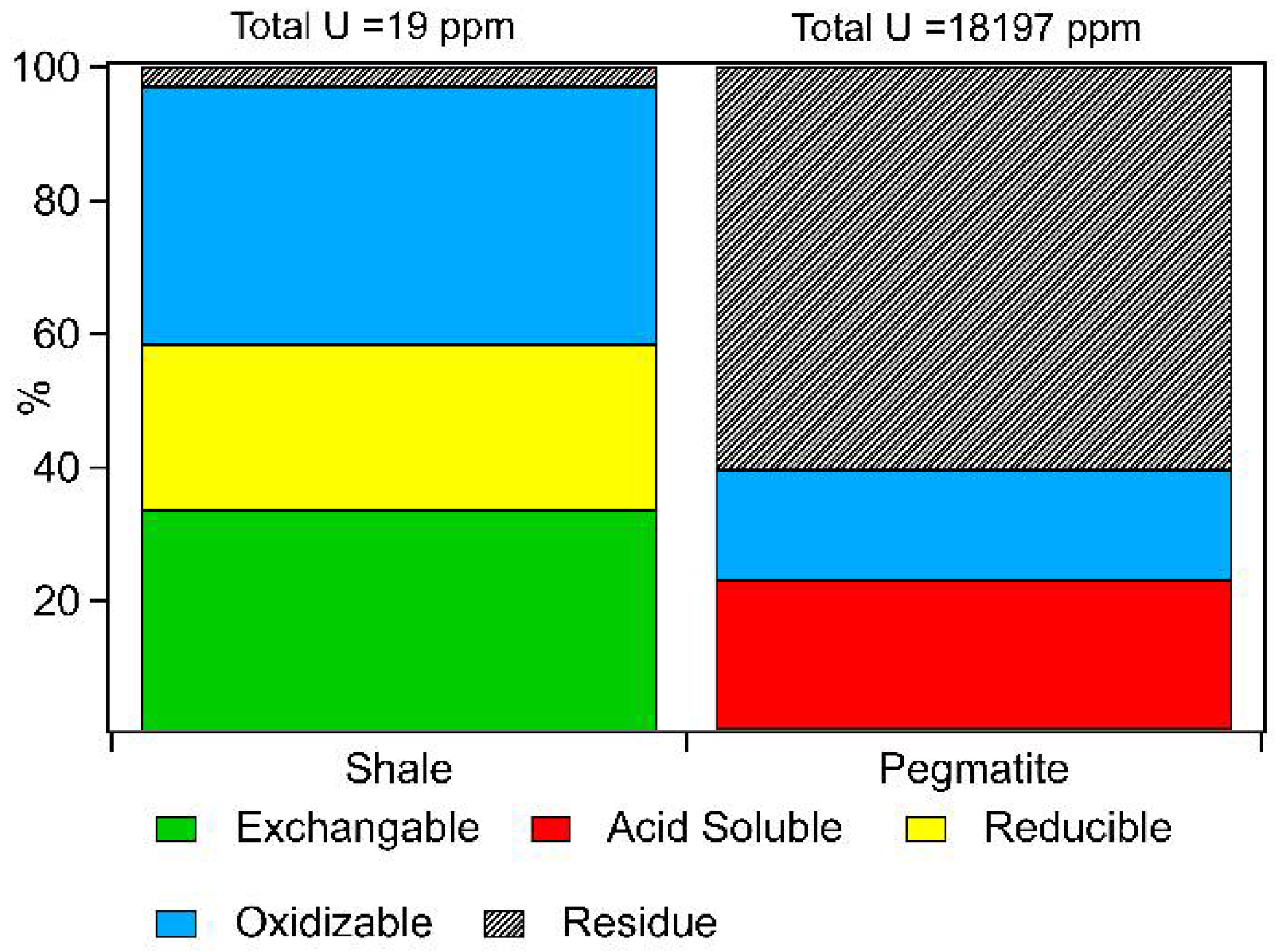
3.2. Sequential Extractions
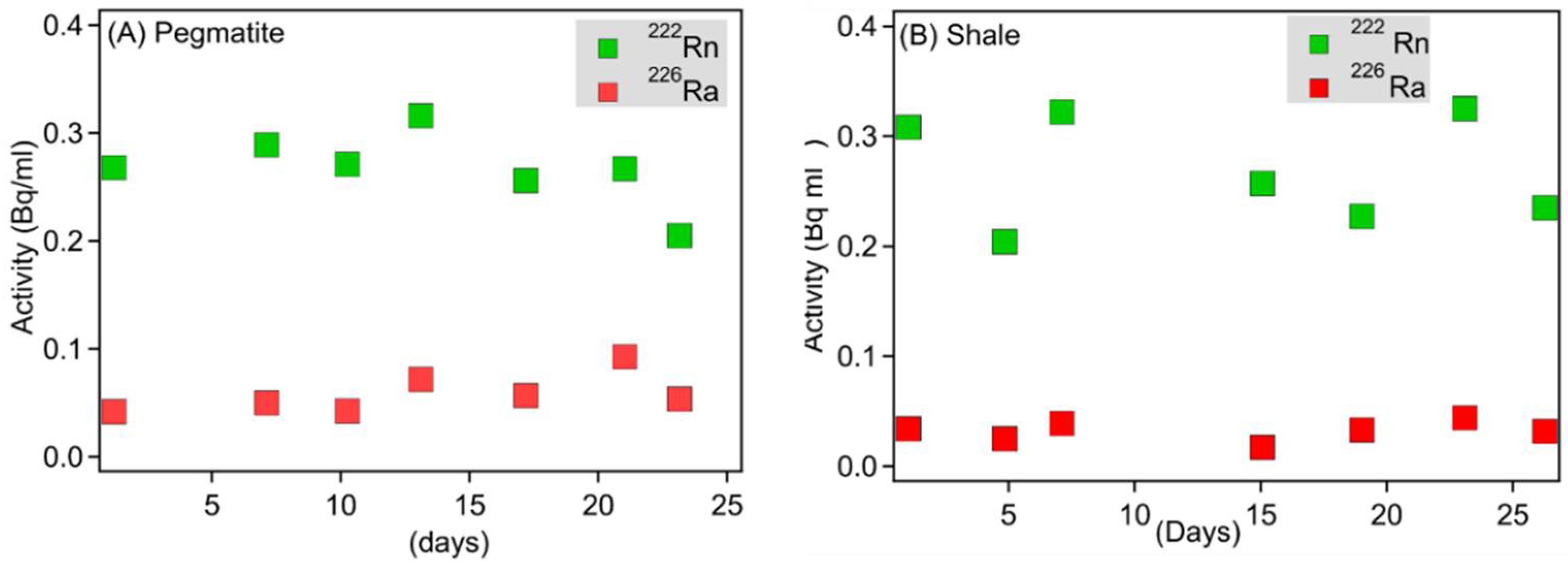
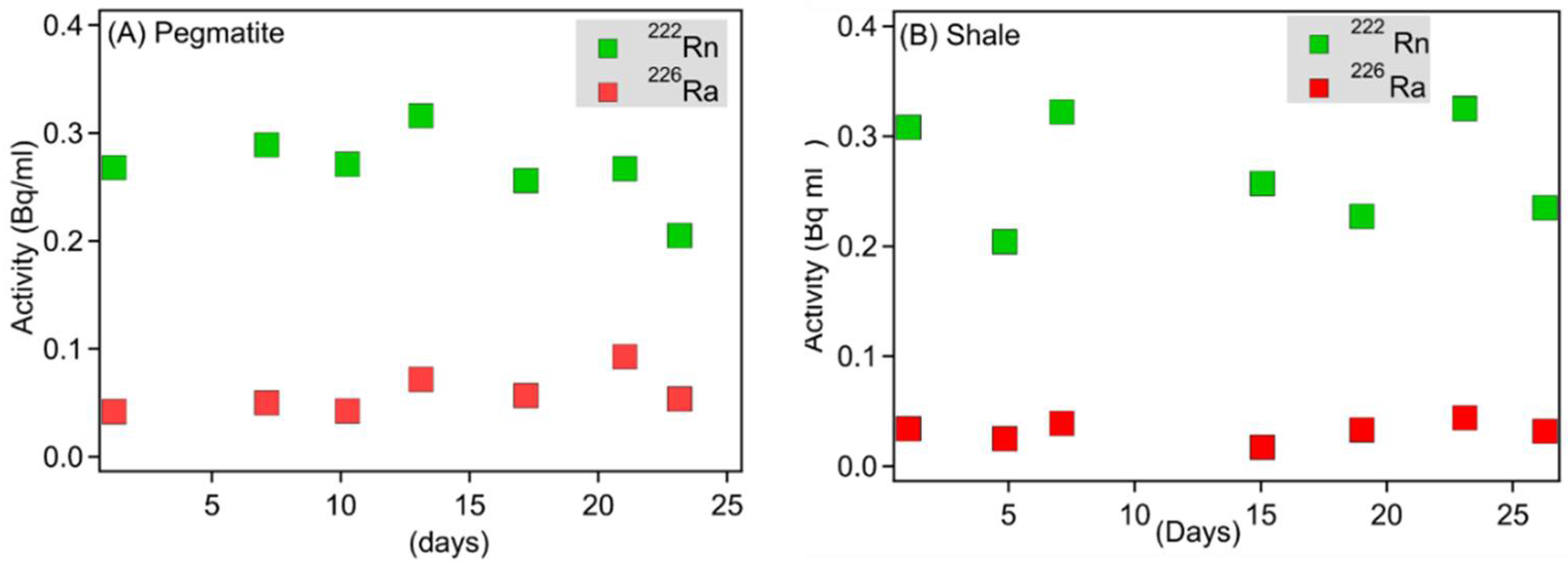
3.3. Porewater Radon Activities within Flow-Through Experiments and Calculated Radon Emanation Coefficients
4. Discussion
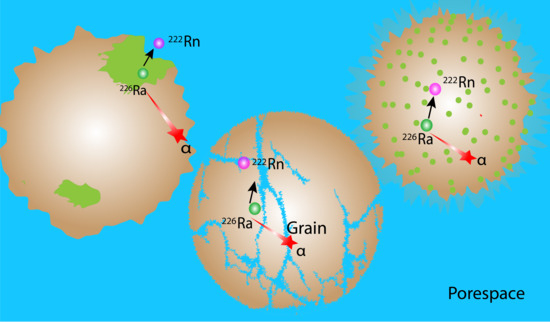
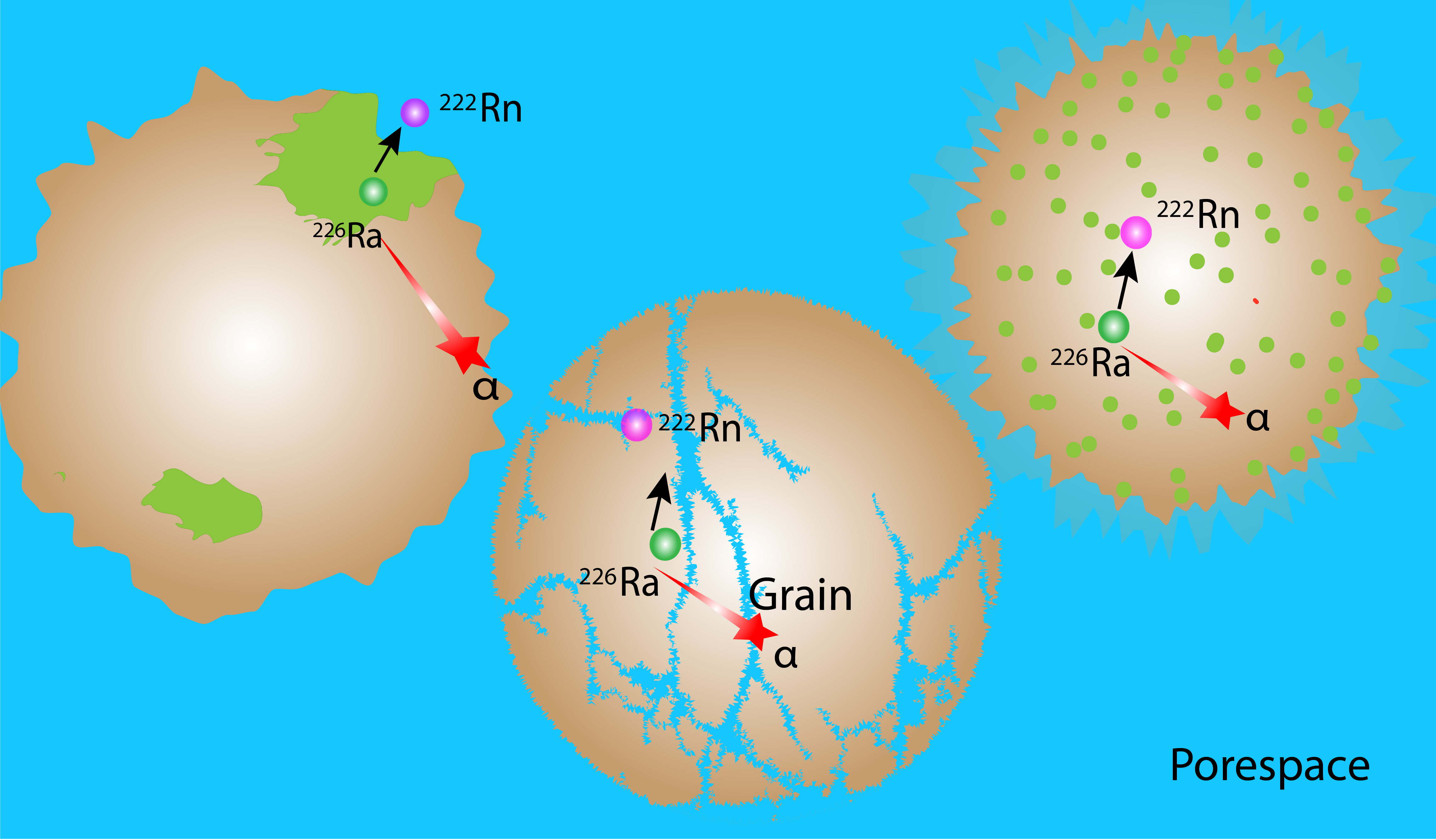
