

Reducing Brain Edema Using Berotralstat, an Inhibitor of Bradykinin, Repurposed as Treatment Adjunct in Glioblastoma

{kind=link}
{kind=link}
Abstract
1. Introduction
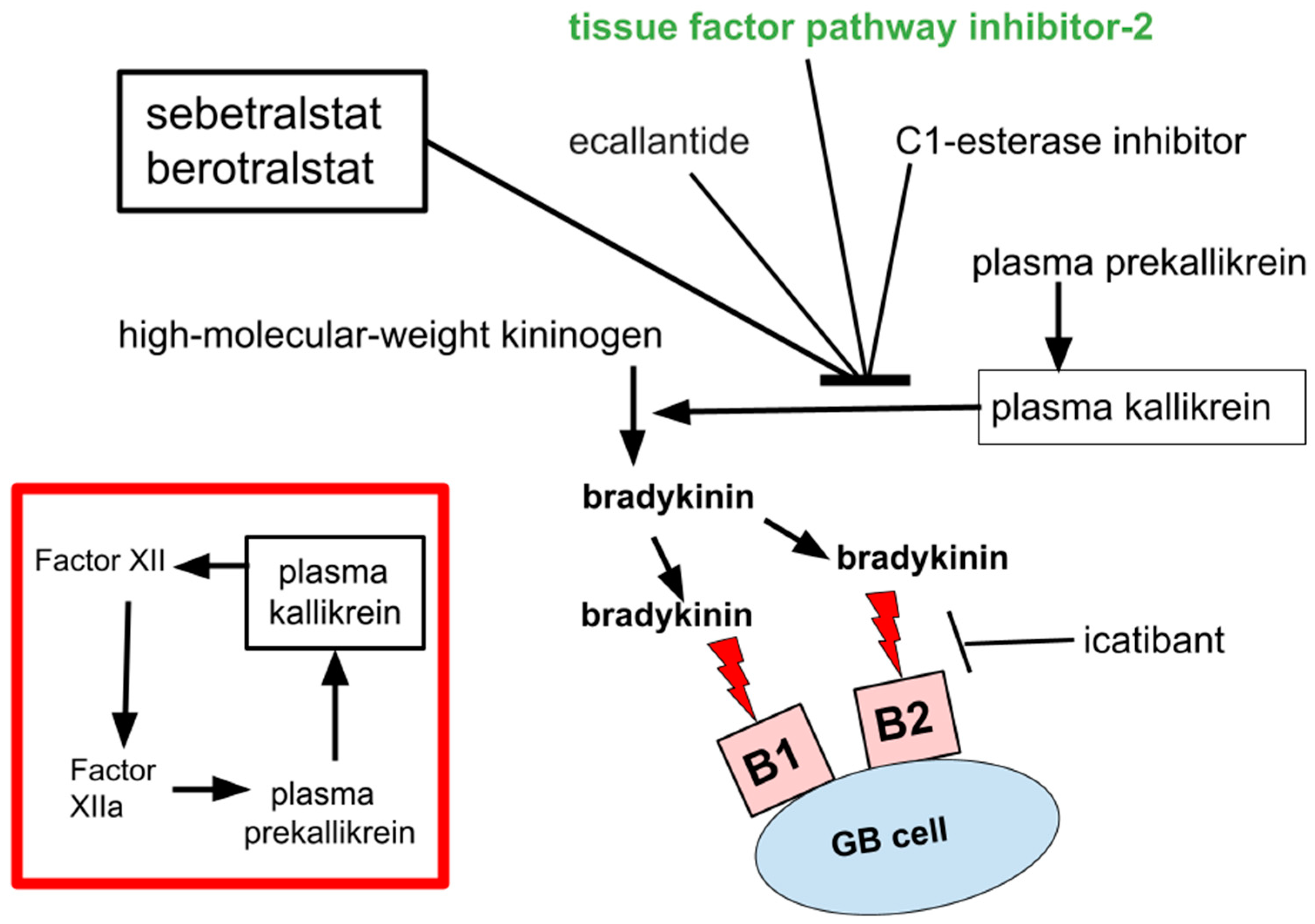
2. Bradykinin
3. Safety of Bradykinin Inhibition
4. Pathogenic Contributions of Bradykinin in GB
- The edema consequent to BBB leakage predisposes that edematous tissue to allow or enhance GB invasion and regrowth [40]. Striking evidence of the importance of restraining bradykinin signaling to the mammalian brain comes from C1 esterase inhibitor knock-down mice, who experience widespread brain edema and generalized cerebral non-malignant glial activation [42].
- GBs’ peritumoral edema itself can, and not rarely does, precipitate death by brain herniation.
- Edema clinically requires dexamethasone use to control it, at least today in 2024.
