Action Potential Firing Induces Sodium Transients in Macroglial Cells of the Mouse Corpus Callosum



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Animals, Tissue Preparation, and Salines
2.2. Sodium Imaging and Electrophysiology
2.3. Immunohistochemistry
2.4. SDS-PAGE and Western Blot
2.5. Data Analysis and Presentation
3. Results
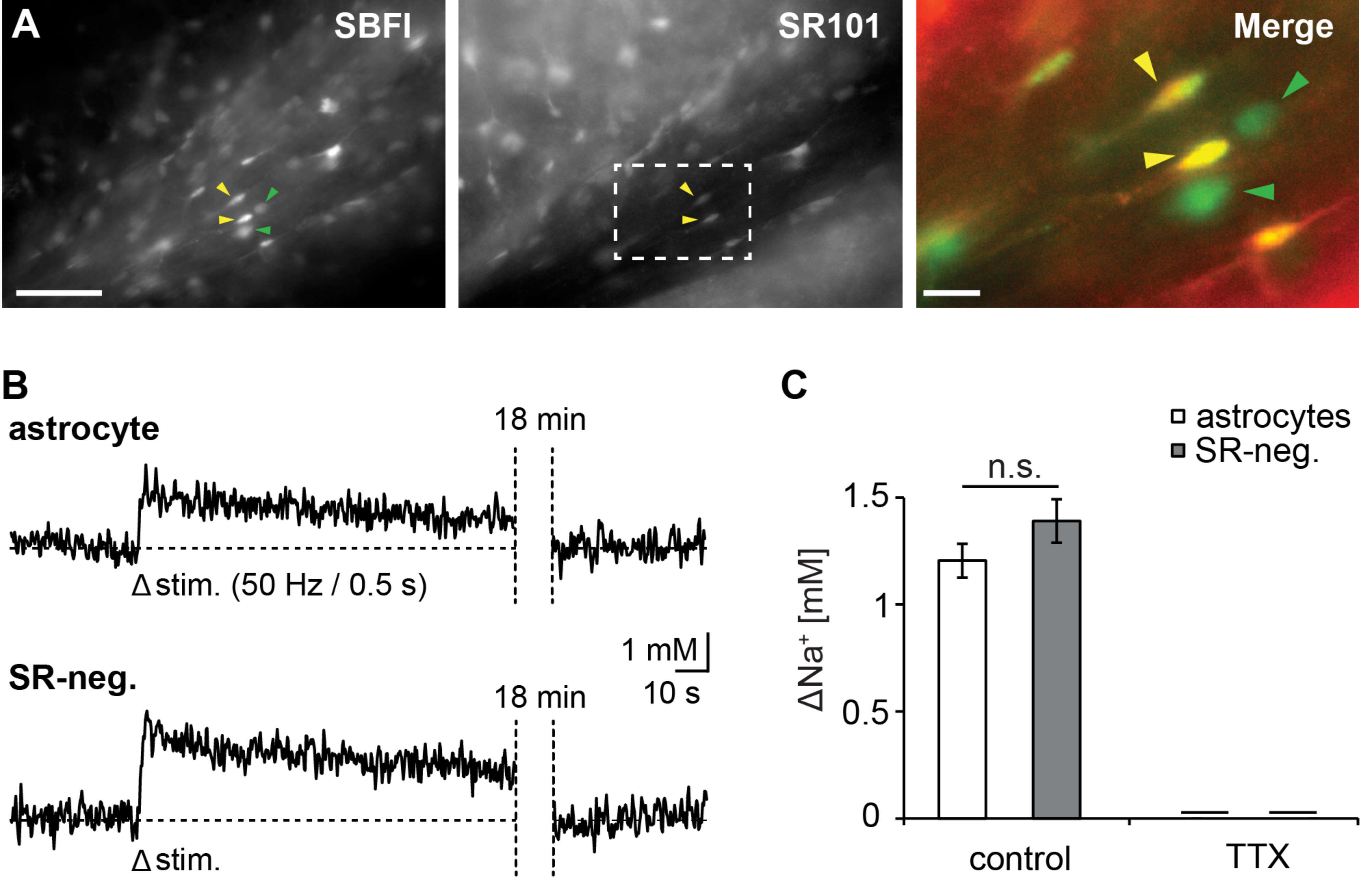
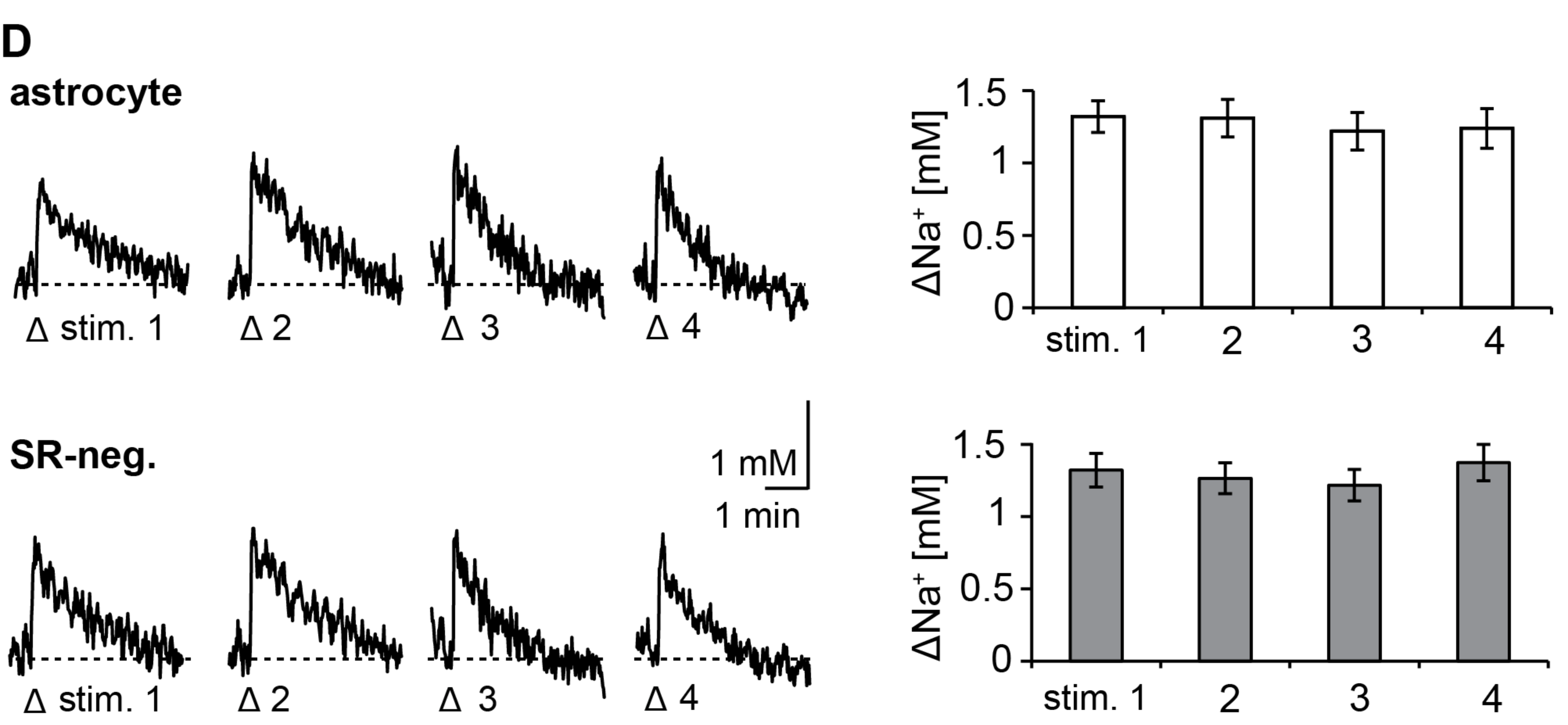
3.1. Action Potentials Evoke Sodium Transients in Macroglial Cells of the Corpus Callosum
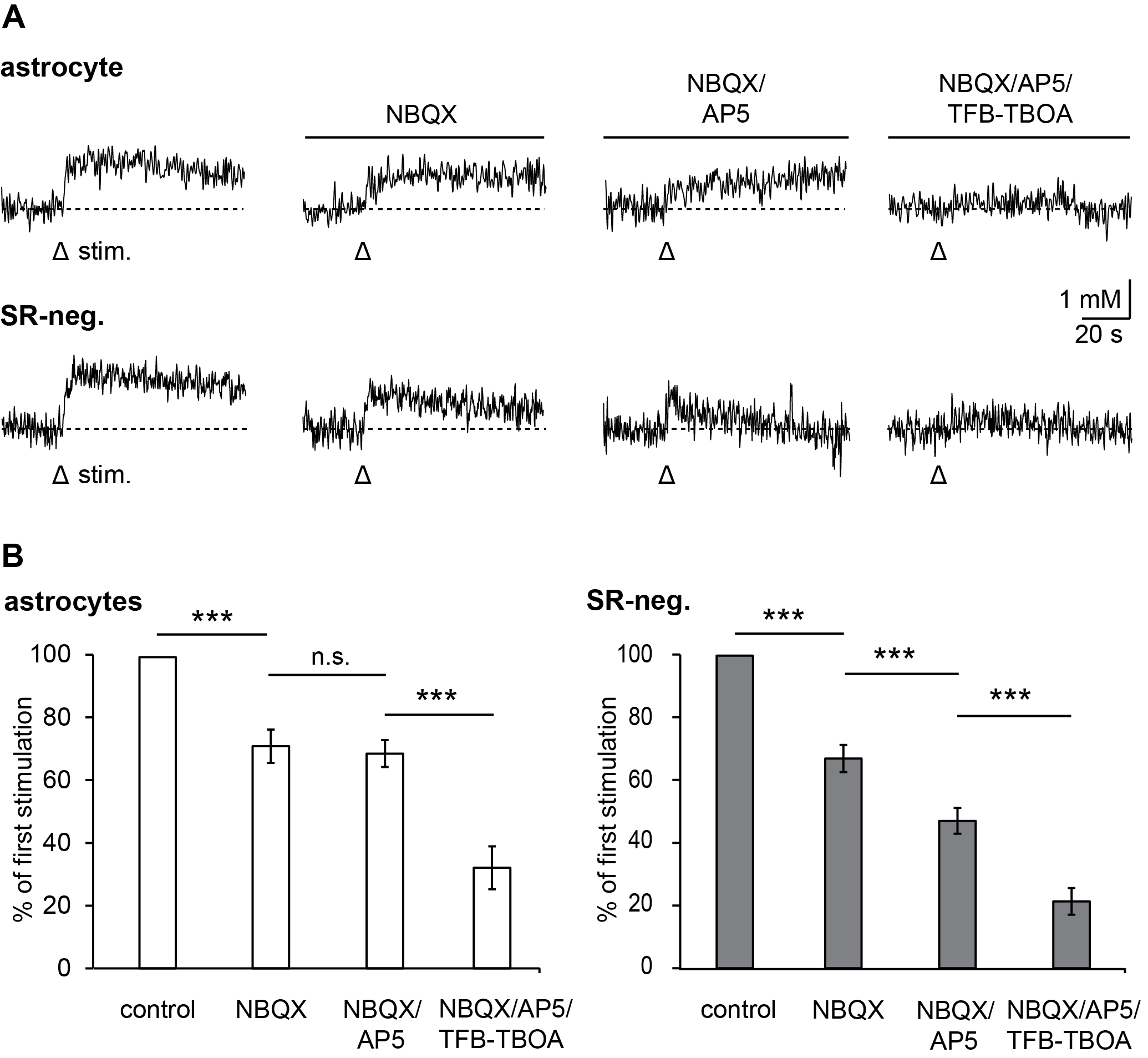
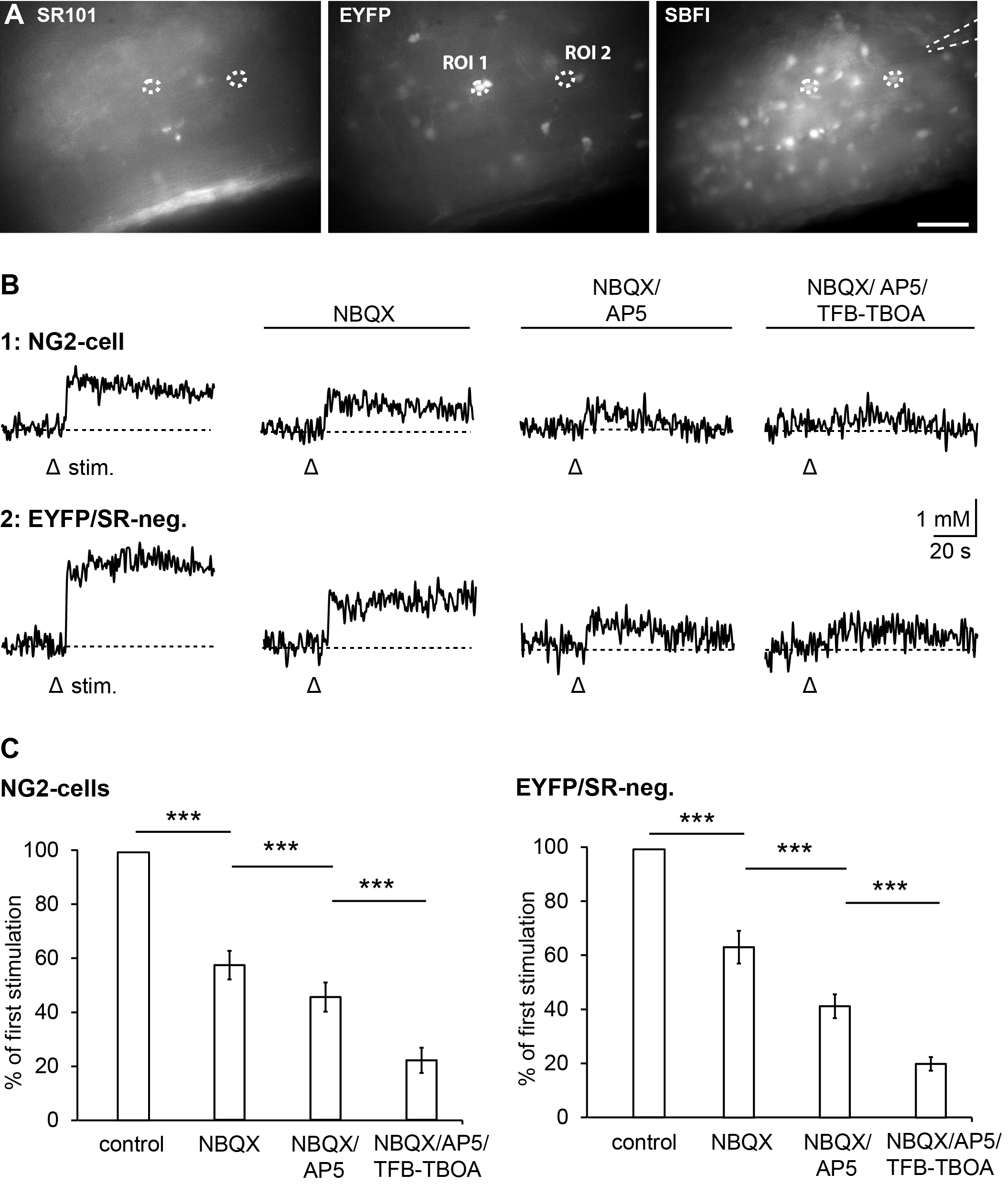
3.2. Pharmacology of Action Potential-Induced Sodium Transients in Macroglial Cells
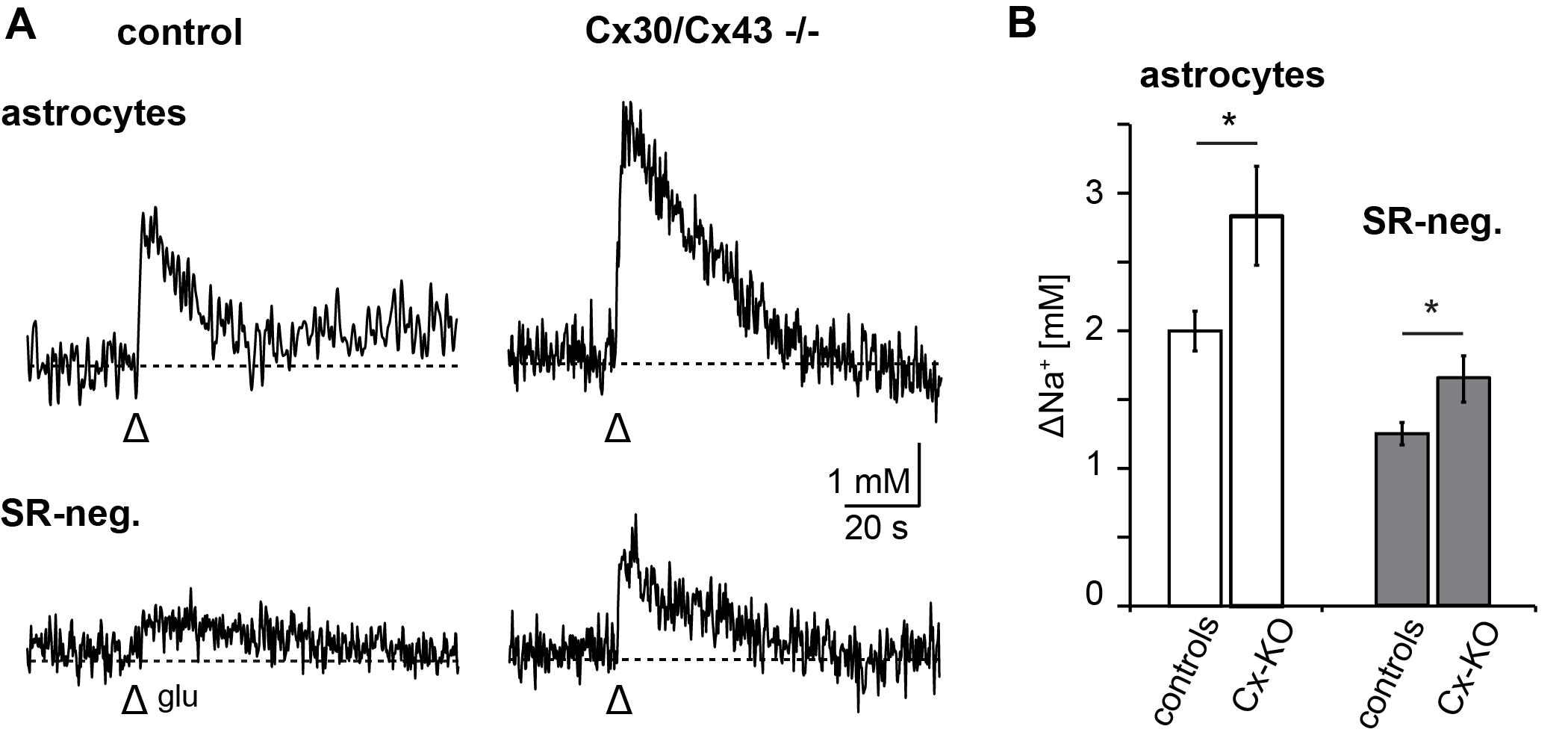
3.3. Relevance of Gap Junctional Coupling
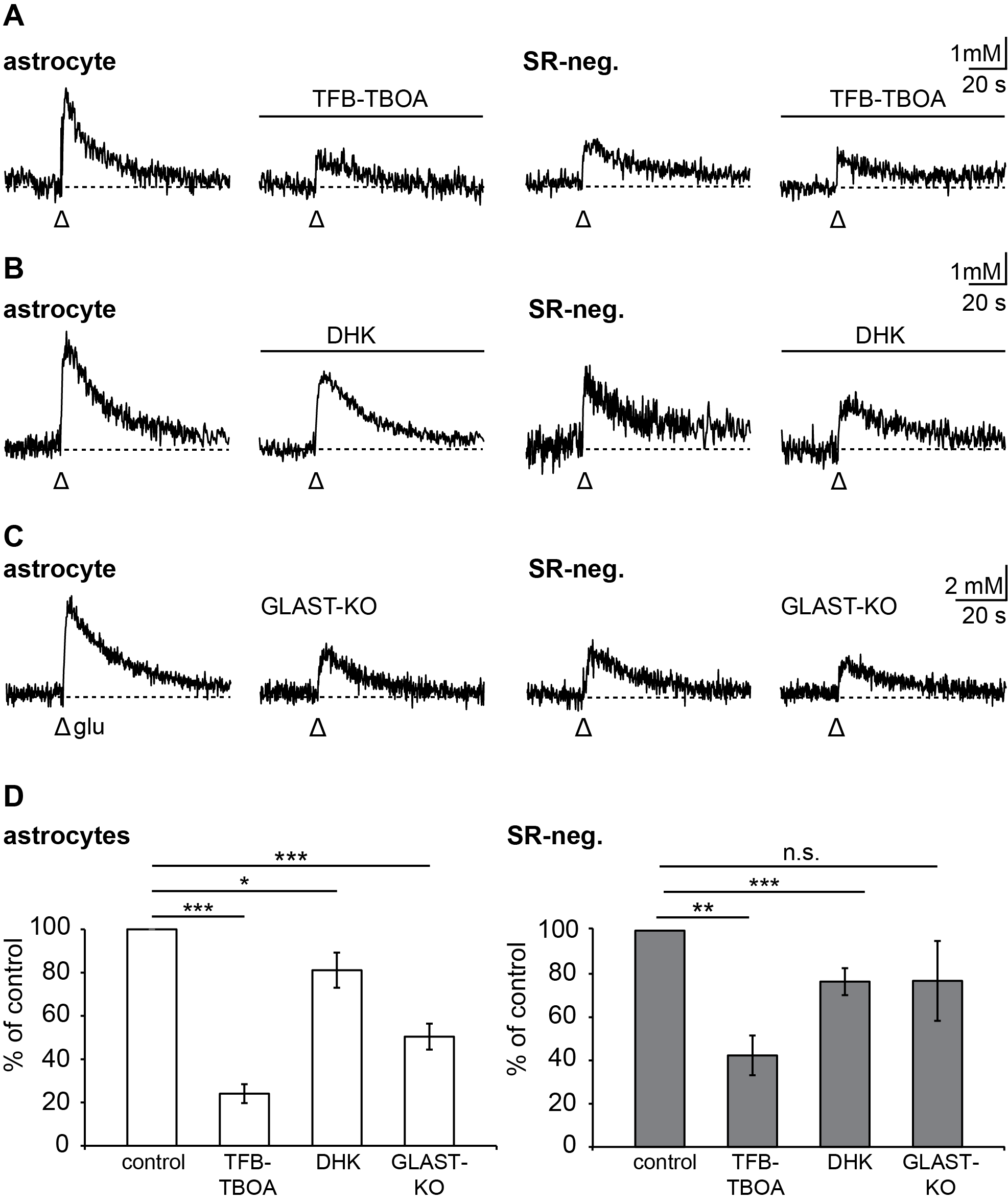
3.4. Relevance of Different Glutamate Transporter Subtypes
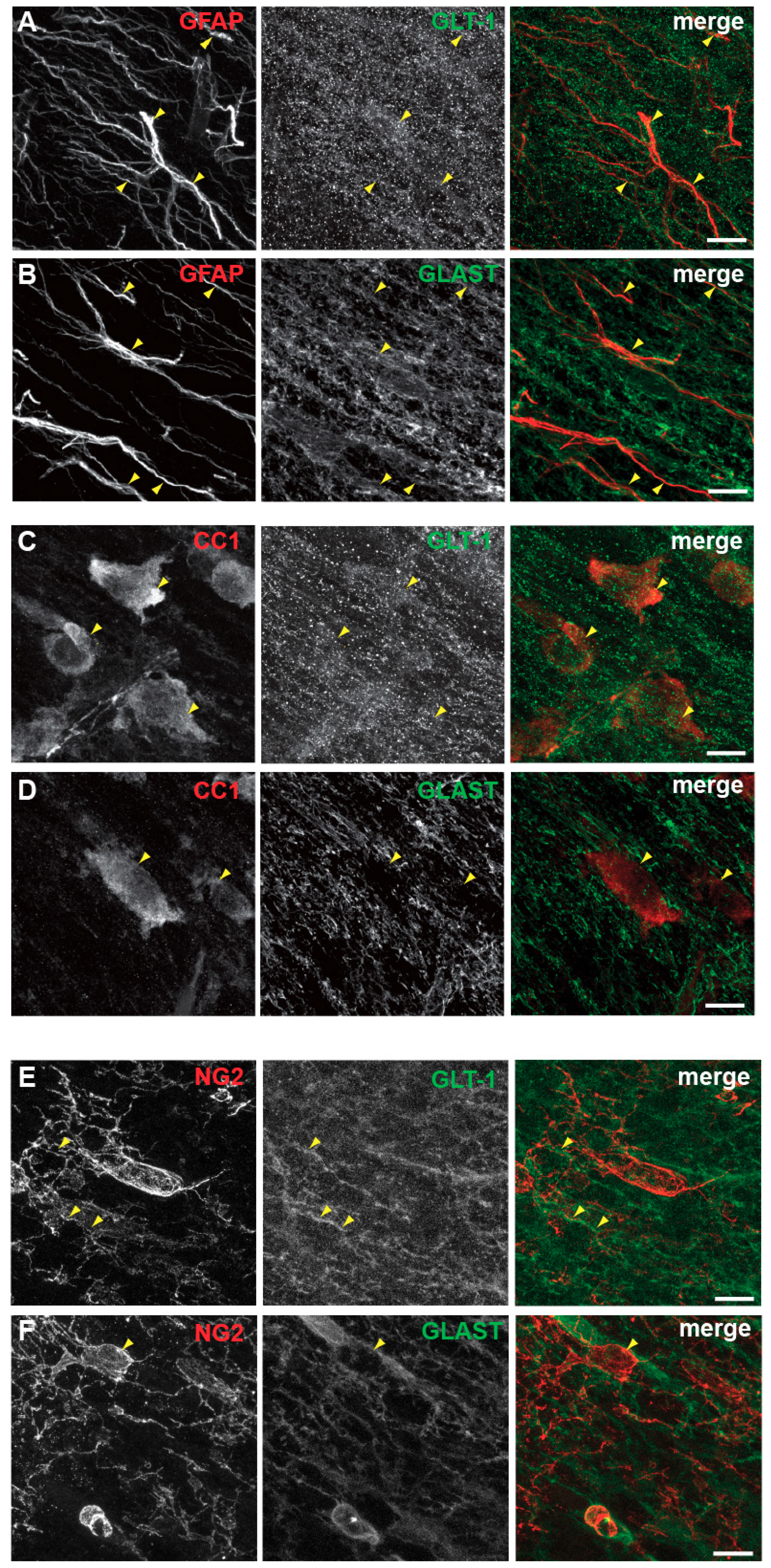
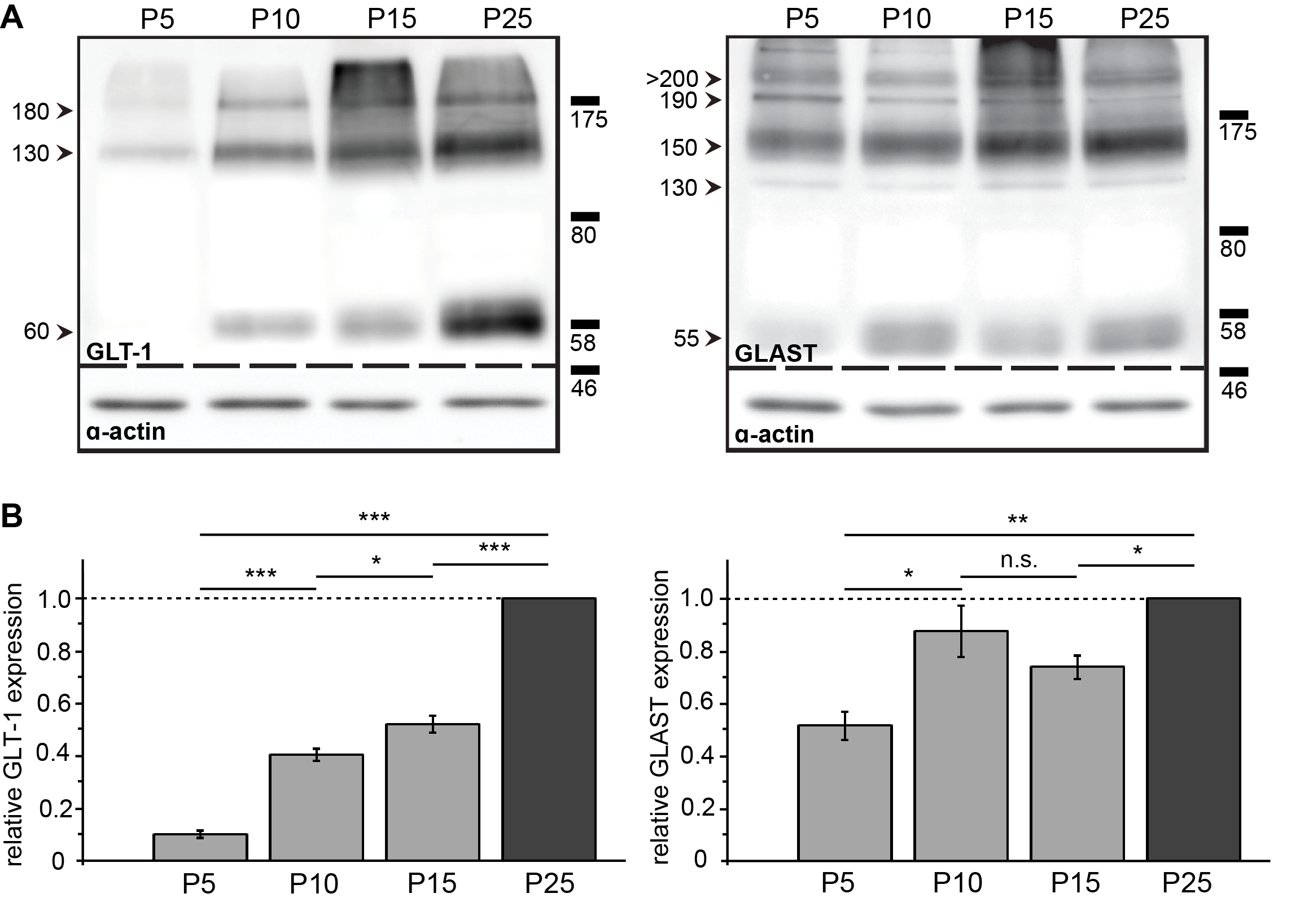
3.5. Spatial and Developmental Expression Profile of Glutamate-Aspartate-Transporter and Glutamate Transporter-1 in Corpus Callosum
4. Discussion
4.1. Action Potentials Induce Sodium Transients in White Matter Glial Cells That Are Related to Glutamate
4.2. Relevance of Different Sodium Influx Pathways
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Rose, C.R.; Verkhratsky, A. Principles of sodium homeostasis and sodium signalling in astroglia. Glia 2016, 64, 1611–1627. [Google Scholar] [CrossRef] [PubMed]
- Kirischuk, S.; Heja, L.; Kardos, J.; Billups, B. Astrocyte sodium signaling and the regulation of neurotransmission. Glia 2016, 64, 1655–1666. [Google Scholar] [CrossRef] [PubMed]
- Chatton, J.Y.; Marquet, P.; Magistretti, P.J. A quantitative analysis of l-glutamate-regulated Na+ dynamics in mouse cortical astrocytes: Implications for cellular bioenergetics. Eur. J. Neurosci. 2000, 12, 3843–3853. [Google Scholar] [CrossRef] [PubMed]
- Langer, J.; Rose, C.R. Synaptically induced sodium signals in hippocampal astrocytes in situ. J. Physiol. 2009, 587, 5859–5877. [Google Scholar] [CrossRef] [PubMed]
- Kirischuk, S.; Kettenmann, H.; Verkhratsky, A. Membrane currents and cytoplasmic sodium transients generated by glutamate transport in bergmann glial cells. Pflugers Arch. 2007, 454, 245–252. [Google Scholar] [CrossRef] [PubMed]
- Bennay, M.; Langer, J.; Meier, S.D.; Kafitz, K.W.; Rose, C.R. Sodium signals in cerebellar Purkinje neurons and Bergmann glial cells evoked by glutamatergic synaptic transmission. Glia 2008, 56, 1138–1149. [Google Scholar] [CrossRef] [PubMed]
- Karus, C.; Mondragao, M.A.; Ziemens, D.; Rose, C.R. Astrocytes restrict discharge duration and neuronal sodium loads during recurrent network activity. Glia 2015, 63, 936–957. [Google Scholar] [CrossRef] [PubMed]
