
Antioxidant System Disturbances, Bioenergetic Disruption, and Glial Reactivity Induced by Methylmalonic Acid in the Developing Rat Brain

 , , , ,
, , , ,  and
and
Abstract
1. Introduction
2. Materials and Methods
2.1. Animals and Reagents
2.2. Intraperitoneal Administration of MMA
2.3. Intracerebroventricular Administration of MMA
2.4. Antioxidant Defenses
2.5. Bioenergetics
2.6. Western Blotting
2.7. Determination of Amino Acids Using LC-MS/MS Analysis
2.8. Statistical Analysis
3. Results
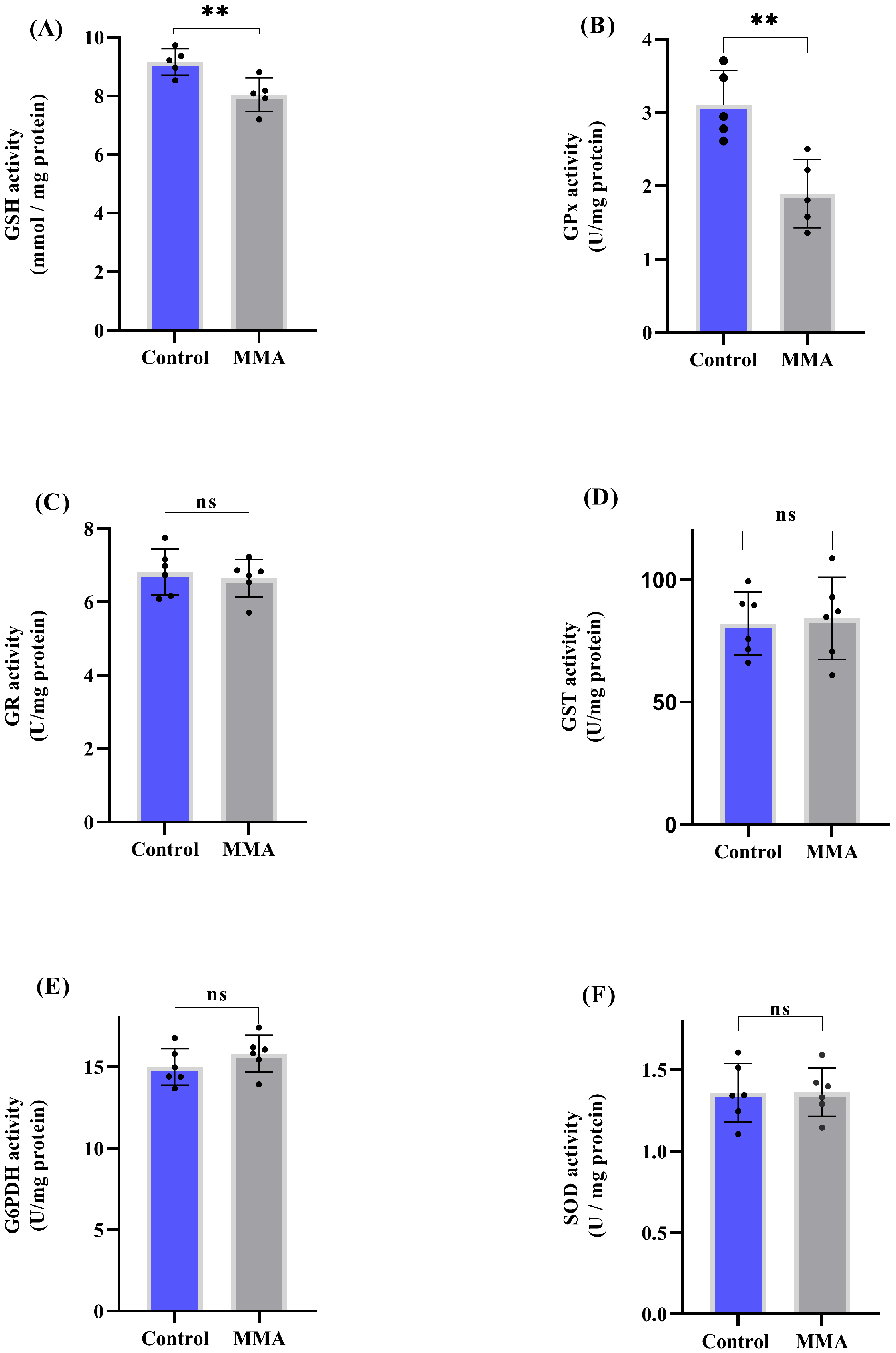
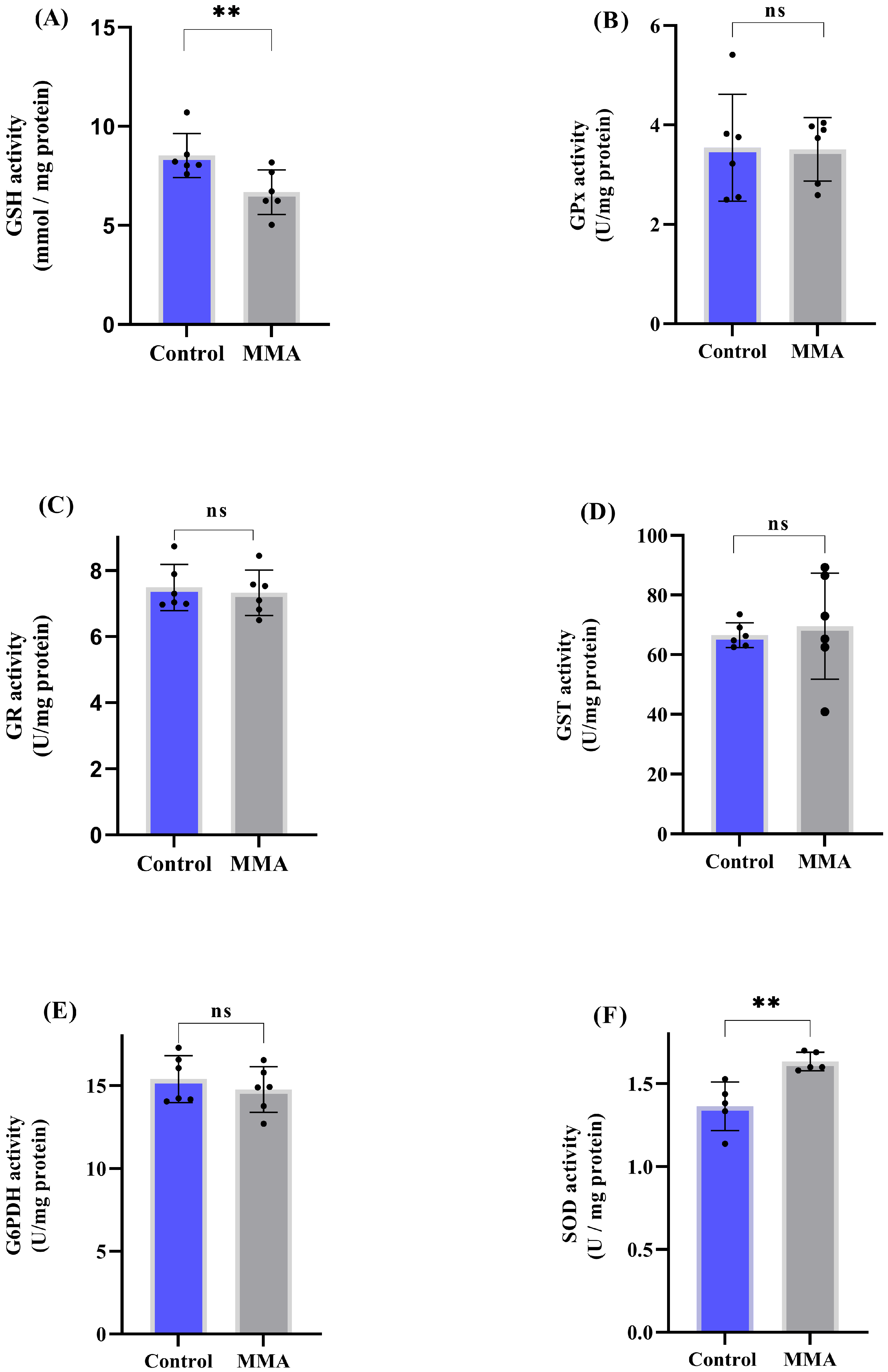
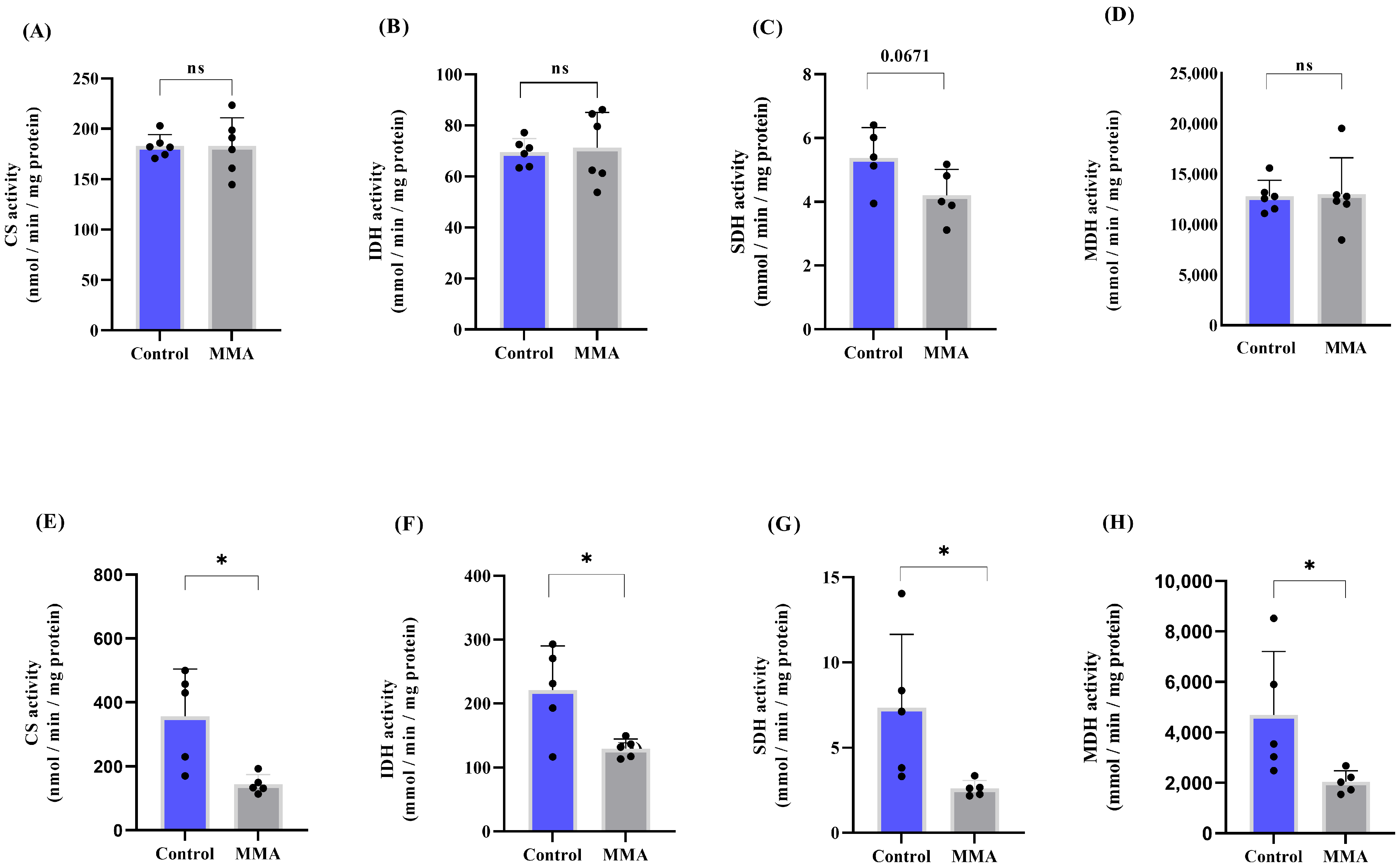
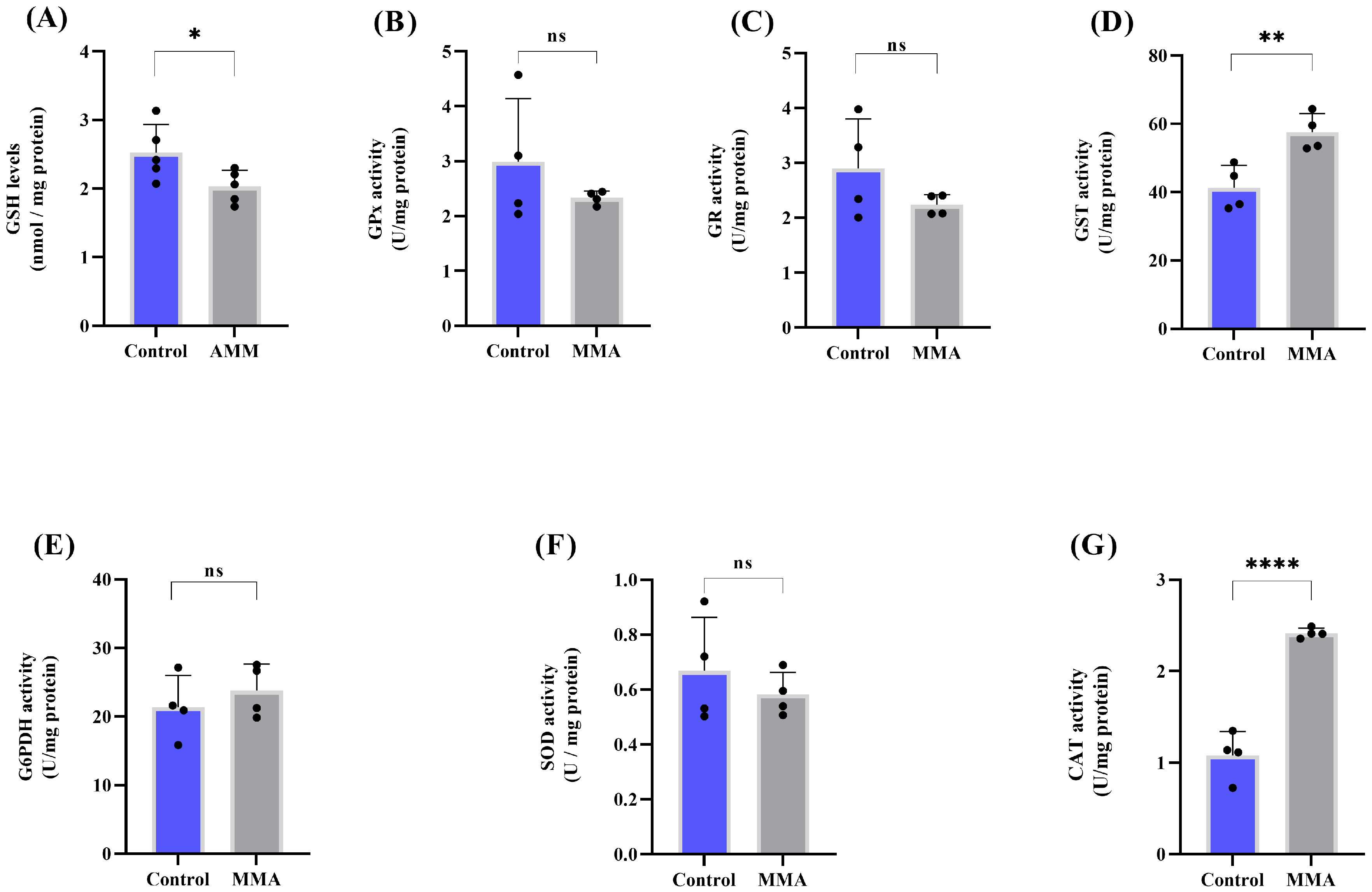
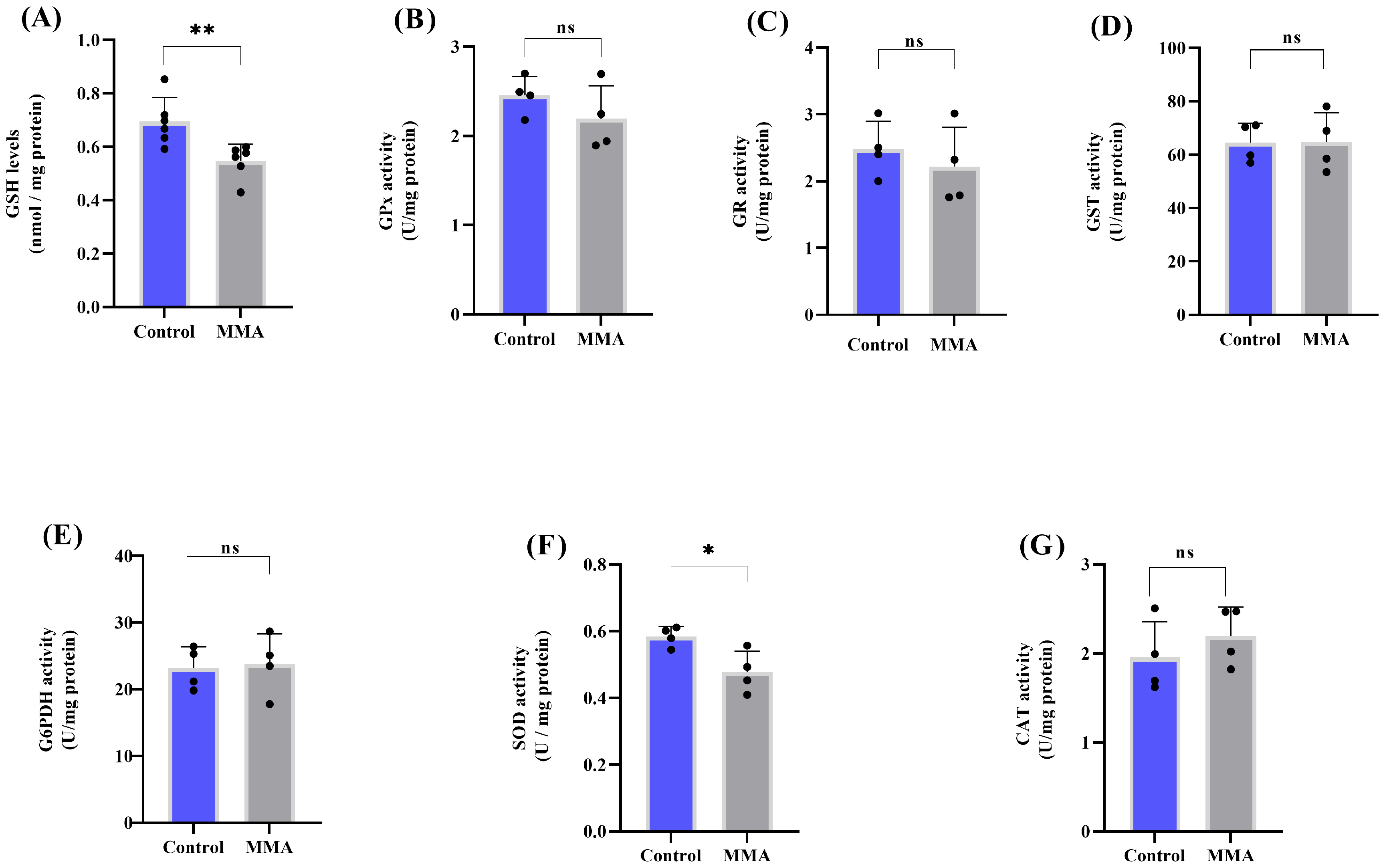
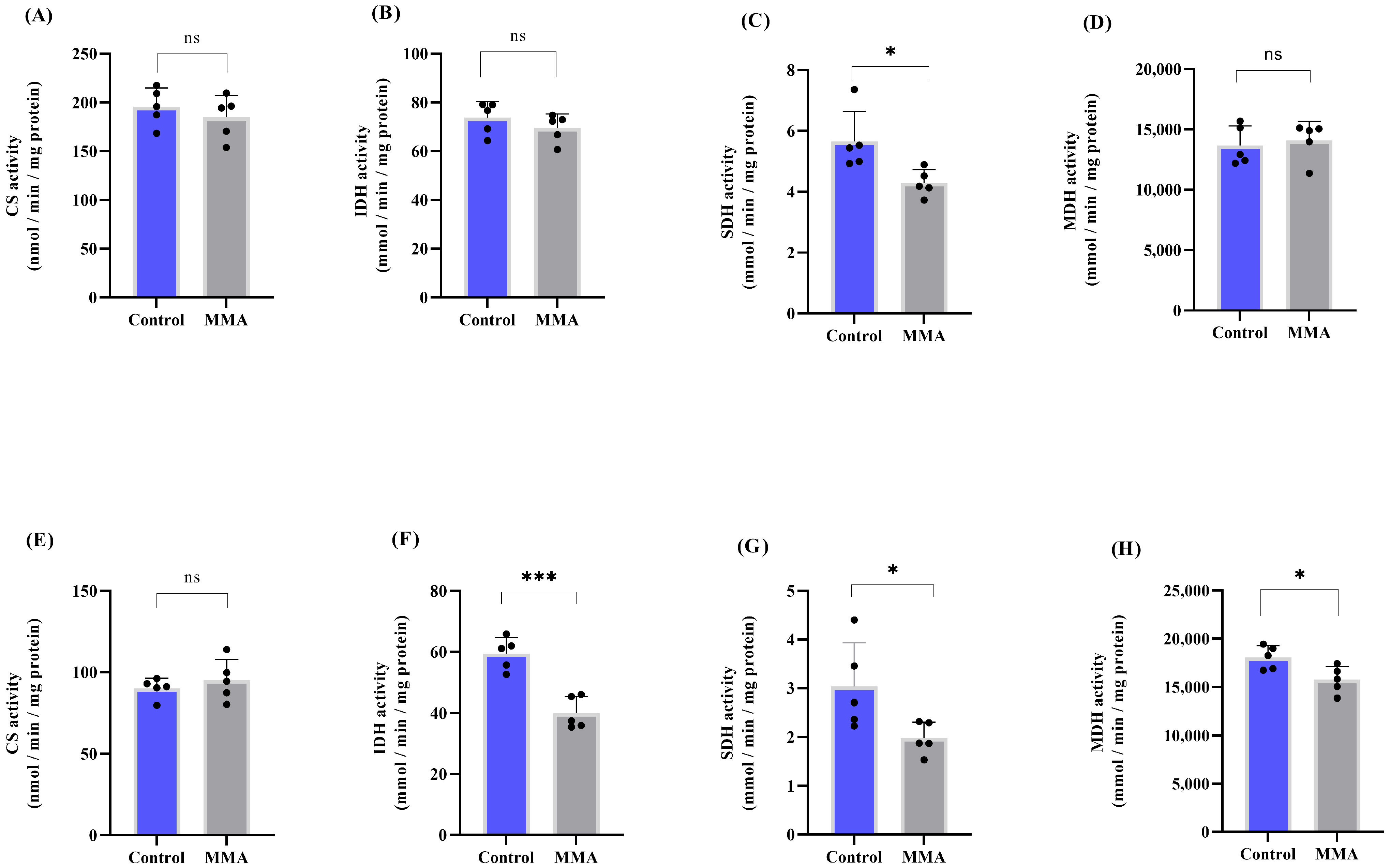
3.1. MMA Administration Disturbs Redox Status and Energy Metabolism in Rat Brain
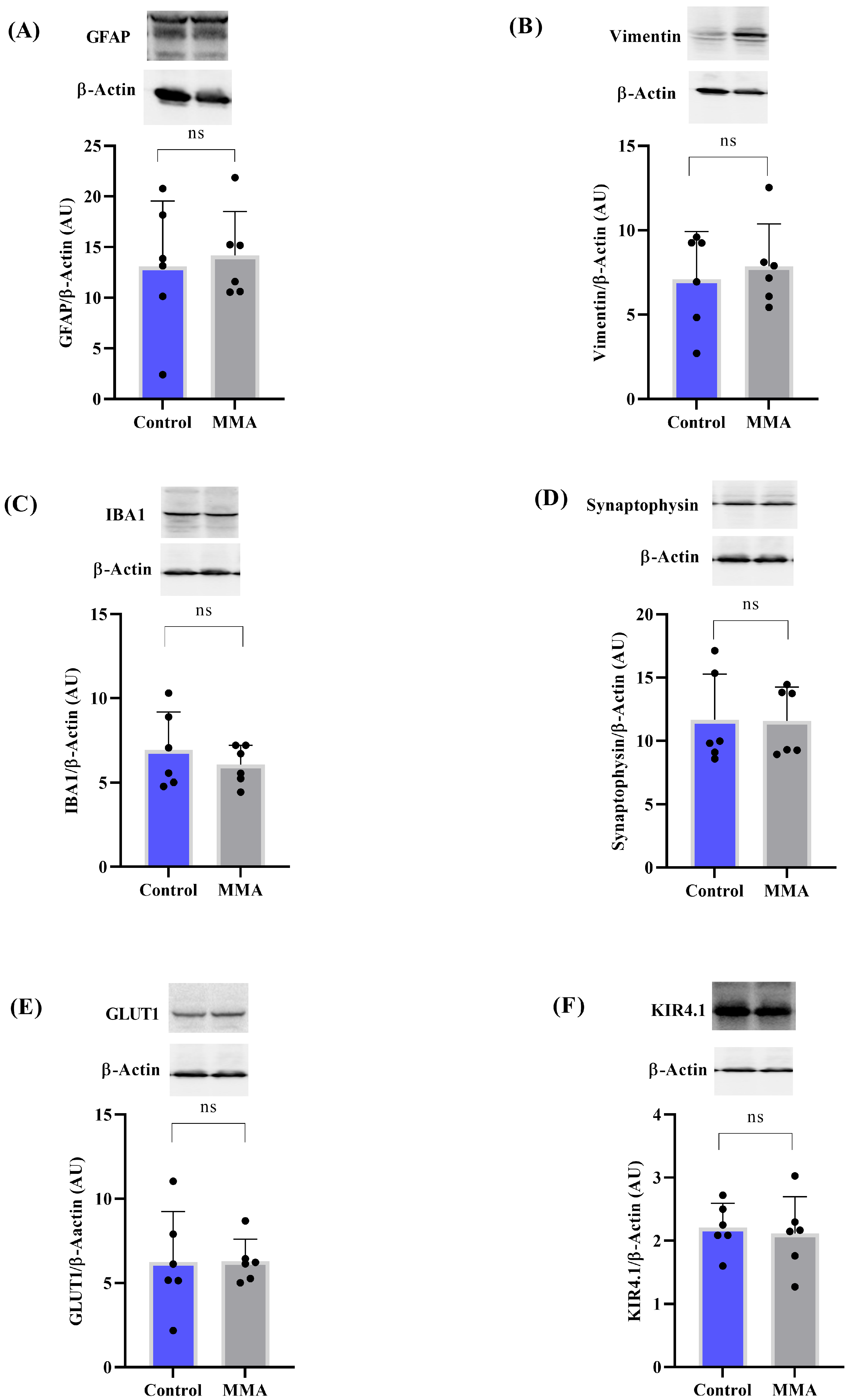
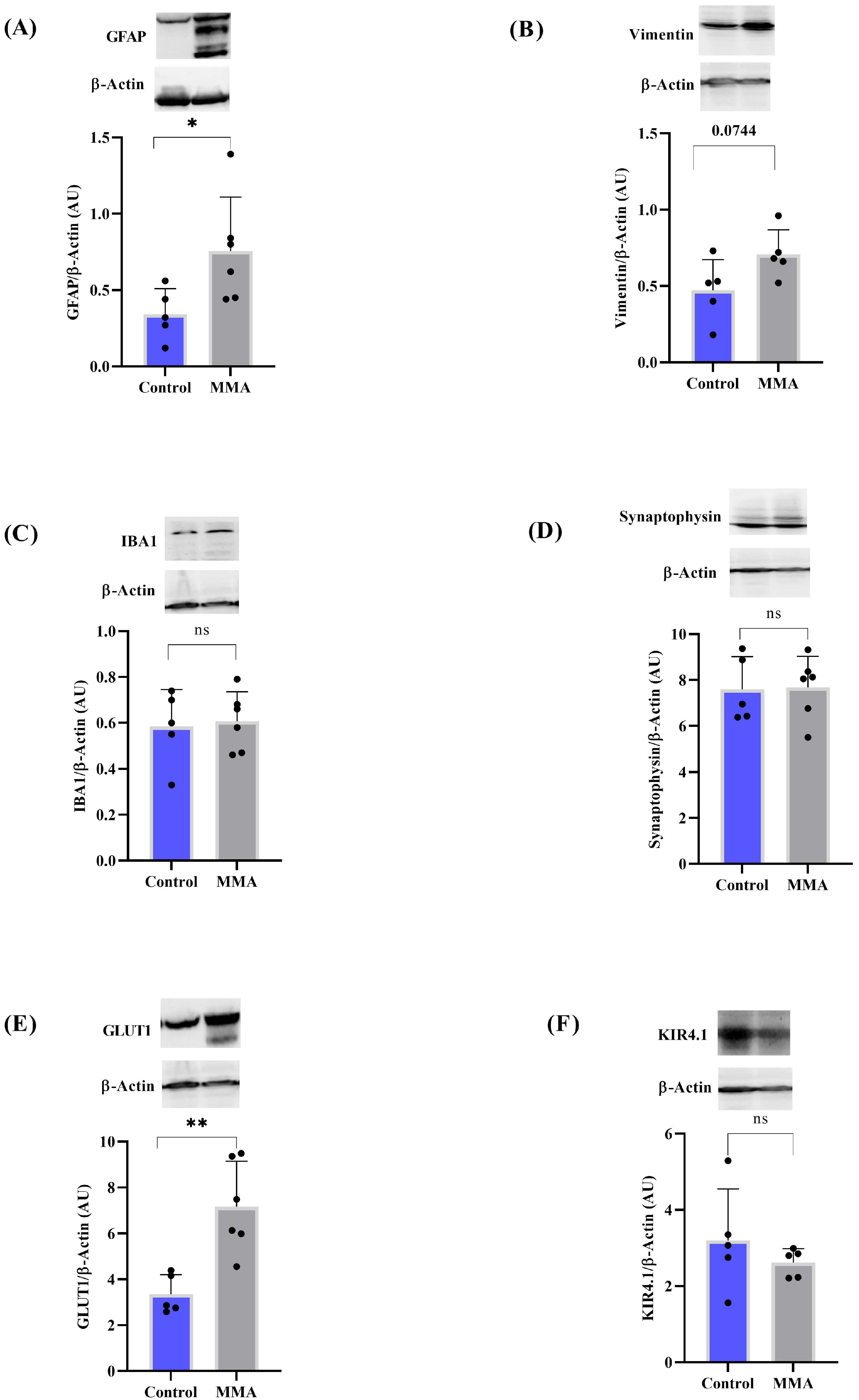
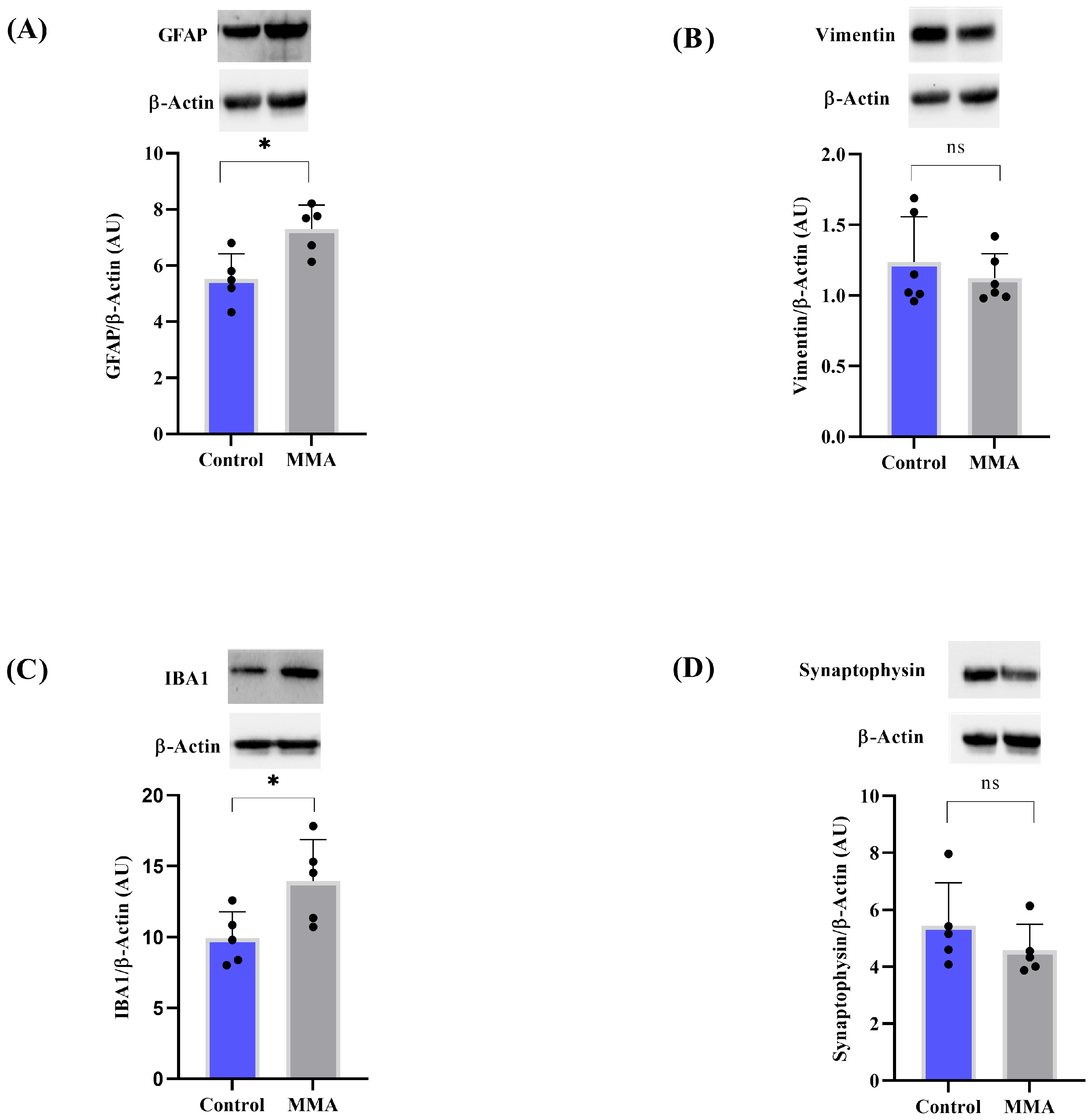
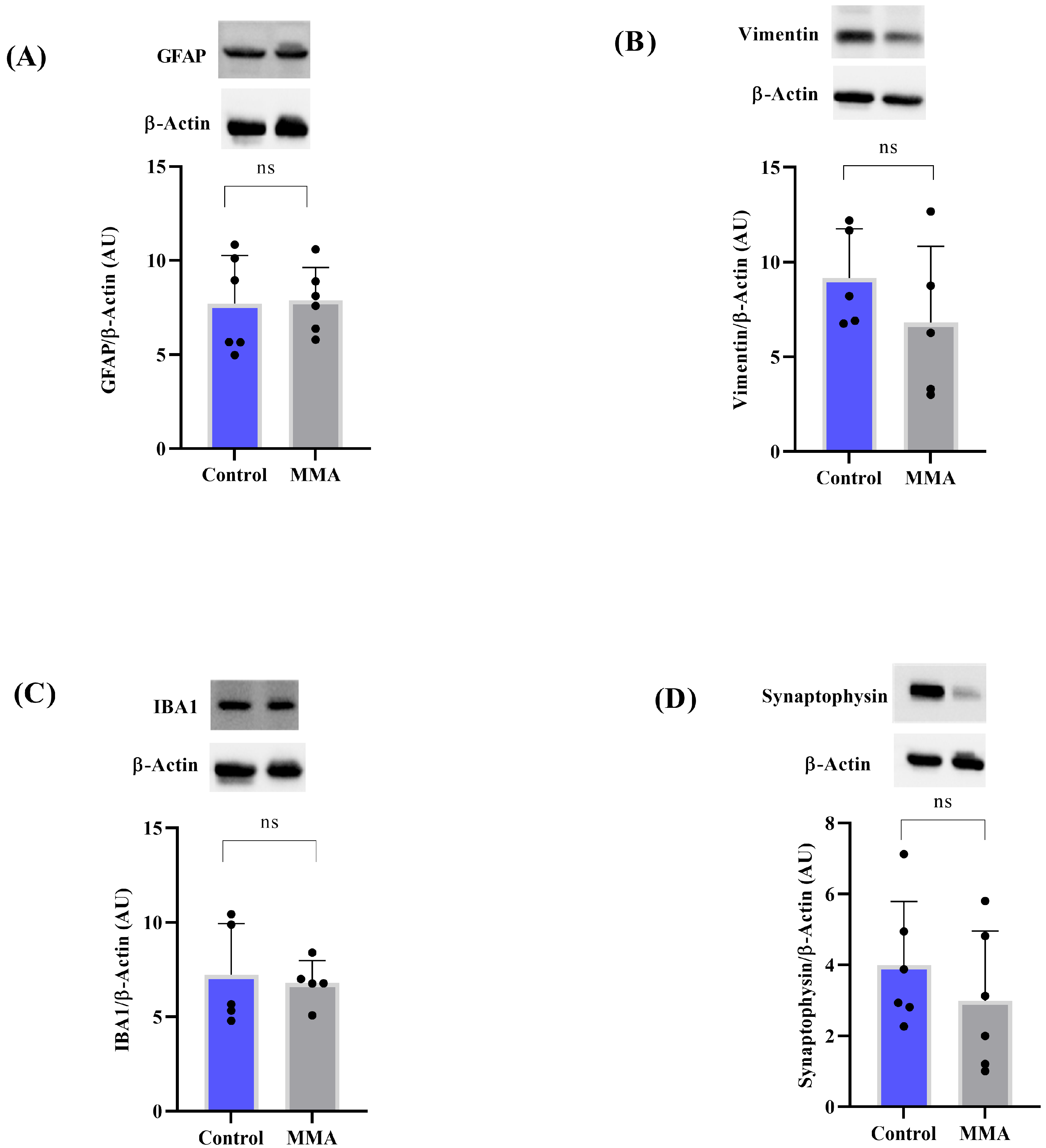
3.2. MMA Induces Glial Reactivity in the Striatum of Rats
3.3. MMA Changes Amino Acid Profile
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| CAC | Citric acid cycle |
| CAT | Catalase |
| CS | Citrate synthase |
| G6PDH | Glucose-6-phosphate dehydrogenase |
| GFAP | Glial fibrillary acid protein |
| GLUT1 | Glucose transporter 1 |
| GPx | Glutathione peroxidase |
| GR | Glutathione reductase |
| GSH | Glutathione |
| GST | Glutathione-S-transferase |
| IBA1 | Ionized calcium-binding adapter molecule 1 |
| IDH | Isocitrate Dehydrogenase |
| KIR4.1 | Inwardly rectifying potassium channel |
| MDH | Malate dehydrogenase |
| MMA | Methylmalonic acid |
| NADPH | Nicotinamide adenine dinucleotide phosphate |
| NMDA | N-methyl-D-aspartate |
| ROS | Reactive oxygen species |
| SDH | Succinate dehydrogenase |
| SOD | Superoxide Dismutase |
References
- Chen, T.; Gao, Y.; Zhang, S.; Wang, Y.; Sui, C.; Yang, L. Methylmalonic acidemia: Neurodevelopment and neuroimaging. Front. Neurosci. 2023, 17, 1110942. [Google Scholar] [CrossRef]
- Manoli, I.; Sloan, J.L.; Venditti, C.P. Isolated Methylmalonic Acidemia. In GeneReviews®; University of Washington: Seattle, WA, USA, 2005. [Google Scholar]
- Head, P.E.; Meier, J.L.; Venditti, C.P. New insights into the pathophysiology of methylmalonic acidemia. J. Inherit. Metab. Dis. 2023, 46, 436–449. [Google Scholar] [CrossRef]
- Jin, L.; Han, X.; He, F.; Zhang, C. Prevalence of methylmalonic acidemia among newborns and the clinical-suspected population: A meta-analysis. J. Matern.-Fetal Neonatal Med. 2021, 35, 8952–8967. [Google Scholar] [CrossRef]
- Fraser, J.L.; Venditti, C.P. Methylmalonic and propionic acidemias: Clinical management update. Curr. Opin. Pediatr. 2016, 28, 682–693. [Google Scholar] [CrossRef]