5. Evidence of Bradykinin Signaling Driving Other Cancers
6. Data on BBB Opening for Drug Delivery Using Bradykinin Agonists
7. Discussion
- (1)
- Dapsone
- (2)
- Celecoxib
- (3)
- SEC
8. Conclusions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Weydt, P.; Möller, T.; Labrakakis, C.; Patt, S.; Kettenmann, H. Neuroligand-triggered calcium signalling in cultured human glioma cells. Neurosci. Lett. 1997, 228, 91–94. [Google Scholar] [CrossRef]
- Stadnicka, I.; Strzałka-Mrozik, B.; Kimsa-Dudek, M.; Kaspera, W.; Plewka, A.; Szopa, W.; Stadnicki, A. Kinin Receptors and Kinin-Related Gene Expression in Astrocytic Brain Tumors. Cancers 2024, 16, 241. [Google Scholar] [CrossRef]
- Farkas, H.; Balla, Z. A review of berotralstat for the treatment of hereditary angioedema. Expert Rev. Clin. Immunol. 2023, 19, 145–153. [Google Scholar] [CrossRef]
- Ohsawa, I.; Honda, D.; Suzuki, Y.; Fukuda, T.; Kohga, K.; Morita, E.; Moriwaki, S.; Ishikawa, O.; Sasaki, Y.; Tago, M.; et al. Oral berotralstat for the prophylaxis of hereditary angioedema attacks in patients in Japan: A phase 3 randomized trial. Allergy 2021, 76, 1789–1799. [Google Scholar] [CrossRef]
- Zuraw, B.; Lumry, W.R.; Johnston, D.T.; Aygören-Pürsün, E.; Banerji, A.; Bernstein, J.A.; Christiansen, S.C.; Jacobs, J.S.; Sitz, K.V.; Gower, R.G.; et al. Oral once-daily berotralstat for the prevention of hereditary angioedema attacks: A randomized, double-blind, placebo-controlled phase 3 trial. J. Allergy Clin. Immunol. 2020, 148, 164–172. [Google Scholar] [CrossRef]
- Davie, R.L.; Edwards, H.J.; Evans, D.M.; Hodgson, S.T.; Stocks, M.J.; Smith, A.J.; Rushbrooke, L.J.; Pethen, S.J.; Roe, M.B.; Clark, D.E.; et al. Sebetralstat (KVD900): A Potent and Selective Small Molecule Plasma Kallikrein Inhibitor Featuring a Novel P1 Group as a Potential Oral On-Demand Treatment for Hereditary Angioedema. J. Med. Chem. 2022, 65, 13629–13644. [Google Scholar] [CrossRef]
- Mutch, P.; Bashir, M.; Jung, B.; Yi, P.; Iverson, M. Absorption, metabolism, and excretion of 14C-sebetralstat (KVD900) following a single oral dose in healthy male participants. Xenobiotica 2022, 52, 707–717. [Google Scholar] [CrossRef]
- Cohn, D.M.; Aygören-Pürsün, E.; Bernstein, J.A.; Farkas, H.; Lumry, W.R.; Maurer, M.; Zanichelli, A.; Iverson, M.; Hao, J.; Smith, M.D.; et al. Evaluation of patient reported outcome measures for on-demand treatment of hereditary angioedema attacks and design of KONFIDENT, a phase 3 trial of sebetralstat. Clin. Transl. Allergy 2023, 13, e12288. [Google Scholar] [CrossRef]
- Aygören-Pürsün, E.; Zanichelli, A.; Cohn, D.M.; Cancian, M.; Hakl, R.; Kinaciyan, T.; Magerl, M.; Martinez-Saguer, I.; Stobiecki, M.; Farkas, H.; et al. An investigational oral plasma kallikrein inhibitor for on-demand treatment of hereditary angioedema: A two-part, randomised, double-blind, placebo-controlled, crossover phase 2 trial. Lancet 2023, 401, 458–469. [Google Scholar] [CrossRef]
- Lima, H.; Zheng, J.; Wong, D.; Waserman, S.; Sussman, G.L. Pathophysiology of bradykinin and histamine mediated angioedema. Front. Allergy 2023, 4, 1263432. [Google Scholar] [CrossRef]
- Arora, H.; Mammi, M.; Patel, N.M.; Zyfi, D.; Dasari, H.R.; Yunusa, I.; Simjian, T.; Smith, T.R.; Mekary, R.A. Dexamethasone and overall survival and progression free survival in patients with newly diagnosed glioblastoma: A meta-analysis. J. Neurooncol. 2024, 166, 17–26. [Google Scholar] [CrossRef]
