
Illustrating the Pathogenesis and Therapeutic Approaches of Epilepsy by Targeting Angiogenesis, Inflammation, and Oxidative Stress

 ,
,
Abstract
1. Introduction
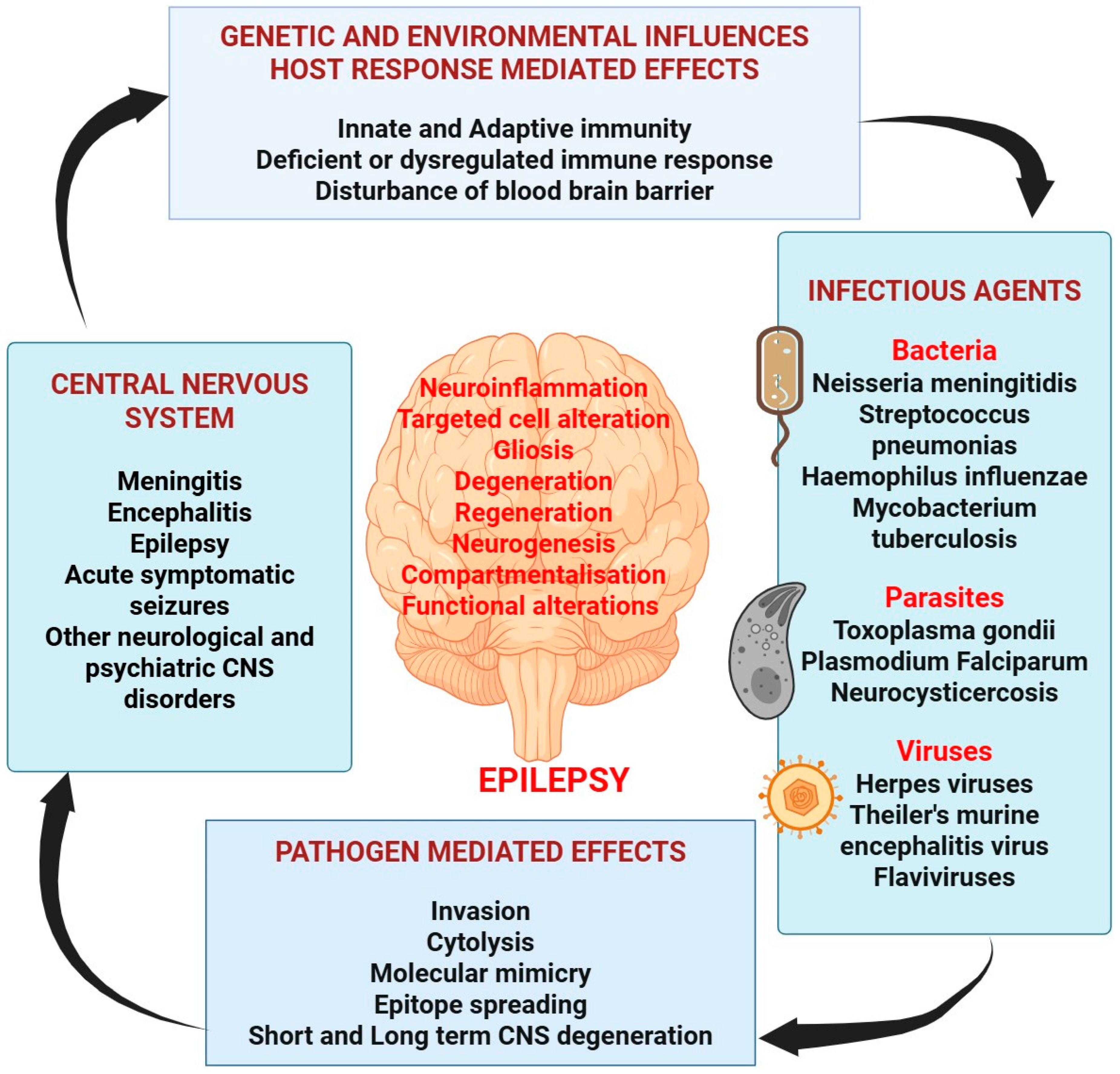
2. Contributing Etiology of Epilepsy
2.1. Genetics and Epilepsy
2.2. Brain Tumor and Brain Injury Leading to Epilepsy
2.3. Infection and Epilepsy
2.4. Stroke and Epilepsy
3. Understanding the Pathogenesis of Epilepsy
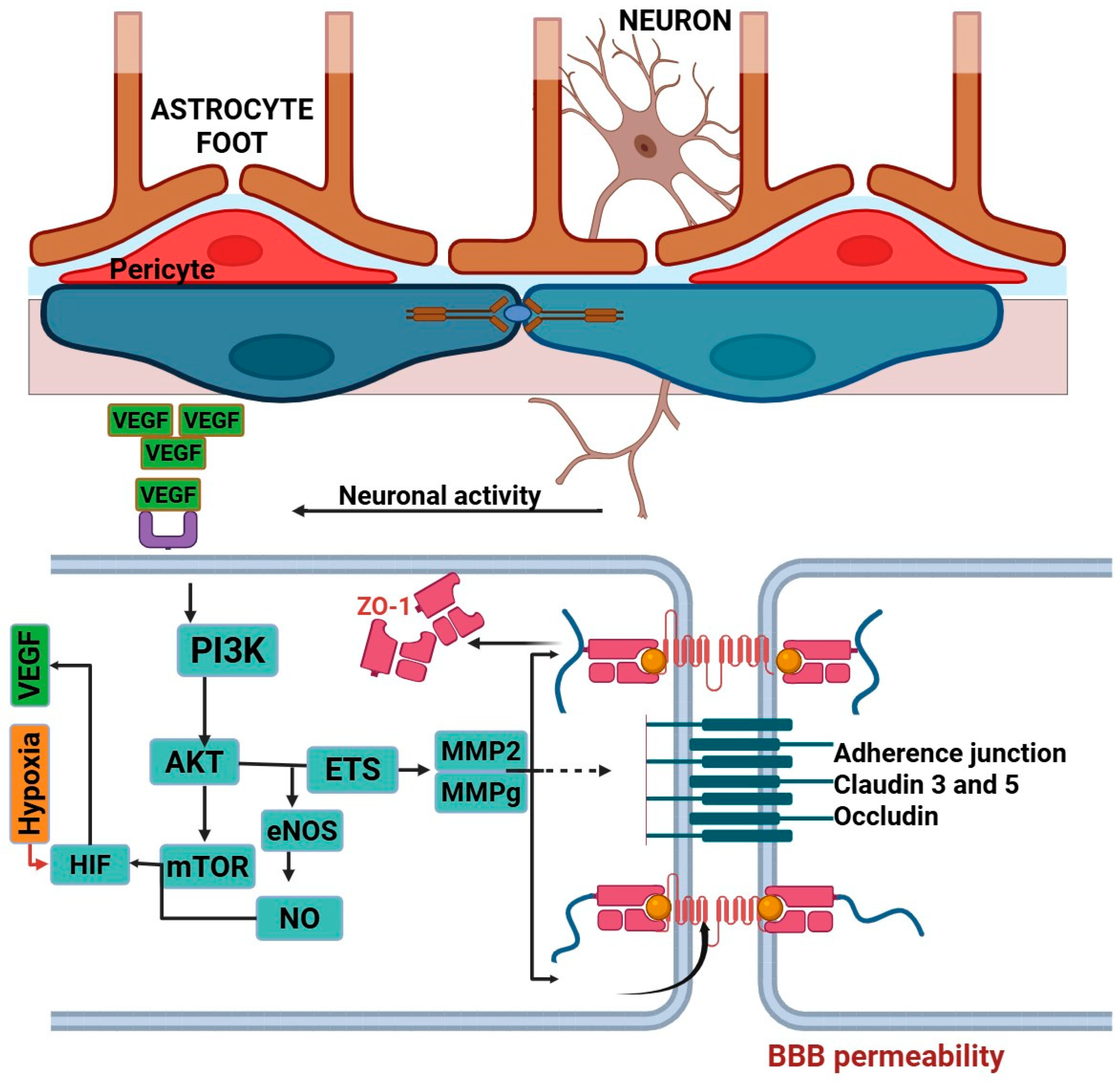
3.1. Role of Angiogenesis in Epilepsy
3.1.1. Vascular Endothelial Growth Factor (VEGF)
3.1.2. PDGF-β
3.1.3. Blood–Brain Barrier
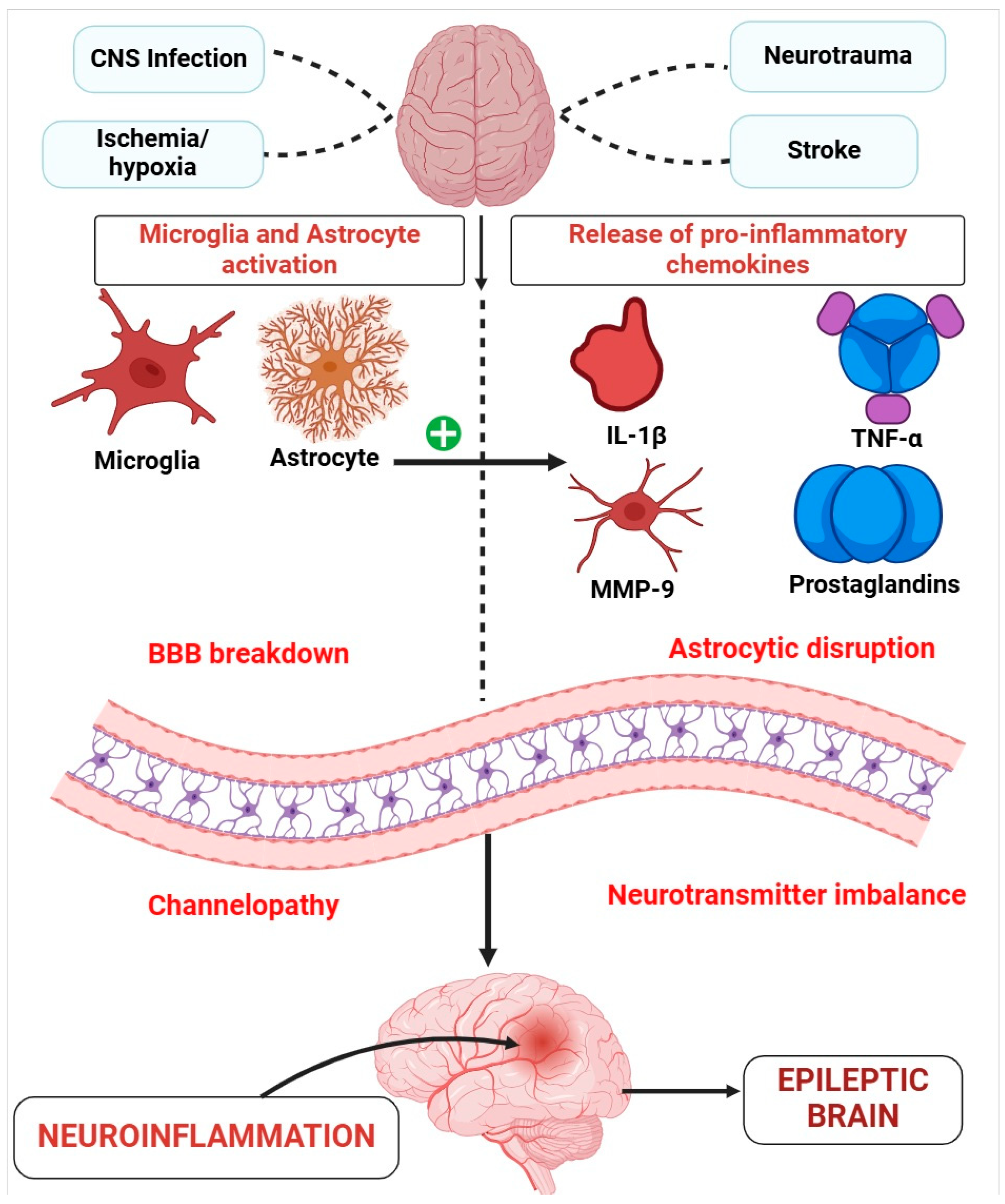
3.2. Role of Inflammation in Epilepsy
3.2.1. Glia, Neuroinflammation, and Epilepsy
3.2.2. Chemokines and Epilepsy
3.2.3. Infections as a Cause of Neuroinflammation in Epilepsy
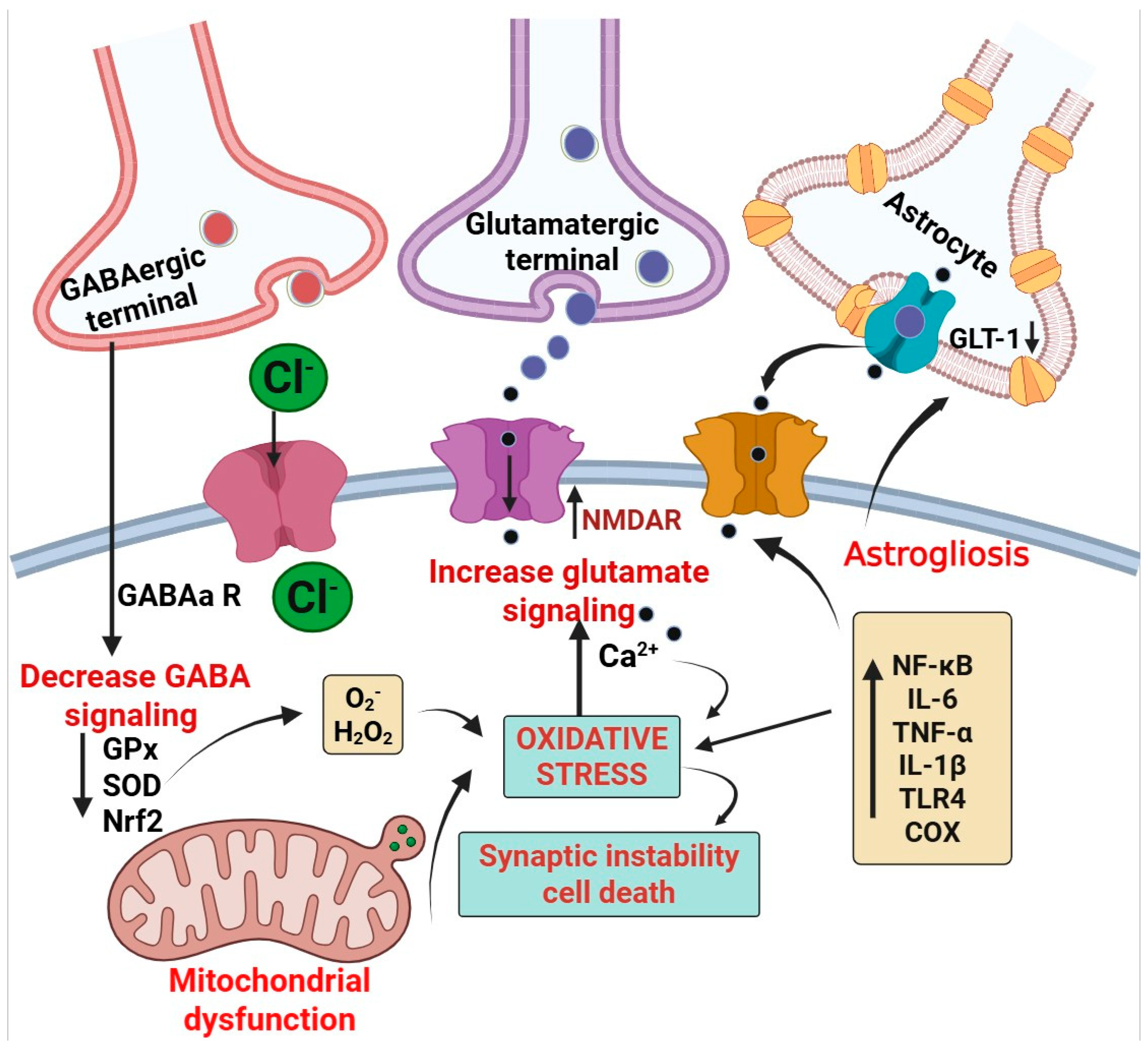
3.3. Role of Oxidative Stress in Epilepsy
3.3.1. Mitochondrial Oxidative Stress and Epilepsy
3.3.2. Excitatory/Inhibitory (E/I) Imbalance: Relevance to Oxidative Stress and Epilepsy
4. Angiogenesis, Oxidative Stress, and Inflammation Targeting Diagnostics
5. Therapeutic Approaches for Epilepsy Targeting Angiogenesis, Inflammation, and Oxidative Stress
5.1. Angiogenesis Targeted Approach for the Treatment of Epilepsy
5.1.1. Sunitinib
5.1.2. Aliskiren
5.1.3. Ephrin-A5/EphA4
5.1.4. Levetiracetam (LEV)
5.2. Inflammation Targeted Approach for the Treatment of Epilepsy
5.3. Oxidative Stress Targeted Approach for the Treatment of Epilepsy

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sr. No. | Drugs | Targeted Seizure | Mechanism of Action | Effects on Certain Markers | References |
|---|---|---|---|---|---|
| Anti-inflammatory therapy | |||||
| 1. | Adalimumab | Partial convulsion accompanied by focal seizure | It is a monoclonal antibody that act as a TNF-α blocker | It declines the TNF-α level during epilepsy and produces anti-inflammatory action | [56] |
| 2. | Anakinra | Infection-mediated febrile seizure | Interleukin-1 receptor blocker | Depletion in IL-1-mediated brain inflammation | [58] |
| 4. | Canakinumab + Anakinra | Generalized tonic clonic seizure | Monoclonal Ab interleukin-1 receptor blocker | Depletion in IL-1-mediated brain inflammation | [59] |
| 5. | Tocilizumab | During acute epilepsy and SE | Monoclonal antibody, which is an interleukin-6 antagonist | It decreases the interleukin-6 | [60] |
| 5. | Minocycline | Drug-resistant seizure | Interleukin-1β suppressant and blocks the stimulation of microglial cells | It declines the interleukin-1β secretion from microglial cells | [61] |
| 6. | Aspirin | Recurrent seizure | COX-PGE2 inhibitor | It promotes hippocampal neurogenesis by blocking the COX-PGE2 pathway | [62] |
| 7. | VX09-765-401 | Partial seizure | Interleukin-1β blocker | No evidence | [63] |
| Antioxidant therapy | |||||
| 8. | N-acetylcysteine | Pentylenetetrazole-induced epilepsy | Reduction of glutathione precursor | Milder disruption of glutathione homeostasis | [64] |
| 9. | Curcumin | Pentylenetetrazole-induced epilepsy | Chelators of metals and elevated level of free radicals | Inflammation-mediating gene transcription is reduced along with elevating the level of superoxide dismutase | [65] |
| 10. | Cannabidiol | Drug tolerance | Adenosine uptake is inhibited and suppresses GRP55 | Degrades the concentration of ROS and a rise in a defense mechanism related to antioxidant properties | [66] |
| 11. | Coenzyme Q10 | Pilocarpine-induced rat model | Elevated level of TCA and enzymes of antioxidants | Deterioration of lipid peroxidation and elevates the level of growth-stimulating factor | [67] |
| 12. | Naringenin | Pilocarpine-induced rat model | Free radicals’ concentration is elevated | Antioxidant enzymes and glutathione concentrations are shown to be enhanced | [68] |
| 13. | Sulforaphane | Status epilepticus | Stimulation of NRF2 pathway | Elevates the level of glutathione and depletes the level of malondialdehyde | [69] |
| 14. | Vitamin E | Refractory epilepsy | Elevates the level of peroxyl radicals | Antioxidant capacity as well as glutathione level is enhanced | [70] |
| Angiogenesis targeted therapy | |||||