- Fernandes, C.G.; Borges, C.G.; Seminotti, B.; Amaral, A.U.; Knebel, L.A.; Eichler, P.; de Oliveira, A.B.; Leipnitz, G.; Wajner, M. Experimental evidence that methylmalonic acid provokes oxidative damage and compromises antioxidant defenses in nerve terminal and striatum of young rats. Cell. Mol. Neurobiol. 2011, 31, 775–785. [Google Scholar] [CrossRef]
- Chandler, R.J.; Zerfas, P.M.; Shanske, S.; Sloan, J.; Hoffmann, V.; DiMauro, S.; Venditti, C.P. Mitochondrial dysfunction in mut methylmalonic acidemia. FASEB J. 2009, 23, 1252–1261. [Google Scholar] [CrossRef]
- Schumann, A.; Brutsche, M.; Havermans, M.; Grünert, S.C.; Kölker, S.; Groß, O.; Hannibal, L. The impact of metabolic stressors on mitochondrial homeostasis in a renal epithelial cell model of methylmalonic aciduria. Sci. Rep. 2023, 13, 7677. [Google Scholar] [CrossRef]
- Melo, D.R.; Kowaltowski, A.J.; Wajner, M.; Castilho, R.F. Mitochondrial energy metabolism in neurodegeneration associated with methylmalonic acidemia. J. Bioenerg. Biomembr. 2011, 43, 39–46. [Google Scholar] [CrossRef]
- Richard, E.; Gallego-Villar, L.; Rivera-Barahona, A.; Oyarzábal, A.; Pérez, B.; Rodríguez-Pombo, P.; Desviat, L.R. Altered Redox Homeostasis in Branched-Chain Amino Acid Disorders, Organic Acidurias, and Homocystinuria. Oxidative Med. Cell. Longev. 2018, 2018, 1246069. [Google Scholar] [CrossRef]
- Semple, B.D.; Blomgren, K.; Gimlin, K.; Ferriero, D.M.; Noble-Haeusslein, L.J. Brain development in rodents and humans: Identifying benchmarks of maturation and vulnerability to injury across species. Prog. Neurobiol. 2013, 106–107, 1–16. [Google Scholar] [CrossRef]
- da Rosa, M.S.; Seminotti, B.; Amaral, A.U.; Fernandes, C.G.; Gasparotto, J.; Moreira, J.C.; Gelain, D.P.; Wajner, M.; Leipnitz, G. Redox homeostasis is compromised in vivo by the metabolites accumulating in 3-hydroxy-3-methylglutaryl-CoA lyase deficiency in rat cerebral cortex and liver. Free Radic. Res. 2013, 47, 1066–1075. [Google Scholar] [CrossRef] [PubMed]
- Paxinos, G.; Watson, C. The Rat Brain in Stereotaxic Coordinates, 7th ed.; Academic Press: Cambridge, MA, USA, 2013. [Google Scholar]
- Browne, R.W.; Armstrong, D. Reduced glutathione and glutathione disulfide. Methods Mol. Biol. 1998, 108, 347–352. [Google Scholar] [PubMed]
- Marklund, S.L. Pyrogallol autooxidation. In Handbook of Methods for Oxygen Radical Research; CRC Press: Boca Raton, FL, USA, 1985; pp. 243–247. [Google Scholar]
- Aebi, H. Catalase in vitro. In Methods in Enzymology; Academic Press: Cambridge, MA, USA, 1984; Volume 105, pp. 121–126. [Google Scholar]