- Shields, L.B.; Shelton, B.J.; Shearer, A.J.; Chen, L.; Sun, D.A.; Parsons, S.; Bourne, T.D.; LaRocca, R.; Spalding, A.C. Dexamethasone administration during definitive radiation and temozolomide renders a poor prognosis in a retrospective analysis of newly diagnosed glioblastoma patients. Radiat. Oncol. 2015, 10, 222. [Google Scholar] [CrossRef]
- Marceau, F.; Bachelard, H.; Bouthillier, J.; Fortin, J.P.; Morissette, G.; Bawolak, M.T.; Charest-Morin, X.; Gera, L. Bradykinin receptors: Agonists, antagonists, expression, signaling, and adaptation to sustained stimulation. Int. Immunopharmacol. 2020, 82, 106305. [Google Scholar] [CrossRef]
- Kazandjieva, J.; Christoff, G. Angioedema as a systemic disease. Clin. Dermatol. 2019, 37, 636–643. [Google Scholar] [CrossRef]
- Margaglione, M.; D‘Apolito, M.; Santocroce, R.; Maffione, A.B. Hereditary angioedema: Looking for bradykinin production and triggers of vascular permeability. Clin. Exp. Allergy 2019, 49, 1395–1402. [Google Scholar] [CrossRef]
- Maas, C.; López-Lera, A. Hereditary Angioedema: Insights into inflammation and allergy. Mol. Immunol. 2019, 112, 378–386. [Google Scholar] [CrossRef]
- Patel, G.; Pongracic, J.A. Hereditary and acquired angioedema. Allergy Asthma Proc. 2019, 40, 441–445. [Google Scholar] [CrossRef]
- Silvani, A.; Gaviani, P.; Lamperti, E.; Botturi, A.; Ferrari, D.; Simonetti, G.; Fariselli, L.; Salmaggi, A. Metabolic, electrolytes disorders and thromboembolic risk in malignant glioma patients. Neurol. Sci. 2011, 32 (Suppl. S2), S229–S231. [Google Scholar] [CrossRef]
- Renné, T.; Stavrou, E.X. Roles of Factor XII in Innate Immunity. Front. Immunol. 2019, 10, 2011. [Google Scholar] [CrossRef]
- Dewhirst, M.W.; Vinuya, R.Z.; Ong, E.T.; Klitzman, B.; Rosner, G.; Secomb, T.W.; Gross, J.F. Effects of bradykinin on the hemodynamics of tumor and granulating normal tissue microvasculature. Radiat. Res. 1992, 130, 345–354. [Google Scholar] [CrossRef]
- Zerrouqi, A.; Pyrzynska, B.; Brat, D.J.; Van Meir, E.G. P14ARF suppresses tumor-induced thrombosis by regulating the tissue factor pathway. Cancer Res. 2014, 74, 1371–1378. [Google Scholar] [CrossRef]
- Yee, P.P.; Wei, Y.; Kim, S.Y.; Lu, T.; Chih, S.Y.; Lawson, C.; Tang, M.; Liu, Z.; Anderson, B.; Thamburaj, K.; et al. Neutrophil-induced ferroptosis promotes tumor necrosis in glioblastoma progression. Nat. Commun. 2020, 11, 5424. [Google Scholar] [CrossRef]
- Schiffer, D.; Mellai, M.; Bovio, E.; Bisogno, I.; Casalone, C.; Annovazzi, L. Glioblastoma niches: From the concept to the phenotypical reality. Neurol. Sci. 2018, 39, 1161–1168. [Google Scholar] [CrossRef]
- Schiffer, D.; Annovazzi, L.; Mellai, M. A comprehensive view of tumor stem cells and their regulation by the microenvironment in glioblastoma. Neurol. Sci. 2017, 38, 527–529. [Google Scholar] [CrossRef]
- Srinivasan, C.; Ritchie, B.; Adatia, A. Berotralstat in hereditary angioedema due to C1 inhibitor deficiency: First real-world evidence from a Canadian center. Front. Immunol. 2024, 15, 1339421. [Google Scholar] [CrossRef]
- Aygören-Pürsün, E.; Bygum, A.; Grivcheva-Panovska, V.; Magerl, M.; Graff, J.; Steiner, U.C.; Fain, O.; Huissoon, A.; Kinaciyan, T.; Farkas, H.; et al. Oral Plasma Kallikrein Inhibitor for Prophylaxis in Hereditary Angioedema. N. Engl. J. Med. 2018, 379, 352–362. [Google Scholar] [CrossRef]
- Johnson, F.; Stenzl, A.; Hofauer, B.; Heppt, H.; Ebert, E.V.; Wollenberg, B.; Lochbaum, R.; Hahn, J.; Greve, J.; Trainotti, S. A Retrospective Analysis of Long-Term Prophylaxis with Berotralstat in Patients with Hereditary Angioedema and Acquired C1-Inhibitor Deficiency-Real-World Data. Clin. Rev. Allergy Immunol. 2023, 65, 354–364. [Google Scholar] [CrossRef]
