Abstract
A number of germline syndromes that predispose affected individuals to develop renal cancer have been described, each with unique manifestations, histopathology, and tumor behavior. Patients tend to present with early onset and/or multifocal tumors. Familiarity with these syndromes helps to identify at-risk patients and recommend genetic screening. Early detection is essential to direct appropriate cancer surveillance protocols for patients and other family members and care strategies that preserve lifelong renal function while minimizing risk of death from metastatic cancer.
Introduction
Hereditary syndromes represent 5% to 8% of all renal cell carcinomas (RCCs) [1]. It is important for urologists to recognize and solicit their features. Early detection facilitates appropriate cancer surveillance protocols and appropriate surgical planning [2], and helps identify other family members at risk. Positive family history, early age of onset, or multifocal tumor should trigger consideration for referral for genetic counseling and screening.
von Hippel-Lindau Disease (VHL)
VHL is caused by inheritance of one inactivated copy of the VHL tumor suppressor gene (TSG), located on chromosome 3 (3p25–26) [3]. Inheritance is autosomal dominant, occurring in 1 in 35 000 births, with estimated prevalence in the United States of 7000 to 8000 people. Affected patients develop renal cancer and cysts, pheochromocytomas or paragangliomas, central nervous system (CNS) hemangioblastomas of the brain or spine, retinal angiomas, neuroendocrine tumors or cysts of the pancreas, and endolymphatic sac tumors of the inner ear or papillary cystadenomas of the epididymis or broad ligament [4]. RCCs in VHL are uniformly clear cell histology (ccRCC). Tumors are multifocal and early onset, and some patients require a first RCC intervention in their 20s [5]. Due to its multiorgan nature of VHL, VHL patients are optimally managed by a multidisciplinary team managing routine retinal and CNS examination and imaging as well as adrenal and pancreatic functional testing. Cancer surveillance with abdominal ultrasound or cross-sectional imaging is recommended biannually starting at age 8. Magnetic resonance imaging (MRI) is preferred, when possible, to minimize risks for repeated radiation exposure. To prevent potentially lethal complications, pheochromocytoma must be excluded or managed prior to renal surgery.
Historically, RCC was the leading cause of death in VHL. Though RCC resection can decrease risk for metastases, bilateral nephrectomy profoundly limits quality and quantity of life. To preserve renal function, a conservative approach of active surveillance until the largest renal tumor reaches 3 cm, followed by nephron-sparing surgery to remove all tumors from that kidney was established [6,7,8]. The safety of this “3 cm rule” for prevention of metastatic disease in VHL patients has been confirmed [9]. More than 80% of VHL patients will develop a recurrent de novo renal tumor within 10 years of resection [10]. The “3 cm rule” can be applied again to trigger a second [11] or even a third or fourth nephron-sparing surgery [12]. Alternatively, cutaneous ablation using radiofrequency, cryotherapy, or microwave therapy has been reported in select patients with comparable functional and oncologic outcomes [13,14]. In 2021, the oral hypoxia-inducible factor 2α (HIF-2α) inhibitor belzutifan [15] was approved by the United States Food and Drug Administration (FDA) for systemic management of VHL associated RCC, CNS, or pancreatic tumors based on a 97% RCC response rate at 21 months. Belzutifan is generally well tolerated, with the most frequent side effects being anemia and fatigue [15].
Birt-Hogg-Dubé Syndrome (BHD)
BHD is an autosomal dominant, familial syndrome [16,17,18] caused by inactivating mutations of folliculin (FLCN) [19,20]. Manifestations include cutaneous fibrofolliculomas (hair follicle tumors) or trichodiscomas. Present in at least 70% of patients by 40 years, these benign skin lesions are raised white papules on the nose and malar region and less commonly on the neck, ears, forehead, or trunk. Pulmonary cysts occur in about 80% and may rupture, with a 30% lifetime risk for spontaneous pneumothorax [21]. All individuals with pathogenic FLCN variants should be considered at risk for developing RCC regardless of family history [19]. Lifetime risk for RCC is 25% to 30%, presenting as early as 20 years, with mean age at diagnosis of 50 years [21,22]. The most characteristic BHD histopathology is a hybrid chromophobe/oncocytic pattern, but chromophobe, clear cell, and papillary RCC have all been described [22,23]. Other potential manifestations include colorectal polyps and cancer, thyroid cancer, and melanoma, though these have not yet been confirmed. Colonoscopy is indicated if there is a family history of colorectal cancer [24].
Affected or at-risk individuals should begin surveillance for RCC with annual MRI at 20 years. Ultrasonography can be used if MRI is unavailable or not tolerated. When a solid renal lesion is detected, it is managed using the “3 cm rule,” followed by nephron-sparing surgery or ablation [23]. Although mouse and cell-based models of FLCN inactivation demonstrate mammalian target of rapamycin (mTOR) pathway activation [25], a clinical trial of mTOR inhibition with topical rapamycin for fibrofolliculomas did not demonstrate therapeutic efficacy [26] and additional study is required.
Hereditary Leiomyomatosis and Renal Cell Carcinoma (HLRCC)
HLRCC was first described 50 years ago as the autosomal dominant familial Reed’s syndrome of cutaneous leiomyomas [27]. Recognition of associated uterine leiomyomas and RCC led to the designation of HLRCC [28]. In 2002, fumarate hydratase (FH) was identified as the causal TSG [ 29]. HLRCC was initially thought to be a rare disease with high penetrance, but recent data suggests that HLRCC may have an incidence as high as 1 in 1000 [30] with highly variable penetrance [31,32]. Cutaneous leiomyomas are observed in 50% to 80% as raised skin papules that may be painful. Uterine leiomyomas are reported in 30% to 80%. Though benign, they can present with heavy vaginal bleeding and early hysterectomy. FH-deficient RCC occurs in 10% to 20% and is notoriously aggressive, with early progression to metastatic disease [31,32,33]. Median age of onset is 36 to 40 years (range, 11–90 years) [17,34] and 7% present under the age of 20. If not detected by screening, RCC presents with symptoms from advanced disease (Figure 1) [33,35]. To date, most RCCs have been unifocal. However, with screening and effective early treatment, metachronous tumors have now been reported. FH-deficient RCC histology is variable and includes type 2 papillary, collecting duct, or tubulocystic morphology [32,35], frequently with two or more growth patterns, and pleiomorphic eosinophilic nucleoli surrounded by a clear halo [32].