- Wendel, A. Glutathione peroxidase. In Methods in Enzymology; Academic Press: Cambridge, MA, USA, 1985; Volume 77, pp. 325–333. [Google Scholar]
- Mannervik, B.; Guthenberg, C. Glutathione transferase (human placenta). In Methods in Enzymology; Academic Press: Cambridge, MA, USA, 1981; Volume 77, pp. 231–235. [Google Scholar] [CrossRef]
- Carlberg, I.; Mannervik, B. Glutathione reductase. In Methods in Enzymology; Academic Press: Cambridge, MA, USA, 1985; Volume 113, pp. 484–490. [Google Scholar]
- Leong, S.F.; Clark, J.B. Regional development of glutamate dehydrogenase in the at brain. J. Neurochem. 1984, 43, 106–111. [Google Scholar] [CrossRef]
- Grings, M.; Moura, A.P.; Parmeggiani, B.; Pletsch, J.T.; Cardoso, G.M.F.; August, P.M.; Matté, C.; Wyse, A.T.S.; Wajner, M.; Leipnitz, G. Bezafibrate prevents mitochondrial dysfunction, antioxidant system disturbance, glial reactivity and neuronal damage induced by sulfite administration in striatum of rats: Implications for a possible therapeutic strategy for sulfite oxidase deficiency. Biochim. Biophys. Acta Mol. Basis Dis. 2017, 1863, 2135–2148. [Google Scholar] [CrossRef]
- Shepherd, D.; Garland, P.B. Citrate synthase from rat liver:[EC 4.1. 3.7 Citrate oxaloacetate-lyase (CoA-acetylating)]. In Methods in Enzymology; Academic Press: Cambridge, MA, USA, 1969; Volume 13, pp. 11–16. [Google Scholar]
- Fischer, J.C.; Ruitenbeek, W.; Berden, J.A.; Trijbels, J.F.; Veerkamp, J.H.; Stadhouders, A.M.; Janssen, A.J. Differential investigation of the capacity of succinate oxidation in human skeletal muscle. Clin. Chim. Acta. 1985, 153, 23–36. [Google Scholar] [CrossRef]
- Kitto, G.B. Intra-and extramitochondrial malate dehydrogenases from chicken and tuna heart:[EC 1.1. 1.37 l-Malate: NAD oxidoreductase]. In Methods in Enzymology; Academic Press: Cambridge, MA, USA, 1969; Volume 13, pp. 106–116. [Google Scholar]
- Lowry, O.H.; Rosebrough, N.J.; Farr, A.L.; Randall, R.J. Protein measurement with the Folin phenol reagent. J. Biol. Chem. 1951, 193, 265–275. [Google Scholar] [CrossRef]
- Silveira, J.A.; Marcuzzo, M.B.; da Rosa, J.S.; Kist, N.S.; Hoffmann, C.I.H.; Carvalho, A.S.; Ribeiro, R.T.; Quincozes-Santos, A.; Netto, C.A.; Wajner, M.; et al. 3-Hydroxy-3-Methylglutaric Acid Disrupts Brain Bioenergetics, Redox Homeostasis, and Mitochondrial Dynamics and Affects Neurodevelopment in Neonatal Wistar Rats. Biomedicines 2024, 12, 1563. [Google Scholar] [CrossRef]
- Glänzel, N.M.; Parmeggiani, B.; Grings, M.; Seminotti, B.; Brondani, M.; Bobermin, L.D.; Ribeiro, C.A.J.; Quincozes-Santos, A.; Vockley, J.; Leipnitz, G. Myelin Disruption, Neuroinflammation, and Oxidative Stress Induced by Sulfite in the Striatum of Rats Are Mitigated by the pan-PPAR agonist Bezafibrate. Cells 2023, 12, 1557. [Google Scholar] [CrossRef]