- Danbolt, N.C. Glutamate uptake. Prog. Neurobiol. 2001, 65, 1–105. [Google Scholar] [CrossRef]
- Rose, C.R.; Ziemens, D.; Untiet, V.; Fahlke, C. Molecular and cellular physiology of sodium-dependent glutamate transporters. Brain Res. Bull. 2018, 136, 13–16. [Google Scholar] [CrossRef] [PubMed]
- Chatton, J.Y.; Magistretti, P.J.; Barros, L.F. Sodium signaling and astrocyte energy metabolism. Glia 2016, 64, 1667–1676. [Google Scholar] [CrossRef] [PubMed]
- Magistretti, P.J.; Allaman, I. Lactate in the brain: From metabolic end-product to signalling molecule. Nat. Rev. Neurosci. 2018, 19, 235–249. [Google Scholar] [CrossRef] [PubMed]
- Kirischuk, S.; Parpura, V.; Verkhratsky, A. Sodium dynamics: Another key to astroglial excitability? Trends Neurosci. 2012, 35, 497–506. [Google Scholar] [CrossRef] [PubMed]
- Parpura, V.; Sekler, I.; Fern, R. Plasmalemmal and mitochondrial Na+-Ca2+ exchange in neuroglia. Glia 2016, 64, 1646–1654. [Google Scholar] [CrossRef] [PubMed]
- Jackson, J.G.; O’Donnell, J.C.; Takano, H.; Coulter, D.A.; Robinson, M.B. Neuronal activity and glutamate uptake decrease mitochondrial mobility in astrocytes and position mitochondria near glutamate transporters. J. Neurosci. 2014, 34, 1613–1624. [Google Scholar] [CrossRef] [PubMed]
- Gerkau, N.J.; Rakers, C.; Durry, S.; Petzold, G.; Rose, C.R. Reverse NCX attenuates cellular sodium loading in metabolically compromised cortex. Cereb. Cortex 2017, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Kukley, M.; Capetillo-Zarate, E.; Dietrich, D. Vesicular glutamate release from axons in white matter. Nat. Neurosci. 2007, 10, 311–320. [Google Scholar] [CrossRef] [PubMed]
- Ziskin, J.L.; Nishiyama, A.; Rubio, M.; Fukaya, M.; Bergles, D.E. Vesicular release of glutamate from unmyelinated axons in white matter. Nat. Neurosci. 2007, 10, 321–330. [Google Scholar] [CrossRef] [PubMed]
- Fruhbeis, C.; Frohlich, D.; Kuo, W.P.; Amphornrat, J.; Thilemann, S.; Saab, A.S.; Kirchhoff, F.; Mobius, W.; Goebbels, S.; Nave, K.A.; et al. Neurotransmitter-triggered transfer of exosomes mediates oligodendrocyte-neuron communication. PLoS Biol. 2013, 11, e1001604. [Google Scholar] [CrossRef] [PubMed]
- Hamilton, N.; Vayro, S.; Wigley, R.; Butt, A.M. Axons and astrocytes release ATP and glutamate to evoke calcium signals in NG2-glia. Glia 2010, 58, 66–79. [Google Scholar] [CrossRef] [PubMed]
- Fern, R.F.; Matute, C.; Stys, P.K. White matter injury: Ischemic and nonischemic. Glia 2014, 62, 1780–1789. [Google Scholar] [CrossRef] [PubMed]
- Domercq, M.; Matute, C. Expression of glutamate transporters in the adult bovine corpus callosum. Brain Res. Mol. Brain Res. 1999, 67, 296–302. [Google Scholar] [CrossRef]