4.1. Chemical Heterogeneity and Its Impact on Radon Emanation
4.2. Role of Microstructure on Emanation
4.3. Predicting Radon Emanation
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Hassan, N.M.; Masahiro, H.; Ishikawa, T.; Sorimachi, A.; Sahoo, S.K.; Tokonami, S.; Masahiro, F. Radon Migration Process and Its Influence Factors. Jpn. J. Health Phys. 2009, 44, 218–231. [Google Scholar] [CrossRef]
- Nazaroff, W.W. Radon transport from soil to air. Rev. Geophys. 1992, 30, 137–160. [Google Scholar] [CrossRef]
- Baskaran, M. Radon: A Tracer for Geological, Geophysical and Geochemical Studies, 1st ed.; Springer: Cham, Switzerland, 2016; pp. 167–188. [Google Scholar]
- Hopke, P.K.; Borak, T.B.; Doull, J.; Cleaver, J.E.; Eckerman, K.F.; Gundersen, L.C.S.; Harley, N.H.; Hess, C.T.; Kinner, N.E.; Kopecky, K.J.; et al. Health Risks Due to Radon in Drinking Water. Environ. Sci. Technol. 2000, 34, 921–926. [Google Scholar] [CrossRef]
- USEPA. Health Risk of Radon. 2004. Available online: http://www.epa.gov/radon/health-risk-radon (accessed on 12 May 2016).