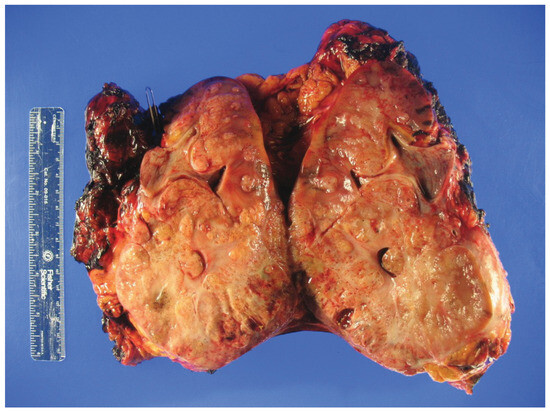
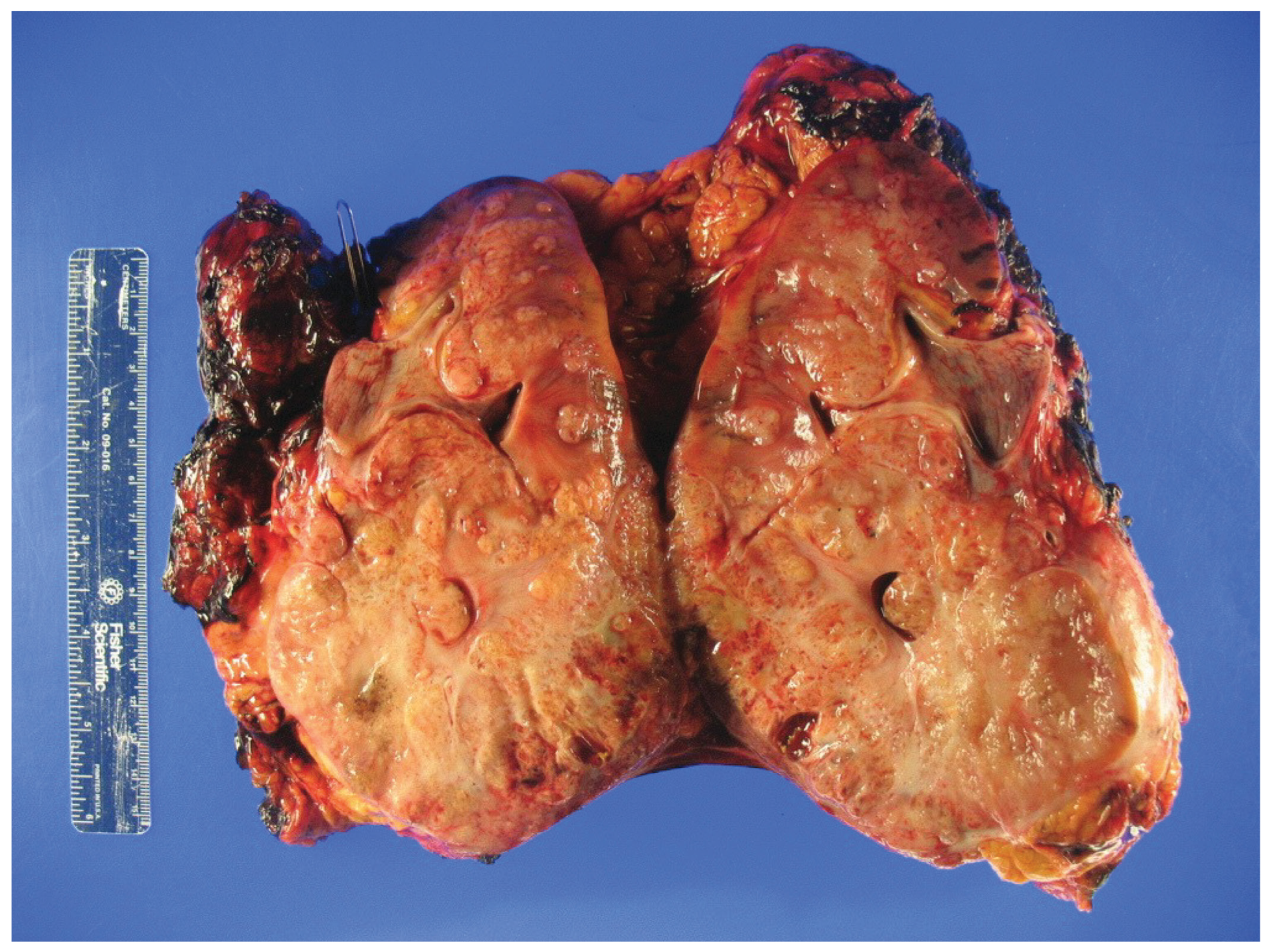
Figure 1.
Gross inspection of a radical nephrectomy specimen showing hereditary leiomyomatosis and renal cell carcinoma (HLRCC) with an infiltrative tumor appearance.
Once a germline FH mutation is identified, all first-degree relatives should be tested—ideally prior to age 10. Any patient with multiple cutaneous leiomyomas, early-onset fibroids, or non-clear cell RCC by the age of 46 should also be considered for testing [36,37,38]. Current management strategy for FH-deficient RCC is early detection with immediate intervention. Annual cross-sectional imaging with MRI (preferred) or computed tomography (CT) is necessary, as renal ultrasound can easily miss a small papillary tumor. Fluorodeoxyglucose positron emission tomography (FDG-PET) imaging may be useful due to the increased metabolic activity of FH-deficient RCC [39]. Tumors may demonstrate solid and cystic elements and are frequently infiltrative in appearance [40]. Once detected, prompt excision with clear margins is required regardless of tumor size. The “3 cm rule” does not apply to HLRCC, and nephron-sparing surgery should be performed only if a negative margin is reasonably possible. Regional retroperitoneal recurrences are common, and consideration should be given to regional node dissection even if the nodes are clinically negative [40].
Efficacy of available systemic therapy for disseminated HLRCC is limited. National Comprehensive Cancer Network (NCCN) guidelines currently recommend bevacizumab plus erlotinib as first-line therapy based on response rates of 70%, with a median progression-free survival (PFS) of 21 months [41]. Outcomes with other tyrosine kinase inhibitors have been variable. One series reported promising partial response rates approaching 50% for sunitinib or cabozantinib [42], but others found that most patients progressed within 6 months [43]. Checkpoint inhibitors have similarly shown mixed outcomes, with reports ranging from no response to complete response [44]. An ongoing trial (NCT03914742) is investigating whether DNA damage repair inhibitors may have therapeutic benefit [45].
Hereditary Papillary Renal Cell Carcinoma (HPRC)
HPRC is a rare autosomal dominant disease (incidence 1 in 500 000) [46], caused by germline-activating mutations of the MET proto-oncogene (chromosome 7). MET in turn activates multiple signaling pathways to promote RCC proliferation and survival [47]. HPRCs are typically multifocal and indolent with an International Society of Urologic Pathologists (ISUP) grade 1 or 2. They demonstrate predominantly papillary/tubulopapillary features with type 1 papillary histology [46], though concurrent ccRCC has been reported [47,48]. The median age of tumor presentation is 41 years [1] with earliest onset at 30 years. Penetrance approaches 100% by the age of 80. Advanced HPRC tumors may present with the classic triad of flank pain, hematuria, and an abdominal mass or, rarely, with lung metastasis [49,50]. No nonrenal manifestations of HPRC have been observed.