| 15. | Anti-VEGF antibody | Kainate-induced seizure in rats | Following kainate-induced seizures, administration of a neutralizing anti-VEGF antibody was performed, and it was found that this completely ceased tight junction protein breakdown and vascularization | Anti-VEGF activity utilized for improving vascularization inside brain | [71] |
| 16. | Angiopoietin-1 | Pilocarpine-induced SE in rats | The suppression of VEGF-induced vascular permeability did not impact VEGF’s neuroprotective effects. In order to reduce vascular anomalies in the epileptic brain, andiopoetin-1 may be used as a therapeutic approach | VEGF is targeted and an angiogenic marker is regulated for the treatment of epilepsy | [72] |
6. Recent Advances Accomplished in the Treatment of Epilepsy Targeting Angiogenesis, Inflammation, and Oxidative Stress
6.1. Drug Repurposed for the Treatment of Epilepsy
6.2. Antiepileptic Drugs in Different Phases of Clinical Trials
7. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Riva, A.; Golda, A.; Balagura, G.; Amadori, E.; Vari, M.S.; Piccolo, G.; Iacomino, M.; Lattanzi, S.; Salpietro, V.; Minetti, C.; et al. New trends and most promising therapeutic strategies for epilepsy treatment. Front. Neurol. 2021, 12, 753753. [Google Scholar] [CrossRef] [PubMed]
- Fiest, K.M.; Sauro, K.M.; Wiebe, S.; Patten, S.B.; Kwon, C.S.; Dykeman, J.; Pringsheim, T.; Lorenzetti, D.L.; Jetté, N. Prevalence and Incidence of Epilepsy: A Systematic Review and Meta-analysis of International Studies. Neurology 2017, 88, 296–303. [Google Scholar] [CrossRef]
- Beghi, E. The epidemiology of epilepsy. Neuroepidemiology 2020, 54, 185–191. [Google Scholar] [CrossRef] [PubMed]
- Sumadewi, K.T.; Harkitasari, S.; Tjandra, D.C. Biomolecular mechanisms of epileptic seizures and epilepsy: A review. Acta Epileptol. 2023, 5, 28. [Google Scholar] [CrossRef]
- Huang, L.; Xiao, W.; Wang, Y.; Li, J.; Gong, J.; Tu, E.; Long, L.; Xiao, B.; Yan, X.; Wan, L. Metabotropic glutamate receptors (mGluRs) in epileptogenesis: An update on abnormal mGluRs signaling and its therapeutic implications. Neural Regen. Res. 2024, 19, 360–368. [Google Scholar] [CrossRef] [PubMed]
- Berendt, M.; Farquhar, R.G.; Mandigers, P.J.J.; Pakozdy, A.; Bhatti, S.F.M.; De Risio, L.; Fischer, A.; Long, S.; Matiasek, K.; Muñana, K.; et al. International veterinary epilepsy task force consensus report on epilepsy definition, classification and terminology in companion animals. BMC Vet. Res. 2015, 11, 182. [Google Scholar] [CrossRef]
- Han, W.; Jiang, L.; Song, X.; Li, T.; Chen, H.; Cheng, L. VEGF modulates neurogenesis and microvascular remodeling in epileptogenesis after status epilepticus in immature rats. Front. Neurol. 2021, 12, 808568. [Google Scholar] [CrossRef]
- Vezzani, A.; Balosso, S.; Ravizza, T. Neuroinflammatory pathways as treatment targets and biomarkers in epilepsy. Nat. Rev. Neurol. 2019, 15, 459–472. [Google Scholar] [CrossRef]
- Perucca, P.; Bahlo, M.; Berkovic, S.F. The genetics of epilepsy. Annu. Rev. Genom. Hum. Genet. 2020, 21, 205–230. [Google Scholar] [CrossRef]
- Kazis, D.; Chatzikonstantinou, S.; Ciobica, A.; Kamal, F.Z.; Burlui, V.; Calin, G.; Mavroudis, I. Epidemiology, risk factors, and biomarkers of post-traumatic epilepsy: A comprehensive overview. Biomedicines 2024, 12, 410. [Google Scholar] [CrossRef]
- Khatab, A.A.; Soliman, M.A.; El-Dabaa, S.S.; Abd El-Naby, S.A. Probable relationship between Toxoplasma gondii and children with cryptogenic epilepsy. Menoufia Med. J. 2020, 33, 157–161. [Google Scholar]
- Mohapatra, L.; Tripathi, A.S.; Prajapati, B.G.; Alka Mishra, D.; Yasir, M.; Maurya, R.K. Granulomatous Amebic Encephalitis: Evolutionary Dynamics, Advances in Diagnostics and Therapeutic Interventions. In Rising Contagious Diseases: Basics, Management, and Treatments; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2024; pp. 378–395. [Google Scholar]
- Lekoubou, A.; Debroy, K.; Kwegyir-Aggrey, A.; Bonilha, L.; Kengne, A.P.; Chinchilli, V.M. Risk models to predict late-onset seizures after stroke: A systematic review. Epilepsy Behav. 2021, 121, 108003. [Google Scholar] [CrossRef]
