

Complex Oxide Nanoparticle Synthesis: Where to Begin to Do It Right?


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Precursor Data Collection Methods
2.2. Experimental Methods
2.2.1. Doped Cerium Oxide Ce0.9(Pr0.025Nd0.025Sm0.025Gd0.025)O2−δ
2.2.2. Doped Lanthanum Manganite (La0.8Ca0.2)(Mn0.8Al0.2)O3−δ (LCMA)
2.3. Characterization Methods
3. Results and Discussion
3.1. Precursor Selection Tools
3.1.1. Availability of Metal Salts
3.1.2. Melting/Decomposition Temperature
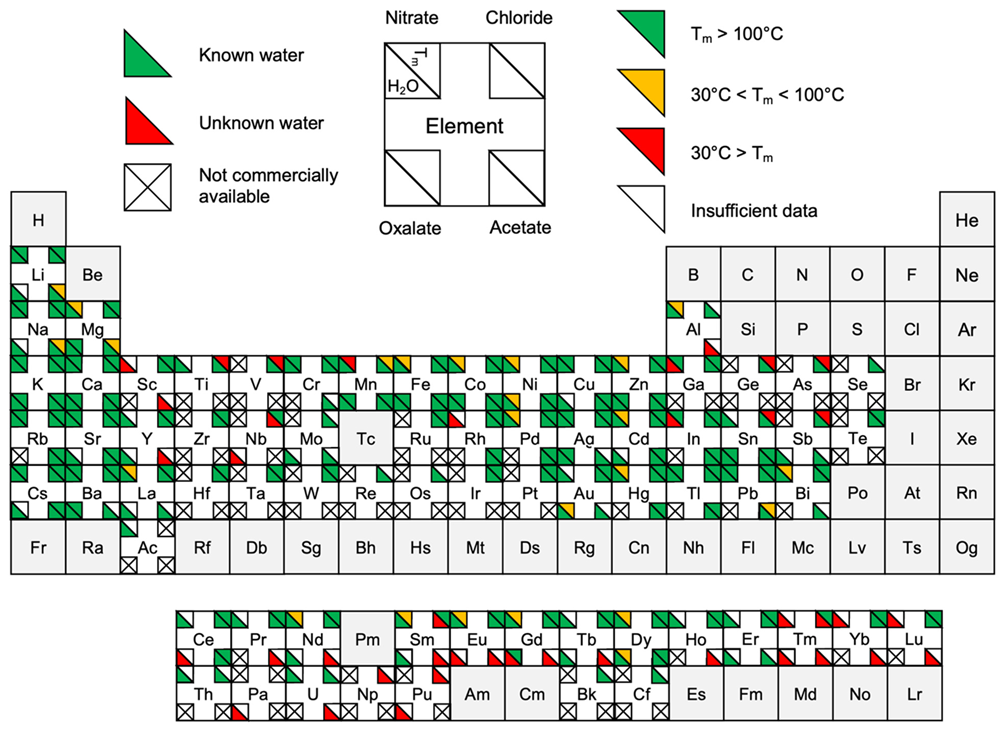
3.1.3. Hydration
3.1.4. Solvent Selection
3.2. Test Cases
3.2.1. Fluorites
3.2.2. Perovskites
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Gnesin, G.G. Revisiting the History of Materials Science on the Origin and Development of Ceramic Technology. Powder Metall. Met. Ceram. 2012, 51, 496–501. [Google Scholar] [CrossRef]
- Advanced Ceramics—Timeline. Science Learning Hub. Available online: https://www.sciencelearn.org.nz/resources/1793-advanced-ceramics-timeline (accessed on 20 June 2022).