- Ahuja, M.; Dorr, A.; Bode, E.; Boulton, A.P.R.; Buckland, M.; Chee, S.; Dalley, C.; Denman, S.; Ekbote, A.; Elkhalifa, S.; et al. Berotralstat for the prophylaxis of hereditary angioedema-Real-world evidence data from the United Kingdom. Allergy 2023, 78, 1380–1383. [Google Scholar] [CrossRef]
- Lu, D.Y.; Leung, Y.M.; Huang, S.M.; Wong, K.L. Bradykinin-induced cell migration and COX-2 production mediated by the bradykinin B1 receptor in glioma cells. J. Cell. Biochem. 2010, 110, 141–150. [Google Scholar] [CrossRef]
- Gallo, V. Lethal migration: The bradykinin story. J. Physiol. 2014, 592, 4805–4806. [Google Scholar] [CrossRef]
- Nicoletti, N.F.; Sénécal, J.; da Silva, V.D.; Roxo, M.R.; Ferreira, N.P.; de Morais, R.L.T.; Pesquero, J.B.; Campos, M.M.; Couture, R.; Morrone, F.B. Primary Role for Kinin B1 and B2 Receptors in Glioma Proliferation. Mol. Neurobiol. 2017, 54, 7869–7882. [Google Scholar] [CrossRef]
- Nicoletti, N.F.; Erig, T.C.; Zanin, R.F.; Pereira, T.C.; Bogo, M.R.; Campos, M.M.; Morrone, F.B. Mechanisms involved in kinin-induced glioma cells proliferation: The role of ERK1/2 and PI3K/Akt pathways. J. Neurooncol. 2014, 120, 235–244. [Google Scholar] [CrossRef]
- Marceau, F.; Rivard, G.E.; Gauthier, J.M.; Binkley, K.E.; Bonnefoy, A.; Boccon-Gibod, I.; Bouillet, L.; Picard, M.; Levesque, G.; Elfassy, H.L.; et al. Measurement of Bradykinin Formation and Degradation in Blood Plasma: Relevance for Acquired Angioedema Associated with Angiotensin Converting Enzyme Inhibition and for Hereditary Angioedema Due to Factor XII or Plasminogen Gene Variants. Front. Med. 2020, 7, 358. [Google Scholar] [CrossRef]
- Seifert, S.; Sontheimer, H. Bradykinin enhances invasion of malignant glioma into the brain parenchyma by inducing cells to undergo amoeboid migration. J. Physiol. 2014, 592, 5109–5127. [Google Scholar] [CrossRef]
- Sun, D.P.; Lee, Y.W.; Chen, J.T.; Lin, Y.W.; Chen, R.M. The Bradykinin-BDKRB1 Axis Regulates Aquaporin 4 Gene Expression and Consequential Migration and Invasion of Malignant Glioblastoma Cells via a Ca2+-MEK1-ERK1/2-NF-κB Mechanism. Cancers 2020, 12, 667. [Google Scholar] [CrossRef]
- Guevara-Lora, I.; Blonska, B.; Faussner, A.; Kozik, A. Kinin-generating cellular model obtained from human glioblastoma cell line U-373. Acta Biochim. Pol. 2013, 60, 299–305. [Google Scholar] [CrossRef]
- Tasiou, A.; Konduri, S.D.; Yanamandra, N.; Dinh, D.H.; Olivero, W.C.; Gujrati, M.; Obeyesekere, M.; Rao, J.S. A novel role of tissue factor pathway inhibitor-2 in apoptosis of malignant human gliomas. Int. J. Oncol. 2001, 19, 591–597. [Google Scholar] [CrossRef]
- Rao, C.N.; Lakka, S.S.; Kin, Y.; Konduri, S.D.; Fuller, G.N.; Mohanam, S.; Rao, J.S. Expression of tissue factor pathway inhibitor 2 inversely correlates during the progression of human gliomas. Clin. Cancer Res. 2001, 7, 570–576. [Google Scholar]
- Easton, A.S.; Abbott, N.J. Bradykinin increases permeability by calcium and 5-lipoxygenase in the ECV304/C6 cell culture model of the blood-brain barrier. Brain Res. 2002, 953, 157–169. [Google Scholar] [CrossRef]
- Kast, R.E.; Burns, T.C.; Halatsch, M.E. Short review of SEC, a potential dexamethasone sparing regimen for glioblastoma: Spironolactone, ecallantide, clotrimazole. Neurochirurgie 2021, 67, 508–515. [Google Scholar] [CrossRef]
- Uchida, M.; Chen, Z.; Liu, Y.; Black, K.L. Overexpression of bradykinin type 2 receptors on glioma cells enhances bradykinin-mediated blood-brain tumor barrier permeability increase. Neurol. Res. 2002, 24, 739–746. [Google Scholar] [CrossRef] [PubMed]
