1. Introduction
Separating complex biological samples into their constituent components is a vital step in studying proteins and other biological components. Electrophoresis is a common separation technique used to isolate charged biomolecules based on their migration under external electric fields [
1], and is used to separate biological components ranging from proteins [
2], DNA, and RNA [
3,
4] to entire cells [
5]. Electrophoresis has been applied via different techniques, most commonly in gel electrophoresis, where analytes are suspended in a polymer matrix [
6]. Capillary electrophoresis is a common alternative, offering greater dissipation of Joule heating, enabling high electric field strengths to be applied [
1], with its primary drawback being its limited volume. Both methods are primarily batch operations, though continuous electrophoretic separation techniques have been reported with Free Flow Electrophoresis, where a sample stream is driven through a separation chamber by pressure, with the electric field applied perpendicular to the flow direction [
7]. As the direction of separation is perpendicular to the flow direction, injection, separation, and collection may take place simultaneously, meaning the system can be operated continuously and achieve a high throughput. Continuous operation may also allow for separation efficiency to be enhanced through techniques such as recycling [
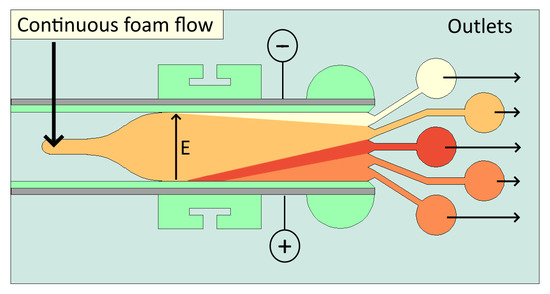
8]. However, large cross-sectional areas and low surface-to-volume ratios make heat dissipation difficult; therefore, Free Flow Electrophoresis is typically limited by Joule heating. Liquid foams may provide an alternative platform for separations, allowing continuous separation similar to Free Flow Electrophoresis, but with the added advantage of enhanced separations due to interactions between the biological molecules and the electrical double layer (EDL) of the charged gas–liquid interfaces.
Liquid foams are composed of gas bubbles suspended in a continuous liquid medium. The lamellae, plateau borders, and nodes of liquid foams effectively provide a network of deformable micro and nano channels, bound by the gas–liquid interfaces instead of solid walls. Thin films between bubbles can have thicknesses in the order of 10 nm [
9]. At this scale interactions with the interface could affect the migration of analytes through the system, steric interactions with the interface, as well as polarization effects and adsorption kinetics affect the motion of molecules. Unique separation techniques have been proposed that take advantage of these effects to modify analyte migration [
10]. Liquid foams could provide a novel platform for electrophoretic separations, providing a cheap and flexible platform with a high interfacial area, without the need for complex and expensive solid channels. Liquid foams may also be pumped similar to liquids [
11], presenting a further opportunity as a platform for continuous electrophoretic separation, while taking advantage of the high surface-to-volume ratio of liquid foams. This approach may effectively take the operation of capillary electrophoresis, but most importantly gives an opportunity to exploit interactions between analytes and charges in the EDL to enhance separation. The foam electrokinetic separation concept was recently demonstrated in batch mode for a charged dye mixture by [
12]. However, measurements were limited to visual observations as direct concentration measurements inside the chip were difficult with batch operation. In addition, the effect of foam flow on separation efficiency needs to be explored for continuous operation. The transition from batch mode to continuous flow operation is not straightforward, and many engineering processes tend to fail during this transition. In this work, a continuously operated foam electrophoresis separation device was designed and constructed, and its operation was demonstrated by separating a dye mixture containing differently charged dyes. The main purpose of this proof-of-concept study is to provide fundamental understanding of a complex dynamical foam system operated in continuous separation mode, where flowing liquid foam is subjected to an external electric field with electrophoresis and chemical reactions at the electrodes continuously changing the system. By using cheap, easily visible dyes, the principles and key operating parameters can be identified and studied. This work directly quantifies dye concentrations at the outlets, which was not possible in the batch operation reported by [
12]. In addition to the transition from batch to continuous mode, this paper presents experimental work detailing the separation current. These findings will be useful in designing further applications, such as the electrophoretic separation of proteins.
3. Results and Discussion
Time lapse recordings were taken during operation of the device to understand flow inside the device and assess separation. Time lapse images are displayed in
Figure 3.
