A Comparison of the Solubility Products of Layered Me(II)–Al(III) Hydroxides Based on Sorption Studies with Ni(II), Zn(II), Co(II), Fe(II), and Mn(II)

Abstract
:1. Introduction
2. Materials and Methods
2.1. Mineral Substrate
2.2. Batch Sorption Experiments
2.3. XAS Analyses
2.4. Thermodynamic Calculations
3. Results and Discussion
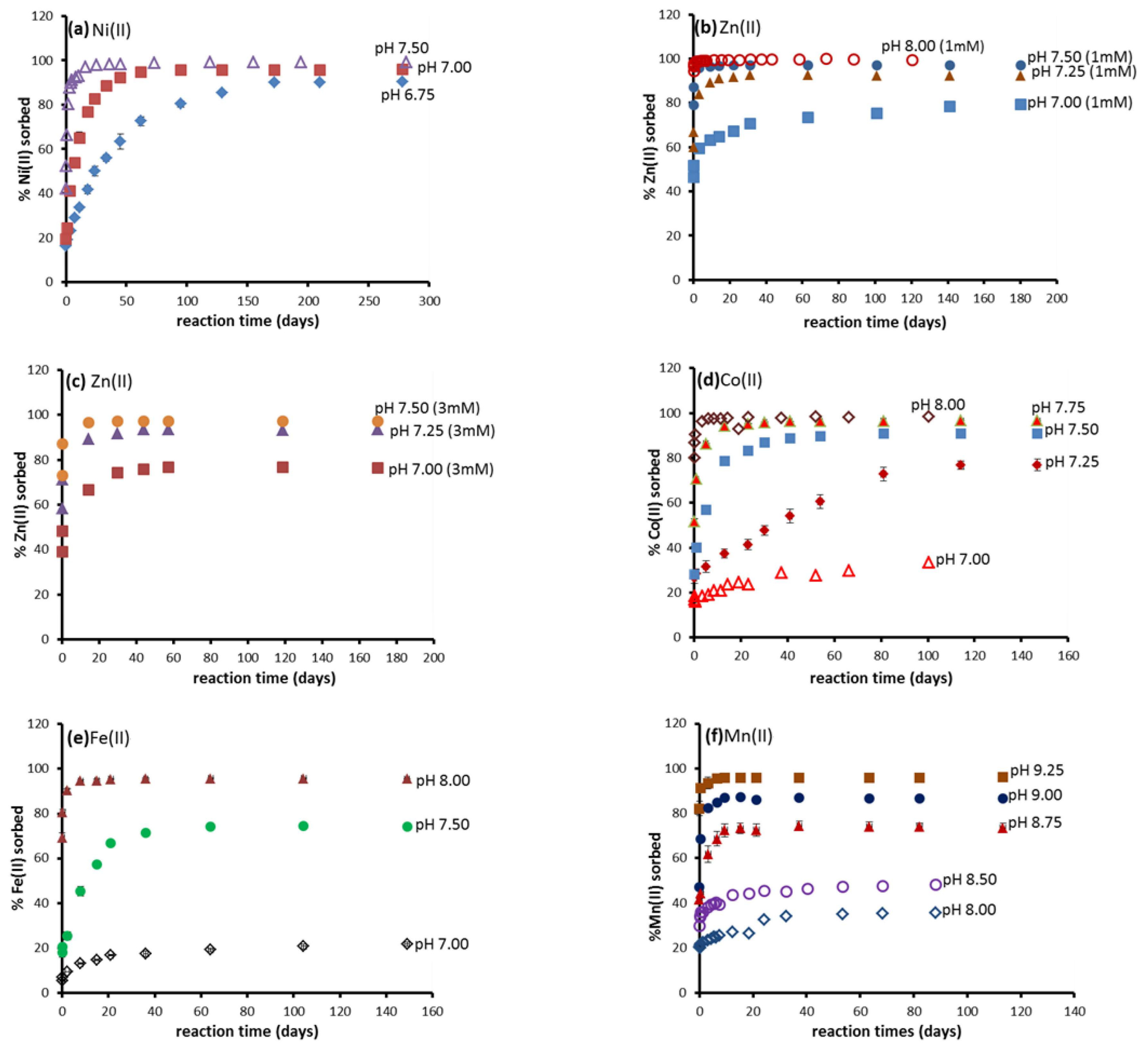
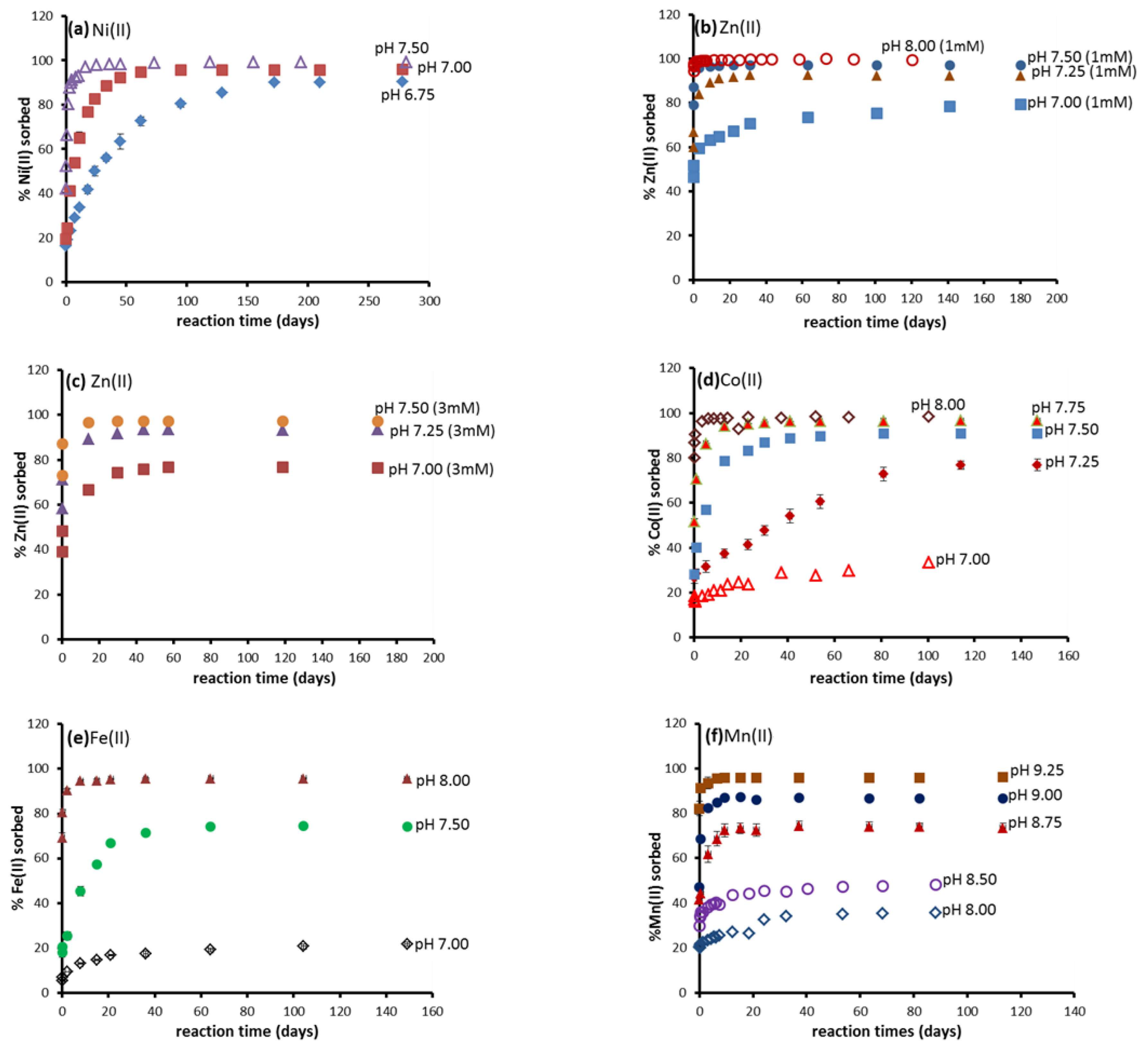
3.1. Batch Kinetic Results
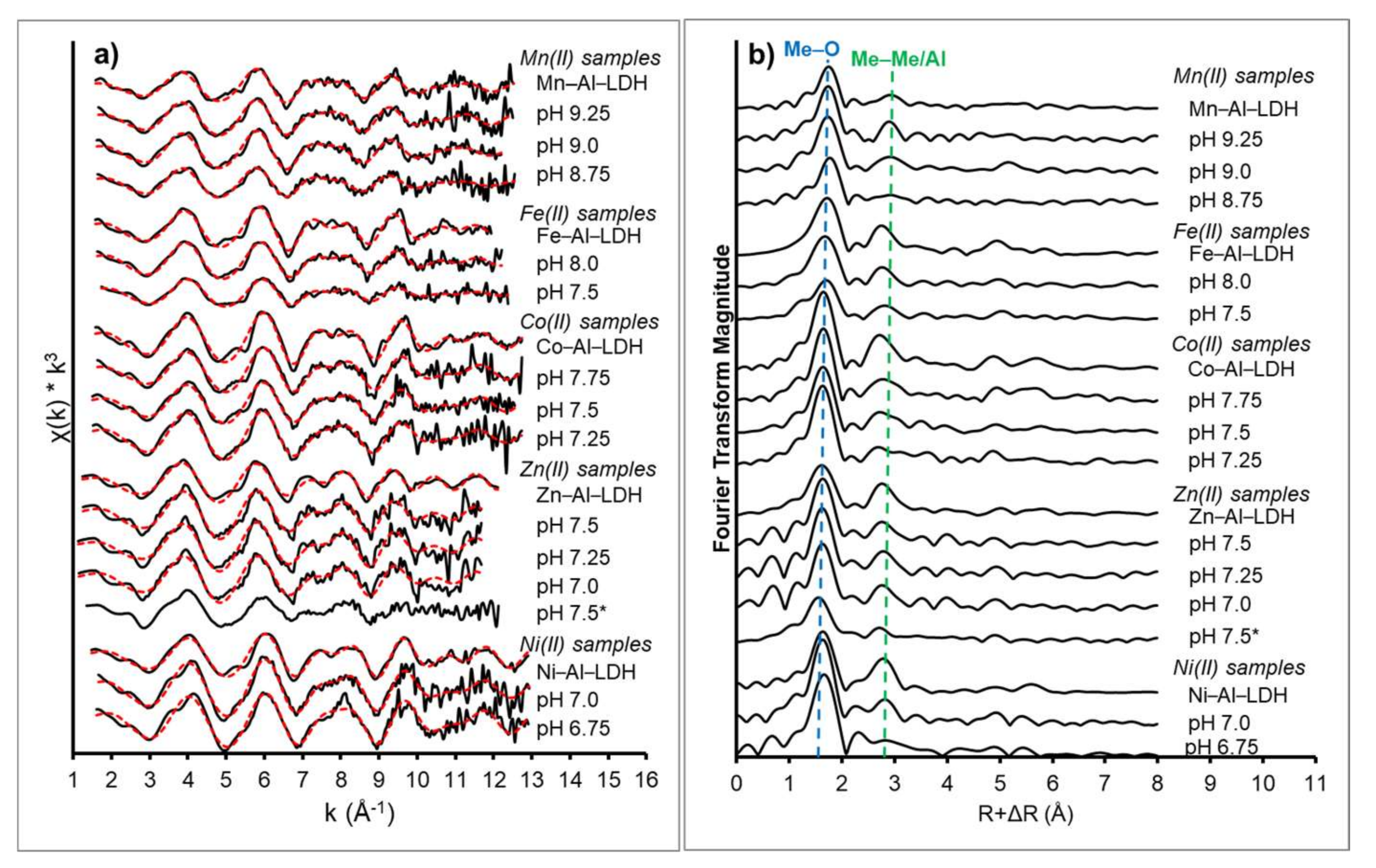
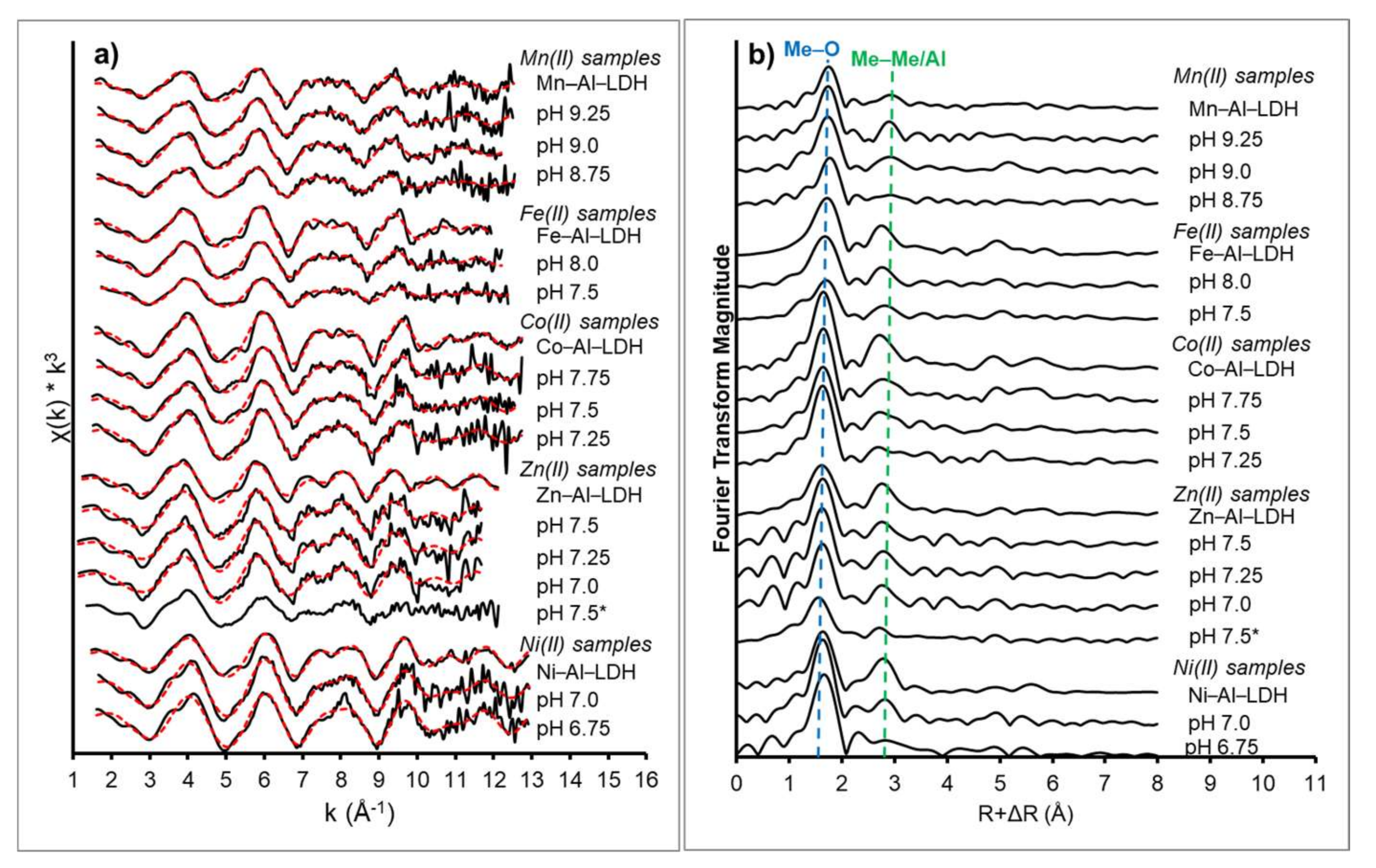
3.2. XAS Data
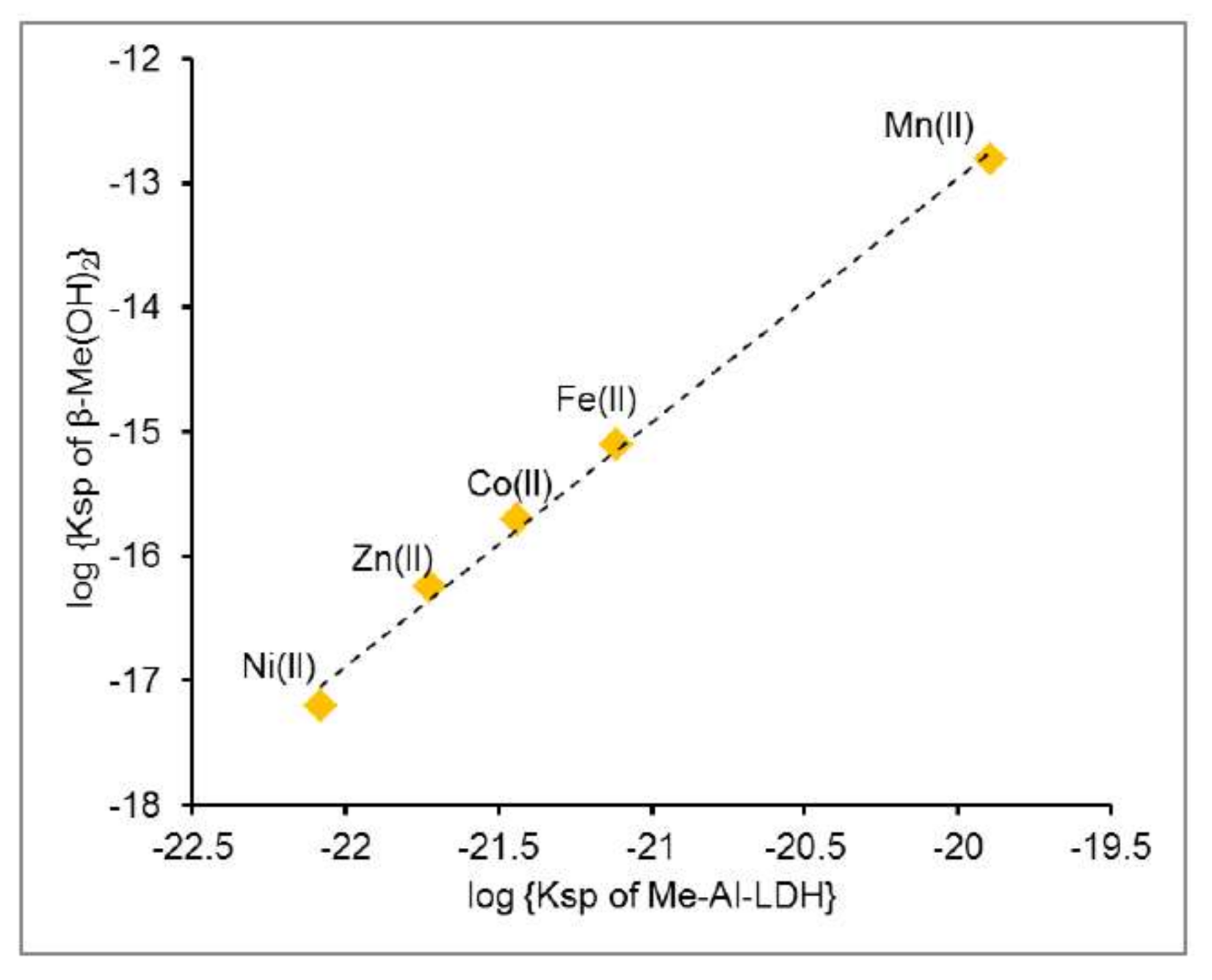
3.3. Ksp Estimates
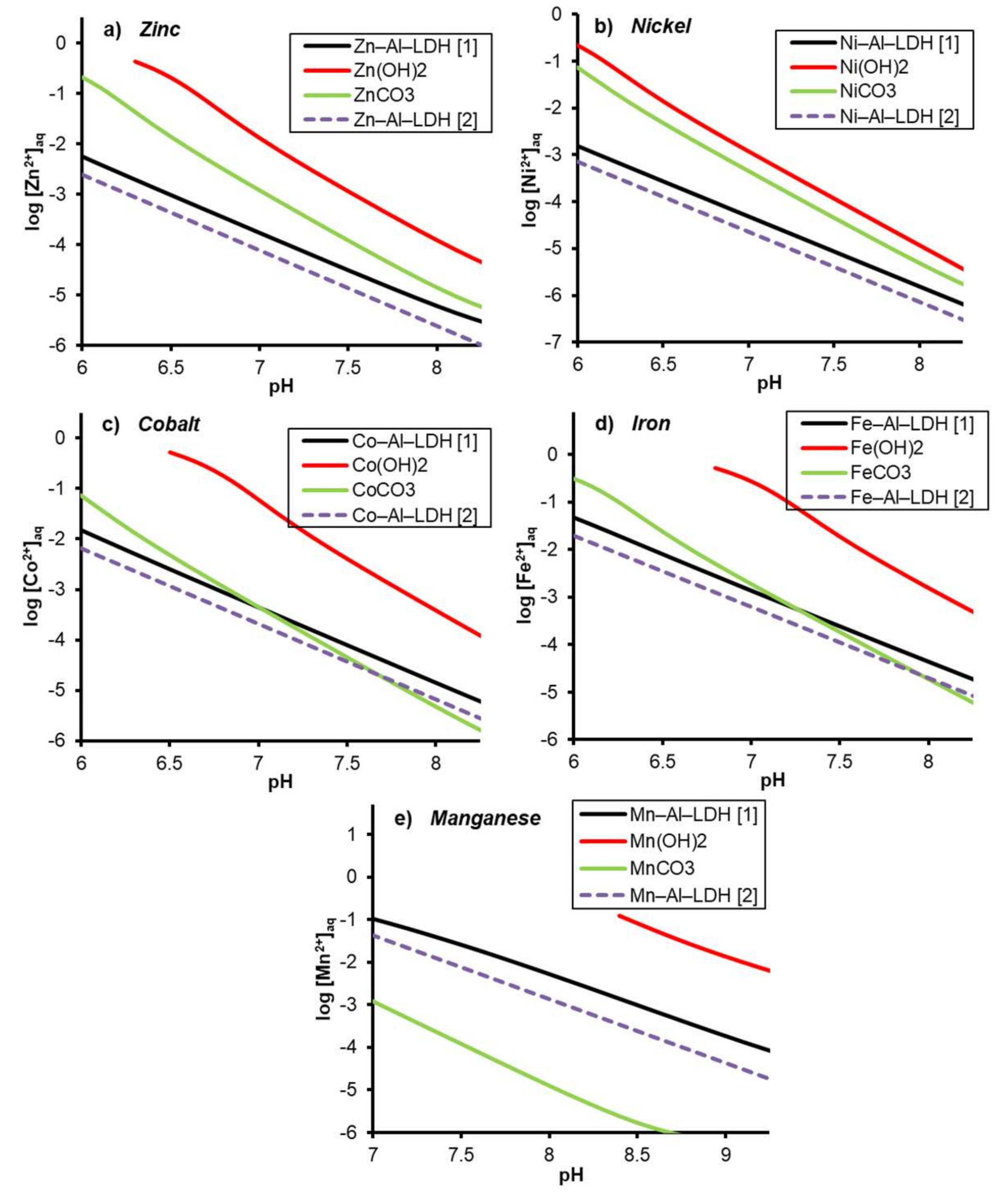
3.4. Me(II)–Al(III)-LDH Stability Relative to That of Other Me(II) Phases
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Sparks, D.L. Toxic metals in the environment: The role of surfaces. Elements 2005, 1, 193–197. [Google Scholar] [CrossRef]
- Sparks, D.L. Environmental Soil Chemistry, 2nd ed.; Academic Press: San Diego, CA, USA, 2003. [Google Scholar]
- McBride, M.B. Environmental Chemistry of Soils; Oxford University Press: New York, NY, USA, 1994. [Google Scholar]
- Scheidegger, A.M.; Sparks, D.L. Kinetics of the formation and the dissolution of nickel surface precipitates on pyrophyllite. Chem. Geol. 1996, 132, 157–164. [Google Scholar] [CrossRef]
- Scheidegger, A.M.; Lamble, G.M.; Sparks, D.L. Spectroscopic evidence for the formation of mixed-cation hydroxide phases upon metal sorption on clays and aluminum oxides. J. Colloid Interface Sci. 1996, 186, 118–128. [Google Scholar] [CrossRef]
- Scheidegger, A.M.; Strawn, D.G.; Lamble, G.M.; Sparks, D.L. The kinetics of mixed Ni-Al hydroxide formation on clay and aluminum oxide minerals: A time-resolved XAFS study. Geochim. Cosmochim. Acta 1998, 62, 2233–2245. [Google Scholar] [CrossRef]
- Towle, S.N.; Bargar, J.R.; Brown, G.E.; Parks, G.A. Surface precipitation of Co.(II)(aq) on Al2O3. J. Colloid Interface Sci. 1997, 187, 62–82. [Google Scholar] [CrossRef] [PubMed]
- Ford, R.G.; Sparks, D.L. The nature of Zn precipitates formed in the presence of pyrophyllite. Environ. Sci. Technol. 2000, 34, 2479–2483. [Google Scholar] [CrossRef]