- Farfara, D.; Feierman, E.; Richards, A.; Revenko, A.S.; MacLeod, R.A.; Norris, E.H.; Strickland, S. Knockdown of circulating C1 inhibitor induces neurovascular impairment, glial cell activation, neuroinflammation, and behavioral deficits. Glia 2019, 67, 1359–1373. [Google Scholar] [CrossRef]
- Liu, Y.S.; Hsu, J.W.; Lin, H.Y.; Lai, S.W.; Huang, B.R.; Tsai, C.F.; Lu, D.Y. Bradykinin B1 receptor contributes to interleukin-8 production and glioblastoma migration through interaction of STAT3 and SP-1. Neuropharmacology 2019, 144, 143–154. [Google Scholar] [CrossRef]
- Albert-Weissenberger, C.; Mencl, S.; Hopp, S.; Kleinschnitz, C.; Sirén, A.L. Role of the kallikrein-kinin system in traumatic brain injury. Front. Cell. Neurosci. 2014, 8, 345. [Google Scholar] [CrossRef]
- Pruneau, D.; Chorny, I.; Benkovitz, V.; Artru, A.; Roitblat, L.; Shapira, Y. Effect of LF 16-0687MS, a new nonpeptide bradykinin B2 receptor antagonist, in a rat model of closed head trauma. J. Neurotrauma 1999, 16, 1057–1065. [Google Scholar] [CrossRef]
- Kaplanski, J.; Pruneau, D.; Asa, I.; Artru, A.A.; Azez, A.; Ivashkova, Y.; Rudich, Z.; Shapira, Y. LF 16-0687 Ms, a bradykinin B2 receptor antagonist, reduces brain edema and improves long-term neurological function recovery after closed head trauma in rats. J. Neurotrauma 2002, 19, 953–964. [Google Scholar] [CrossRef] [PubMed]
- Chao, H.; Liu, Y.; Lin, C.; Xu, X.; Li, Z.; Bao, Z.; Fan, L.; Tao, C.; Zhao, L.; Liu, Y.; et al. Activation of bradykinin B2 receptor induced the inflammatory responses of cytosolic phospholipase A2 after the early traumatic brain injury. Biochim. Biophys. Acta Mol. Basis Dis. 2018, 1864 Pt B, 2957–2971. [Google Scholar] [CrossRef]
- Schulz, J.; Plesnila, N.; Eriskat, J.; Stoffel, M.; Pruneau, D.; Baethmann, A. LF16-0687 a novel non-peptide bradykinin B2 receptor antagonist reduces vasogenic brain edema from a focal lesion in rats. Acta Neurochir. Suppl. 2000, 76, 137–139. [Google Scholar] [CrossRef]
- Vereb, G., Jr.; Szöllösi, J.; Mátyus, L.; Balázs, M.; Hyun, W.C.; Feuerstein, B.G. Depletion of intracellular calcium stores facilitates the influx of extracellular calcium in platelet derived growth factor stimulated A172 glioblastoma cells. Cytometry 1996, 24, 64–73. [Google Scholar] [CrossRef]
- Kim, D.; Cho, S.H.; Kim, J.S.; Jo, S.H.; Lee, S.J.; Kim, K.T.; Choi, S.Y. Human astrocytic bradykinin B(2) receptor modulates zymosan-induced cytokine expression in 1321N1 cells. Peptides 2010, 31, 101–107. [Google Scholar] [CrossRef] [PubMed]
- Montana, V.; Sontheimer, H. Bradykinin promotes the chemotactic invasion of primary brain tumors. J. Neurosci. 2011, 31, 4858–4867. [Google Scholar] [CrossRef] [PubMed]
- Cuddapah, V.A.; Turner, K.L.; Seifert, S.; Sontheimer, H. Bradykinin-induced chemotaxis of human gliomas requires the activation of KCa3.1 and ClC-3. J. Neurosci. 2013, 33, 1427–1440. [Google Scholar] [CrossRef] [PubMed]
- Shen, C.K.; Huang, B.R.; Charoensaensuk, V.; Yang, L.Y.; Tsai, C.F.; Liu, Y.S.; Lu, D.Y.; Yeh, W.L.; Lin, C. Bradykinin B1 Receptor Affects Tumor-Associated Macrophage Activity and Glioblastoma Progression. Antioxidants 2023, 12, 1533. [Google Scholar] [CrossRef] [PubMed]
- Naro, G.R.; Noverati, N.; Craig, T. The Role of C1-Esterase Inhibitors in the Management of Vasogenic Edema in Glioblastoma. Case Rep. Med. 2020, 2020, 7981609. [Google Scholar] [CrossRef] [PubMed]