- Tanaka, T.; Ihara, M.; Fukuma, K.; Mishra, N.K.; Koepp, M.J.; Guekht, A.; Ikeda, A. Pathophysiology, diagnosis, prognosis, and prevention of poststroke epilepsy: Clinical and research implications. Neurology 2024, 102, e209450. [Google Scholar] [CrossRef] [PubMed]
- Castro-Torres, R.D.; Ureña-Guerrero, M.E.; Morales-Chacón, L.M.; Lorigados-Pedre, L.; Estupiñan-Díaz, B.; Rocha, L.; Orozco-Suárez, S.; Rivera-Cervantes, M.C.; Alonso-Vanegas, M.; Beas-Zárate, C. New Aspects of VEGF, GABA, and Glutamate Signaling in the Neocortex of Human Temporal Lobe Pharmacoresistant Epilepsy Revealed by RT-qPCR Arrays. J. Mol. Neurosci. 2020, 70, 916–929. [Google Scholar] [CrossRef]
- Meijer, W.C.; Gorter, J.A. Role of blood–brain barrier dysfunction in the development of poststroke epilepsy. Epilepsia 2024, 65, 2519–2536. [Google Scholar] [CrossRef]
- Ogaki, A.; Ikegaya, Y.; Koyama, R. Vascular abnormalities and the role of vascular endothelial growth factors in the epileptic brain. Front. Pharmacol. 2020, 11, 20. [Google Scholar] [CrossRef] [PubMed]
- Ureña-Guerrero, M.E.; Castañeda-Cabral, J.L.; Rivera-Cervantes, M.C.; Macias-Velez, R.J.; Jarero-Basulto, J.J.; Gudiño-Cabrera, G.; Beas-Zárate, C. Neuroprotective and neurorestorative effects of Epo and VEGF: Perspectives for new therapeutic approaches to neurological diseases. Curr. Pharm. Des. 2020, 26, 1263–1276. [Google Scholar] [CrossRef]
- Guo, D.; Zhang, B.; Han, L.; Rensing, N.R.; Wong, M. Cerebral vascular and blood brain–barrier abnormalities in a mouse model of epilepsy and tuberous sclerosis complex. Epilepsia 2024, 65, 483–496. [Google Scholar] [CrossRef]
- Yamanaka, G.; Takata, F.; Kataoka, Y.; Kanou, K.; Morichi, S.; Dohgu, S.; Kawashima, H. The neuroinflammatory role of pericytes in epilepsy. Biomedicines 2021, 9, 759. [Google Scholar] [CrossRef]
- Kyyriäinen, J.; Ekolle Ndode-Ekane, X.; Pitkänen, A. Dynamics of PDGFRβ expression in different cell types after brain injury. Glia 2017, 65, 322–341. [Google Scholar] [CrossRef]
- Löscher, W.; Friedman, A. Structural, molecular, and functional alterations of the blood-brain barrier during epileptogenesis and epilepsy: A cause, consequence, or both? Int. J. Mol. Sci. 2020, 21, 591. [Google Scholar] [CrossRef] [PubMed]
- Meng, F.; Yao, L. The role of inflammation in epileptogenesis. Acta Epileptol. 2020, 2, 15. [Google Scholar] [CrossRef]
- Shen, W.; Pristov, J.B.; Nobili, P.; Nikolić, L. Can glial cells save neurons in epilepsy? Neural Regen. Res. 2023, 18, 1417–1422. [Google Scholar]
- Chen, P.; Chen, F.; Zhou, B. Understanding the role of glia-neuron communication in the pathophysiology of epilepsy: A review. J. Integr. Neurosci. 2022, 21, 102. [Google Scholar] [CrossRef] [PubMed]
- Wolinski, P.; Ksiazek-Winiarek, D.; Glabinski, A. Cytokines and neurodegeneration in epileptogenesis. Brain Sci. 2022, 12, 380. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Nagib, M.M.; Yasmen, N.; Sluter, M.N.; Littlejohn, T.L.; Yu, Y.; Jiang, J. Neuroinflammatory mediators in acquired epilepsy: An update. Inflamm. Res. 2023, 72, 683–701. [Google Scholar] [CrossRef]
- Löscher, W.; Howe, C.L. Molecular mechanisms in the genesis of seizures and epilepsy associated with viral infection. Front. Mol. Neurosci. 2022, 15, 870868. [Google Scholar] [CrossRef]
- Vezzani, A.; Ravizza, T.; Bedner, P.; Aronica, E.; Steinhäuser, C.; Boison, D. Astrocytes in the initiation and progression of epilepsy. Nat. Rev. Neurol. 2022, 18, 707–722. [Google Scholar] [CrossRef]
- Iwasa, K.; Yamamoto, S.; Yagishita, S.; Maruyama, K.; Yoshikawa, K. Excitotoxicity-induced prostaglandin D2 production induces sustained microglial activation and delayed neuronal death. J. Lipid Res. 2017, 58, 649–655. [Google Scholar] [CrossRef]
- Mukhtar, I. Inflammatory and immune mechanisms underlying epileptogenesis and epilepsy: From pathogenesis to treatment target. Seizure 2020, 82, 65–79. [Google Scholar] [CrossRef]
- Khan, S. Epilepsy; An Insight into Epileptogenic Potential of Infections and Antibiotics. Biomed. J. Sci. Tech. Res. 2023, 53, 44533–44540. [Google Scholar] [CrossRef]
- Tan, T.H.; Perucca, P.; O’Brien, T.J.; Kwan, P.; Monif, M. Inflammation, ictogenesis, and epileptogenesis: An exploration through human disease. Epilepsia 2021, 62, 303–324. [Google Scholar] [CrossRef] [PubMed]