- Goursaud, S.; Kozlova, E.N.; Maloteaux, J.M.; Hermans, E. Cultured astrocytes derived from corpus callosum or cortical grey matter show distinct glutamate handling properties. J. Neurochem. 2009, 108, 1442–1452. [Google Scholar] [CrossRef] [PubMed]
- Lundgaard, I.; Osorio, M.J.; Kress, B.T.; Sanggaard, S.; Nedergaard, M. White matter astrocytes in health and disease. Neuroscience 2014, 276, 161–173. [Google Scholar] [CrossRef] [PubMed]
- Arranz, A.M.; Hussein, A.; Alix, J.J.; Perez-Cerda, F.; Allcock, N.; Matute, C.; Fern, R. Functional glutamate transport in rodent optic nerve axons and glia. Glia 2008, 56, 1353–1367. [Google Scholar] [CrossRef] [PubMed]
- Moshrefi-Ravasdjani, B.; Hammel, E.L.; Kafitz, K.W.; Rose, C.R. Astrocyte sodium signalling and panglial spread of sodium signals in brain white matter. Neurochem. Res. 2017, 42, 2505–2518. [Google Scholar] [CrossRef] [PubMed]
- Butt, A.M.; Fern, R.F.; Matute, C. Neurotransmitter signaling in white matter. Glia 2014, 62, 1762–1779. [Google Scholar] [CrossRef] [PubMed]
- Micu, I.; Plemel, J.R.; Caprariello, A.V.; Nave, K.A.; Stys, P.K. Axo-myelinic neurotransmission: A novel mode of cell signalling in the central nervous system. Nat. Rev. Neurosci. 2018, 19, 49–58. [Google Scholar] [CrossRef] [PubMed]
- Hamilton, N.; Vayro, S.; Kirchhoff, F.; Verkhratsky, A.; Robbins, J.; Gorecki, D.C.; Butt, A.M. Mechanisms of ATP- and glutamate-mediated calcium signaling in white matter astrocytes. Glia 2008, 56, 734–749. [Google Scholar] [CrossRef] [PubMed]
- Bernstein, M.; Lyons, S.A.; Moller, T.; Kettenmann, H. Receptor-mediated calcium signalling in glial cells from mouse corpus callosum slices. J. Neurosci. Res. 1996, 46, 152–163. [Google Scholar] [CrossRef]
- Schipke, C.G.; Boucsein, C.; Ohlemeyer, C.; Kirchhoff, F.; Kettenmann, H. Astrocyte Ca2+ waves trigger responses in microglial cells in brain slices. FASEB J. 2002, 16, 255–257. [Google Scholar] [CrossRef] [PubMed]
- Hamilton, N.; Hubbard, P.S.; Butt, A.M. Effects of glutamate receptor activation on NG2-glia in the rat optic nerve. J. Anat. 2009, 214, 208–218. [Google Scholar] [CrossRef] [PubMed]
- Bergles, D.E.; Jabs, R.; Steinhäuser, C. Neuron-glia synapses in the brain. Brain Res. Rev. 2010, 63, 130–137. [Google Scholar] [CrossRef] [PubMed]
- Dimou, L.; Gallo, V. NG2-glia and their functions in the central nervous system. Glia 2015, 63, 1429–1451. [Google Scholar] [CrossRef] [PubMed]
- Close, B.; Banister, K.; Baumans, V.; Bernoth, E.M.; Bromage, N.; Bunyan, J.; Erhardt, W.; Flecknell, P.; Gregory, N.; Hackbarth, H.; et al. Recommendations for euthanasia of experimental animals: Part 2. DGXT of the european commission. Lab. Anim. 1997, 31, 1–32. [Google Scholar] [CrossRef] [PubMed]