Genetic screening should be considered for any individual who has a known family history of HPRC or who develops type 1 papillary RCC prior to age 45 or multi-focal papillary tumors [51]. Testing requires bidirectional DNA sequencing. To date, all identified mutations reside in 4 of 21 MET exons [47,52,53,54]. Because papillary tumors are frequently isoechoic and missed by ultrasound, routine cross-sectional imaging with CT or MRI is recommended every 2 years [55,56]. Tumors are characteristically hypovascular with enhancement of 10–30 Hounsfield units. Management of RCC follows the “3 cm rule,” with nephron-sparing surgery as in VHL [55]. Good preservation of renal function is documented for partial nephrectomy even with more than 20 tumors [57]. Several agents targeting the MET pathway have been studied for potential efficacy against HPRC [47]. Foretinib, a multikinase inhibitor of MET, VEFGR2, RON, AXL, and TIE-2 receptors, showed responses in 50% of patients with germline HPRC [52]. Other MET-targeting agents including cabozantinib are being explored [58], potentially opening new therapeutic options for these patients.
Tuberous Sclerosis Complex (TSC)
TSC is a multiorgan syndrome with autosomal dominant inheritance. Estimated incidence is 1 in 6000 to 10 000 new births. Although penetrance in TSC is over 95%, there is significant variability in the disease phenotype, including rare cases with no overt manifestations [59]. Dermatologic (facial angiofibromas, hypomelanotic macules, fibrous cephalic plaques, shagreen patches, and ungual fibromas) and CNS manifestations (cognitive/ behavioral impairment or structural issues leading to epilepsy or nodules of the ventricular walls) present in 90% in early childhood [60]. Renal manifestations include angiomyolipomas (AMLs) in nearly 70% and RCC in 2% to 4% [61]. A pathogenic alteration can be found in the genes for TSC (TSC1-hamartin and TSC2- tuberin) in approximately 80% with both somatic and germline mosaicism described [62]. Up to 50% are de novo alterations [63]. TSC2 germline defects are associated with greater disease burden and severity than TSC1 [64].
AMLs (mesenchymal tumors of the PEComa family) are typically bilateral and multifocal. They develop in late childhood at a median age of 16.9 years [65]. Bulky AMLs may merge, making it difficult to distinguish clear boundaries between lesions. The majority are benign, consisting of vascular, smooth muscle, and fatty elements. A rare but important variant, epithelioid AML, is composed of epithelioid cells with minimal to no fat. Epithelioid AMLs may grow rapidly and are more prone to necrosis and hemorrhage and progression, with distant metastases observed in up to 33% in multiple series (Figure 2). Screening recommendations include baseline abdominal imaging with CT or MRI at diagnosis and then every 1 to 3 years [66]. AMLs are usually easily identified by the presence of macroscopic fat, though fat-poor lesions may require confirmatory biopsy. Ultrasound does not adequately assess tumor size or presence of fat. Contrast-enhanced MRI should be coordinated with brain MRI whenever possible to minimize number of procedures under sedation. With prospective surveillance, more than 80% of TSC AMLs are identified prior to onset of symptoms or hemorrhage [65]. They can be safely observed until they reach 4 cm, at which time embolization or resection is indicated. Epithelioid variants should be considered for earlier resection given their risk for malignant behavior. Alternatively, everolimus is approved for the medical management of AML and has also shown efficacy in the setting of metastatic epithelioid AML [67].
Figure 2.
Epithelioid variant angiomyolipoma (AML) in a patient with tuberous sclerosis complex (TSC) who had failed two attempts at angioembolization of large vessel (coil seen).
TSC RCCs occur at a median age of 28 years (range, 7– 59) [61]. Perhaps due to routine surveillance imaging, the majority are small (median size, 2.9 cm), incidental [61], and localized. Nearly 50% are multifocal [61,68]. Our understanding of TSC RCC has evolved as more cases have been reported. Initially, histology was believed to be similar to sporadic RCC, including ccRCC [63] molecularly distinct from VHL [69]. In recent years, common morphologic patterns have emerged including chromophobe and hybrid oncocytic/chromophobe tumors (HOCT), RCC with smooth muscle stroma (also known as renal angiomyoadenomatous tumors), and eosinophilic unclassified variants [61,68]. Notably, the latter are reminiscent of sporadic eosinophilic solid cystic RCC (ESC RCC), which harbors somatic mTOR/ TSC mutations. There are limited data to suggest that TSC RCC should be treated differently than other forms of RCC. However, renal preservation should be prioritized with use of partial nephrectomy or ablation when feasible.
PTEN hamartoma tumor syndrome (PHTS)
PHTS is a spectrum of autosomal dominant disorders caused by germline mutations in the PTEN TSG. The best recognized form is Cowden syndrome (CS), but others include Bannayan-Riley-Ruvalcaba syndrome (BRRS), Proteus-like syndrome, and macrocephaly with autism and/or learning disability. Major manifestations of CS include mucocutaneous papillomatous papules and trichilemmomas, macrocephaly, multinodular goiter, follicular adenomas of the thyroid, and increased lifetime risks for breast (>80%), nonmedullary thyroid (~35%), and endometrial cancers (~30%) [70,71]. RCC, most commonly papillary and chromophobe subtypes, occurs in 15% to 24%, with median age at diagnosis of 50 years [70,72,73]. CS RCC is usually unilateral [73], with reported age at onset as early as 20 years. At least one manifestation of CS will be evident in 90% by age 30 years [74].
When clinical diagnostic criteria for Cowden syndrome are met [74], diagnosis of PHTS is confirmed by germline testing for a PTEN pathogenic variant [75]. PTEN is now included on many multigene cancer testing panels. A positive test should trigger testing of at-risk relatives. Comprehensive PHTS surveillance protocols are recommended starting at age 18 years [74,76]. For women, annual mammography or breast MRI starts at age 30 years and endometrial cancer screening at age 35 years. Prophylactic mastectomy may be offered. Annual thyroid examinations and biannual dermatologic assessments begin at time of diagnosis and regular colonoscopy at 35 years. Biannual RCC screening is started at 20 years [77]. Treatment of PHTS RCC is the same as for sporadic RCC, and available data suggest that the “3 cm rule” can be applied.
BAP1 Tumor Predisposition Syndrome (BAP1-TPDS)
BAP1-TPDS is an autosomal dominant, multiorgan cancer syndrome caused by germline loss or mutation of BAP1, which is located on 3p21.1 and frequently codeleted with VHL in ccRCC [78]. BAP1 codes for ubiquitin carboxyl-terminal hydrolase BAP1, a nuclear deubiquitinating enzyme involved in chromatin remodeling and repair of double-stranded DNA breaks [79]. The prevalence of germline BAP1-TPDS is unknown, but it is estimated to represent 1% to 1.5% of all ccRCCs and nearly 20% of patients who develop both RCC and uveal melanoma [80]. Nonrenal BAP1-TPDS manifestations include pigmented skin lesions (BAP1-inactivated melanocytic tumors), aggressive uveal melanoma with high risk for metastasis and poor survival, and early-onset malignant mesothelioma (MM) of the pleura or peritoneum [81]. Less commonly, cutaneous melanoma, basal cell carcinoma, meningioma, cholangiocarcinoma, and breast cancer are observed. Penetrance is variable but high, with at least one tumor observed in nearly 90% of affected individuals [81].