- Borowicz-Reutt, K.K.; Czuczwar, S.J. Role of oxidative stress in epileptogenesis and potential implications for therapy. Pharmacol. Rep. 2020, 72, 1218–1226. [Google Scholar] [CrossRef]
- Mishra, D.; Mohapatra, L.; Tripathi, A.S.; Paswan, S.K. The influential responsibility of sirtuins in senescence and associated diseases: A review. J. Biochem. Mol. Toxicol. 2024, 38, e23812. [Google Scholar] [CrossRef]
- Lopatina, O.L.; Malinovskaya, N.A.; Komleva, Y.K.; Gorina, Y.V.; Shuvaev, A.N.; Olovyannikova, R.Y.; Belozor, O.S.; Belova, O.A.; Higashida, H.; Salmina, A.B. Excitation/inhibition imbalance and impaired neurogenesis in neurodevelopmental and neurodegenerative disorders. Rev. Neurosci. 2019, 30, 807–820. [Google Scholar] [CrossRef]
- Yang, N.; Guan, Q.W.; Chen, F.H.; Xia, Q.X.; Yin, X.X.; Zhou, H.H.; Mao, X.Y. Antioxidants targeting mitochondrial oxidative stress: Promising neuroprotectants for epilepsy. Oxidative Med. Cell. Longev. 2020, 2020, 6687185. [Google Scholar] [CrossRef] [PubMed]
- Ein Shoka, A.A.; Dessouky, M.M.; El-Sayed, A.; Hemdan, E.E. EEG seizure detection: Concepts, techniques, challenges, and future trends. Multimed. Tools Appl. 2023, 82, 42021–42051. [Google Scholar] [CrossRef]
- Reshma, S.; Megha, K.B.; Amir, S.; Rukhiya, S.; Mohanan, P.V. Blood brain barrier-on-a-chip to model neurological diseases. J. Drug Deliv. Sci. Technol. 2023, 80, 104174. [Google Scholar] [CrossRef]
- Werry, E.L.; Bright, F.M.; Piguet, O.; Ittner, L.M.; Halliday, G.M.; Hodges, J.R.; Kiernan, M.C.; Loy, C.T.; Kril, J.J.; Kassiou, M. Recent developments in TSPO PET imaging as a biomarker of neuroinflammation in neurodegenerative disorders. Int. J. Mol. Sci. 2019, 20, 3161. [Google Scholar] [CrossRef] [PubMed]
- Lach, P.; Klus, W.; Zajdel, K.; Szeleszczuk, A.; Komorowska, E.; Burda, K.; Kurowski, P. Neuroinflammation in epilepsy—Diagnostics and therapeutic perspectives. Curr. Pharmacol. Rep. 2022, 8, 31–35. [Google Scholar] [CrossRef]
- Yang, W.; Liu, R.; Yin, X.; Wu, K.; Yan, Z.; Wang, X.; Fan, G.; Tang, Z.; Li, Y.; Jiang, H. Novel near-infrared fluorescence probe for bioimaging and evaluating superoxide anion fluctuations in ferroptosis-mediated epilepsy. Anal. Chem. 2023, 95, 12240–12246. [Google Scholar] [CrossRef]
- Shao, C.; Yuan, J.; Liu, Y.; Qin, Y.; Wang, X.; Gu, J.; Chen, G.; Zhang, B.; Liu, H.K.; Zhao, J.; et al. Epileptic brain fluorescent imaging reveals apigenin can relieve the myeloperoxidase-mediated oxidative stress and inhibit ferroptosis. Proc. Natl. Acad. Sci. USA 2020, 117, 10155–10164. [Google Scholar] [CrossRef] [PubMed]
- Si, M.; Lv, L.; Shi, Y.; Li, Z.; Zhai, W.; Luo, X.; Zhang, L.; Qian, Y. Activatable Dual-Optical Molecular Probe for Bioimaging Superoxide Anion in Epilepsy. Anal. Chem. 2024, 96, 4632–4638. [Google Scholar] [CrossRef] [PubMed]
- Ponisio, M.R.; Zempel, J.M.; Day, B.K.; Eisenman, L.N.; Miller-Thomas, M.M.; Smyth, M.D.; Hogan, R.E. The role of SPECT and PET in epilepsy. Am. J. Roentgenol. 2021, 216, 759–768. [Google Scholar] [CrossRef]
- Liu, X.; Zhang, Y.; Zhao, Y.; Zhang, Q.; Han, F. The neurovascular unit dysfunction in the molecular mechanisms of epileptogenesis and targeted therapy. Neurosci. Bull. 2024, 40, 621–634. [Google Scholar] [CrossRef] [PubMed]
- Reiss, Y.; Bauer, S.; David, B.; Devraj, K.; Fidan, E.; Hattingen, E.; Liebner, S.; Melzer, N.; Meuth, S.G.; Rosenow, F.; et al. The neurovasculature as a target in temporal lobe epilepsy. Brain Pathol. 2023, 33, e13147. [Google Scholar] [CrossRef]
- Lui, A.; Vanleuven, J.; Perekopskiy, D.; Liu, D.; Xu, D.; Alzayat, O.; Elgokhy, T.; Do, T.; Gann, M.; Martin, R.; et al. FDA-approved kinase inhibitors in preclinical and clinical trials for neurological disorders. Pharmaceuticals 2022, 15, 1546. [Google Scholar] [CrossRef]
- Starace, V.; Battista, M.; Brambati, M.; Cavalleri, M.; Bertuzzi, F.; Amato, A.; Lattanzio, R.; Bandello, F.; Cicinelli, M.V. The role of inflammation and neurodegeneration in diabetic macular edema. Ther. Adv. Ophthalmol. 2021, 13, 25158414211055963. [Google Scholar] [CrossRef]
- Villapol, S.; Janatpour, Z.C.; Affram, K.O.; Symes, A.J. The renin angiotensin system as a therapeutic target in traumatic brain injury. Neurotherapeutics 2023, 20, 1565–1591. [Google Scholar] [CrossRef]