- Yu, S.-H. Hydrothermal/Solvothermal Processing of Advanced Ceramic Materials. J. Ceram. Soc. Japn. 2001, 109, S65–S75. [Google Scholar] [CrossRef]
- Choi, H.G.; Jung, Y.H.; Kim, D.K. Solvothermal Synthesis of Tungsten Oxide Nanorod/Nanowire/Nanosheet. J. Am. Ceram. Soc. 2005, 88, 1684–1686. [Google Scholar] [CrossRef]
- Spiridigliozzi, L.; Ferone, C.; Cioffi, R.; Accardo, G.; Frattini, D.; Dell’Agli, G. Entropy-Stabilized Oxides Owning Fluorite Structure Obtained by Hydrothermal Treatment. Materials 2020, 13, 558. [Google Scholar] [CrossRef]
- Byrappa, K. Novel Hydrothermal Solution Routes of Advanced High Melting Nanomaterials Processing. J. Ceram. Soc. Japn. 2009, 117, 236–244. [Google Scholar] [CrossRef]
- Chilibon, I.; Marat-Mendes, J.N. Ferroelectric Ceramics by Sol–Gel Methods and Applications: A Review. J. Sol.-Gel. Sci. Technol. 2012, 64, 571–611. [Google Scholar] [CrossRef]
- Huang, Z.; Han, W.; Feng, Z.; Qi, J.; Wu, D.; Wei, N.; Tang, Z.; Zhang, Y.; Duan, J.; Lu, T. The Effects of Precipitants on Co-Precipitation Synthesis of Yttria-Stabilized Zirconia Nanocrystalline Powders. J. Sol.-Gel. Sci. Technol. 2019, 90, 359–368. [Google Scholar] [CrossRef]
- Colomban, P. Chemical Preparation Routes and Lowering the Sintering Temperature of Ceramics. Ceramics 2020, 3, 312–339. [Google Scholar] [CrossRef]
- Kolthoff, I.M. Theory of Coprecipitation. The Formation and Properties of Crystalline Precipitates. J. Phys. Chem. 1932, 36, 860–881. [Google Scholar] [CrossRef]
- Sinkó, K.; Szabó, G.; Zrínyi, M. Liquid-Phase Synthesis of Cobalt Oxide Nanoparticles. J. Nanosci. Nanotech. 2011, 11, 4127–4135. [Google Scholar] [CrossRef]
- Tavakoli, H.; Sarraf-Mamoory, R.; Zarei, A. Inverse Co-Precipitation Synthesis of Copper Chromite Nanoparticles. Iran. J. Chem. Chem. Eng.-Int. Engl. Ed. 2016, 35, 51–55. [Google Scholar]
- Dong, H.; Koenig, G. A Review on Synthesis and Engineering of Crystal Precursors Produced Via Coprecipitation for Multicomponent Lithium-Ion Battery Cathode Materials. CrystEngComm 2019, 22, 1514–1530. [Google Scholar] [CrossRef]
- Merlin, J.J.; Rajan, S.S. A Review on Synthesis and Preparation of Nanoparticles. J. Cancer Sci. Treat. 2020, 2, 87–89. [Google Scholar]
- Jian, G.; Xu, Y.; Lai, L.-C.; Wang, C.R.; Zachariah, M. Mn3O4 Hollow Spheres for Lithium-Ion Batteries with High Rate and Capacity. J. Mater. Chem. A 2014, 2, 4627–4632. [Google Scholar] [CrossRef]
- Dale, G.E.; Dalmas, D.A.; Gallegos, M.J.; Jackman, K.R.; Kelsey, C.T.I.; May, I.; Reilly, S.D.; Stange, G.M. 99Mo Separation from High-Concentration Irradiated Uranium Nitrate and Uranium Sulfate Solutions. Ind. Eng. Chem. Res. 2012, 51, 13319–13322. [Google Scholar] [CrossRef]
- Andersson, S.; Nitsche, H.; Sudowe, R. Berkelium Nitrate Complex Formation Using a Solvent Extraction Technique. Radiochim. Acta 2006, 94, 59–61. [Google Scholar] [CrossRef]