RCC occurs in 10% of BAP1-TPDS patients and is often bilateral and multifocal [55,82]. Tumors are predominantly ccRCC, though papillary and chromophobe have been reported [55,81]. Median age at onset is 47 years. BAP1-TPDS RCC is typically high grade with poor clinical outcome, and the “3 cm rule” may not be appropriate. Close surveillance of affected individuals with early excision is recommended until the syndrome is better characterized [82].
Genetic consultation and screening should be offered to any individual with a personal or family history of two or more BAP1-TPDS tumors [83], with testing of at-risk relatives if positive. Current guidelines for affected individuals require annual fundus and full body dermatologic examination annually starting at 2 years, MRI of the brain every other year starting at age 18, and MRI of the chest, abdomen, pelvis, and breast every other year starting at age 30 with annual mammography starting at age 40 [84]. Once a renal tumor is identified, however, annual abdominal imaging is advised due to the documented rapid growth rate of BAP1-TPDS RCC [85].
Succinate Dehydrogenase (SDH)-Deficient RCC
SDH-deficient RCC is a rare autosomal dominant syndrome caused by germline mutations of the succinate dehydrogenase complex (SDHA: 5p15.33; SDHB: 1p36.13; SDHC: 1q23.3; and SDHD: 11q23) [86].
In addition to early-onset RCC, affected individuals may develop paragangliomas (commonly of the head and neck), pheochromocytoma, wild-type (negative for mutations of the KIT and PDGFRA genes) gastrointestinal stromal tumor (GIST), and, less commonly, prolactin-secreting pituitary adenoma [55,82]. Renal masses may be multifocal and bilateral and are morphologically distinct from other RCCs [87]. Germline SDH defects can be detected in up to 15% of all pheochromocytomas and paragangliomas and 1% to 1.5% of all RCCs [55,88].
SDH is a complex enzyme composed of four subunits: SDHA, SDHB, SDHC, and SDHD. It is often referred to as Mitochondrial Complex II and is anchored on the inner mitochondrial membrane where it participates in both the Krebs cycle (oxidation of succinate to fumarate) and the electron transport chain [89]. Loss of SDH function leads to accumulation of succinate and a metabolic shift to aerobic glycolysis [82]. Most cases of SDH-deficient RCC involve germline mutations of SDHB, though all subunits have been reported [55].
SDH-deficient RCCs are tan to brown with well-circumscribed “pushing” margins [90]. Cystic features are common. Tumors have a distinctive histologic appearance of cuboidal cells with inconspicuous nucleoli arranged in nests or tubules, with eosinophilic cytoplasm containing vacuoles and cytoplasmic inclusions [55,86]. Although the majority are low grade, high-grade SDH-deficient RCCs are aggressive [86,91] and known for early metastasis [82]. At-risk individuals should be screened annually with abdominal MRI or CT. The “3 cm rule” does not apply, and prompt excision regardless of size is recommended using nephron-sparing surgery where possible.
Hereditary Hyperparathyroidism Jaw Tumor Syndrome (HPT-JT)
HPT-JT is a syndrome of parathyroid adenoma and cancer, benign ossifying fibromas of the jawbone, and renal and uterine cancers. It commonly presents as early-onset primary hyperparathyroidism, with an estimated penetrance of 65% by age 50 [92]. Inheritance is autosomal dominant and conferred by germline mutations of the CDC73 gene, which encodes the nuclear protein parafibromin. Renal manifestations include renal cysts and tumors, with both ccRCC and Wilm’s tumor reported [93]. Though there are currently no consensus guidelines, renal screening by abdominal ultrasound every 5 years has been recommended for at-risk individuals [55].
Conclusion
Hereditary RCC syndromes are a diverse group with varying penetrance, histology, and clinical behavior. Although hereditary RCCs are uncommon, most urologists will encounter them, and it is important to remain vigilant, take an appropriately detailed history, make use of genetic counselors when indicated, and select a surveillance and management strategy that addresses the tumor biology and lifelong risk for recurrence.
Conflicts of Interest
None declared.
Abbreviations
| AML | angiomyolipoma |
| BAP1-TPDS BAP1 | tumor predisposition syndrome |
| BHD | Birt-Hogg-Dubé syndrome |
| ccRCC | clear cell renal cell carcinoma |
| CNS | central nervous system |
| CS | Cowden syndrome |
| CT | computed tomography |
| ESC RCC | eosinophilic solid cystic RCC |
| FH | fumarate hydratase |
| HLRCC | hereditary leiomyomatosis and renal cell carcinoma |
| MRI | magnetic resonance imaging |
| mTOR | mammalian target of rapamycin |
| PHTS PTEN | hamartoma tumor syndrome |
| RCC | renal cell carcinoma |
| TSC | tuberous sclerosis complex |
| TSG | tumor suppressor gene |
| VHL | von Hippel-Lindau disease |
References
- Shuch, B.; Vourganti, S.; Ricketts, C.J.; Middleton, L.; Peterson, J.; Merino, M.J.; et al. Defining early-onset kidney cancer: implications for germline and somatic mutation testing and clinical management. J Clin Oncol. 2014, 32, 431–437. [Google Scholar] [CrossRef] [PubMed]
- Gomella, P.T.; Linehan, W.M.; Ball, M.W. Precision Surgery and Kidney Cancer: Knowledge of Genetic Alterations Influences Surgical Management. Genes (Basel). 2021, 12. [Google Scholar] [CrossRef]
- Latif, F.; Tory, K.; Gnarra, J.; Yao, M.; Duh, F.M.; Orcutt, M.L.; et al. Identification of the von Hippel-Lindau disease tumor suppressor gene. Science. 1993, 260, 1317–1320. [Google Scholar] [CrossRef] [PubMed]
- Linehan, W.M.; Schmidt, L.S.; Crooks, D.R.; Wei, D.; Srinivasan, R.; Lang, M.; et al. The metabolic basis of kidney cancer. Cancer Discov. 2019, 9, 1006–1021. [Google Scholar] [CrossRef] [PubMed]
- Lonser, R.R.; Glenn, G.M.; Walther, M.; Chew, E.Y.; Libutti, S.K.; Linehan, W.M.; et al. von Hippel-Lindau disease. Lancet. 2003, 361, 2059–2067. [Google Scholar] [CrossRef]