- Yu, H.S.; Lin, T.H.; Tang, C.H. Bradykinin enhances cell migration in human prostate cancer cells through B2 receptor/PKCδ/c-Src dependent signaling pathway. Prostate 2013, 73, 89–100. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.; Ye, Y.; Zhang, X.; Song, J. Bradykinin stimulates IL-6 production and cell invasion in colorectal cancer cells. Oncol. Rep. 2014, 32, 1709–1714. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.; Sun, J.; Liu, G.; Fu, Y.; Zhang, X. Bradykinin Promotes Cell Proliferation, Migration, Invasion, and Tumor Growth of Gastric Cancer through ERK Signaling Pathway. J. Cell. Biochem. 2017, 118, 4444–4453. [Google Scholar] [CrossRef]
- Chan, D.; Gera, L.; Stewart, J.; Helfrich, B.; Verella-Garcia, M.; Johnson, G.; Baron, A.; Yang, J.; Puck, T.; Bunn, P., Jr. Bradykinin antagonist dimer, CU201, inhibits the growth of human lung cancer cell lines by a “biased agonist” mechanism. Proc. Natl. Acad. Sci. USA 2002, 99, 4608–4613. [Google Scholar] [CrossRef] [PubMed]
- Eller-Borges, R.; Rodrigues, E.G.; Teodoro, A.C.S.; Moraes, M.S.; Arruda, D.C.; Paschoalin, T.; Curcio, M.F.; da Costa, P.E.; Do Nascimento, I.R.; Calixto, L.A.; et al. Bradykinin promotes murine melanoma cell migration and invasion through endogenous production of superoxide and nitric oxide. Nitric Oxide 2023, 132, 15–26. [Google Scholar] [CrossRef]
- Yang, W.H.; Chang, J.T.; Hsu, S.F.; Li, T.M.; Cho, D.Y.; Huang, C.Y.; Fong, Y.C.; Tang, C.H. Bradykinin enhances cell migration in human chondrosarcoma cells through BK receptor signaling pathways. J. Cell. Biochem. 2010, 109, 82–92. [Google Scholar] [CrossRef]
- Sethi, T.; Rozengurt, E. Multiple neuropeptides stimulate clonal growth of small cell lung cancer: Effects of bradykinin, vasopressin, cholecystokinin, galanin, and neurotensin. Cancer Res. 1991, 51, 3621–3623. [Google Scholar] [PubMed]
- Woll, P.J.; Rozengurt, E. Multiple neuropeptides mobilise calcium in small cell lung cancer: Effects of vasopressin, bradykinin, cholecystokinin, galanin and neurotensin. Biochem. Biophys. Res. Commun. 1989, 164, 66–73. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Zhang, B.; Huang, Y.; Yao, W.; Tao, F.; Chen, Y. Novel Bradykinin Receptor Inhibitors Inhibit Proliferation and Promote the Apoptosis of Hepatocellular Carcinoma Cells by Inhibiting the ERK Pathway. Molecules 2021, 26, 3915. [Google Scholar] [CrossRef] [PubMed]
- Jutras, S.; Bachvarova, M.; Keita, M.; Bascands, J.L.; Mes-Masson, A.M.; Stewart, J.M.; Gera, L.; Bachvarov, D. Strong cytotoxic effect of the bradykinin antagonist BKM-570 in ovarian cancer cells--analysis of the molecular mechanisms of its antiproliferative action. FEBS J. 2010, 277, 5146–5160. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Akaike, T.; Hayashida, K.; Miyamoto, Y.; Nakagawa, T.; Miyakawa, K.; Müller-Esterl, W.; Maeda, H. Identification of bradykinin receptors in clinical cancer specimens and murine tumor tissues. Int. J. Cancer 2002, 98, 29–35. [Google Scholar] [CrossRef] [PubMed]
- Deepak, K.; Roy, P.K.; Kola, P.; Mukherjee, B.; Mandal, M. An overview of kinin mediated events in cancer progression and therapeutic applications. Biochim. Biophys. Acta Rev. Cancer 2022, 1877, 188807. [Google Scholar] [CrossRef] [PubMed]
- Venkataramani, V.; Yang, Y.; Schubert, M.C.; Reyhan, E.; Tetzlaff, S.K.; Wißmann, N.; Botz, M.; Soyka, S.J.; Beretta, C.A.; Pramatarov, R.L.; et al. Glioblastoma hijacks neuronal mechanisms for brain invasion. Cell 2022, 185, 2899–2917.e31. [Google Scholar] [CrossRef] [PubMed]
- Rodríguez-Massó, S.R.; Erickson, M.A.; Banks, W.A.; Ulrich, H.; Martins, A.H. The Bradykinin B2 Receptor Agonist (NG291) Causes Rapid Onset of Transient Blood-Brain Barrier Disruption without Evidence of Early Brain Injury. Front. Neurosci. 2021, 15, 791709. [Google Scholar] [CrossRef] [PubMed]