- Karram, K.; Goebbels, S.; Schwab, M.; Jennissen, K.; Seifert, G.; Steinhauser, C.; Nave, K.A.; Trotter, J. NG2-expressing cells in the nervous system revealed by the NG2-EYFP-knockin mouse. Genesis 2008, 46, 743–757. [Google Scholar] [CrossRef] [PubMed]
- Wallraff, A.; Kohling, R.; Heinemann, U.; Theis, M.; Willecke, K.; Steinhauser, C. The impact of astrocytic gap junctional coupling on potassium buffering in the hippocampus. J. Neurosci. 2006, 26, 5438–5447. [Google Scholar] [CrossRef] [PubMed]
- Watase, K.; Hashimoto, K.; Kano, M.; Yamada, K.; Watanabe, M.; Inoue, Y.; Okuyama, S.; Sakagawa, T.; Ogawa, S.-i.; Kawashima, N.; et al. Motor discoordination and increased susceptibility to cerebellar injury in glast mutant mice. Eur. J. Neurosci. 1998, 10, 976–988. [Google Scholar] [CrossRef] [PubMed]
- Kafitz, K.W.; Meier, S.D.; Stephan, J.; Rose, C.R. Developmental profile and properties of sulforhodamine 101-labeled glial cells in acute brain slices of rat hippocampus. J. Neurosci. Methods 2008, 169, 84–92. [Google Scholar] [CrossRef] [PubMed]
- Meier, S.D.; Kovalchuk, Y.; Rose, C.R. Properties of the new fluorescent Na+ indicator corona green: Comparison with SBFI and confocal Na+ imaging. J. Neurosci. Methods 2006, 155, 251–259. [Google Scholar] [CrossRef] [PubMed]
- Langer, J.; Stephan, J.; Theis, M.; Rose, C.R. Gap junctions mediate intercellular spread of sodium between hippocampal astrocytes in situ. Glia 2012, 60, 239–252. [Google Scholar] [CrossRef] [PubMed]
- Towbin, H.; Staehelin, T.; Gordon, J. Electrophoretic transfer of proteins from polyacrylamide gels to nitrocellulose sheets: Procedure and some applications. Proc. Natl. Acad. Sci. USA 1979, 76, 4350–4354. [Google Scholar] [CrossRef] [PubMed]
- Burnette, W.N. “Western blotting”: Electrophoretic transfer of proteins from sodium dodecyl sulfate—Polyacrylamide gels to unmodified nitrocellulose and radiographic detection with antibody and radioiodinated protein a. Anal. Biochem. 1981, 112, 195–203. [Google Scholar] [CrossRef]
- Schreiner, A.E.; Durry, S.; Aida, T.; Stock, M.C.; Rüther, U.; Tanaka, K.; Rose, C.R.; Kafitz, K.W. Laminar and subcellular heterogeneity of GLAST and GLT-1 immunoreactivity in the developing postnatal mouse hippocampus. J. Comp. Neurol. 2014, 522, 204–224. [Google Scholar] [CrossRef] [PubMed]
- Tsukada, S.; Iino, M.; Takayasu, Y.; Shimamoto, K.; Ozawa, S. Effects of a novel glutamate transporter blocker, (2s, 3s)-3-[3-[4-(trifluoromethyl)benzoylamino]benzyloxy]aspartate (TFB-TBOA), on activities of hippocampal neurons. Neuropharmacology 2005, 48, 479–491. [Google Scholar] [CrossRef] [PubMed]