- Duffey, B.G.; Choyke, P.L.; Glenn, G.; Grubb, R.L.; Venzon, D.; Linehan, W.M.; et al. The relationship between renal tumor size and metastases in patients with von Hippel-Lindau disease. J Urol. 2004, 172, 63–65. [Google Scholar] [CrossRef] [PubMed]
- Walther, M.M.; Choyke, P.L.; Glenn, G.; Lyne, J.C.; Rayford, W.; Venzon, D.; et al. Renal cancer in families with hereditary renal cancer: prospective analysis of a tumor size threshold for renal parenchymal sparing surgery. J Urol. 1999, 161, 1475–1479. [Google Scholar] [CrossRef] [PubMed]
- Walther, M.M.; Choyke, P.L.; Weiss, G.; Manolatos, C.; Long, J.; Reiter, R.; et al. Parenchymal sparing surgery in patients with hereditary renal cell carcinoma. J Urol. 1995, 153 (3 Pt 2), 913–916. [Google Scholar]
- Herring, J.C.; Enquist, E.G.; Chernoff, A.; Linehan, W.M.; Choyke, P.L.; Walther, M.M. Parenchymal sparing surgery in patients with hereditary renal cell carcinoma: 10-year experience. J Urol. 2001, 165, 777–781. [Google Scholar] [CrossRef]
- Steinbach, F.; Novick, A.C.; Zincke, H.; Miller, D.P.; Williams, R.D.; Lund, G.; et al. Treatment of renal cell carcinoma in von Hippel-Lindau disease: a multicenter study. J Urol. 1995, 153, 1812–1816. [Google Scholar] [CrossRef]
- Johnson, A.; Sudarshan, S.; Liu, J.; Linehan, W.M.; Pinto, P.A.; Bratslavsky, G. Feasibility and outcomes of repeat partial nephrectomy. J Urol. 2008, 180, 89–93, discussion. [Google Scholar] [CrossRef] [PubMed]
- Bratslavsky, G.; Liu, J.J.; Johnson, A.D.; Sudarshan, S.; Choyke, P.L.; Linehan, W.M.; et al. Salvage partial nephrectomy for hereditary renal cancer: feasibility and outcomes. J Urol. 2008, 179, 67–70. [Google Scholar] [CrossRef]
- Carrion, D.M.; Linares-Espinos, E.; Rios Gonzalez, E.; Bazan, A.A.; Alvarez-Maestro, M.; Martinez-Pineiro, L. Invasive management of renal cell carcinoma in von Hippel-Lindau disease. Cent European J Urol. 2020, 73, 167–172. [Google Scholar]
- Park, B.K.; Kim, C.K.; Park, S.Y.; Shen, S.H. Percutaneous radiofrequency ablation of renal cell carcinomas in patients with von Hippel Lindau disease: indications, techniques, complications, and outcomes. Acta Radiol. 2013, 54, 418–427. [Google Scholar] [CrossRef] [PubMed]
- Jonasch, E.; Donskov, F.; Iliopoulos, O.; Rathmell, W.K.; Narayan, V.K.; Maughan, B.L.; et al. Belzutifan for Renal Cell Carcinoma in von Hippel- Lindau Disease. N Engl J Med. 2021, 385, 2036–2046. [Google Scholar] [CrossRef] [PubMed]
- Birt, A.R.; Hogg, G.R.; Dube, W.J. Hereditar y multiple fibrofolliculomas with trichodiscomas and acrochordons. Arch Dermatol. 1977, 113, 1674–1677. [Google Scholar] [CrossRef] [PubMed]
- Menko, F.H.; Maher, E.R.; Schmidt, L.S.; Middelton, L.A.; Aittomaki, K.; Tomlinson, I.; et al. Hereditary leiomyomatosis and renal cell cancer (HLRCC): renal cancer risk, surveillance and treatment. Fam Cancer. 2014, 13, 637–644. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, L.S.; Linehan, W.M. Molecular genetics and clinical features of Birt-Hogg-Dube syndrome. Nat Rev Urol. 2015, 12, 558–569. [Google Scholar] [CrossRef] [PubMed]
- Matsumoto, K.; Lim, D.; Pharoah, P.D.; Maher, E.R.; Marciniak, S.J. A systematic review assessing the existence of pneumothorax-only variants of FLCN. Implications for lifelong surveillance of renal tumours. Eur J Hum Genet. 2021, 29, 1595–1600. [Google Scholar] [CrossRef]
- Nickerson, M.L.; Warren, M.B.; Toro, J.R.; Matrosova, V.; Glenn, G.; Turner, M.L.; et al. Mutations in a novel gene lead to kidney tumors, lung wall defects, and benign tumors of the hair follicle in patients with the Birt-Hogg-Dube syndrome. Cancer Cell. 2002, 2, 157–164. [Google Scholar] [CrossRef]
- Houweling, A.C.; Gijezen, L.M.; Jonker, M.A.; van Doorn, M.B.; Oldenburg, R.A.; van Spaendonck-Zwarts, K.Y.; et al. Renal cancer and pneumothorax risk in Birt-Hogg-Dube syndrome; an analysis of 115 FLCN mutation carriers from 35 BHD families. Br J Cancer. 2011, 105, 1912–1919. [Google Scholar] [CrossRef] [PubMed]
- Benusiglio, P.R.; Giraud, S.; Deveaux, S.; Mejean, A.; Correas, J.M.; Joly, D.; et al. Renal cell tumour characteristics in patients with the Birt-Hogg- Dube cancer susceptibility syndrome: a retrospective, multicentre study. Orphanet J Rare Dis. 2014, 9, 163. [Google Scholar] [CrossRef]
- Stamatakis, L.; Metwalli, A.R.; Middelton, L.A.; Marston Linehan, W. Diagnosis and management of BHD-associated kidney cancer. Fam Cancer. 2013, 12, 397–402. [Google Scholar] [CrossRef] [PubMed]
- Sattler, E.C.; Syunyaeva, Z.; Reithmair, M.; Dempke, W.; Steinlein, O.K. Colorectal cancer risk in families with Birt-Hogg-Dube syndrome increased. Eur J Cancer. 2021, 151, 168–174. [Google Scholar] [CrossRef] [PubMed]
- Baba, M.; Furihata, M.; Hong, S.B.; Tessarollo, L.; Haines, D.C.; Southon, E.; et al. Kidney-targeted Birt-Hogg-Dube gene inactivation in a mouse model: Erk1/2 and Akt-mTOR activation, cell hyperproliferation, and polycystic kidneys. J Natl Cancer Inst. 2008, 100, 140–154. [Google Scholar] [CrossRef] [PubMed]
- Gijezen, L.M.; Vernooij, M.; Martens, H.; Oduber, C.E.; Henquet, C.J.; Starink, T.M.; et al. Topical rapamycin as a treatment for fibrofolliculomas in Birt- Hogg-Dube syndrome: a double-blind placebo-controlled randomized split-face trial. PLoS One. 2014, 9, e99071. [Google Scholar] [CrossRef]
- Reed, W.B.; Walker, R.; Horowitz, R. Cutaneous leiomyomata with uterine leiomyomata. Acta Derm Venereol. 1973, 53, 409–416. [Google Scholar] [CrossRef] [PubMed]
- Launonen, V.; Vierimaa, O.; Kiuru, M.; Isola, J.; Roth, S.; Pukkala, E.; et al. Inherited susceptibility to uterine leiomyomas and renal cell cancer. Proc Natl Acad Sci U S A. 2001, 98, 3387–3392. [Google Scholar] [CrossRef] [PubMed]