- Ford, R.G.; Scheinost, A.C.; Scheckel, K.G.; Sparks, D.L. The link between clay mineral weathering and the stabilization of Ni surface precipitates. Environ. Sci. Technol. 1999, 33, 3140–3144. [Google Scholar] [CrossRef]
- Scheinost, A.C.; Ford, R.G.; Sparks, D.L. The role of Al in the formation of secondary Ni precipitates on pyrophyllite, gibbsite, talc, and amorphous silica: A DRS study. Geochim. Cosmochim. Acta 1993, 63, 3193–3203. [Google Scholar] [CrossRef]
- Scheinost, A.C.; Sparks, D.L. Formation of layered single- and double-metal hydroxide precipitates at the mineral/water interface: A multiple-scattering XAFS analysis. J. Colloid Interface Sci. 2000, 223, 167–178. [Google Scholar] [CrossRef] [PubMed]
- Thompson, H.A.; Parks, G.A.; Brown, G.E. Dynamic interactions of dissolution, surface adsorption, and precipitation in an aging cobalt(II)-clay-water system. Geochim. Cosmochim. Acta 1999, 63, 1767–1779. [Google Scholar] [CrossRef]
- Elzinga, E.J.; Sparks, D.L. Nickel sorption mechanisms in a pyrophyllite-montmorillonite mixture. J. Colloid Interface Sci. 1999, 213, 506–512. [Google Scholar] [CrossRef] [PubMed]
- Elzinga, E.J.; Sparks, D.L. Reaction condition effects on nickel sorption mechanisms in illite-water suspensions. Soil Sci. Soc. Am. J. 2001, 65, 94–101. [Google Scholar] [CrossRef]
- Scheckel, K.G.; Scheinost, A.C.; Ford, R.G.; Sparks, D.L. Stability of layered Ni hydroxide surface precipitates—A dissolution kinetics study. Geochim. Cosmochim. Acta 2000, 64, 2727–2735. [Google Scholar] [CrossRef]
- Scheckel, K.G.; Sparks, D.L. Kinetics of the formation and dissolution of Ni precipitates in a gibbsite/amorphous silica mixture. J. Colloid Interface Sci. 2000, 229, 222–229. [Google Scholar] [CrossRef] [PubMed]
- Boyle-Wight, E.J.; Katz, L.E.; Hayes, K.F. Macroscopic studies of the effects of selenate and selenite on cobalt sorption to γ-Al2O3. Environ. Sci. Technol. 2002, 36, 1212–1218. [Google Scholar] [CrossRef] [PubMed]