- Dejanovic, B.; Sheng, M.; Hanson, J.E. Targeting synapse function and loss for treatment of neurodegenerative diseases. Nat. Rev. Drug Discov. 2024, 23, 23–42. [Google Scholar] [CrossRef]
- Gildea, H.K.; Liddelow, S.A. Mechanisms of astrocyte aging in reactivity and disease. Mol. Neurodegener. 2025, 20, 21. [Google Scholar] [CrossRef]
- Seminotti, B.; Grings, M.; Tucci, P.; Leipnitz, G.; Saso, L. Nuclear Factor Erythroid-2-Related Factor 2 Signaling in the Neuropathophysiology of Inherited Metabolic Disorders. Front. Cell. Neurosci. 2021, 15, 785057. [Google Scholar] [CrossRef] [PubMed]
- Zemniaçak, Â.B.; Ribeiro, R.T.; Pinheiro, C.V.; de Azevedo Cunha, S.; Tavares, T.Q.; Castro, E.T.; Leipnitz, G.; Wajner, M.; Amaral, A.U. In Vivo Intracerebral Administration of α-Ketoisocaproic Acid to Neonate Rats Disrupts Brain Redox Homeostasis and Promotes Neuronal Death, Glial Reactivity, and Myelination Injury. Mol. Neurobiol. 2024, 61, 2496–2513. [Google Scholar] [CrossRef] [PubMed]
- Wongkittichote, P.; Cunningham, G.; Summar, M.L.; Pumbo, E.; Forny, P.; Baumgartner, M.R.; Chapman, K.A. Tricarboxylic acid cycle enzyme activities in a mouse model of methylmalonic aciduria. Mol. Genet. Metab. 2019, 128, 444–451. [Google Scholar] [CrossRef]
- Zhang, Y.; Brasher, A.L.; Park, N.R.; Taylor, H.A.; Kavazis, A.N.; Hood, W.R. High activity before breeding improves reproductive performance by enhancing mitochondrial function and biogenesis. J. Exp. Biol. 2018, 221 Pt 7, jeb177469. [Google Scholar] [CrossRef]
- Schild, L.; Jaroscakova, I.; Lendeckel, U.; Wolf, G.; Keilhoff, G. Neuronal nitric oxide synthase controls enzyme activity pattern of mitochondria and lipid metabolism. FASEB J. 2005, 20, 145–147. [Google Scholar] [CrossRef]
- Okun, J.G.; Hörster, F.; Farkas, L.M.; Feyh, P.; Hinz, A.; Sauer, S.; Hoffmann, G.F.; Unsicker, K.; Mayatepek, E.; Kölker, S. Neurodegeneration in methylmalonic aciduria involves inhibition of complex II and the tricarboxylic acid cycle, and synergistically acting excitotoxicity. J. Biol. Chem. 2002, 277, 14674–14680. [Google Scholar] [CrossRef]
- Hadrava Vanova, K.; Kraus, M.; Neuzil, J.; Rohlena, J. Mitochondrial complex II and reactive oxygen species in disease and therapy. Redox Rep. Commun. Free Radic. Res. 2020, 25, 26–32. [Google Scholar] [CrossRef]
- Brusque, A.M.; Borba Rosa, R.; Schuck, P.F.; Dalcin, K.B.; Ribeiro, C.A.; Silva, C.G.; Wannmacher, C.M.; Dutra-Filho, C.S.; Wyse, A.T.; Briones, P.; et al. Inhibition of the mitochondrial respiratory chain complex activities in rat cerebral cortex by methylmalonic acid. Neurochem. Int. 2002, 40, 593–601. [Google Scholar] [CrossRef]