- Smith, R.C.; Hoilien, N.; Dykstra, C.; Campbell, S.A.; Roberts, J.T.; Gladfelter, W.L. Chemical Vapor Deposition of TixSi1–XO2 Films: Precursor Chemistry Impacts Films Composition. Chem. Vap. Depos. 2003, 9, 79–86. [Google Scholar] [CrossRef]
- Kim, D.-H.; Park, T.-H.; Bae, S.-E.; Lee, N.; Kim, J.-Y.; Cho, Y.-H.; Yeon, J.-W.; Song, K. Electrochemical Preparation and Spectroelectrochemical Study of Neptunium Chloride Complexes in LiCl–KCl Eutectic Melts. J. Radioanal. Nucl. Chem. 2016, 308, 31–36. [Google Scholar] [CrossRef]
- Homma-Takeda, S.; Terada, Y.; Nakata, A.; Sahoo, S.K.; Yoshida, S.; Ueno, S.; Inoue, M.; Iso, H.; Ishikawa, T.; Konishi, T.; et al. Elemental Imaging of Kidneys of Adult Rats Exposed to Uranium Acetate. Nucl. Instrum. Methods Phys. Res. Sect. B Beam Interact. Mater. Atoms 2009, 267, 2167–2170. [Google Scholar] [CrossRef]
- Galley, S.S.; Gaggioli, C.A.; Zeller, M.; Celis-Barros, C.; Albrecht-Schmitt, T.E.; Gagliardi, L.; Bart, S.C. Evidence of Alpha Radiolysis in the Formation of a Californium Nitrate Complex. Chem. Eur. J. 2020, 26, 8885–8888. [Google Scholar] [CrossRef]
- Murali, A.; Sohn, H.Y. Plasma-Assisted Chemical Vapor Synthesis of Indium Tin Oxide (ITO) Nanopowder and Hydrogen-Sensing Property of ITO Thin Film. Mater. Res. Express 2018, 5, 065045. [Google Scholar] [CrossRef]
- Abrão, A.; de Freitas, A.A.; de Carvalho, F.M.S. Preparation of Highly Pure Thorium Nitrate via Thorium Sulfate and Thorium Peroxide. J. Alloys Compd. 2001, 323–324, 53–56. [Google Scholar] [CrossRef]
- Yoshihara, K.; Kanno, M.; Mukaibo, T. Preparation of Uranium Disulfide from Uranium Chloride. J. Nucl. Sci. Technol. 1967, 4, 578–581. [Google Scholar] [CrossRef][Green Version]
- Stroud, M.; Salazar, R.; Sandoval, A.; Garcia, V.; Danis, J.; Abney, K.; Hatler, V. Purification of Aqueous Plutonium Chloride Solutions via Precipitation and Washing. In Abstracts of Paper of the American Chemical Society; American Chemical Society: Washington, DC, USA, 2003; Volume 226, p. U633. [Google Scholar]
- Wenzel, A.W.; Pietri, C.E. Purification of Plutonium Sulfate Tetrahydrate by Recrystallization and Ion Exchange. Anal. Chem. 1963, 35, 1324–1325. [Google Scholar] [CrossRef]
- Corbey, J.; Sweet, L.; Sinkov, S.; Reilly, D.; Parker, C.; Lonergan, J.; Johnson, T. Quantitative Microstructural Characterization of Plutonium Oxalate Auto-Degradation and Evidence for PuO2 Nanocrystal Formation. Eur. J. Inorg. Chem. 2021, 2021, 3277–3291. [Google Scholar] [CrossRef]
- Young, J.P.; Shaw, R.W.; Webb, O.F. Radioactive Origin of Emissions Observed from Uranium Compounds and Their Silica Cells. Inorg. Chem. 1999, 38, 5192–5194. [Google Scholar] [CrossRef]
- Muxart, R.; Vernois, J.; Guillaum, R.; Arapakis, H. Studies on Protactinium Oxalate. Bull. Soc. Chim. Fr. 1967, 8, 2890. [Google Scholar]