- Li, Y.; Su, P.; Chen, Y.; Nie, J.; Yuan, T.F.; Wong, A.H.; Liu, F. The Eph receptor A4 plays a role in demyelination and depression-related behavior. J. Clin. Investig. 2022, 132, e152187. [Google Scholar] [CrossRef]
- Contreras-García, I.J.; Cárdenas-Rodríguez, N.; Romo-Mancillas, A.; Bandala, C.; Zamudio, S.R.; Gómez-Manzo, S.; Hernández-Ochoa, B.; Mendoza-Torreblanca, J.G.; Pichardo-Macías, L.A. Levetiracetam mechanisms of action: From molecules to systems. Pharmaceuticals 2022, 15, 475. [Google Scholar] [CrossRef] [PubMed]
- Webb, C.A.; Wanbon, R.; Otto, E.D. Levetiracetam for status epilepticus in adults: A systematic review. Can. J. Hosp. Pharm. 2022, 75, 46. [Google Scholar] [CrossRef]
- Lagarde, S.; Villeneuve, N.; Trébuchon, A.; Kaphan, E.; Lepine, A.; McGonigal, A.; Roubertie, A.; Barthez, M.A.; Trommsdorff, V.; Lefranc, J.; et al. Anti–tumor necrosis factor alpha therapy (adalimumab) in Rasmussen’s encephalitis: An open pilot study. Epilepsia 2016, 57, 956–966. [Google Scholar] [CrossRef]
- Wiciński, M.; Puk, O.; Malinowski, B. Cenobamate: Neuroprotective potential of a new antiepileptic drug. Neurochem. Res. 2021, 46, 439–446. [Google Scholar] [CrossRef]
- Berman, E.; Noyman, I.; Medvedovsky, M.; Ekstein, D.; Eyal, S. Not your usual drug-drug interactions: Monoclonal antibody–based therapeutics may interact with antiseizure medications. Epilepsia 2022, 63, 271–289. [Google Scholar] [CrossRef] [PubMed]
- Łukawski, K.; Czuczwar, S.J. Oxidative stress and neurodegeneration in animal models of seizures and epilepsy. Antioxidants 2023, 12, 1049. [Google Scholar] [CrossRef]
- Lai, Y.C.; Muscal, E.; Wells, E.; Shukla, N.; Eschbach, K.; Hyeong Lee, K.; Kaliakatsos, M.; Desai, N.; Wickström, R.; Viri, M.; et al. Anakinra usage in febrile infection related epilepsy syndrome: An international cohort. Ann. Clin. Transl. Neurol. 2020, 7, 2467–2474. [Google Scholar] [CrossRef] [PubMed]
- Costagliola, G.; Depietri, G.; Michev, A.; Riva, A.; Foiadelli, T.; Savasta, S.; Bonuccelli, A.; Peroni, D.; Consolini, R.; Marseglia, G.L.; et al. Targeting inflammatory mediators in epilepsy: A systematic review of its molecular basis and clinical applications. Front. Neurol. 2022, 13, 741244. [Google Scholar] [CrossRef]
- Cantarín-Extremera, V.; Jiménez-Legido, M.; Duat-Rodríguez, A.; García-Fernández, M.; Ortiz-Cabrera, N.V.; Ruiz-Falcó-Rojas, M.L.; González-Gutiérrez-Solana, L. Tocilizumab in pediatric refractory status epilepticus and acute epilepsy: Experience in two patients. J. Neuroimmunol. 2020, 340, 577142. [Google Scholar] [CrossRef]
- Singh, T.; Thapliyal, S.; Bhatia, S.; Singh, V.; Singh, M.; Singh, H.; Kumar, A.; Mishra, A. Reconnoitering the transformative journey of minocycline from an antibiotic to an antiepileptic drug. Life Sci. 2022, 293, 120346. [Google Scholar] [CrossRef]
- Zhu, K.; Hu, M.; Yuan, B.; Liu, J.X.; Liu, Y. Aspirin attenuates spontaneous recurrent seizures in the chronically epileptic mice. Neurol. Res. 2017, 39, 744–757. [Google Scholar] [CrossRef] [PubMed]
- Vezzani, A.; Rüegg, S. Introduction to the 2nd meeting on Immunity and Inflammation in Epilepsy (IIE 2016). Epilepsia 2017, 58, 7–10. [Google Scholar] [CrossRef]
- Egilmez, C.B.; Pazarlar, B.A.; Erdogan, M.A.; Erbas, O. N-acetyl cysteine: A new look at its effect on PTZ-induced convulsions. Epilepsy Res. 2023, 193, 107144. [Google Scholar] [CrossRef]
- Rehman, S.; Ray, A. Neuroprotective medicinal plants: Focus on Curcuma longa. EC Pharmacol. Toxicol. 2020, 8, 16–30. [Google Scholar]
- Massey, S.; Quigley, A.; Rochfort, S.; Christodoulou, J.; Van Bergen, N.J. Cannabinoids and Genetic Epilepsy Models: A Review with Focus on CDKL5 Deficiency Disorder. Int. J. Mol. Sci. 2024, 25, 10768. [Google Scholar] [CrossRef] [PubMed]
- Pradhan, N.; Singh, C.; Singh, A. Coenzyme Q10 is a mitochondrial restorer for various brain disorders. Naunyn-Schmiedeberg’s Arch. Pharmacol. 2021, 394, 2197–2222. [Google Scholar] [CrossRef]
- Shrivastava, A.; Gupta, J.K.; Goyal, M.K. Flavonoids and antiepileptic drugs: A comprehensive review on their neuroprotective potentials. J. Med. Pharm. Allied Sci. 2022, 11, 4179–4186. [Google Scholar] [CrossRef]