- Boyle-Wight, E.J.; Katz, L.E.; Hayes, K.F. Spectroscopic studies of the effects of selenate and selenite on cobalt sorption to γ-Al2O3. Environ. Sci. Technol. 2002, 36, 1219–1225. [Google Scholar] [CrossRef] [PubMed]
- Charlet, L.; Manceau, A. Evidence for the neoformation of clays upon sorption of Co.(II) and Ni(II) on silicates. Geochim. Cosmochim. Acta 1994, 58, 2577–2582. [Google Scholar] [CrossRef]
- Trainor, T.P.; Brown, G.E.; Parks, G.A. Adsorption and precipitation of aqueous Zn(II) on alumina powders. J. Colloid Interface Sci. 2000, 231, 359–372. [Google Scholar] [CrossRef] [PubMed]
- Roberts, D.R.; Ford, R.G.; Sparks, D.L. Kinetics and mechanisms of Zn complexation on metal oxides using EXAFS spectroscopy. J. Colloid Interface Sci. 2003, 263, 364–376. [Google Scholar] [CrossRef]
- Li, W.; Livi, K.J.T.; Xu, W.; Siebecker, M.G.; Wang, Y.; Phillips, B.L.; Spaks, D.L. Formation of crystalline Zn-Al layered double hydroxide precipitates on γ-alumina: The role of mineral dissolution. Environ. Sci. Technol. 2012, 46, 11670–11677. [Google Scholar] [CrossRef] [PubMed]
- Eick, M.J.; Fendorf, S.E. Reaction sequence of nickel(II) with kaolinite: Mineral dissolution and surface complexation and precipitation. Soil Sci. Soc. Am. J. 1998, 62, 1257–1267. [Google Scholar] [CrossRef]
- Siebecker, M.; Li, W.; Khalid, S.; Sparks, D.L. Real time Q-EXAFS spectroscopy measures rapid precipitate formation at the mineral-water interface. Nat. Commun. 2014, 5, 5003. [Google Scholar] [CrossRef] [PubMed]
- Siebecker, M.; Li, W.; Sparks, D.L. The important role of layered double hydroxides in soil chemical processes and remediation: What we have learned over the past 20 years. In Advances in Agronomy; Sparks, D.L., Ed.; Academic Press: New York, NY, USA, 2007; Volume 147, p. 5003. [Google Scholar] [CrossRef]
- Elzinga, E.J. Formation of layered Fe(II)-Al(III)-hydroxides during reaction of Fe(II) with aluminum oxide. Environ. Sci. Technol. 2012, 46, 4894–4901. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.; Elzinga, E.J. Formation of layered Fe(II)-hydroxides during Fe(II) sorption onto clay and metal-oxide substrates. Environ. Sci. Technol. 2014, 48, 4937–4945. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.; Elzinga, E.J. Macroscopic and spectroscopic assessment of the co-sorption of Fe(II) with As(III) and As(V) on Al-oxide. Environ. Sci. Technol. 2015, 49, 13369–13377. [Google Scholar] [CrossRef] [PubMed]