- de Mattos-Dutra, A.; Meirelles, R.; Bevilaqua da Rocha, B.; Kommers, T.; Wofchuk, S.T.; Wajner, M.; Pessoa-Pureur, R. Methylmalonic and propionic acids increase the in vitro incorporation of 32P into cytoskeletal proteins from cerebral cortex of young rats through NMDA glutamate receptors. Brain Res. 2000, 856, 111–118. [Google Scholar] [CrossRef]
- Ribeiro, L.R.; Della-Pace, I.D.; de Oliveira Ferreira, A.P.; Funck, V.R.; Pinton, S.; Bobinski, F.; de Oliveira, C.V.; da Silva Fiorin, F.; Duarte, M.M.; Furian, A.F.; et al. Chronic administration of methylmalonate on young rats alters neuroinflammatory markers and spatial memory. Immunobiology 2013, 218, 1175–1183. [Google Scholar] [CrossRef]
- Royes, L.F.; Fighera, M.R.; Furian, A.F.; Oliveira, M.S.; da Silva, L.G.; Malfatti, C.R.; Schneider, P.H.; Braga, A.L.; Wajner, M.; Mello, C.F. Creatine protects against the convulsive behavior and lactate production elicited by the intrastriatal injection of methylmalonate. Neuroscience 2003, 118, 1079–1090. [Google Scholar] [CrossRef] [PubMed]
- Greene, J.G.; Greenamyre, J.T. Bioenergetics and glutamate excitotoxicity. Prog. Neurobiol. 1996, 48, 613–634. [Google Scholar] [CrossRef] [PubMed]
- Head, P.E.; Myung, S.; Chen, Y.; Schneller, J.L.; Wang, C.; Duncan, N.; Hoffman, P.; Chang, D.; Gebremariam, A.; Gucek, M.; et al. Aberrant methylmalonylation underlies methylmalonic acidemia and is attenuated by an engineered sirtuin. Sci. Transl. Med. 2022, 14, eabn4772. [Google Scholar] [CrossRef]
- Anzmann, A.F.; Pinto, S.; Busa, V.; Carlson, J.; McRitchie, S.; Sumner, S.; Pandey, A.; Vernon, H.J. Multi-omics studies in cellular models of methylmalonic acidemia and propionic acidemia reveal dysregulation of serine metabolism. Biochim. Biophys. Acta Mol. Basis Dis. 2019, 1865, 165538. [Google Scholar] [CrossRef]
- Denley, M.C.S.; Straub, M.S.; Marcionelli, G.; Güra, M.A.; Penton, D.; Delvendahl, I.; Poms, M.; Vekeriotaite, B.; Cherkaoui, S.; Conte, F.; et al. Mitochondrial dysfunction drives a neuronal exhaustion phenotype in methylmalonic aciduria. Commun. Biol. 2025, 8, 410. [Google Scholar] [CrossRef]
- Fernstrom, J.D.; Fernstrom, M.H. Tyrosine, phenylalanine, and catecholamine synthesis and function in the brain. J. Nutr. 2007, 137, 1539S–1548S. [Google Scholar] [CrossRef] [PubMed]
- Linssen, A.M.; Riedel, W.J.; Sambeth, A. Effects of tyrosine/phenylalanine depletion on electrophysiological correlates of memory in healthy volunteers. J. Psychopharmacol. 2011, 25, 230–238. [Google Scholar] [CrossRef]