The two dyes exhibit different dynamic behaviours when subjected to an external electric field. As rhodamine B is neutral, it is not affected by electrophoresis, and its concentration is expected to remain unchanged throughout the entire domain of the foam (Fauvel et al., 2022). The charge of fluorescein depends on the local pH; when pH is greater than 6.7 fluorescein is dianionic, between pH 6.7 and 4.3 fluorescein is monoanionic, between pH 4.3 and 2.1 it is neutrally charged, and below pH 2.1 it is cationic [
20,
24]. Upon entry to the device, the foam is at pH 7.4; therefore, fluorescein is negatively charged, and will initially migrate towards the anode (bottom electrode). This is observed in
Figure 3 as a band of dark orange forming along the anode, gradually widening between the inlet and the outlets. When an external electric field is applied, electrochemical reactions cause pH changes within the foam [
25], establishing a pH gradient across the device despite limited buffer solution, i.e., pH drops near the anode and raises near the cathode. The longer the foam remains inside the device, the more the pH gradient extends away from the electrodes, leading to uniform pH at the inlet and to a non-uniform pH at the outlets. This gradient causes widening of the dark orange fluorescein band at the anode, as pH drops below 4.3 and the fluorescein converts to its neutral form, halting electrophoretic migration. Direct visualisation of pH changes is provided in the
Supplementary Materials: Figure S1. This corresponds with observations recorded in [
12] for stagnant foam. Further visualisation of dye transport may be seen in the white band forming next to the cathode at around 20 min. This appears to be a region where the fluorescein concentration has dropped significantly, apparently leaving only rhodamine B, resulting in a white colour. According to the time lapse recordings, the residence time of a single bubble inside the device under an external electric field of 2500 V/m is estimated to be 10 min ± 30 s. Residence time is expected to vary depending on the rate of foam collapse inside the device and the foam flowrate.
It is important to consider liquid flow inside the foam, and the effects of charged interfaces on the charged dye molecules. SDS molecules adsorb onto the air–liquid interfaces, causing it to be negatively charged. The presence of non-ionic Triton X-100 is expected to decrease the negative charge in comparison to a pure SDS foam. Measurements of the zeta potential of the gas–liquid interfaces may provide useful data on how the introduction of non-ionic surfactant affects electroosmotic flows inside the system. Positive counterions are attracted to the negative interfaces, establishing the EDL. Under the influence of an external electric field, the positive counterions in the EDL are drawn towards the cathode by the resulting Coulomb force, causing fluid adjacent to the EDL to experience electroosmotic flow (EOF) towards the cathode. Given the initial average bubble size of 11 µm, with a standard deviation of 4.25 µm, and neglecting any foam collapse inside the device, the surface area of the air–liquid interface is estimated to be 0.41 m2, compared to the maximum possible surface area of the solid–liquid interfaces of 0.0007 m2. From this, it can be concluded that the electroosmotic flow at air–liquid interfaces dominates over that of solid–liquid interfaces, which may possess differing surface charges to the air–liquid interfaces. Fluid transported by EOF eventually reaches a solid wall at the electrode and a “backflow” develops as fluid moves away from the wall through the thicker regions of the film where electroosmotic flow is weaker, namely the plateau borders, away from the gas–liquid interfaces. As fluorescein is negatively charged in the bulk, it is expected to be positioned away from the gas–liquid interfaces and hence will be affected by the backflow. The magnitude of EOF and backflow will depend on the interfacial charge established by the surfactants. As a mixture of ionic/non-ionic surfactant was used, the magnitude of EOF is expected to be lower than that of pure SDS foam, but greater compared to that of pure Triton X-100 foam. Further investigation into the effect of surfactant blends on electroosmotic flow is outside the scope of this study but worth consideration in future work. The use of charged dye mixtures as models for separation provides an opportunity to understand this complex system using simple visual aids and will be useful in extending separations to future applications.
Foam collapse is another important aspect to consider when evaluating foam-based separation applications. Despite efforts to mitigate foam collapse, pockets of untimely collapse are observed within the device, with foam collapse being more noticeable at longer timescales (typically after 20–30 min from the initial foam formation). Collapse inside the separation chamber could lead to unwanted mixing of already separated components and could reduce separation efficiency. Collapse at the outlet channels may lead to outlets being blocked, visible after 10 min near the outlet in
Figure 3. However, if foam collapse could be accounted for and timed accordingly, it may be possible to collapse after passing through the separation chamber and into the collection channels. In addition, foam rheology and foam flow can have significant effects on the behaviour of the system. Local topological rearrangements of individual bubbles, or T1 events, may also contribute to mixing, and may occur when the yield stress surpasses the limit defined by the plasticity of the foam [
25]. This may place constraints on what volumetric flowrates could be used, as well as foam formulation, bubble size [
26], and liquid fraction [
27]. Setting the volumetric foam flowrate too low may result in long residence times and untimely foam collapse, whereas setting it too high may incur unacceptable levels of mixing due to the extra stresses. This aspect of the operation is still unexplored and presents an opportunity for in-depth analysis and further development.