- Haugeto, O.; Ullensvang, K.; Levy, L.M.; Chaudhry, F.A.; Honore, T.; Nielsen, M.; Lehre, K.P.; Danbolt, N.C. Brain glutamate transporter proteins form homomultimers. J. Biol. Chem. 1996, 271, 27715–27722. [Google Scholar] [CrossRef] [PubMed]
- Peacey, E.; Miller, C.C.; Dunlop, J.; Rattray, M. The four major N- and C-terminal splice variants of the excitatory amino acid transporter GLT-1 form cell surface homomeric and heteromeric assemblies. Mol. Pharmacol. 2009, 75, 1062–1073. [Google Scholar] [CrossRef] [PubMed]
- Nimmerjahn, A.; Kirchhoff, F.; Kerr, J.N.; Helmchen, F. Sulforhodamine 101 as a specific marker of astroglia in the neocortex in vivo. Nat. Methods 2004, 1, 31–37. [Google Scholar] [CrossRef] [PubMed]
- Uwechue, N.M.; Marx, M.C.; Chevy, Q.; Billups, B. Activation of glutamate transport evokes rapid glutamine release from perisynaptic astrocytes. J. Physiol. 2012, 590, 2317–2331. [Google Scholar] [CrossRef] [PubMed]
- Lamy, C.M.; Chatton, J.Y. Optical probing of sodium dynamics in neurons and astrocytes. Neuroimage 2011, 58, 572–578. [Google Scholar] [CrossRef] [PubMed]
- Rose, C.R.; Chatton, J.Y. Astrocyte sodium signaling and neuro-metabolic coupling in the brain. Neuroscience 2016, 323, 121–134. [Google Scholar] [CrossRef] [PubMed]
- Ballanyi, K.; Kettenmann, H. Intracellular Na+ activity in cultured mouse oligodendrocytes. J. Neurosci. Res. 1990, 26, 455–460. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Kintner, D.B.; Jones, M.; Matsuda, T.; Baba, A.; Kiedrowski, L.; Sun, D. Ampa-mediated excitotoxicity in oligodendrocytes: Role for Na+-K+-Cl- co-transport and reversal of Na+/Ca2+ exchanger. J. Neurochem. 2007, 102, 1783–1795. [Google Scholar] [CrossRef] [PubMed]
- Tong, X.P.; Li, X.Y.; Zhou, B.; Shen, W.; Zhang, Z.J.; Xu, T.L.; Duan, S. Ca2+ signaling evoked by activation of Na+ channels and Na+/Ca2+ exchangers is required for GABA-induced NG2 cell migration. J. Cell Biol. 2009, 186, 113–128. [Google Scholar] [CrossRef] [PubMed]
- Baranauskas, G.; David, Y.; Fleidervish, I.A. Spatial mismatch between the Na+ flux and spike initiation in axon initial segment. Proc. Natl. Acad. Sci. USA 2013, 110, 4051–4056. [Google Scholar] [CrossRef] [PubMed]
- Miyazaki, K.; Ross, W.N. Simultaneous sodium and calcium imaging from dendrites and axons. eNeuro 2015, 2. [Google Scholar] [CrossRef] [PubMed]
- Ona-Jodar, T.; Gerkau, N.J.; Sara Aghvami, S.; Rose, C.R.; Egger, V. Two-photon Na+ imaging reports somatically evoked action potentials in rat olfactory bulb mitral and granule cell neurites. Front. Cell. Neurosci. 2017, 11, 50. [Google Scholar] [CrossRef] [PubMed]