- Starcher, A.N.; Elzinga, E.J.; Sparks, D.L. Formation of a mixed Fe(II)-Zn-Al layered hydroxide: Effects of Zn co-sorption on Fe(II) layered hydroxide formation and kinetics. Chem. Geol. 2017, 464, 46–56. [Google Scholar] [CrossRef]
- Starcher, A.N.; Li, W.; Kukkadapu, R.K.; Elzinga, E.J.; Sparks, D.L. Fe(II) sorption on pyrophyllite: Effect of structural Fe(III) (impurity) in pyrophyllite on nature of layered double hydroxide (LDH) secondary mineral formation. Chem. Geol. 2016, 439, 152–160. [Google Scholar] [CrossRef]
- Elzinga, E.J.; Zhu, Y. Co-sorption of aqueous Fe(II) and Mn(II) in anoxic aluminum-oxide suspensions. In Proceedings of the 249th ACS National Meeting & Exposition, Denver, CO, USA, 22–24 March 2015. Abstract GEOC-114. [Google Scholar]
- Taylor, R.M. The rapid formation of crystalline double hydroxy salts and other compounds by controlled hydrolysis. Clay Miner. 1984, 19, 591–603. [Google Scholar] [CrossRef]
- Evans, D.G.; Slade, R.C.T. Structural aspects of layered double hydroxides. Struct. Bond. 2006, 119, 1–87. [Google Scholar]
- Bravo-Suarez, J.J.; Paez-Mozo, E.; Oyama, S.T. Review of the synthesis of layered double hydroxides: A thermodynamic approach. Quim. Nova 2004, 27, 60–614. [Google Scholar] [CrossRef]
- Cavani, F.; Trifiro, F.; Vaccari, A. Hydrotalcite type anionic clays: Preparation, properties and applications. Catal. Today 1991, 11, 173–301. [Google Scholar] [CrossRef]
- Roberts, D.R.; Scheidegger, A.M.; Sparks, D.L. Kinetics of mixed Ni-Al precipitate formation on a soil clay fraction. Environ. Sci. Technol. 1999, 33, 3749–3754. [Google Scholar] [CrossRef]
- Juillot, F.; Morin, G.; Ildefonse, P.; Trainor, T.P.; Benedetti, M.; Galoisy, L.; Calas, G.; Brown, G.E. Occurrence of Zn/Al hydrotalcite in smelter-impacted soils from Northern France: Evidence from EXAFS spectroscopy and chemical extractions. Am. Mineral. 2003, 88, 509–526. [Google Scholar] [CrossRef]