- Sarin, H.; Kanevsky, A.S.; Fung, S.H.; Butman, J.A.; Cox, R.W.; Glen, D.; Reynolds, R.; Auh, S. Metabolically stable bradykinin B2 receptor agonists enhance transvascular drug delivery into malignant brain tumors by increasing drug half-life. J. Transl. Med. 2009, 7, 33. [Google Scholar] [CrossRef]
- Emerich, D.F.; Dean, R.L.; Osborn, C.; Bartus, R.T. The development of the bradykinin agonist labradimil as a means to increase the permeability of the blood-brain barrier: From concept to clinical evaluation. Clin. Pharmacokinet. 2001, 40, 105–123. [Google Scholar] [CrossRef]
- Côté, J.; Savard, M.; Neugebauer, W.; Fortin, D.; Lepage, M.; Gobeil, F. Dual kinin B1 and B2 receptor activation provides enhanced blood-brain barrier permeability and anticancer drug delivery into brain tumors. Cancer Biol. Ther. 2013, 14, 806–811. [Google Scholar] [CrossRef] [PubMed]
- Goldman, M.; Lucke-Wold, B.; Martinez-Sosa, M.; Katz, J.; Mehkri, Y.; Valisno, J.; Quintin, S. Steroid utility, immunotherapy, and brain tumor management: An update on conflicting therapies. Explor. Target Antitumor Ther. 2022, 3, 659–675. [Google Scholar] [CrossRef] [PubMed]
- Ricciardolo, F.L.M.; Folkerts, G.; Folino, A.; Mognetti, B. Bradykinin in asthma: Modulation of airway inflammation and remodelling. Eur. J. Pharmacol. 2018, 827, 181–188. [Google Scholar] [CrossRef] [PubMed]
- Qin, L.J.; Gu, Y.T.; Zhang, H.; Xue, Y.X. Bradykinin-induced blood-tumor barrier opening is mediated by tumor necrosis factor-alpha. Neurosci. Lett. 2009, 450, 172–175. [Google Scholar] [CrossRef] [PubMed]
- Kast, R.E. Research Supporting a Pilot Study of Metronomic Dapsone during Glioblastoma Chemoirradiation. Med. Sci. 2021, 9, 12. [Google Scholar] [CrossRef] [PubMed]
- Kast, R.E. The OSR9 Regimen: A New Augmentation Strategy for Osteosarcoma Treatment Using Nine Older Drugs from General Medicine to Inhibit Growth Drive. Int. J. Mol. Sci. 2023, 24, 15474. [Google Scholar] [CrossRef]
- Kast, R.E. IPIAD—An augmentation regimen added to standard treatment of pancreatic ductal adenocarcinoma using already-marketed repurposed drugs irbesartan, pyrimethamine, itraconazole, azithromycin, and dapsone. Oncoscience 2024, 11, 15–31. [Google Scholar] [CrossRef]
- Kast, R.E.; Scheuerle, A.; Wirtz, C.R.; Karpel-Massler, G.; Halatsch, M.E. The rationale of targeting neutrophils with dapsone during glioblastoma treatment. Anticancer Agents Med. Chem. 2011, 11, 756–761. [Google Scholar] [CrossRef]
- Stanimirovic, D.; Satoh, K. Inflammatory mediators of cerebral endothelium: A role in ischemic brain inflammation. Brain Pathol. 2000, 10, 113–126. [Google Scholar] [CrossRef]
- Schruefer, R.; Lutze, N.; Schymeinsky, J.; Walzog, B. Human neutrophils promote angiogenesis by a paracrine feedforward mechanism involving endothelial interleukin-8. Am. J. Physiol. Heart Circ. Physiol. 2005, 288, H1186–H1192. [Google Scholar] [CrossRef]
- Kajdácsi, E.; Veszeli, N.; Mező, B.; Jandrasics, Z.; Kőhalmi, K.V.; Ferrara, A.L.; Cervenak, L.; Varga, L.; Farkas, H. Pathways of Neutrophil Granulocyte Activation in Hereditary Angioedema with C1 Inhibitor Deficiency. Clin. Rev. Allergy Immunol. 2021, 60, 383–395. [Google Scholar] [CrossRef]
- Kenne, E.; Rasmuson, J.; Renné, T.; Vieira, M.L.; Müller-Esterl, W.; Herwald, H.; Lindbom, L. Neutrophils engage the kallikrein-kinin system to open up the endothelial barrier in acute inflammation. FASEB J. 2019, 33, 2599–2609. [Google Scholar] [CrossRef]