- Tanner, A.B. Radon migration in the ground: A supplementary review. In Natural Radiation Environment III: Proceedings of a Symposium Held at Houston, Texas, 23–28 April 1978, Volume 1 (DOE Symposium Series 51); U.S. Department of Energy: Washington, DC, USA, 1980; pp. 5–56. [Google Scholar]
- Bollhöfer, A.; Storm, J.; Martin, P.; Tims, S. Geographic variability in radon exhalation at a rehabilitated uranium mine in the Northern Territory, Australia. Environ. Monit. Assess. 2006, 114, 313–330. [Google Scholar] [CrossRef] [PubMed]
- Kiro, Y.; Weinstein, Y.; Starinsky, A.; Yechieli, Y. Application of radon and radium isotopes to groundwater flow dynamics: An example from the Dead Sea. Chem. Geol. 2015, 411, 155–171. [Google Scholar] [CrossRef]
- Genereux, D.P.; Hemond, F. Naturally Occurring Radon 222 as a Tracer for Streamflow Generation: Steady State Methodology and Field Example. Water Resour. Res. 1990, 26, 3065–3075. [Google Scholar]
- Vinson, D.S.; Tagma, T.; Bouchaou, L.; Dwyer, G.S.; Warner, N.R.; Vengosh, A. Occurrence and mobilization of radium in fresh to saline coastal groundwater inferred from geochemical and isotopic tracers (Sr, S, O, H, Ra, Rn). Appl. Geochem. 2013, 38, 161–175. [Google Scholar] [CrossRef]
- Liu, S.C.; Mcafee, J.R.; Cicerone, R.J. Radon 222 and Tropospheric Vertical Transport. J. Geophys. Res. 1984, 89, 7291–7297. [Google Scholar] [CrossRef]
- Kritz, M.A.; Le Roulley, J.-C.; Danielsen, E.F. The China Clipper—Fast advective transport of radon-rich air from the Asian boundary layer to the upper troposphere near California. Tellus B 1990, 42, 46–61. [Google Scholar] [CrossRef]
- Fleischer, R.L. Theory of alpha recoil effects on radon release and isotopic disequilibrium. Geochim. Cosmochim. Acta 1983, 47, 779–784. [Google Scholar] [CrossRef]
- Dyck, W. Radon-222 emanations from a uranium deposit. Econ. Geol. 1968, 63, 288–291. [Google Scholar] [CrossRef]
- Fleischer, R.L. Alpha-recoil damage and solution effects in minerals: Uranium isotopic disequilibrium and radon release. Geochim. Cosmochim. Acta 1982, 46, 2191–2201. [Google Scholar] [CrossRef]
- Rama; Moore, W.S. Mechanism of transport of U-Th series radioisotopes from solids into ground water. Geochim. Cosmochim. Acta 1984, 48, 395–399. [Google Scholar] [CrossRef]
- Semkow, T.M. Recoil-emanation theory applied to radon release from mineral grains. Geochim. Cosmochim. Acta 1989, 54, 425–440. [Google Scholar] [CrossRef]
- Hussain, N. Supply rates of natural U-TH series radionuclides from aquifer solids into groundwater. Geophys. Res. Lett. 1995, 22, 1521–1524. [Google Scholar] [CrossRef]
- Damkjaer, A.; Korsbech, U. Measurement of the emanation of radon-222 from Danish soils. Sci. Total Environ. 1985, 45, 343–350. [Google Scholar] [CrossRef]
- Megumi, K.; Mamuro, T. Emanation and Exhalation of Radon and Thoron Gases from Soil Particles. J. Geophys. Res. 1974, 79, 3–6. [Google Scholar] [CrossRef]
- Sakoda, A.; Ishimori, Y.; Yamaoka, K. A comprehensive review of radon emanation measurements for mineral, rock, soil, mill tailing and fly ash. Appl. Radiat. Isot. 2011, 69, 1422–1435. [Google Scholar] [CrossRef] [PubMed]