- Kremer, C.B. The Purification of Thorium Chloride Octahydrate. J. Am. Chem. Soc. 1942, 64, 1009–1010. [Google Scholar] [CrossRef]
- Smith, S.J.; Huang, B.; Liu, S.; Liu, Q.; Olsen, R.E.; Boerio-Goates, J.; Woodfield, B.F. Synthesis of Metal Oxide Nanoparticles via a Robust “Solvent-Deficient” Method. Nanoscale 2015, 7, 144–156. [Google Scholar] [CrossRef]
- Akrami, S.; Edalati, P.; Fuji, M.; Edalati, K. High-Entropy Ceramics: Review of Principles, Production and Applications. Mater. Sci. Eng. R Rep. 2021, 146, 100644. [Google Scholar] [CrossRef]
- Tseng, K.-P.; Yang, Q.; McCormack, S.J.; Kriven, W.M. High-Entropy, Phase-Constrained, Lanthanide Sesquioxide. J. Am. Ceram. Soc. 2020, 103, 569–576. [Google Scholar] [CrossRef]
- Alemayehu, A.; Zakharanka, A.; Tyrpekl, V. Homogeneous Precipitation of Lanthanide Oxalates. ACS Omega 2022, 7, 12288–12295. [Google Scholar] [CrossRef] [PubMed]
- Ollendorff, W.; Weigel, F. The Crystal Structure of Some Lanthanide Oxalate Decahydrates, Ln2(C2O4)3·10H2O, with Ln = La, Ce, Pr, and Nd. Inorg. Nucl. Chem. Lett. 1969, 5, 263–269. [Google Scholar] [CrossRef]
- Alemayehu, A.; Zákutná, D.; Kohúteková, S.; Tyrpekl, V. Transition between Two Solid-Solutions: Effective and Easy Way for Fine Ce1−xGdxO2−x/2 Powders Preparation. J. Am. Ceram. Soc. 2022, 105, 4621–4631. [Google Scholar] [CrossRef]
- Wang, D.; Belharouak, I.; Zhou, G.; Amine, K. Synthesis of Lithium and Manganese-Rich Cathode Materials via an Oxalate Co-Precipitation Method. J. Electrochem. Soc. 2013, 160, A3108. [Google Scholar] [CrossRef]
- Pelosato, R.; Cristiani, C.; Dotelli, G.; Latorrata, S.; Ruffo, R.; Zampori, L. Co-Precipitation in Aqueous Medium of La0.8Sr0.2Ga0.8Mg0.2O3−δ via Inorganic Precursors. J. Power Sources 2010, 195, 8116–8123. [Google Scholar] [CrossRef]
- Mullen, M.R.; Spirig, J.V.; Hoy, J.; Routbort, J.L.; Singh, D.; Dutta, P.K. Development of Nanosized Lanthanum Strontium Aluminum Manganite as Electrodes for Potentiometric Oxygen Sensor. Sens. Actuators B Chem. 2014, 203, 670–676. [Google Scholar] [CrossRef]









Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gager, E.; Halbert, W.; Nino, J.C. Complex Oxide Nanoparticle Synthesis: Where to Begin to Do It Right? Ceramics 2022, 5, 1019-1034. https://doi.org/10.3390/ceramics5040073
Gager E, Halbert W, Nino JC. Complex Oxide Nanoparticle Synthesis: Where to Begin to Do It Right? Ceramics. 2022; 5(4):1019-1034. https://doi.org/10.3390/ceramics5040073
Chicago/Turabian StyleGager, Elizabeth, William Halbert, and Juan C. Nino. 2022. "Complex Oxide Nanoparticle Synthesis: Where to Begin to Do It Right?" Ceramics 5, no. 4: 1019-1034. https://doi.org/10.3390/ceramics5040073
APA StyleGager, E., Halbert, W., & Nino, J. C. (2022). Complex Oxide Nanoparticle Synthesis: Where to Begin to Do It Right? Ceramics, 5(4), 1019-1034. https://doi.org/10.3390/ceramics5040073