- Kempuraj, D.; Dourvetakis, K.D.; Cohen, J.; Valladares, D.S.; Joshi, R.S.; Kothuru, S.P.; Anderson, T.; Chinnappan, B.; Cheema, A.K.; Klimas, N.G.; et al. Neurovascular unit, neuroinflammation and neurodegeneration markers in brain disorders. Front. Cell. Neurosci. 2024, 18, 1491952. [Google Scholar] [CrossRef]
- de Souza Almeida, R.R.; Bobermin, L.D.; Parmeggiani, B.; Wartchow, K.M.; Souza, D.O.; Gonçalves, C.A.; Wajner, M.; Leipnitz, G.; Quincozes-Santos, A. Methylmalonic acid induces inflammatory response and redox homeostasis disruption in C6 astroglial cells: Potential glioprotective roles of melatonin and resveratrol. Amino Acids 2022, 54, 1505–1517. [Google Scholar] [CrossRef]
- Olivera, S.; Fernandez, A.; Latini, A.; Rosillo, J.C.; Casanova, G.; Wajner, M.; Cassina, P.; Barbeito, L. Astrocytic proliferation and mitochondrial dysfunction induced by accumulated glutaric acidemia I (GAI) metabolites: Possible implications for GAI pathogenesis. Neurobiol. Dis. 2008, 32, 528–534. [Google Scholar] [CrossRef]
- Olivera-Bravo, S.; Ribeiro, C.A.; Isasi, E.; Trías, E.; Leipnitz, G.; Díaz-Amarilla, P.; Woontner, M.; Beck, C.; Goodman, S.I.; Souza, D.; et al. Striatal neuronal death mediated by astrocytes from the Gcdh-/- mouse model of glutaric acidemia type I. Hum. Mol. Genet. 2015, 24, 4504–4515. [Google Scholar] [CrossRef]
- Li, Q.; Jin, H.; Liu, Y.; Rong, Y.; Yang, T.; Nie, X.; Song, W. Determination of Cytokines and Oxidative Stress Biomarkers in Cognitive Impairment Induced by Methylmalonic Acidemia. Neuroimmunomodulation 2021, 28, 178–186. [Google Scholar] [CrossRef]
- Glänzel, N.M.; Grings, M.; da Rosa-Junior, N.T.; de Carvalho, L.M.C.; Mohsen, A.W.; Wipf, P.; Wajner, M.; Vockley, J.; Leipnitz, G. The mitochondrial-targeted reactive species scavenger JP4-039 prevents sulfite-induced alterations in antioxidant defenses, energy transfer, and cell death signaling in striatum of rats. J. Inherit. Metab. Dis. 2021, 44, 481–491. [Google Scholar] [CrossRef]
- da Rosa, M.S.; da Rosa-Junior, N.T.; Parmeggiani, B.; Glänzel, N.M.; de Moura Alvorcem, L.; Ribeiro, R.T.; Grings, M.; Wajner, M.; Leipnitz, G. 3-Hydroxy-3-Methylglutaric Acid Impairs Redox and Energy Homeostasis, Mitochondrial Dynamics, and Endoplasmic Reticulum-Mitochondria Crosstalk in Rat Brain. Neurotox. Res. 2020, 37, 314–325. [Google Scholar] [CrossRef] [PubMed]
- Seminotti, B.; Amaral, A.U.; Ribeiro, R.T.; Rodrigues, M.D.N.; Colín-González, A.L.; Leipnitz, G.; Santamaría, A.; Wajner, M. Oxidative Stress, Disrupted Energy Metabolism, and Altered Signaling Pathways in Glutaryl-CoA Dehydrogenase Knockout Mice: Potential Implications of Quinolinic Acid Toxicity in the Neuropathology of Glutaric Acidemia Type I. Mol. Neurobiol. 2016, 53, 6459–6475. [Google Scholar] [CrossRef] [PubMed]