- Voegelin, A.; Kretzschmar, R. Formation and dissolution of single and mixed Zn and Ni precipitates in soil: Evidence from column experiments and extended X-ray absorption fine structure spectroscopy. Environ. Sci. Technol. 2005, 39, 5311–5318. [Google Scholar] [CrossRef] [PubMed]
- Voegelin, A.; Pfister, S.; Scheinost, A.C.; Marcus, M.A.; Kretzschmar, R. Changes in zinc speciation in field soil after contamination with zinc oxide. Environ. Sci. Technol. 2005, 39, 6616–6623. [Google Scholar] [CrossRef] [PubMed]
- Peltier, E.; Allada, R.; Navrotsky, A.; Sparks, D.L. Nickel solubility and precipitation in soils: A thermodynamic study. Clays Clay Miner. 2006, 54, 153–164. [Google Scholar] [CrossRef]
- Peltier, E.; Van Der Lelie, D.; Sparks, D.L. Formation and stability of Ni-Al hydroxide phases in soils. Environ. Sci. Technol. 2010, 44, 302–308. [Google Scholar] [CrossRef] [PubMed]
- Jacquat, O.; Voegelin, A.; Villard, A.; Marcus, M.A.; Kretzschmar, R. Formation of Zn-rich phyllosilicate, Zn-layered double hydroxide and hydrozincite in contaminated calcareous soils. Geochim. Cosmochim. Acta 2008, 72, 5037–5054. [Google Scholar] [CrossRef]
- Nachtegaal, M.; Marcus, M.A.; Sonke, J.E.; Vangronsveld, J.; Livi, K.J.T.; Van Der Lelie, D.; Sparks, D.L. Effects of in situ remediation on the speciation and bioavailability of zinc in a smelter contaminated soil. Geochim. Cosmochim. Acta 2005, 69, 4649–4664. [Google Scholar] [CrossRef]
- McNear, D.H.; Chaney, R.L.; Sparks, D.L. The effects of soil type and chemical treatment on nickel speciation in refinery enriched soils: A multi-technique investigation. Geochim. Cosmochim. Acta 2007, 71, 2190–2208. [Google Scholar] [CrossRef]
- Shi, Z.; Peltier, E.; Sparks, D.L. Kinetics of Ni sorption in soils: Roles of soil organic matter and Ni precipitation. Environ. Sci. Technol. 2012, 46, 2212–2219. [Google Scholar] [CrossRef] [PubMed]
- Johnson, C.A.; Glasser, F.P. Hydrotalcite-like minerals (M2Al(OH)5(CO3)0.5.H2O, where M = Mg, Zn, Co., Ni) in the environment: Synthesis, characterization and thermodynamic stability. Clays Clay Miner. 2003, 51, 1–8. [Google Scholar] [CrossRef]