- Breitner, D.; Arvela, H.; Hellmuth, K.-H.; Renvall, T. Effect of moisture content on emanation at different grain size fractions—A pilot study on granitic esker sand sample. J. Environ. Qual. 2010, 101, 1002–1006. [Google Scholar] [CrossRef] [PubMed]
- Sun, H.; Furbish, D.J. Moisture content effect on radon emanation in porous media. J. Contam. Hydrol. 1995, 18, 239–255. [Google Scholar] [CrossRef]
- Stajic, J.M.; Nikezic, D. Theoretical calculation of radon emanation fraction. Nucl. Instrum. Methods Phys. Res. Sect. B 2014, 336, 19–25. [Google Scholar] [CrossRef]
- Arvela, H.; Holmgren, O.; Hänninen, P. Effect of soil moisture on seasonal variation in indoor radon concentration: Modelling and measurements in 326 Finnish houses. Radiat. Protect. Dosim. 2016, 168, 277–290. [Google Scholar] [CrossRef] [PubMed]
- Hellmuth, K.-H.; Siitari-Kauppi, M.; Arvela, H.; Lindberg, A.; Fonteneau, L.; Sardini, P. Radon emanation from fresh, altered and disturbed granitic rock characterized by 14 C-PMMA impregnation and autoradiography. Appl. Radiat. Isot. 2017, 127, 195–208. [Google Scholar] [CrossRef] [PubMed]
- Eakin, M.; Brownlee, S.J.; Baskaran, M.; Barbero, L. Mechanisms of radon loss from zircon; microstructural controls on emanation and diffusion. Geochim. Cosmochim. Acta 2016, 184, 212–226. [Google Scholar] [CrossRef]
- Morawska, L.; Phillips, C.R. Dependence of the radon emanation coefficient on radium distribution and internal structure of the material. Geochim. Cosmochim. Acta 1993, 57, 1783–1797. [Google Scholar] [CrossRef]
- Sun, H.; Semkow, T.M. Mobilization of thorium, radium and radon radionuclides in ground water by successive alpha-recoils. J. Hydrol. 1998, 205, 126–136. [Google Scholar] [CrossRef]
- Luo, S.; Ku, T.L.; Roback, R.; Murrell, M.; McLing, T.L. In-situ radionuclide transport and preferential groundwater flows at INEEL (Idaho): Decay-series disequilibrium studies. Geochim. Cosmochim. Acta 2000, 64, 867–881. [Google Scholar] [CrossRef]
- Sakoda, A.; Nishiyama, Y.; Hanamoto, K.; Ishimori, Y.; Yamamoto, Y.; Kataoka, T.; Kawabe, A.; Yamaoka, K. Differences of natural radioactivity and radon emanation fraction among constituent minerals of rock or soil. Appl. Radiat. Isot. 2010, 68, 1180–1184. [Google Scholar] [CrossRef] [PubMed]
- Taylor, P.; Sasaki, T.; Gunji, Y.; Okuda, T. Mathematical Modeling of Radon Emanation. J. Nucl. Sci. Technol. 2004, 41, 142–151. [Google Scholar]
- Haquin, G.; Yungrais, Z.; Ilzycer, D.; Zafrir, H.; Weisbrod, N. Detailed effects of particle size and surface area on Rn-222 emanation of a phosphate rock. J. Environ. Radioact. 2017, 180, 77–81. [Google Scholar] [CrossRef] [PubMed]
- Bateman, H. The solution of a system of differential equations occurring in the theory of radio-active transformations. Proc. Camb. Philos. Soc. 1910, 15, 423–427. [Google Scholar]
- USEPA. Method 3052: Microwave Assisted Acid Digestion of Siliceous and Organically Based Matrices; US Environmental Protection Agency: Washington, DC, USA, 1996.
- Giles, J.R. Pond Radium-226 Concentrations and Corrections. 1998. Available online: https://inldigitallibrary.inl.gov/PRR/81164.pdf (accessed on 23 April 2018).