- Vannucci, S.J. Developmental expression of GLUT1 and GLUT3 glucose transporters in rat brain. J. Neurochem. 1994, 62, 240–246. [Google Scholar] [CrossRef]
- Thieren, L.; Zanker, H.S.; Droux, J.; Dalvi, U.; Wyss, M.T.; Waag, R.; Germain, P.-L.; von Ziegler, L.M.; Looser, Z.J.; Hösli, L.; et al. Astrocytic GLUT1 deletion in adult mice enhances glucose metabolism and resilience to stroke. Nat. Commun. 2025, 16, 4190. [Google Scholar] [CrossRef] [PubMed]
- Almeida, A.; Jimenez-Blasco, D.; Bolaños, J.P. Cross-talk between energy and redox metabolism in astrocyte-neuron functional cooperation. Essays Biochem. 2023, 67, 17–26. [Google Scholar] [CrossRef]
- Luciani, A.; Schumann, A.; Berquez, M.; Chen, Z.; Nieri, D.; Failli, M.; Debaix, H.; Festa, B.P.; Tokonami, N.; Raimondi, A.; et al. Impaired mitophagy links mitochondrial disease to epithelial stress in methylmalonyl-CoA mutase deficiency. Nat. Commun. 2020, 11, 970. [Google Scholar] [CrossRef]
- Brustovetsky, N.; Brustovetsky, T.; Purl, K.J.; Capano, M.; Crompton, M.; Dubinsky, J.M. Increased Susceptibility of Striatal Mitochondria to Calcium-Induced Permeability Transition. J. Neurosci. 2003, 23, 4858–4867. [Google Scholar] [CrossRef]
- Pickrell, A.M.; Fukui, H.; Wang, X.; Pinto, M.; Moraes, T.C. The Striatum Is Highly Susceptible to Mitochondrial Oxidative Phosphorylation Dysfunctions. J. Neurosci. 2011, 31, 9895–9904. [Google Scholar] [CrossRef] [PubMed]
- Reig, R.; Silberberg, G. Distinct. Corticostriatal and Intracortical Pathways Mediate Bilateral Sensory Responses in the Striatum. Cereb. Cortex 2016, 26, 4405–4415. [Google Scholar] [CrossRef] [PubMed]
- Halliwell, B.; Gutteridge, J.M.C. Free Radicals in Biology and Medicine, 5th ed.; Oxford Academic: Oxford, UK, 2015. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cerebral Cortex | Striatum | |||||
|---|---|---|---|---|---|---|
| AMINO ACID | Control (ng/mg) | MMA (ng/mg) | p Value | Control (ng/mg) | MMA (ng/mg) | p Value |
| Threonine | 4.05 ± 0.47 | 4.93 ± 2.91 | 0.3862 | 3.93 ± 0.85 | 4.14 ± 0.49 | 0.6037 |
| Tryptophan | 5.22 ± 1.05 | 4.05 ± 1.10 | 0.3386 | 5.00 ± 1.24 | 4.62 ± 0.70 | 0.5202 |
| Valine | 1.65 ± 0.34 | 1.77 ± 0.44 | 0.6146 | 1.45 ± 0.23 | 1.55 ± 0.36 | 0.5956 |
| Tyrosine | 100 ± 20.2 | 105 ± 32.6 | 0.7740 | 91.0 ± 15.9 | 87.1 ± 12.4 | 0.0472 * |
| Serine | 1.34 ± 0.08 | 1.38 ± 0.40 | 0.7843 | 1.23 ± 0.21 | 1.43 ± 0.15 | 0.0492 * |
| Proline | 0.62 ± 0.10 | 0.84 ± 0.72 | 0.5460 | 0.60 ± 0.14 | 0.62 ± 0.12 | 0.7249 |
| Methionine | 3.30 ± 0.54 | 3.64 ± 1.23 | 0.6298 | 3.72 ± 0.70 | 3.60 ± 0.63 | 0.7751 |
| Lysine | 0.62 ± 0.15 | 0.72 ± 0.27 | 0.5818 | 0.63 ± 0.16 | 0.62 ± 0.09 | 0.9634 |