To provide quantitative analysis of the separation, the concentration of fluorescein and rhodamine B relative to their initial concentration at each outlet stream is presented in
Figure 4. Outlets are numbered such that outlet 1 is closest to the anode and outlet 5 is closest to the cathode. To analyse the effect of electric field strength, the experiment is repeated multiple times, varying electric field strength between 1500 and 3000 V/m.
Rhodamine B concentration only varies between 96% and 106% of the inlet concentration at all electric field strengths, which means it remains largely unaffected by the electric field as shown in
Figure 4b. As rhodamine B is neutral, its concentration is expected to remain uniform across the device. Rhodamine B concentrations slightly differing from the initial concentrations may be caused by the absorption of rhodamine B into the agar gel coated at each electrode, which becomes visible as a pink stain on the surface of the gel during experiments. However, this would initially be expected to cause a drop in rhodamine B concentration in samples 1 and 5. The variation may potentially be caused by the foams structure, and how electroosmotic flows develop within, as well as the decay of the foam. Due to the difference in scale, electroosmotic flow is expected to dominate within the thin film lamellae between bubbles, while pressure driven backflow is expected to dominate within the thicker plateau borders away from the outlets [
28,
29,
30,
31]. Rhodamine B is neither attracted nor repelled by the charged interface, and is expected to be present throughout the foam, and so the electroosmotic flow and backflow will act to mix the rhodamine B throughout the device, potentially leading to uneven concentrations upon eventual foam collapse.
The variation of fluorescein dye at the outlets is shown in
Figure 4a. In general, fluorescein concentration increases from the cathode to the anode, as negatively charged fluorescein is transported towards the anode by electrophoresis. Interestingly, for all electric field strengths less than 3000 V/m, the peak in fluorescein concentration is observed at outlet 3, halfway between the anode and the cathode. The fluorescein concentration achieved at this point is 1.49 times the original concentration, at an electric field strength of 1500 V/m. This mirrors observations made in [
12] where fluorescein concentrates away from the electrodes. This is a consequence of the acidic front that develops from the anode, causing fluorescein to transition to its neutral form, stopping electrophoretic migration. This results in a fluorescein concentration peak a certain distance away from the anode, and this distance decreases as the external electric field strength increases. An anomaly to this is the high fluorescein concentration in outlet 1 at 1500 V/m. At lower voltages, the pH gradient is expected to develop slower, as it must first permeate through the agar gel layer before reaching the liquid foam. If fluorescein can migrate to the anode before this pH front reaches the liquid foam it may cause an increase in concentration in outlet 1. The formation of this pH gradient over time is an important factor when designing a system for a specific application and will require further investigation. At 3000 V/m, a different trend is observed, where fluorescein concentration increases linearly from the cathode to the anode, and no concentration peak is observed at the midpoint of the device. This suggests fluorescein can concentrate at the anode without being hindered by the pH gradient, most likely due to the greater electric field strength and faster electrophoretic migration overtaking the formation of the pH gradient. Interestingly, fluorescein concentration is below the inlet concentration for all points at 3000 V/m, which suggests a fraction of fluorescein get absorbed into the agar gel at the anode and is being removed from the system. In these experiments, full separation is not achieved, as the concentration gradient at the outlet takes time to establish.
Figure 3 shows the colour intensity at the outlet changing over time between 15 min and 35 min. Between these times a white band forms near the cathode as fluorescein concentration is reduced. The delayed formation of this band is likely to be related to the formation of the pH gradient across the device.
To investigate the effects of foam flowrate on separation, the experiment was repeated by varying the foam volumetric flowrate between 4.71 µL/s and 7.85 µL/s. The electric field strength was kept constant at 2500 V/m. The results are displayed in
Figure 5. Rhodamine B concentrations show no deviation from the trend shown in
Figure 4b and are not shown here.