- Tessier, A.; Campbell, P.G.C.; Bisson, M. Sequential Extraction Procedure for the Speciation of Particulate Trace Metals. Anal. Chem. 1979, 51, 844–851. [Google Scholar] [CrossRef]
- Rauret, G.; López-Sánchez, J.F.; Sahuquillo, A.; Rubio, R.; Davidson, C.; Ure, A.; Quevauviller, P. Improvement of the BCR three step sequential extraction procedure prior to the certification of new sediment and soil reference materials. J. Environ. Monit. 1999, 1, 57–61. [Google Scholar] [CrossRef] [PubMed]
- Ziegler, J.F.; Ziegler, M.D.; Biersack, J.P. SRIM—The stopping and range of ions in matter (2010). Nucl. Instrum. Methods Phys. Res. Sect. B 2010, 268, 1818–1823. [Google Scholar] [CrossRef]
- Chalmers, G.R.; Bustin, R.M.; Power, I.M. Characterization of gas shale pore systems by porosimetry, pycnometry, surface area, and field emission scanning electron microscopy/transmission electron microscopy image analyses: Examples from the Barnett, Woodford, Haynesville, Marcellus, and Doig units. AAPG Bull. 2012, 96, 1099–1119. [Google Scholar]
- Chermak, J.A.; Schreiber, M.E. Mineralogy and trace element geochemistry of gas shales in the United States: Environmental implications. Int. J. Coal Geol. 2014, 126, 32–44. [Google Scholar] [CrossRef]
- Sardini, P.; Angileri, A.; Descostes, M.; Duval, S.; Oger, T.; Patrier, P.; Rividi, N.; Siitari-Kauppi, M.; Toubon, H.; Donnard, J. Quantitative autoradiography of alpha particle emission in geo-materials using the BeaverTM system. Nucl. Instrum. Methods Phys. Res. Sect. A 2016, 833, 15–22. [Google Scholar] [CrossRef]
- Degering, D.; Krüger, F.; Scheiber, J.; Wolfgramm, M.; Köhler, M. Radionuclide Release in Geothermal Aquifers—The Role of Alpha Recoil. In Proceedings of the 2015 World Geothermal Congress, Melbourne, Australia, 19–25 April 2015. [Google Scholar]
- Weber, W.J.; Ewing, R.C.; Catlow, C.R.A.; de la Rubia, T.D.; Hobbs, L.W.; Kinoshita, C.; Matzke, H.; Motta, A.T.; Nastasi, M.; Salje, E.K.H.; et al. Radiation effects in crystalline ceramics for the immobilization of high-level nuclear waste and plutonium. J. Mater. Res. 1998, 13, 1434–1484. [Google Scholar] [CrossRef]
- Bloch, S.; Key, R.M. Modes of Formation of Anomalously High Radioactivity in Oil-Field Brines: Geologic Notes. AAPG Bull. 1953, 65, 154–159. [Google Scholar]
- Bower, W.R.; Pearce, C.I.; Droop, G.T.R.; Mosselmans, J.F.W.; Geraki, K.; Pattrick, R.A.D. Radiation damage from long-term alpha particle bombardment of silicates—A microfocus XRD and Fe K-edge XANES study. Mineral. Mag. 2015, 79, 1455–1466. [Google Scholar] [CrossRef]
- Lumpkin, G.R.; Ewing, R.C. Alpha-decay damage in minerals of the pyrochlore group. Phys. Chem. Miner. 1988, 16, 2–20. [Google Scholar] [CrossRef]
- Ames, L.L.; McGarrah, J.E.; Walker, B.A.; Salter, P.F. Uranium and radium sorption on amorphous ferric oxyhydroxide. Chem. Geol. 1983, 40, 135–148. [Google Scholar] [CrossRef]
- Gascoyne, M. The Environmental Behaviour of Radium; IAEA: Vienna, Austria, 1990. [Google Scholar]
- Sajih, M.; Bryan, N.D.; Livens, F.R.; Vaughan, D.J.; Descostes, M.; Phrommavanh, V.; Nos, J.; Morris, K. Adsorption of radium and barium on goethite and ferrihydrite: A kinetic and surface complexation modelling study. Geochim. Cosmochim. Acta 2014, 146, 150–163. [Google Scholar] [CrossRef]