- Regelink, I.C.; Temminghoff, E.J.M. Ni adsorption and Ni-Al LDH precipitation in a sandy aquifer: An experimental and mechanistic modeling study. Environ. Pollut. 2011, 159, 716–721. [Google Scholar] [CrossRef] [PubMed]
- Boclair, J.W.; Braterman, P.S. Layered double hydroxides. 1. Relative stabilities of layered double hydroxides and their simple counterparts. Chem. Mater. 1999, 11, 298–302. [Google Scholar] [CrossRef] [PubMed]
- Allada, R.K.; Peltier, E.; Navrotsky, A.; Casey, W.H.; Johnson, C.A.; Berbeco, H.T.; Sparks, D.L. Calorimetric determination of the enthalpies of formation of hydrotalcite-like solids and their use in the geochemical modeling of metals in natural waters. Clays Clay Miner. 2006, 54, 409–417. [Google Scholar] [CrossRef]
- Allada, R.K.; Navrotsky, A.; Berbeco, H.T.; Casey, W.H. Thermochemistry and aqueous solubilities of hydrotalcite-like solids. Science 2002, 296, 721–723. [Google Scholar] [CrossRef] [PubMed]
- Carrier, X.; Marceau, E.; Lambert, J.F.; Che, M. Transformations of gamma-alumina in aqueous suspensions. 1. Alumina chemical weathering studies as a function of pH. J. Colloid Interface Sci. 2007, 308, 429–437. [Google Scholar]
- Wijnja, H.; Schulthess, C.P. ATR-FTIR and DRIFT spectroscopy of carbonate species at the aged gamma-Al2O3/water interface. Spectrochim. Acta Part A 1999, 55, 861–872. [Google Scholar]
- Ressler, T. WinXAS: A new software package not only for the analysis of energy-dispersive XAS data. J. Phys. IV 1997, 7, C2-269. [Google Scholar] [CrossRef]
- Ankudinov, A.L.; Rehr, J.J. Relativistic calculations of spin-dependent X-ray-absorption spectra. Phys. Rev. B 1997, 56, R1712–R1715. [Google Scholar] [CrossRef]
- Ravel, B.; Newville, M. ATHENA, ARTEMIS, HEPHAESTUS: Data analysis for X-ray absorption spectroscopy using IFEFFIT. J. Synchrotron Radiat. 2005, 12, 537–541. [Google Scholar] [CrossRef] [PubMed]
- Huminicki, D.M.C.; Hawthorne, F.C. The crystal structure of nikischerite, NaFe2+6Al3(SO4)2(OH)18(H2O)12 a mineral from the shigaite group. Can. Mineral. 2003, 41, 79–82. [Google Scholar] [CrossRef]