Increasing the flow rate from 4.71 to 6.28 µL/s shows little change in fluorescein concentrations, following the same trend, and with concentrations falling within error margins of each other, suggesting no significant effect. However, at 7.85 µL/s the trend changes, showing significantly more fluorescein in outlets 4 and 5. This suggests that greater flowrate does not allow fluorescein sufficient resident time to migrate, and so the concentration pattern does not fully develop, leading to a wider distribution of fluorescein across the device. This suggests that there may be an optimum flowrate, which allows the separation pattern to fully develop, while allowing the greatest possible throughput with least foam collapse. From all the experiments, the highest fluorescein concentration recovered was 1.75 times the initial concentration from outlet 1 under 1500 V/m electric field at a volumetric flowrate of 4.71 µm/s. This concentration is relatively low compared to similar experiments conducted using free flow zone electrophoresis. [
32] reported total separation of rhodamine and fluorescein, with a residence time of 3.3 s under an electric field of 25,000 V/m. [
33] reported successful separation and concentration of fluorescein from eosin G using free flow isotachophoresis, with a residence time of 93.7 s and an electric field strength of 35,000 V/m. There are two key differences between the two aforementioned studies and the work presented here. First, the width of the inlet stream relative to the separation chamber is small in our study, as opposed to evenly dispersed foam throughout the entire space before separation in the previous work. Second, the electric field strengths used in our study are substantially lower than the previous work mentioned, and so will have a lesser heating effect, reducing risk of damage to heat sensitive analytes. Ref. [
34] reports partial separation and concentration of alexa fluor 591 and fluorescein through free flow isotachophoresis, noting the fluorescein concentration increased by 1.5-fold during the process. All approaches reported in the Literature and in this study have their own merits and limitations, but the suitability of a separation technique for a specific application depends on the analyte properties.
Although full separation is not demonstrated here, an interesting concept is presented. It may be possible to recycle specific outlet fractions, and continuously pass them through the device multiple times to enhance complete separation. This approach is not possible with conventional batch operated electrophoresis systems. Similarly, it may be possible to recycle output from the first 15–30 min of operation, where the separation pattern has not fully formed, further enhancing separation [
8]. While recycling may offer enhanced separation, untimely foam decay near the outlets may cause unwanted mixing. This may be improved in future trials by tuning surfactant formulations, electric field strength, and foam flowrate. In this study, it was noted that some channels may become effectively blocked, with foam being redirected towards other channels, causing unwanted mixing of the separated fractions. Therefore, outlet design also requires further consideration in future designs to avoid blockages and efficient removal.
The separation current can provide valuable insights into the power consumption of the device, heat dissipation, electrolysis processes occurring at the electrodes, and reproducibility of the separation process. To measure the electrical current through the foam, the experiment was repeated using an electric field strength of 2500 V/m and a volumetric flowrate of 6.28 µL/s. This experiment was repeated four times and the average current over time is presented in
Figure 6.
The electric current profile across the device can be split into three main regions. Region I corresponds to the period of time in which the device is filled with foam, and the current rises accordingly, as more foam becomes available to conduct the current. At around 6 min, the device is completely full of foam, where maximum current is reached. Then region II begins, where the current gradually drops, likely due to some foam collapse inside the device. Conductance may be used to give an indication of the liquid fraction within the device [
35]. However, existing empirical relationships utilise alternating current [
36] to negate electrochemical reactions at the electrodes. As this setup uses direct current (DC), such relationships do not hold, as the conductance of the foam will be affected by the electrochemical reactions. As a result of this, region III develops, where the current gradually rises with time and plateaus. During this stage, foam conductance increases slightly due to additional ions released by the electrochemical reactions and balances the decrease in conductance due to foam collapse. These ionic changes within the system were confirmed by pH measurements shown in the
Supplementary Materials, Figure S1. However, this region experiences prominent fluctuations compared to regions I and II due to random foam collapse. The greatest variation between each individual run is seen in region III, but all runs exhibit similar trends, indicating reasonable reproducibility. The foam supply was refreshed at 10, 20, and 30 min during these runs. While the current profile trend changes after the 10-min mark, no noticeable change was observed after 20 and 30 min. This suggests that foam syringe changeover causes negligible effects on the experiment, and that foam decay is primarily driven by other factors, rather than the foam collapsing inside the syringe, outside of the device.
According to the electric current measurements, the device consumes 0.035 W of power on average for this separation. Assuming most of the power is used in heating the liquid foam, a temperature rise up to 8 °C can be expected. However, the heating effects of applied electric fields on liquid foam are known to be inhomogeneous [
37]. As such, further study is required to investigate the local temperature changes caused by application of an electric field on continuously flowing foam. Of particular interest is the potential difference in temperature between the lamellae and plateau borders inside the foam, as this determines the dynamics of the whole system.





 ,
, 








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}