- Tomlinson, I.P.; Alam, N.A.; Rowan, A.J.; Barclay, E.; Jaeger, E.E.; Kelsell, D.; et al. Germline mutations in FH predispose to dominantly inherited uterine fibroids, skin leiomyomata and papillary renal cell cancer. Nat Genet. 2002, 30, 406–410. [Google Scholar]
- Shuch, B.; Li, S.; Risch, H.; Bindra, R.S.; McGillivray, P.D.; Gerstein, M. Estimation of the carrier frequency of fumarate hydratase alterations and implications for kidney cancer risk in hereditary leiomyomatosis and renal cancer. Cancer. 2020, 126, 3657–3666. [Google Scholar] [CrossRef]
- Forde, C.; Lim, D.H.K.; Alwan, Y.; Burghel, G.; Butland, L.; Cleaver, R.; et al. Hereditary Leiomyomatosis and Renal Cell Cancer: Clinical, Molecular, and Screening Features in a Cohort of 185 Affected Individuals. Eur Urol Oncol. 2019. [Google Scholar] [CrossRef] [PubMed]
- Muller, M.; Ferlicot, S.; Guillaud-Bataille, M.; Le Teuff, G.; Genestie, C.; Deveaux, S.; et al. Reassessing the clinical spectrum associated with hereditary leiomyomatosis and renal cell carcinoma syndrome in French FH mutation carriers. Clin Genet. 2017, 92, 606–615. [Google Scholar] [CrossRef] [PubMed]
- Grubb, R.L., 3rd; Franks, M.E.; Toro, J.; Middelton, L.; Choyke, L.; Fowler, S.; et al. Hereditary leiomyomatosis and renal cell cancer: a syndrome associated with an aggressive form of inherited renal cancer. J Urol. 2007, 177, 2074–2079; discussion 9–80. [Google Scholar] [CrossRef]
- Chen, Y.B.; Brannon, A.R.; Toubaji, A.; Dudas, M.E.; Won, H.H.; Al-Ahmadie, H.A.; et al. Hereditary leiomyomatosis and renal cell carcinoma syndrome-associated renal cancer: recognition of the syndrome by pathologic features and the utility of detecting aberrant succination by immunohistochemistry. Am J Surg Pathol. 2014, 38, 627–637. [Google Scholar] [CrossRef] [PubMed]
- Merino, M.J.; Torres-Cabala, C.; Pinto, P.; Linehan, W.M. The morphologic spectrum of kidney tumors in hereditary leiomyomatosis and renal cell carcinoma (HLRCC) syndrome. Am J Surg Pathol. 2007, 31, 1578–1585. [Google Scholar] [CrossRef] [PubMed]
- Bratslavsky, G.; Mendhiratta, N.; Daneshvar, M.; Brugarolas, J.; Ball, M.W.; Metwalli, A.; et al. Genetic risk assessment for hereditary renal cell carcinoma: Clinical consensus statement. Cancer. 2021, 127, 3957–3966. [Google Scholar] [CrossRef] [PubMed]
- Joseph, N.M.; Solomon, D.A.; Frizzell, N.; Rabban, J.T.; Zaloudek, C.; Garg, K. Morphology and Immunohistochemistry for 2SC and FH aid in detection of fumarate hydratase gene aberrations in uterine leiomyomas from young patients. Am J Surg Pathol. 2015, 39, 1529–1539. [Google Scholar] [CrossRef] [PubMed]
- Kopp, R.P.; Stratton, K.L.; Glogowski, E.; Schrader, K.A.; Rau-Murthy, R.; Russo, P.; et al. Utility of prospective pathologic evaluation to inform clinical genetic testing for hereditary leiomyomatosis and renal cell carcinoma. Cancer. 2017, 123, 2452–2458. [Google Scholar] [CrossRef]
- Yamasaki, T.; Tran, T.A.; Oz, O.K.; Raj, G.V.; Schwarz, R.E.; Deberardinis, R.J.; et al. Exploring a glycolytic inhibitor for the treatment of an FH-deficient type-2 papillary RCC. Nat Rev Urol. 2011, 8, 165–171. [Google Scholar] [CrossRef] [PubMed]
- Nikolovski, I.; Carlo, M.I.; Chen, Y.B.; Vargas, H.A. Imaging features of fumarate hydratase-deficient renal cell carcinomas: a retrospective study. Cancer Imaging. 2021, 21, 24. [Google Scholar] [CrossRef]
- Srinivasan, R.; Gurram, S.; Al Harthy, M.A.; Singer, E.A.; Sidana, A.; Shuch, B.; et al. Results from a phase II study of bevacizumab and erlotinib in subjects with advanced hereditary leiomyomatosis and renal cell cancer (HLRCC) or sporadic papillary renal cell cancer. J Clin Oncol. 2020, 38, 5004. [Google Scholar] [CrossRef]
- Carril-Ajuria, L.; Colomba, E.; Cerbone, L.; Romero-Ferreiro, C.; Crouzet, L.; Laguerre, B.; et al. Response to systemic therapy in fumarate hydratase- deficient renal cell carcinoma. Eur J Cancer. 2021, 151, 106–114. [Google Scholar] [CrossRef] [PubMed]
- Gleeson, J.P.; Nikolovski, I.; Dinatale, R.; Zucker, M.; Knezevic, A.; Patil, S.; et al. Comprehensive Molecular Characterization and Response to Therapy in Fumarate Hydratase-Deficient Renal Cell Carcinoma. Clin Cancer Res. 2021, 27, 2910–2919. [Google Scholar] [CrossRef]
- Iribe, Y.; Furuya, M.; Shibata, Y.; Yasui, M.; Funahashi, M.; Ota, J.; et al. Complete response of hereditary leiomyomatosis and renal cell cancer (HLRCC)-associated renal cell carcinoma to nivolumab and ipilimumab combination immunotherapy by: a case report. Fam Cancer. 2021, 20, 75–80. [Google Scholar] [CrossRef] [PubMed]
- Sulkowski, P.L.; Sundaram, R.K.; Oeck, S.; Corso, C.D.; Liu, Y.; Noorbakhsh, S.; et al. Krebs-cycle-deficient hereditary cancer syndromes are defined by defects in homologous-recombination DNA repair. Nat Genet 2018, 50, 1086–1092. [Google Scholar] [CrossRef]
- Zbar, B.; Tory, K.; Merino, M.; Schmidt, L.; Glenn, G.; Choyke, P.; et al. Hereditary papillary renal cell carcinoma. J Urol. 1994, 151, 561–566. [Google Scholar] [CrossRef]
- Schmidt, L.; Duh, F.M.; Chen, F.; Kishida, T.; Glenn, G.; Choyke, P.; et al. Germline and somatic mutations in the tyrosine kinase domain of the MET proto- oncogene in papillary renal carcinomas. Nat Genet. 1997, 16, 68–73. [Google Scholar] [CrossRef]
- Ferlicot, S.; Just, P.A.; Comperat, E.; Rouleau, E.; Tissier, F.; Vaessen, C.; et al. Clear cell and papillary renal cell carcinomas in hereditary papillary renal cell carcinoma (HPRCC) syndrome: a case report. Diagn Pathol. 2021, 16, 107. [Google Scholar] [CrossRef]
- Choyke, P.L.; Glenn, G.M.; Walther, M.M.; Zbar, B.; Linehan, W.M. Hereditary renal cancers. Radiology. 2003, 226, 33–46. [Google Scholar] [CrossRef]
- Lubensky, I.A.; Schmidt, L.; Zhuang, Z.; Weirich, G.; Pack, S.; Zambrano, N.; et al. Hereditary and sporadic papillary renal carcinomas with c-met mutations share a distinct morphological phenotype. Am J Pathol. 1999, 155, 517–526. [Google Scholar] [CrossRef]
- PCGE, B. Hereditary Papillary Renal Carcinoma (PDQ®)2021.