- Kole, M.H.; Ilschner, S.U.; Kampa, B.M.; Williams, S.R.; Ruben, P.C.; Stuart, G.J. Action potential generation requires a high sodium channel density in the axon initial segment. Nat. Neurosci. 2008, 11, 178–186. [Google Scholar] [CrossRef] [PubMed]
- Dietzel, I.; Heinemann, U.; Hofmeier, G.; Lux, H.D. Stimulus-induced changes in extracellular Na+ and Cl- concentration in relation to changes in the size of the extracellular space. Exp. Brain Res. 1982, 46, 73–84. [Google Scholar] [CrossRef] [PubMed]
- Deng, W.; Rosenberg, P.A.; Volpe, J.J.; Jensen, F.E. Calcium-permeable AMPA/kainate receptors mediate toxicity and preconditioning by oxygen-glucose deprivation in oligodendrocyte precursors. Proc. Natl. Acad. Sci. USA 2003, 100, 6801–6806. [Google Scholar] [CrossRef] [PubMed]
- Karadottir, R.; Cavelier, P.; Bergersen, L.H.; Attwell, D. NMDA receptors are expressed in oligodendrocytes and activated in ischaemia. Nature 2005, 438, 1162–1166. [Google Scholar] [CrossRef] [PubMed]
- Salter, M.G.; Fern, R. Nmda receptors are expressed in developing oligodendrocyte processes and mediate injury. Nature 2005, 438, 1167–1171. [Google Scholar] [CrossRef] [PubMed]
- Matute, C.; Alberdi, E.; Domercq, M.; Sanchez-Gomez, M.V.; Perez-Samartin, A.; Rodriguez-Antiguedad, A.; Perez-Cerda, F. Excitotoxic damage to white matter. J. Anat. 2007, 210, 693–702. [Google Scholar] [CrossRef] [PubMed]
- Micu, I.; Ridsdale, A.; Zhang, L.; Woulfe, J.; McClintock, J.; Brantner, C.A.; Andrews, S.B.; Stys, P.K. Real-time measurement of free Ca2+ changes in CNS myelin by two-photon microscopy. Nat. Med. 2007, 13, 874–879. [Google Scholar] [CrossRef] [PubMed]
- Gallo, V.; Zhou, J.M.; McBain, C.J.; Wright, P.; Knutson, P.L.; Armstrong, R.C. Oligodendrocyte progenitor cell proliferation and lineage progression are regulated by glutamate receptor-mediated k+ channel block. J. Neurosci. 1996, 16, 2659–2670. [Google Scholar] [CrossRef] [PubMed]
- Yuan, X.; Eisen, A.M.; McBain, C.J.; Gallo, V. A role for glutamate and its receptors in the regulation of oligodendrocyte development in cerebellar tissue slices. Development 1998, 125, 2901–2914. [Google Scholar] [PubMed]
- Nagy, B.; Hovhannisyan, A.; Barzan, R.; Chen, T.J.; Kukley, M. Different patterns of neuronal activity trigger distinct responses of oligodendrocyte precursor cells in the corpus callosum. PLoS Biol. 2017, 15, e2001993. [Google Scholar] [CrossRef] [PubMed]
- Funfschilling, U.; Supplie, L.M.; Mahad, D.; Boretius, S.; Saab, A.S.; Edgar, J.; Brinkmann, B.G.; Kassmann, C.M.; Tzvetanova, I.D.; Mobius, W.; et al. Glycolytic oligodendrocytes maintain myelin and long-term axonal integrity. Nature 2012, 485, 517–521. [Google Scholar] [CrossRef] [PubMed]
