Abstract
Follicular lymphoma (FL) is the second most common lymphoma in Western countries. FL is characterized by being incurable, usually having an indolent clinical course with frequent relapses, and an eventual patient’s death or transformation to Diffuse Large B-cell Lymphoma. The immune response and the tumoral immune microenvironment, including FOXP3+Tregs, PD-1+TFH cells, TNFRSF14 (HVEM), and BTLA play a role in the pathogenesis. We aimed to analyze the gene expression of FL by Artificial Intelligence (machine learning, deep learning), to identify genes associated with the prognosis of the patients and with the microenvironment in terms of overall survival (OS). A series of 184 cases of the GSE16131 dataset was analyzed by multilayer perceptron (MLP) and radial basis function (RBF) neural networks. In the analysis, MLP and RBF had a synergistic effect. From an initial set of 22,215 genes probes, a final set of 43 genes was highlighted. These 43 genes predicted the OS and correlated with the immune microenvironment: in a multivariate Cox analysis, 18 genes were associated with a poor prognosis (namely, MED8, KRT19, CDC40, SLC24A2, PRB1, KIAA0100, EVA1B, KLK10, TMEM70, BTN2A3P, TRPM4, MED6, FRYL, CBFA2T2, RANBP9, BNIP2, PTP4A2 and ALDH1L1) and 25 genes were associated with a good prognosis of the patients. Gene set enrichment analysis (GSEA) confirmed these findings and showed a typical sinusoidal-like shape. Some of the most relevant genes for poor OS were EVA1B, KRT19, BTN2A3P, KLK10, TRPM4, TMEM70, and SLC24A2 (hazard risk = from 1.7 to 4.3, p < 0.005) and for good OS, these were TDRD12 and ZNF230 (HR = 0.34 and 0.28, p < 0.001). EVA1B, KRT19, BTN2AP3, KLK10, and TRPM4 also associated with M2-like macrophage markers including CD163, MRC1 (CD206), and IL10 in the core enrichment for dead OS outcome by GSEA and to poor OS by Kaplan–Meier with Log rank test. The scientific literature showed that some of these genes also play a role in other types of cancer. In conclusion, by Artificial Intelligence, we have identified new biomarkers with prognostic relevance in FL.
1. Introduction
Predictive neural networks are usually the chosen tool for many data-mining applications because they can handle complex processes, such as the analysis of gene expression data of human pathological conditions. The multilayer perceptron (MLP) and radial basis function (RBF) networks are supervised methods and their architecture is feedforward [1,2]. The choice between MLP or RBF depends on the data and the level of complexity that you seek to uncover. In general, the MLP procedure can find more complex relationships, while the RBF is generally faster [3,4,5].
Lymphomas are a type of hematologic neoplasia of the immune system. The 2016 revision of the World Health Organization classification of lymphoid neoplasms classifies the entity of follicular lymphoma into the Mature B-cell neoplasms (as a part of the non-Hodgkin lymphomas), which includes other relevant subtypes such as the chronic lymphocytic leukemia/small lymphocytic lymphoma, splenic marginal zone lymphoma, plasma cell myeloma, extranodal marginal zone lymphoma of mucosa-associated lymphoid tissue (MALT lymphoma), mantle cell lymphoma, and diffuse large b-cell lymphoma, among others [6]. All these tumors are originated from B-lymphocytes in a defined stage of B-cell maturation and differentiation. Lymphocytes are one of the five types of the white blood cells. B-lymphocytes are produced in the bone marrow; they have a B-cell receptor specific for a specific antigen, differentiate into plasma cells, and secrete antibodies or become memory cells. Mantle cell lymphoma and diffuse large b-cell lymphoma behave aggressively. On the other hand, chronic lymphocytic leukemia and marginal zone lymphomas are relatively benign. Follicular lymphoma (FL) is a heterogeneous clinicopathological entity derived from germinal center B-lymphocytes, and it is one of most common non-Hodgkin lymphomas in Western countries. Therefore, it is clinically relevant [6,7].
Follicular lymphoma is characterized by the translocation (14;18) that results in BCL2 overexpression due to the IGH/BCL2 fusion gene. BCL2 is an oncogene with anti-apoptosis function. Molecular genetics using next-generation sequencing (NGS) has shown recurrent mutations of several genes including KMT2D, EZH2, ARID1A, EP300, MEF2B, and FOXO1. The prognosis of follicular lymphoma is variable, and to date, not many biological markers have been identified to predict the prognosis of the patients [7]. The Follicular Lymphoma International Prognostic Index (FLIPI) analyzes five adverse prognostic factors to identify risk groups with significantly different overall survival. The FLIPI includes the following variables: (1) Age >60 years; (2) Stage III or IV; (3) Hemoglobin level <12.0 g/dL; (4) Number of involved nodal areas >4; and (5) Serum lactate dehydrogenase level greater than the upper limit of normal. In addition to the FLIPI, gene expression analysis highlighted the role of the tumoral immune microenvironment. An immune response type 1, rich in T cells, was associated to good prognosis. In the context of the immune response type 1, FOXP3+regulatory T lymphocytes (Tregs) and PD-1+follicular T helper cells (TFH cells) are also included. Conversely, an immune response type 2, rich in macrophages, correlated to a poor prognosis; in this context, high TNFRSF14 and low BTLA are also included [8,9,10,11,12,13].