- Gou, W.X.; Ji, J.F.; Li, W. An EXAFS investigation of the mechanism of competitive sorption between Co.(II) and Ni(II) at gamma-alumina/solution interface. Acta Geochim. 2017, 36, 462–464. [Google Scholar] [CrossRef]
- Gustaffson, J.P. Visual MINTEQ Chemical Equilibrium Model Version 3.1; Stockholm Royal Institute of Technology (KTH): Stockholm, Sweden, 2017. [Google Scholar]
- Shannon, R.D. Revised effective ionic radii and systematic studies of interatomic distances in halides and chalcogenides. Acta Cryst. 1976, A32, 751–767. [Google Scholar] [CrossRef]
- Brown, T.; LeMay, H.E.; Bursten, B. Chemistry: The Central Science, 8th ed.; Prentice-Hall: Upper Saddle River, NJ, USA, 2002. [Google Scholar]
- Borch, T.; Kretzschmar, R.; Kappler, A.; Van Cappellen, P.; Ginder-Vogel, M.; Voegelin, A.; Campbell, K. Biogeochemical redox processes and their impact on contaminant dynamics. Environ. Sci. Technol. 2010, 44, 15–23. [Google Scholar] [CrossRef] [PubMed]
- Weber, K.A.; Achenbach, L.A.; Coates, J.D. Microorganisms pumping iron: Anaerobic microbial iron oxidation and reduction. Nat. Rev. Microbiol. 2006, 4, 752–764. [Google Scholar] [CrossRef] [PubMed]
- Lovley, D.R.; Phillips, E.J.P. Organic matter mineralization with reduction of ferric iron in anaerobic sediments. Appl. Environ. Microbiol. 1986, 51, 683–689. [Google Scholar] [PubMed]
- Baedecker, M.J.; Cozzarelli, I.M. The determination and fate of unstable constituents of contaminated groundwater. In Groundwater Contamination and Analysis at Hazardous Waste Sites; Lesage, S., Jackson, R.E., Eds.; Marcel Dekker, Inc.: New York, NY, USA, 1992; p. 425. [Google Scholar]
- Kirk, G. The Biogeochemistry of Submerged Soils; John Wiley & Sons, Ltd.: Chichester, UK, 2004. [Google Scholar]
- Ponnamperuma, F.N. The chemistry of submerged soils. Adv. Agron. 1972, 24, 29–96. [Google Scholar]
- Hem, J.D. Study and Interpretation of the Chemical Characteristics of Natural Water, 3rd ed.; United States Government Printing Office: Washington, DC, USA, 1985.