- Choueiri, T.K.; Vaishampayan, U.; Rosenberg, J.E.; Logan, T.F.; Harzstark, A.L.; Bukowski, R.M.; et al. Phase II and biomarker study of the dual MET/VEGFR2 inhibitor foretinib in patients with papillary renal cell carcinoma. J Clin Oncol. 2013, 31, 181–186. [Google Scholar] [CrossRef]
- Duh, F.M.; Scherer, S.W.; Tsui, L.C.; Lerman, M.I.; Zbar, B.; Schmidt, L. Gene structure of the human MET proto-oncogene. Oncogene. 1997, 15, 1583–1586. [Google Scholar] [CrossRef] [PubMed]
- Park, M.; Dean, M.; Kaul, K.; Braun, M.J.; Gonda, M.A.; Vande Woude, G. Sequence of MET protooncogene cDNA has features characteristic of the tyrosine kinase family of growth-factor receptors. Proc Natl Acad Sci U S A. 1987, 84, 6379–6383. [Google Scholar] [CrossRef]
- Carlo, M.I.; Hakimi, A.A.; Stewart, G.D.; Bratslavsky, G.; Brugarolas, J.; Chen, Y.B.; et al. Familial kidney cancer: implications of new syndromes and molecular insights. Eur Urol. 2019, 76, 754–764. [Google Scholar] [CrossRef] [PubMed]
- Kiuru, M.; Kujala, M.; Aittomaki, K. Inherited forms of renal cell carcinoma. Scand J Surg. 2004, 93, 103–111. [Google Scholar] [CrossRef] [PubMed]
- Fadahunsi, A.T.; Sanford, T.; Linehan, W.M.; Pinto, P.A.; Bratslavsky, G. Feasibility and outcomes of partial nephrectomy for resection of at least 20 tumors in a single renal unit. J Urol. 2011, 185, 49–53. [Google Scholar] [CrossRef]
- Martinez Chanza, N.; Xie, W.; Asim Bilen, M.; Dzimitrowicz, H.; Burkart, J.; Geynisman, D.M.; et al. Cabozantinib in advanced non-clear-cell renal cell carcinoma: a multicentre, retrospective, cohort study. Lancet Oncol. 2019, 20, 581. [Google Scholar] [CrossRef]
- Osborne, J.P.; Jones, A.C.; Burley, M.W.; Jeganathan, D.; Young, J.; O'Callaghan, F.J.; et al. Non-penetrance in tuberous sclerosis. Lancet. 2000, 355, 1698. [Google Scholar] [CrossRef]
- Curatolo, P.; Bombardieri, R.; Jozwiak, S. Tuberous sclerosis. Lancet. 2008, 372, 657–668. [Google Scholar] [CrossRef]
- Yang, P.; Cornejo, K.M.; Sadow, P.M.; Cheng, L.; Wang, M.; Xiao, Y.; et al. Renal cell carcinoma in tuberous sclerosis complex. Am J Surg Pathol. 2014, 38, 895–909. [Google Scholar] [CrossRef]
- Verhoef, S.; Bakker, L.; Tempelaars, A.M.; Hesseling-Janssen, A.L.; Mazurczak, T.; Jozwiak, S.; et al. High rate of mosaicism in tuberous sclerosis complex. Am J Hum Genet. 1999, 64, 1632–1637. [Google Scholar] [CrossRef] [PubMed]
- Crino, P.B.; Nathanson, K.L.; Henske, E.P. The tuberous sclerosis complex. N Engl J Med. 2006, 355, 1345–1356. [Google Scholar] [CrossRef] [PubMed]
- Sancak, O.; Nellist, M.; Goedbloed, M.; Elfferich, P.; Wouters, C.; Maat- Kievit, A.; et al. Mutational analysis of the TSC1 and TSC2 genes in a diagnostic setting: genotype--phenotype correlations and comparison of diagnostic DNA techniques in Tuberous Sclerosis Complex. Eur J Hum Genet. 2005, 13, 731–741. [Google Scholar] [CrossRef]
- Kingswood, J.C.; Belousova, E.; Benedik, M.P.; Carter, T.; Cottin, V.; Curatolo, P.; et al. Renal angiomyolipoma in patients with tuberous sclerosis complex: findings from the TuberOus SClerosis registry to increase disease Awareness. Nephrol Dial Transplant. 2019, 34, 502–508. [Google Scholar] [CrossRef] [PubMed]
- Northrup, H.; Aronow, M.E.; Bebin, E.M.; Bissler, J.; Darling, T.N.; de Vries, P.J.; et al. Updated international tuberous sclerosis complex diagnostic criteria and surveillance and management recommendations. Pediatr Neurol. 2021, 123, 50–66. [Google Scholar] [CrossRef]
- Guo, G.; Gu, L.; Zhang, X. Everolimus in invasive malignant renal epithelioid angiomyolipoma. Front Oncol. 2020, 10, 610858. [Google Scholar] [CrossRef]
- Guo, J.; Tretiakova, M.S.; Troxell, M.L.; Osunkoya, A.O.; Fadare, O.; Sangoi, A.R.; et al. Tuberous sclerosis-associated renal cell carcinoma: a clinicopathologic study of 57 separate carcinomas in 18 patients. Am J Surg Pathol. 2014, 38, 1457–1467. [Google Scholar] [CrossRef] [PubMed]
- Duffy, K.; Al-Saleem, T.; Karbowniczek, M.; Ewalt, D.; Prowse, A.H.; Henske, E.P. Mutational analysis of the von hippel lindau gene in clear cell renal carcinomas from tuberous sclerosis complex patients. Mod Pathol. 2002, 15, 205–210. [Google Scholar] [CrossRef]
- Hendricks, L.A.J.; Hoogerbrugge, N.; Schuurs-Hoeijmakers, J.H.M.; Vos, J.R. A review on age-related cancer risks in PTEN hamartoma tumor syndrome. Clin Genet. 2021, 99, 219–225. [Google Scholar] [CrossRef]
- Tan, M.H.; Mester, J.L.; Ngeow, J.; Rybicki, L.A.; Orloff, M.S.; Eng, C. Lifetime cancer risks in individuals with germline PTEN mutations. Clin Cancer Res. 2012, 18, 400–407. [Google Scholar] [CrossRef] [PubMed]
- Mester, J.L.; Zhou, M.; Prescott, N.; Eng, C. Papillary renal cell carcinoma is associated with PTEN hamartoma tumor syndrome. Urology. 2012, 79, 1187–e1. [Google Scholar] [CrossRef] [PubMed]
- Shuch, B.; Ricketts, C.J.; Vocke, C.D.; Komiya, T.; Middelton, L.A.; Kauffman, E.C.; et al. Germline PTEN mutation Cowden syndrome: an underappreciated form of hereditary kidney cancer. J Urol. 2013, 190, 1990–1998. [Google Scholar] [CrossRef] [PubMed]
- Yehia, L.; Eng CPTENHamartoma Tumor Syndrome In: Adam, M.P.; Ardinger, H.H.; Pagon, R.A.; Wallace, S.E.; Bean, L.J.H.; Gripp, K.W.; et al., eds. GeneReviews((R)). Seattle (WA)1993.