- Saab, A.S.; Tzvetavona, I.D.; Trevisiol, A.; Baltan, S.; Dibaj, P.; Kusch, K.; Mobius, W.; Goetze, B.; Jahn, H.M.; Huang, W.; et al. Oligodendroglial NMDA receptors regulate glucose import and axonal energy metabolism. Neuron 2016, 91, 119–132. [Google Scholar] [CrossRef] [PubMed]
- Meyer, N.; Richter, N.; Fan, Z.; Siemonsmeier, G.; Pivneva, T.; Jordan, P.; Steinhauser, C.; Semtner, M.; Nolte, C.; Kettenmann, H. Oligodendrocytes in the mouse corpus callosum maintain axonal function by delivery of glucose. Cell Rep. 2018, 22, 2383–2394. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.; Morrison, B.M.; Li, Y.; Lengacher, S.; Farah, M.H.; Hoffman, P.N.; Liu, Y.; Tsingalia, A.; Jin, L.; Zhang, P.W.; et al. Oligodendroglia metabolically support axons and contribute to neurodegeneration. Nature 2012, 487, 443–448. [Google Scholar] [CrossRef] [PubMed]
- Baraban, M.; Koudelka, S.; Lyons, D.A. Ca2+ activity signatures of myelin sheath formation and growth in vivo. Nat. Neurosci. 2018, 21, 19–23. [Google Scholar] [CrossRef] [PubMed]
- Krasnow, A.M.; Ford, M.C.; Valdivia, L.E.; Wilson, S.W.; Attwell, D. Regulation of developing myelin sheath elongation by oligodendrocyte calcium transients in vivo. Nat. Neurosci. 2018, 21, 24–28. [Google Scholar] [CrossRef] [PubMed]
- Brown, A.M.; Sickmann, H.M.; Fosgerau, K.; Lund, T.M.; Schousboe, A.; Waagepetersen, H.S.; Ransom, B.R. Astrocyte glycogen metabolism is required for neural activity during aglycemia or intense stimulation in mouse white matter. J. Neurosci. Res. 2005, 79, 74–80. [Google Scholar] [CrossRef] [PubMed]
- Brown, A.M.; Ransom, B.R. Astrocyte glycogen and brain energy metabolism. Glia 2007, 55, 1263–1271. [Google Scholar] [CrossRef] [PubMed]
- Regan, M.R.; Huang, Y.H.; Kim, Y.S.; Dykes-Hoberg, M.I.; Jin, L.; Watkins, A.M.; Bergles, D.E.; Rothstein, J.D. Variations in promoter activity reveal a differential expression and physiology of glutamate transporters by glia in the developing and mature CNS. J. Neurosci. 2007, 27, 6607–6619. [Google Scholar] [CrossRef] [PubMed]
- DeSilva, T.M.; Kabakov, A.Y.; Goldhoff, P.E.; Volpe, J.J.; Rosenberg, P.A. Regulation of glutamate transport in developing rat oligodendrocytes. J. Neurosci. 2009, 29, 7898–7908. [Google Scholar] [CrossRef] [PubMed]
- Hanson, E.; Armbruster, M.; Cantu, D.; Andresen, L.; Taylor, A.; Danbolt, N.C.; Dulla, C.G. Astrocytic glutamate uptake is slow and does not limit neuronal nmda receptor activation in the neonatal neocortex. Glia 2015, 63, 1784–1796. [Google Scholar] [CrossRef] [PubMed]
- Morrison, B.M.; Lee, Y.; Rothstein, J.D. Oligodendroglia: Metabolic supporters of axons. Trends Cell Biol. 2013, 23, 644–651. [Google Scholar] [CrossRef] [PubMed]