- Folbergrová, J.; Ješina, P.; Otáhal, J. Protective effect of sulforaphane on oxidative stress and mitochondrial dysfunction associated with status epilepticus in immature rats. Mol. Neurobiol. 2023, 60, 2024–2035. [Google Scholar] [CrossRef]
- Yang, M.T.; Chou, I.C.; Wang, H.S. Role of vitamins in epilepsy. Epilepsy Behav. 2023, 139, 109062. [Google Scholar] [CrossRef]
- Shoja, A.; Sani, M.; Balaghirad, N.; Jafary, H.; Sagharichi, M.; Alipour, M.A.; Nazerian, Y.; Moghaddam, M.H.; Bayat, A.H.; Ashraf, H.; et al. Intrahippocampal transplantation of dental pulp stem cells improved memory function and reduced neuroinflamma-tion-induced cell death in the rat’s model of seizure. Res. Sq. 2023. [Google Scholar] [CrossRef]
- Jackson, L.; Eldahshan, W.; Fagan, S.C.; Ergul, A. Within the brain: The renin angiotensin system. Int. J. Mol. Sci. 2018, 19, 876. [Google Scholar] [CrossRef] [PubMed]
- Sanz, P.; Rubio, T.; Garcia-Gimeno, M.A. Neuroinflammation and Epilepsy: From Pathophysiology to Therapies Based on Repurposing Drugs. Int. J. Mol. Sci. 2024, 25, 4161. [Google Scholar] [CrossRef] [PubMed]
- Parreira, G.M.; de Oliveira, A.C.; de Oliveira Guarnieri, L.; Vieira, R.P. Drug Repurposing in CNS and Clinical Trials: Recent Achievements and Perspectives Focusing on Epilepsy and Related Comorbidities. In Frontiers in Clinical Drug Research-CNS and Neurological Disorders; Bentham Science Publishers Singapore Pte. Ltd.: Singapore, 2024; Volume 12, p. 171. [Google Scholar]
- Wang, L.; Ding, J.; Zhu, C.; Guo, B.; Yang, W.; He, W.; Li, X.; Wang, Y.; Li, W.; Wang, F.; et al. Semaglutide attenuates seizure severity and ameliorates cognitive dysfunction by blocking the NLR family pyrin domain containing 3 inflammasome in pentylenetetrazole-kindled mice. Int. J. Mol. Med. 2021, 48, 219. [Google Scholar] [CrossRef]
- Pawlik, M.J.; Miziak, B.; Walczak, A.; Konarzewska, A.; Chrościńska-Krawczyk, M.; Albrecht, J.; Czuczwar, S.J. Selected molecular targets for antiepileptogenesis. Int. J. Mol. Sci. 2021, 22, 9737. [Google Scholar] [CrossRef]
- Vishnoi, S.; Raisuddin, S.; Parvez, S. Glutamate excitotoxicity and oxidative stress in epilepsy: Modulatory role of melatonin. J. Environ. Pathol. Toxicol. Oncol. 2016, 35, 365–374. [Google Scholar] [CrossRef] [PubMed]
- Cumbres-Vargas, I.M.; Zamudio, S.R.; Pichardo-Macías, L.A.; Ramírez-San Juan, E. Thalidomide attenuates epileptogenesis and seizures by decreasing brain inflammation in lithium pilocarpine rat model. Int. J. Mol. Sci. 2023, 24, 6488. [Google Scholar] [CrossRef] [PubMed]
- Rayadurg, V.; Muthuchellappan, R.; Rao, U. Volatile anesthetic for the control of posthypoxic refractory myoclonic status. Indian J. Crit. Care Med. Peer-Rev. Off. Publ. Indian Soc. Crit. Care Med. 2016, 20, 485. [Google Scholar] [CrossRef]
- Mohammadi, E.; Nikbakht, F.; Barati, M.; Roghani, M.; Vazifekhah, S.; Khanizadeh, A.M.; Heidari, Z. Protective effect of N-acetyl cysteine on the mitochondrial dynamic imbalance in temporal lobe epilepsy: Possible role of mTOR. Neuropeptides 2022, 96, 102294. [Google Scholar] [CrossRef]
- Mohamed, A.M.; Ali, D.A.; Kolieb, E.; Abdelaziz, E.Z. Ceftriaxone and selenium mitigate seizures and neuronal injury in pentylenetetrazole-kindled rats: Oxidative stress and inflammatory pathway. Int. Immunopharmacol. 2023, 120, 110304. [Google Scholar] [CrossRef]
- Madireddy, S.; Madireddy, S. Therapeutic strategies to ameliorate neuronal damage in epilepsy by regulating oxidative stress, mitochondrial dysfunction, and neuroinflammation. Brain Sci. 2023, 13, 784. [Google Scholar] [CrossRef]
- Klein, P.; Friedman, A.; Hameed, M.Q.; Kaminski, R.M.; Bar-Klein, G.; Klitgaard, H.; Koepp, M.; Jozwiak, S.; Prince, D.A.; Rotenberg, A.; et al. Repurposed molecules for antiepileptogenesis: Missing an opportunity to prevent epilepsy? Epilepsia 2020, 61, 359–386. [Google Scholar] [CrossRef] [PubMed]
- Sun Yat-Sen Memorial Hospital of Sun Yat-Sen University. Ketogenic Diet in Pediatric Intractable Epilepsy. ClinicalTrials.gov Identifier NCT06310954. Available online: https://clinicaltrials.gov/study/NCT06310954 (accessed on 25 March 2024).