| Hydroxyproline | 0.77 ± 0.09 | 0.88 ± 0.24 | 0.9747 | 0.49 ± 0.10 | 0.68 ± 0.11 | 0.7827 |
| Histidine | 19.1 ± 5.12 | 20.8 ± 8.40 | 0.3893 | 20.0 ± 4.28 | 22.4 ± 2.57 | 0.2770 |
| Leucine | 5.63 ± 0.83 | 6.19 ± 1.88 | 0.5810 | 6.55 ± 1.27 | 6.38 ± 1.60 | 0.8328 |
| Isoleucine | 2.65 ± 0.46 | 2.87 ± 0.71 | 0.5334 | 2.49 ± 0.37 | 2.52 ± 0.50 | 0.8908 |
| Glutamine | 26.9 ± 8.90 | 29.0 ± 14.4 | 0.8524 | 24.2 ± 6.07 | 25.0 ± 4.11 | 0.7827 |
| Alanine | 4.60 ± 0.53 | 3.92 ± 0.74 | 0.7120 | 3.20 ± 0.36 | 3.67 ± 0.56 | 0.3739 |
| Arginine | 4.76 ± 1.24 | 5.44 ± 2.73 | 0.2816 | 6.46 ± 1.04 | 6.61 ± 1.34 | 0.9289 |
| Asparagine | 0.70 ± 0.12 | 0.66 ± 0.22 | 0.7263 | 0.56 ± 0.09 | 0.60 ± 0.07 | 0.3584 |
| Phenylalanine | 349 ± 60.4 | 384 ± 68.9 | 0.3659 | 354 ± 75.8 | 356 ± 66.5 | 0.9684 |
| Glycine | 1.37 ± 0.20 | 1.52 ± 0.34 | 0.9065 | 1.15 ± 0.16 | 1.32 ± 0.14 | 0.0087 |
| Glutamic acid | 77.8 ± 16.4 | 80.6 ± 20.4 | 0.7963 | 54.3 ± 7.53 | 65.0 ± 6.14 | 0.0217 * |
| Aspartic acid | 42.4 ± 7.78 | 40.7 ± 12.2 | 0.7781 | 38.6 ± 7.39 | 43.9 ± 8.34 | 0.2739 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dalpizolo, C.A.; de Andrade Silveira, J.; Marcuzzo, M.B.; Gayger-Dias, V.; Da Silva, V.-F.; Pinheiro, C.V.; Santos, B.P.d.; de Oliveira, T.F.; Gonçalves, C.-A.; Leipnitz, G. Antioxidant System Disturbances, Bioenergetic Disruption, and Glial Reactivity Induced by Methylmalonic Acid in the Developing Rat Brain. Neuroglia 2025, 6, 25. https://doi.org/10.3390/neuroglia6030025
Dalpizolo CA, de Andrade Silveira J, Marcuzzo MB, Gayger-Dias V, Da Silva V-F, Pinheiro CV, Santos BPd, de Oliveira TF, Gonçalves C-A, Leipnitz G. Antioxidant System Disturbances, Bioenergetic Disruption, and Glial Reactivity Induced by Methylmalonic Acid in the Developing Rat Brain. Neuroglia. 2025; 6(3):25. https://doi.org/10.3390/neuroglia6030025
Chicago/Turabian StyleDalpizolo, Cristiano Antonio, Josyane de Andrade Silveira, Manuela Bianchin Marcuzzo, Vitor Gayger-Dias, Vanessa-Fernanda Da Silva, Camila Vieira Pinheiro, Bruno Pereira dos Santos, Tiago Franco de Oliveira, Carlos-Alberto Gonçalves, and Guilhian Leipnitz. 2025. "Antioxidant System Disturbances, Bioenergetic Disruption, and Glial Reactivity Induced by Methylmalonic Acid in the Developing Rat Brain" Neuroglia 6, no. 3: 25. https://doi.org/10.3390/neuroglia6030025
APA StyleDalpizolo, C. A., de Andrade Silveira, J., Marcuzzo, M. B., Gayger-Dias, V., Da Silva, V.-F., Pinheiro, C. V., Santos, B. P. d., de Oliveira, T. F., Gonçalves, C.-A., & Leipnitz, G. (2025). Antioxidant System Disturbances, Bioenergetic Disruption, and Glial Reactivity Induced by Methylmalonic Acid in the Developing Rat Brain. Neuroglia, 6(3), 25. https://doi.org/10.3390/neuroglia6030025