- Hirrlinger, J.; Nave, K.A. Adapting brain metabolism to myelination and long-range signal transduction. Glia 2014, 62, 1749–1761. [Google Scholar] [CrossRef] [PubMed]
- Borges, K.; Kettenmann, H. Blockade of K+ channels induced by AMPA/kainate receptor activation in mouse oligodendrocyte precursor cells is mediated by Na+ entry. J. Neurosci. Res. 1995, 42, 579–593. [Google Scholar] [CrossRef] [PubMed]
- Knutson, P.; Ghiani, C.A.; Zhou, J.M.; Gallo, V.; McBain, C.J. K+ channel expression and cell proliferation are regulated by intracellular sodium and membrane depolarization in oligodendrocyte progenitor cells. J. Neurosci. 1997, 17, 2669–2682. [Google Scholar] [CrossRef] [PubMed]
- Schroder, W.; Seifert, G.; Huttmann, K.; Hinterkeuser, S.; Steinhauser, C. AMPA receptor-mediated modulation of inward rectifier K+ channels in astrocytes of mouse hippocampus. Mol. Cell. Neurosci. 2002, 19, 447–458. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Lozada, Z.; Waggener, C.T.; Kim, K.; Zou, S.; Knapp, P.E.; Hayashi, Y.; Ortega, A.; Fuss, B. Activation of sodium-dependent glutamate transporters regulates the morphological aspects of oligodendrocyte maturation via signaling through calcium/calmodulin-dependent kinase IIβ’s actin-binding/-stabilizing domain. Glia 2014, 62, 1543–1558. [Google Scholar] [CrossRef] [PubMed]
- Friess, M.; Hammann, J.; Unichenko, P.; Luhmann, H.J.; White, R.; Kirischuk, S. Intracellular ion signaling influences myelin basic protein synthesis in oligodendrocyte precursor cells. Cell Calcium 2016, 60, 322–330. [Google Scholar] [CrossRef] [PubMed]








© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Moshrefi-Ravasdjani, B.; Ziemens, D.; Pape, N.; Färfers, M.; Rose, C.R. Action Potential Firing Induces Sodium Transients in Macroglial Cells of the Mouse Corpus Callosum. Neuroglia 2018, 1, 106-125. https://doi.org/10.3390/neuroglia1010009
Moshrefi-Ravasdjani B, Ziemens D, Pape N, Färfers M, Rose CR. Action Potential Firing Induces Sodium Transients in Macroglial Cells of the Mouse Corpus Callosum. Neuroglia. 2018; 1(1):106-125. https://doi.org/10.3390/neuroglia1010009
Chicago/Turabian StyleMoshrefi-Ravasdjani, Behrouz, Daniel Ziemens, Nils Pape, Marcel Färfers, and Christine R. Rose. 2018. "Action Potential Firing Induces Sodium Transients in Macroglial Cells of the Mouse Corpus Callosum" Neuroglia 1, no. 1: 106-125. https://doi.org/10.3390/neuroglia1010009
APA StyleMoshrefi-Ravasdjani, B., Ziemens, D., Pape, N., Färfers, M., & Rose, C. R. (2018). Action Potential Firing Induces Sodium Transients in Macroglial Cells of the Mouse Corpus Callosum. Neuroglia, 1(1), 106-125. https://doi.org/10.3390/neuroglia1010009