- Hospital, School of Medicine, Zhejiang University. Neural Autoantibody Prevalence in New-Onset Focal Seizures of Unknown Etiology. ClinicalTrials.gov Identifier: NCT06388161. Available online: https://clinicaltrials.gov/ct2/show/NCT06388161 (accessed on 25 March 2024).
- University of California, Davis. Imaging of Neuro-Inflammation and the Risk for Post-Traumatic Epilepsy. ClinicalTrials.gov Identifier NCT03999164. Available online: https://clinicaltrials.gov/study/NCT03999164 (accessed on 25 March 2024).
- Mostafa, B. Clinical Study Evaluating Safety of Pentoxifylline and Celecoxib in Patients with Grand-Mal Epilepsy Treated by Phenytoin Monotherapy. ClinicalTrials.gov Identifier NCT05637086. Updated 26 January 2024. Available online: https://clinicaltrials.gov/study/NCT05637086 (accessed on 20 July 2024).
- Yurtdaş, D.G. Effects of Low Glycemic Index Diet in Children with Drug-Resistant Epilepsy. ClinicalTrials.gov Identifier NCT06432231. Izmir Katip Celebi University. 2024. Available online: https://clinicaltrials.gov/study/NCT06432231 (accessed on 15 July 2024).
- University Hospital, Montpellier. Serum Profile of Inflammatory Factors, Immune and Angiogenic in Temporal Lobe Epilepsy. ClinicalTrials.gov Identifier NCT01563627. Available online: https://clinicaltrials.gov/study/NCT01563627 (accessed on 15 July 2024).
- Li, F. Xiangya Hospital of Central South University. Exploring the Preventive Effect of Mitochondrial Protective Agent Idebenone on Post-Stroke Epilepsy. ClinicalTrials.gov Identifier NCT05987397. ClinicalTrials.gov. Updated 10 August 2023. Available online: https://clinicaltrials.gov/ct2/show/NCT05987397 (accessed on 7 November 2024).
- Washington University School of Medicine. Sodium-Glucose Cotransporter-2 Inhibitors: A Potential Novel Treatment for Epilepsy. ClinicalTrials.gov Identifier NCT05512130. ClinicalTrials.gov. Updated 24 August 2022. Available online: https://clinicaltrials.gov/ct2/show/NCT05512130 (accessed on 7 November 2024).
| Diagnostic Method | Level of Evidence | Application | References |
|---|---|---|---|
| Diagnosis of angiogenesis-associated pathological implications in epilepsy | |||
| Synchrotron radiation-based imaging | This technique allows for high-resolution, three-dimensional imaging of microvessels within the hippocampal area, highlighting considerable modifications in vascular architecture subsequent to seizure occurrences. Empirical observations have suggested an increase in vascular density together with activation of angiogenic factors like VEGF in individuals with temporal lobe epilepsy (TLE), reflecting a correlation with seizure frequency. | Super-resolution-based inverse light penetration computed imaging (SR-based ILPCI) offers systematic and intricate representations of cerebrovascular architecture at the micron scale, achieved without the utilization of contrast-enhancing substances. | [39] |
| Angiogenesis diagnostic chip | An extensive variety of genes, revealed through experimental investigations as being differentially expressed in tissues that undergo collateral development due to arterial occlusion, allows for the identification of single-nucleotide polymorphisms (SNPs) and other epigenetic alterations, including changes in DNA methylation patterns. | The detection of abnormally low or high differential expressions of any combination of the candidate genes can be accomplished in tissues such as peripheral blood cells. The recognition of SNPs, alongside shifts in DNA methylation patterns and differences in expression levels corresponding to one or more of the candidate genes, implies an underlying genetic predisposition toward inferior compared to superior collateral development. | [40] |
| Diagnosis of neuroinflammation in epilepsy | |||
| Positron emission tomography (PET) | PET imaging, notably using translocator protein (TSPO) ligands such as [11C] DPA713, has highlighted an elevated uptake in epileptogenic zones, thereby suggesting the manifestation of neuroinflammation. Investigations reveal that 85.1% of patients diagnosed with focal epilepsy showed an increased accumulation of [11C] DPA713 in their lesions. | Neuroimaging approaches assist in the exact localization of epileptogenic foci, particularly in situations of treatment resistance when conventional magnetic resonance imaging (MRI) may overlook abnormalities. An increase in TSPO binding has been correlated with microglial activation, thereby implying the significant role of inflammation in the pathophysiology of seizures. | [41] |
| Electroencephalogram | EEG conducted within twenty-four hours of a seizure has a greater chance of detecting epileptogenic disturbances than one performed later. On the other hand, any reduction in the EEG baseline activity that takes place up to forty-eight hours after a seizure may be momentary and must be cautiously treated. | In particular for infants, the test should be completed within twenty-four hours of an episode. | [42] |
| Brain CT scan | Performed even on those with focal seizures or neurological complications that have been found to be epileptogenic, and related anomalies can be seen in up to half of adults and up to one-third of children. | When structural damage is indicated or when it is difficult to pinpoint the cause of the episode, a CT scan is mandatory. Among many other aspects, structural lesions include spatial traumas, neuronal hypoxia, neurological damage, and post-traumatic implications that may be indicated by prolonged consciousness loss and/or post-ictal impairments. | [42] |
| Examination of CSF | In a bid to rule out a brain infection, a CSF examination is usually performed whenever a febrile seizure is occurring and associated by meningeal indicators. Despite the absence of meningeal inflammation indications, the CSF may be abnormal. | Examining the CSF is only prescribed if a brain complication is suspected in adults and children, except in the case of infants younger than six months. Examination of the CSF is generally not suggested if the patient is not suffering from high fever. | [42] |
| Diagnosis of oxidative stress markers in epilepsy | |||
| Near-infrared fluorescence probes | A novel molecular sensor has been developed for the accurate monitoring of superoxide anions, oxygen anion (O2•−), within the framework of ferroptosis-associated epilepsy, demonstrating exceptional sensitivity and specificity. This probe has been proficiently employed in numerous epilepsy models, generating valuable insights regarding the oscillations of O2•− throughout seizure occurrences. | This activatable dual-optical molecular probe enables concurrent fluorescence imaging and chemiluminescence detection of O2•− within neuronal cells, thereby enhancing the understanding of oxidative stress mechanisms in epilepsy. | [43] |
| Two-photon fluorescence probe (named HCP) | The authors have developed a robust two-photon fluorescence probe, termed HCP, which allows for the real-time observation of endogenous HClO signals produced by myeloperoxidase (MPO) in the brains of kainic acid (KA)-induced epileptic murine models, in which the chlorination of the quinolone fluorophore mediated by HClO results in an amplified fluorescence response. | Notably, by leveraging HCP, the investigators have additionally established a high-throughput screening approach to rapidly identify potential antiepileptic compounds focused on reducing MPO-mediated oxidative stress. | [44] |
| Activable dual optical molecular probe | Superoxide anion (O2•−) in epilepsy may be precisely imaged thanks to the activatable molecular probe CL-SA, opening the door to a better comprehension of the disease’s processes and possible treatment options. | Overall, CL-SA gives us a useful instrument for O2•− chemical and biological research, encouraging the study of O2•− variations in epilepsy and offering a trustworthy way to investigate epilepsy diagnosis and treatment. | [45] |