- Rashid, M.A.; Leonard, J.D. Modifications in the solubility and precipitation behavior of various metals as a result of their interaction with sedimentary humic acid. Chem. Geol. 1973, 11, 89–97. [Google Scholar] [CrossRef]
- Fredrickson, J.K.; Gorby, Y.A. Environmental processes mediated by iron-reducing bacteria. Curr. Opin. Biotechnol. 1996, 7, 287–294. [Google Scholar] [CrossRef]
- Burton, E.A.; Walter, L.M. The role of pH in phosphate inhibition of calcite and aragonite precipitation rates in seawater. Geochim. Cosmochim. Acta 1990, 54, 797–808. [Google Scholar] [CrossRef]
- Sanchez-Roman, M.; Puente-Sanchez, F.; Parro, V.; Amils, R. Nucleation of Fe-rich phosphates and carbonates on microbial cells and exopolymeric substances. Front. Microbiol. 2015, 6, 1024. [Google Scholar] [CrossRef] [PubMed]
- Nachtegaal, M.; Sparks, D.L. Nickel sequestration in a kaolinite-humic acid complex. Environ. Sci. Technol. 2003, 37, 529–534. [Google Scholar] [CrossRef] [PubMed]
- Yamaguchi, N.U.; Scheinost, A.C.; Sparks, D.L. Influence of gibbsite surface area and citrate on Ni sorption mechanisms at pH 7.5. Clays Clay Miner. 2002, 50, 784–790. [Google Scholar] [CrossRef]
- Yamaguchi, N.U.; Scheinost, A.C.; Sparks, D.L. Surface-induced nickel hydroxide precipitation in the presence of citrate and salicylate. Soil Sci. Soc. Am. J. 2001, 65, 729–736. [Google Scholar] [CrossRef]
- Zhu, Y.; Liu, J.J.; Goswami, O.; Rouff, A.A.; Elzinga, E.J. Effects of humic substances on Fe(II) sorption onto aluminum oxide and clay. Geochem. Trans. 2018, 19, 3. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Source * | LDH Phase | Ksp Value ** | Synthesis Method | Ksp Measurement | Notes | |
|---|---|---|---|---|---|---|
| Boclair & Braterman [48] | Mn2/3Al1/3(OH)2Cl1/3 | 1.12 × 10−18 | Titration (2h) of 2:1 mixed Me(II):Al(III) solution to alkaline pH with base | Calculated from base consumption in titration curve | Ksp values were not corrected for effects of ionic strength and aqueous complexation | |
| Co2/3Al1/3(OH)2Cl1/3 | 8.19 × 10−20 | |||||
| Ni2/3Al1/3(OH)2Cl1/3 | 5.45 × 10−20 | |||||
| Zn2/3Al1/3(OH)2Cl1/3 | 3.31 × 10−20 | |||||
| Johnson & Glasser [46] | Co2/3Al1/3(OH)2(CO3)1/6 | 4.23 × 10−21 | Aging of alkaline 2:1 Me(II):Al(III) carbonate solutions at 70–80 °C for 1 wk | Calculated from solution chemistry of LDH suspensions following 147-411 d of equilibration. | Suspensions were prepared by resuspending washed and dried LDH material in carbonate solutions following synthesis. | |
| Ni2/3Al1/3(OH)2(CO3)1/6 | 4.75 × 10−22 | |||||
| Zn2/3Al1/3(OH)2(CO3)1/6 | 8.58 × 10−22 | |||||
| Peltier et al. [40] | Ni2/3Al1/3(OH)2(NO3)1/3 | 7.08 × 10−23 | Room-temperature controlled hydrolysis method of Taylor (1984) | Calculated from solid phase enthalpies measured with calorimetry | LDH material was freeze-dried before calorimetry measurements | |
| Ni2/3Al1/3(OH)2(CO3)1/6 | 1.58 × 10−24 | |||||
| Ni2/3Al1/3(OH)2(SO4)1/6 | 1.62 × 10−25 | |||||
| Allada et al. [49] | Ni2/3Al1/3(OH)2(CO3)1/6 | 1.74 × 10−26 | As in Johnson and Glasser (2003) | Calculated from solid phase enthalpies measured with calorimetry | LDH material was freeze-dried before calorimetry measurements | |
| Zn2/3Al1/3(OH)2(CO3)1/6 | 5.37 × 10−25 | |||||
| Regelink & Temminghoff [47] | Ni2/3Al1/3(OH)2(CO3)1/6 | 8.58 × 10−23 | Aging of 2:1 Ni(II):Al(III) carbonate solutions at pH 8 for 15–21 d | Calculated from the solution chemistry of the LDH synthesis suspensions | Additionally calculated Ksp without accounting for anion activity (equation 3 in text) | |
| Zhu & Elzinga [27] | Fe2/3Al1/3(OH)2(Cl)1/3 | 9.43 × 10−22 # | Fe(II) sorption onto Al-oxide at circumneutral pH | Calculated from solution chemistry of equilibrated sorption samples | Methods and calculations are identical to those of the current study | |
| 2.41 × 10−21 # |
| Me(II) | Me(II)–Al(III)–LDH | β–Me(OH)2 | |||
|---|---|---|---|---|---|
| With Anion Activitiy * | Without Anion Activity * | ||||
| Ksp (gibbsite) ** | Ksp (bayerite) ** | K’sp (gibbsite) ** | K’sp (bayerite) ** | Ksp *** | |
| Ni(II) | 8.43(±0.1) × 10−23 | 2.16(±0.02) × 10−22 | 2.00(±0.02) × 10−22 | 5.11(±0.04) × 10−22 | 10−17.21 |
| Zn(II) | 1.89(±0.05) × 10−22 | 4.81(±0.12) × 10−22 | 4.46(±0.11) × 10−22 | 1.14(±0.03) × 10−21 | 10−16.25 |
| Co(II) | 3.58(±0.13) × 10−22 | 9.13(±0.34) × 10−22 | 8.48(±0.30) × 10−22 | 2.16(±0.08) × 10−21 | 10−15.71 |
| Fe(II) | 7.55(±0.06) × 10−22 | 1.93(±0.02) × 10−21 | 1.79(±0.01) × 10−21 | 4.55(±0.04) × 10−21 | 10−15.11 |
| Mn(II) | 1.27(±0.02) × 10−20 | 3.23(±0.04) × 10−20 | 3.00(±0.04) × 10−20 | 7.65(±0.10) × 10−20 | 10−12.81 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bhattacharya, L.; Elzinga, E.J. A Comparison of the Solubility Products of Layered Me(II)–Al(III) Hydroxides Based on Sorption Studies with Ni(II), Zn(II), Co(II), Fe(II), and Mn(II). Soil Syst. 2018, 2, 20. https://doi.org/10.3390/soilsystems2020020
Bhattacharya L, Elzinga EJ. A Comparison of the Solubility Products of Layered Me(II)–Al(III) Hydroxides Based on Sorption Studies with Ni(II), Zn(II), Co(II), Fe(II), and Mn(II). Soil Systems. 2018; 2(2):20. https://doi.org/10.3390/soilsystems2020020
Chicago/Turabian StyleBhattacharya, Lasita, and Evert J. Elzinga. 2018. "A Comparison of the Solubility Products of Layered Me(II)–Al(III) Hydroxides Based on Sorption Studies with Ni(II), Zn(II), Co(II), Fe(II), and Mn(II)" Soil Systems 2, no. 2: 20. https://doi.org/10.3390/soilsystems2020020