- Ngeow, J.; Liu, C.; Zhou, K.; Frick, K.D.; Matchar, D.B.; Eng, C. Detecting Germline PTEN Mutations among at-risk patients with cancer: an age- and sex-specific cost-effectiveness analysis. J Clin Oncol. 2015, 33, 2537–2544. [Google Scholar] [CrossRef]
- Tischkowitz, M.; Colas, C.; Pouwels, S.; Hoogerbrugge, N.; Group, P.G.D.; European Reference Network, G. Cancer Surveillance Guideline for individuals with PTEN hamartoma tumour syndrome. Eur J Hum Genet. 2020, 28, 1387–1393. [Google Scholar] [CrossRef] [PubMed]
- Kim, R.H.; Wang, X.; Evans, A.J.; Campbell, S.C.; Nguyen, J.K.; Farncombe, K.M.; et al. Early-onset renal cell carcinoma in PTEN harmatoma tumour syndrome. NPJ Genom Med. 2020, 5, 40. [Google Scholar] [CrossRef] [PubMed]
- Pena-Llopis, S.; Vega-Rubin-de-Celis, S.; Liao, A.; Leng, N.; Pavia-Jimenez, A.; Wang, S.; et al. BAP1 loss defines a new class of renal cell carcinoma. Nat Genet. 2012, 44, 751–759. [Google Scholar] [CrossRef]
- Brugarolas, J. PBRM1 and BAP1 as novel targets for renal cell carcinoma. Cancer J. 2013, 19, 324–332. [Google Scholar] [CrossRef]
- Shao, Y.F.; DeBenedictis, M.; Yeaney, G.; Singh, A.D. Germ Line BAP1 Mutation in Patients with Uveal Melanoma and Renal Cell Carcinoma. Ocul Oncol Pathol. 2021, 7, 340–345. [Google Scholar] [CrossRef]
- Pilarski, R.; Carlo, M.; Cebulla, C.; Abdel-Rahman, M. BAP1 Tumor predisposition syndrome. In: Adam MP, Ardinger HH, Pagon RA, Wallace SE, Bean LJH, Gripp KW, et al., editors. GeneReviews((R)). Seattle (WA)1993.
- Schmidt, L.S.; Linehan, W.M. Genetic predisposition to kidney cancer. Semin Oncol. 2016, 43, 566–574. [Google Scholar] [CrossRef]
- Chau, C.; van Doorn, R.; van Poppelen, N.M.; van der Stoep, N.; Mensenkamp, A.R.; Sijmons, R.H.; et al. Families with BAP1-Tumor Predisposition Syndrome in The Netherlands: path to Identification and a Proposal for Genetic Screening Guidelines. Cancers (Basel). 2019, 11. [Google Scholar] [CrossRef]
- Carbone, M.; Pass, H.I.; Ak, G.; Alexander, H.R., Jr.; Baas, P.; Baumann, F.; et al. Medical and Surgical Care of Patients With Mesothelioma and Their Relatives Carrying Germline BAP1 Mutations. J Thorac Oncol. 2022, 17, 873–889. [Google Scholar] [CrossRef] [PubMed]
- Ball, M.W.; An, J.Y.; Gomella, P.T.; Gautam, R.; Ricketts, C.J.; Vocke, C.D.; et al. Growth rates of genetically defined renal tumors: implications for active surveillance and intervention. J Clin Oncol. 2020, 38, 1146–1153. [Google Scholar] [CrossRef] [PubMed]
- Tsai, T.H.; Lee, W.Y. Succinate dehydrogenase-deficient renal cell carcinoma. Arch Pathol Lab Med. 2019, 143, 643–647. [Google Scholar] [CrossRef] [PubMed]
- Gill, A.J.; Pachter, N.S.; Chou, A.; Young, B.; Clarkson, A.; Tucker, K.M.; et al. Renal tumors associated with germline SDHB mutation show distinctive morphology. Am J Surg Pathol. 2011, 35, 1578–1585. [Google Scholar] [CrossRef] [PubMed]
- Aggarwal, R.K.; Luchtel, R.A.; Machha, V.; Tischer, A.; Zou, Y.; Pradhan, K.; et al. Functional succinate dehydrogenase deficiency is a common adverse feature of clear cell renal cancer. Proc Natl Acad Sci U S A. 2021, 118. [Google Scholar] [CrossRef] [PubMed]
- Milionis, V.; Goutas, D.; Vlachodimitropoulos, D.; Katsoulas, N.; Kyriazis, I.D.; Liatsikos, E.N.; et al. SDH-deficient renal cell carcinoma: a case report associated with a novel germline mutation. Clin Case Rep. 2021, 9, e04605. [Google Scholar] [CrossRef] [PubMed]
- Wilczek, Y.; Sachdeva, A.; Turner, H.; Veeratterapillay, R. SDH-deficient renal cell carcinoma: a clinicopathological analysis highlighting the role of genetic counselling. Ann R Coll Surg Engl. 2021, 103, e20-e2. [Google Scholar] [CrossRef] [PubMed]
- Kuroda, N.; Yorita, K.; Nagasaki, M.; Harada, Y.; Ohe, C.; Jeruc, J.; et al. Review of succinate dehydrogenase-deficient renal cell carcinoma with focus on clinical and pathobiological aspects. Pol J Pathol. 2016, 67, 3–7. [Google Scholar] [CrossRef]
- van der Tuin, K.; Tops, C.M.J.; Adank, M.A.; Cobben, J.M.; Hamdy, N.A.T.; Jongmans, M.C.; et al. CDC73-Related Disorders: Clinical Manifestations and Case Detection in Primary Hyperparathyroidism. J Clin Endocrinol Metab. 2017, 102, 4534–4540. [Google Scholar] [CrossRef]
- Iacobone, M.; Carnaille, B.; Palazzo, F.F.; Vriens, M. Hereditary hyperparathyroidism--a consensus report of the European Society of Endocrine Surgeons (ESES). Langenbecks Arch Surg. 2015, 400, 867–886. [Google Scholar] [CrossRef] [PubMed]
This is an open access article under the terms of a license that permits non-commercial use, provided the original work is properly cited. © 2022 The Authors. Société Internationale d'Urologie Journal, published by the Société Internationale d'Urologie, Canada.