| Other diagnostic tools | Neuropsychological assessments using FMRI and SPECT cannot identify a first seizure episode from follow ups, according to the research, but can be proven effective in detection of seizure afterwards and can be based on case-by-case evidence. Neuroinflammation can be visualized properly using such techniques. | The use of neuropsychological testing, functional MRI, SPECT, and PET scans in patients who have had their first epileptic seizure is not often advised, but it is possible in some circumstances. Lesions can be properly visualized using these techniques, which can help in early diagnosis of epilepsy and other associated neurological disorders. | [46] |
| Drugs | Therapeutic Target | Clinically Used | Mechanism of Action in Epilepsy | References |
|---|---|---|---|---|
| Semaglutide | Inflammation and oxidative stress | Diabetes Mellitus | Blocking the NLR family pyrin domain-containing 3 inflammasome. It reduces oxidative stress via modulating oxidative markers. | [75] |
| Celecoxib | Inflammation | NSAIDs | Blocking of the cyclooxygenase 2 and HMGB1/TLR-4 pathways occurs. | [76] |
| Melatonin | Oxidative stress | Dietary supplement | Melatonin substantially scavenges reactive oxygen species, including hydroxyl radicals, peroxy radicals, peroxynitrite anions, and superoxide radicals, and concurrently stimulates the synthesis of superoxide dismutase and glutathione peroxidase, recognized as highly effective antioxidant enzymes. | [77] |
| Losartan | Angiogenesis | Antihypertensive | Blocks angiotensin II receptors and has shown promise as an AEG drug. | [76] |
| Thalidomide | Angiogenesis and inflammation | Sedative and tranquilizer | In order to prevent apoptosis, vascular damage, and neuronal injury in the brain, thalidomide and its derivatives demonstrated action via activating the GABAergic system. Thalidomide also lowers IL-1β, TNF-α, and IL-6 levels. | [78] |
| Isoflurane | Vasoprotective | Anesthetics | Isoflurane acts via altering thalamocortical pathways and amplifying inhibitory postsynaptic GABAA receptor-mediated currents. Additionally, it causes dose-dependent cerebral vasodilation, which raises intracranial pressure and cerebral blood flow. | [79] |
| N-Acetylcysteine | Oxidative stress | Antioxidant | NAC acts via GSH depletion under oxidative stress that is averted by tissue replenishment. The direct scavenging of ROS in brain tissues takes place due to providing a sulfhydryl group. | [80] |
| Ceftriaxone | Oxidative stress | β-lactam antibiotic | Ceftriaxone-mediated restoration of GLT-1 expression and extracellular glutamate clearance also increases the activity of the system xc−antiporter, and ceftriaxone upregulates the expression of the antiporter directly via increased nuclear Nrf2 levels. These actions increase intracellular GSH and decrease oxidative stress. | [81] |
| Atorvastatin | Inflammation | Antihyperlipidemic | Stations have anti-inflammatory effects, reducing brain penetration by monocytes and lymphocytes, reducing production of IL-1β, tumor necrosis factor (TNF), interferon-γ, and IL-6, and increasing production of IL-10, it has a free radical quenching effect, and impacts neurosteroid synthesis, all mechanisms relevant to epileptogenesis. However, only a small number of preclinical studies have evaluated antiepileptogenesis of statins, with mixed results. | [82] |
| Fingolimod | Inflammation and oxidative stress | Multiple sclerosis | The anti-inflammatory and antioxidant attributes of fingolimod (1 mg/kg ip) have been tied to the mitigation of social deficits, cognitive impairments, neuronal degeneration, and neuroinflammatory responses. Furthermore, it has been linked to the mitigation of the tripling of hippocampal levels of COX-2 and TNF. | [83] |
| Clinical Trial ID | Intervention/Treatment | Summary of Study | Status | References |
|---|---|---|---|---|
| NCT06310954 | Ketogenic diet | Aims to evaluate the efficacy of a ketogenic diet in treating pediatric intractable epilepsy and to explore its relationship with changes in inflammatory markers. | Recruiting | [84] |
| NCT06388161 | Detectable autoimmune antibodies targeting inflammation | Detectable serum neural autoantibodies, such as NMDAR, AMPAR1, AMPAR2, LON5, DPPX, GAD65, mGluR5, and MOG, will be evaluated for focal epileptic seizure. | Recruiting | [85] |
| NCT03999164 | Drug: [18F] DPA-714 positron emission tomography scanning | Patients undergo PET scans of the brain at two weeks and two months after injury to measure neuroinflammation. The results of the PET scans will be analyzed and correlated with the risk of post-traumatic epilepsy. | Phase I | [86] |
| NCT05637086 | Pentoxifylline (400 mg) and Celecoxib (200 mg) | They studied the pathogenesis of epilepsy involving multiple processes, including genetics, oxidative stress, ion channels, neuroinflammation, and cellular damage, through autophagy and apoptosis. | Phase II | [87] |
| NCT06432231 | Low glycemic index diet treatment | The goal of this clinical trial was to evaluate the effectiveness of a low glycemic index diet (LGID) on seizure frequency, oxidative stress markers, and quality of life in children with drug-resistant epilepsy. They measured malondialdehyde and paraoxonase enzyme activity | Completed | [88] |
| NCT01563627 | Blood sampling for drug resistance biomarkers. Device used: magnetic resonance imaging | The researchers demonstrated through their clinical and experimental analyses that the dysfunction of the blood–brain barrier (BBB) significantly contributes to the mechanisms underlying epileptogenesis and the occurrence of drug resistance, particularly in relation to inflammatory mediators, immune responses, and angiogenic factors in temporal lobe epilepsy. | Completed | [89] |
| NCT05987397 | Idebenone 30 mg for 14 days | Idebenone is to be administered for continuous 14 days in the post-stroke epileptic patients. The proportion of patients possessing epilepsy after stroke would be measured at different timeframes, i.e., at enrollment, after 24 weeks, and after 48 weeks, respectively. | Phase 4 | [90] |
| NCT05512130 | Empagliflozin (25 mg daily for two weeks) | Sodium-glucose cotransporter-2 inhibitors (SGLT2i), such as empagliflozin, have become important additions to the armamentarium for treating type 2 diabetes. SGLT2i decrease blood sugar by causing glucosuria, and they induce mild ketosis. These actions raise the possibility that SGLT2i can replace the MAD and LGIT as epilepsy treatments. | [91] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mohapatra, L.; Mishra, D.; Tripathi, A.S.; Parida, S.K.; Palei, N.N. Illustrating the Pathogenesis and Therapeutic Approaches of Epilepsy by Targeting Angiogenesis, Inflammation, and Oxidative Stress. Neuroglia 2025, 6, 26. https://doi.org/10.3390/neuroglia6030026
Mohapatra L, Mishra D, Tripathi AS, Parida SK, Palei NN. Illustrating the Pathogenesis and Therapeutic Approaches of Epilepsy by Targeting Angiogenesis, Inflammation, and Oxidative Stress. Neuroglia. 2025; 6(3):26. https://doi.org/10.3390/neuroglia6030026
Chicago/Turabian StyleMohapatra, Lucy, Deepak Mishra, Alok Shiomurti Tripathi, Sambit Kumar Parida, and Narahari N. Palei. 2025. "Illustrating the Pathogenesis and Therapeutic Approaches of Epilepsy by Targeting Angiogenesis, Inflammation, and Oxidative Stress" Neuroglia 6, no. 3: 26. https://doi.org/10.3390/neuroglia6030026
APA StyleMohapatra, L., Mishra, D., Tripathi, A. S., Parida, S. K., & Palei, N. N. (2025). Illustrating the Pathogenesis and Therapeutic Approaches of Epilepsy by Targeting Angiogenesis, Inflammation, and Oxidative Stress. Neuroglia, 6(3), 26. https://doi.org/10.3390/neuroglia6030026