- Porcelli, D.; Swarzenski, P.W. The Behavior of U- and Th-series Nuclides in Groundwater. Rev. Miner. Geochem. 2003, 52, 317–361. [Google Scholar] [CrossRef]
- Reynolds, B.C.; Wasserburg, G.J.; Baskaran, M. The transport of U- and Th-series nuclides in sandy confined aquifers. Geochim. Cosmochim. Acta 2003, 67, 1955–1972. [Google Scholar] [CrossRef]
- Sturchio, N.C.; Bohlke, J.K.; Markun, F.J. Radium isotope geochemistry of thermal waters, Yellowstone National Park, Wyoming, USA. Geochim. Cosmochim. Acta 1993, 57, 1203–1214. [Google Scholar] [CrossRef]
- Chen, M.; Kocar, B. Radium Sorption to Iron (hydr)oxides, Pyrite, and Montmorillonite: Implications for Ra Mobility. Environ. Sci. Technol. 2018, 52, 4023–4030. [Google Scholar] [CrossRef] [PubMed]
- Tamamura, S.; Takada, T.; Tomita, J.; Nagao, S.; Fukushi, K.; Yamamoto, M. Salinity dependence of 226Ra adsorption on montmorillonite and kaolinite. J. Radioanal. Nucl. Chem. 2014, 299, 569–575. [Google Scholar] [CrossRef]
- Ames, L.L.; McGarrah, J.E.; Walker, B.A. Sorption of Trace Constitutents From Aqueous Solution onto Secondary Minerals. II Radium. Clays Clay Miner. 1983, 31, 335–342. [Google Scholar] [CrossRef]
- Tachi, Y.; Shibutani, T.; Sato, H.; Yui, M. Experimental and modeling studies on sorption and diffusion of radium in bentonite. J. Contam. Hydrol. 2001, 47, 171–186. [Google Scholar] [CrossRef]
- Ishimori, Y.; Lange, K.; Martin, P.; Mayya, Y.; Phaneuf, M. Measurement and Calculation of Radon Releases from NORM Residues; IAEA: Vienna, Austria, 2013. [Google Scholar]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Rn Emanation Co-Efficient () | Bulk Uranium Concentration | |
|---|---|---|
| Shale | 62 ± 10% | 19 ± 0.7 ppm |
| Pegmatite | 0.2 ± 0.03% | 18197 ± 560 ppm |
| Mineral | Surface Area (m2 g−1) | Bulk Density (g cm−3) | Recoil Range (nm) a | Emanation Coefficient (Predicted) b | Emanation Coefficient (Measured) c |
|---|---|---|---|---|---|
| Quart (low S.A.) | 0.04 | 2.65 | 38.4 | 0.001 | 0.046 |
| Quartz (high S.A.) | 2.00 | 2.65 | 38.4 | 0.05 | 0.40 |
| Microcline | 27.91 | 1.00 | 35.7 | 0.25 | 0.27 |
| Goethite (high S.A.) | 86.4 | 3.00 | 27.9 | 1.81 | 0.43 |
| Goethite (low S.A.) d | 20 | 3.00 | 27.9 | 0.42 | 0.43 |
| Thorite | 1.00 | 5.35 | 27.1 | 0.04 | - |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mehta, N.; Kocar, B.D. Deciphering and Predicting Microscale Controls on Radon Production in Soils, Sediments and Rock. Soil Syst. 2018, 2, 30. https://doi.org/10.3390/soilsystems2020030
Mehta N, Kocar BD. Deciphering and Predicting Microscale Controls on Radon Production in Soils, Sediments and Rock. Soil Systems. 2018; 2(2):30. https://doi.org/10.3390/soilsystems2020030
Chicago/Turabian StyleMehta, Neha, and Benjamin D. Kocar. 2018. "Deciphering and Predicting Microscale Controls on Radon Production in Soils, Sediments and Rock" Soil Systems 2, no. 2: 30. https://doi.org/10.3390/soilsystems2020030
APA StyleMehta, N., & Kocar, B. D. (2018). Deciphering and Predicting Microscale Controls on Radon Production in Soils, Sediments and Rock. Soil Systems, 2(2), 30. https://doi.org/10.3390/soilsystems2020030