- Ricciardolo, F.L.; Sorbello, V.; Benedetto, S.; Defilippi, I.; Sabatini, F.; Robotti, A.; van Renswouw, D.C.; Bucca, C.; Folkerts, G.; De Rose, V. Bradykinin- and lipopolysaccharide-induced bradykinin B2 receptor expression, interleukin 8 release and “nitrosative stress” in bronchial epithelial cells BEAS-2B: Role for neutrophils. Eur. J. Pharmacol. 2012, 694, 30–38. [Google Scholar] [CrossRef]
- Kast, R.E. Adding high-dose celecoxib to increase effectiveness of standard glioblastoma chemoirradiation. Ann. Pharm. Fr. 2021, 79, 481–488. [Google Scholar] [CrossRef]
- Dean, P.T.; Hooks, S.B. Pleiotropic effects of the COX-2/PGE2 axis in the glioblastoma tumor microenvironment. Front. Oncol. 2023, 12, 1116014. [Google Scholar] [CrossRef]
- Wang, X.; Chen, Y.; Zhang, S.; Zhang, L.; Liu, X.; Zhang, L.; Li, X.; Chen, D. Co-expression of COX-2 and 5-LO in primary glioblastoma is associated with poor prognosis. J. Neurooncol. 2015, 125, 277–285. [Google Scholar] [CrossRef]
- Shono, T.; Tofilon, P.J.; Bruner, J.M.; Owolabi, O.; Lang, F.F. Cyclooxygenase-2 expression in human gliomas: Prognostic significance and molecular correlations. Cancer Res. 2001, 61, 4375–4381. [Google Scholar]
- Hara, A.; Okayasu, I. Cyclooxygenase-2 and inducible nitric oxide synthase expression in human astrocytic gliomas: Correlation with angiogenesis and prognostic significance. Acta Neuropathol. 2004, 108, 43–48. [Google Scholar] [CrossRef]
- Pistolesi, S.; Boldrini, L.; Gisfredi, S.; Ursino, S.; Alì, G.; Nuti, S.; De Ieso, K.; Pieracci, N.; Parenti, G.; Fontanini, G. Expression of cyclooxygenase-2 and its correlation with vasogenic brain edema in human intracranial meningiomas. Cancer Investig. 2007, 25, 555–562. [Google Scholar] [CrossRef]
- Chen, L.; Niu, Q.; Gao, C.; Du, F. Celecoxib treatment alleviates cerebral injury in a rat model of post-traumatic epilepsy. PeerJ 2023, 11, e16555. [Google Scholar] [CrossRef]
- Badie, B.; Schartner, J.M.; Hagar, A.R.; Prabakaran, S.; Peebles, T.R.; Bartley, B.; Lapsiwala, S.; Resnick, D.K.; Vorpahl, J. Microglia cyclooxygenase-2 activity in experimental gliomas: Possible role in cerebral edema formation. Clin. Cancer Res. 2003, 9, 872–877. [Google Scholar] [PubMed]
- Chu, K.; Jeong, S.W.; Jung, K.H.; Han, S.Y.; Lee, S.T.; Kim, M.; Roh, J.K. Celecoxib induces functional recovery after intracerebral hemorrhage with reduction of brain edema and perihematomal cell death. J. Cereb. Blood Flow Metab. 2004, 24, 926–933. [Google Scholar] [CrossRef] [PubMed]
- Park, H.K.; Lee, S.H.; Chu, K.; Roh, J.K. Effects of celecoxib on volumes of hematoma and edema in patients with primary intracerebral hemorrhage. J. Neurol. Sci. 2009, 279, 43–46. [Google Scholar] [CrossRef] [PubMed]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kast, R.E. Reducing Brain Edema Using Berotralstat, an Inhibitor of Bradykinin, Repurposed as Treatment Adjunct in Glioblastoma. Neuroglia 2024, 5, 223-233. https://doi.org/10.3390/neuroglia5030016
Kast RE. Reducing Brain Edema Using Berotralstat, an Inhibitor of Bradykinin, Repurposed as Treatment Adjunct in Glioblastoma. Neuroglia. 2024; 5(3):223-233. https://doi.org/10.3390/neuroglia5030016
Chicago/Turabian StyleKast, Richard E. 2024. "Reducing Brain Edema Using Berotralstat, an Inhibitor of Bradykinin, Repurposed as Treatment Adjunct in Glioblastoma" Neuroglia 5, no. 3: 223-233. https://doi.org/10.3390/neuroglia5030016
APA StyleKast, R. E. (2024). Reducing Brain Edema Using Berotralstat, an Inhibitor of Bradykinin, Repurposed as Treatment Adjunct in Glioblastoma. Neuroglia, 5(3), 223-233. https://doi.org/10.3390/neuroglia5030016