The purpose of this work was to re-analyze the gene expression data of a robust series of 184 cases of follicular lymphoma using an Artificial Intelligence approach in order to prove that our methodological approach was feasible and to identify new genes that were associated to the prognosis of the patients. We searched for genes that correlated with the survival outcome (dead/alive) as well as other clinicopathological variables included in the FLIPI and the immune microenvironment (immune response 2 versus 1). This work is significant because to our knowledge, the use of Artificial Intelligence is a novel approach. We found a new set of 43 genes associated with the prognosis of the follicular lymphoma patients.
2. Materials and Methods
2.1. Statistical Analysis
All the statistical analysis was performed using both R programming language with R version 3.6.3 (2020-02-29) and RStudio (version 1.3.959), and IBM SPSS (version 25; IBM, Armonk, NY, USA). The analysis was performed following the manufacturers’ instructions that can be found at the following webpages: (1) https://www.r-project.org/, (2) https://rstudio.com/, and (3) https://www.ibm.com/support/pages/node/618179 [3].
Comparisons between groups were performed with crosstabulations and Chi-square tests that included the Pearson Chi-square, Likelihood ratio, and the Fisher’s exact test when required; non-parametric tests included the two-independent-samples test (the Mann–Whitney U test). Survival analysis was performed with the Kaplan–Meier with Log rank, Breslow, and Tarone–Ware tests; and the Cox regression in the univariate analysis (enter method) as well as in the multivariate analysis (backward conditional method). A p value of less than 0.05 was considered as statistically significant. The definition of overall survival was the standard.
2.2. Follicular Lymphoma Gene Expression Dataset
The gene expression omnibus (GEO) series GSE16131 of follicular lymphoma [14] was downloaded from the National Center for Biotechnology Information (NCBI) website (https://www.ncbi.nlm.nih.gov/geo/). The samples corresponded to follicular lymphoma cases from lymph nodes; fresh-frozen tumor-biopsy specimens were from 184 untreated patients. The series was last updated in 10 August 2018.
The RNA had been extracted from the biopsy specimens (Fast track 2.0 mRNA isolation kit, extraction method: oligo dt cellulose; Invitrogen, Carlsbad, CA, USA). Biotinylated cRNA had been prepared according to the standard Affymetrix protocol from 1 microg mRNA (Expression analysis technical manual, 2001, Affymetrix). The RNA had been examined for gene expression with the use of Affymetrix U133A and U133B microarrays. Following fragmentation, 20 micrograms of cRNA had been hybridized for 16 hours at 45C on U133A/B GeneChips. GeneChips had been washed and stained in the Affymetrix Fluidics Station 400. Scanning had been performed by the Affymetrix 3000 Scanner. Calculation method: The data had been analyzed with Microarray suite version 5.0 (MAS 5.0) using Affymetrix default analysis settings and global scaling as the normalization method. The trimmed mean target intensity of each array was arbitrarily set to 500. The data were normalized and log2 transformed.
Definition of the immune response type 1 and 2 signatures in follicular lymphoma as originally described by Dave SS et al. published in N Engl J Med 2004 [12]. From the total number of genes of the chip, a set of 3299 genes were identified as being associated with survival. The genes that were associated with good prognosis (1568 genes) and poor prognosis (1731 genes) were hierarchically clustered separately. Five gene signatures were found among the genes predicting good prognosis, and five signatures were found among the genes predicting poor prognosis. Within each signature, the expression levels of the component genes were averaged, resulting in 10 signature averages associated with each sample. By multivariate survival analysis, it was noted that a combination of two signatures provided the better prediction. These two signatures were later named immune-response 1 and immune-response 2 based on the known function of the constituent genes. The coefficients in the final model were derived from the Cox model applied to the training set. For each sample, the final model generated a survival-predictor score according to the formula: survival-predictor score = (2.71 × immune-response 2 signature average) − (2.36 × immune-response 1 signature average) [12].
The genes of the immune-response 1 signature were ACTN1, ATP8B2, BIN2, C1RL, C6orf37, C9orf52, CD7, CD8B1, DDEF2, DKFZP566G1424, DKFZP761D1624, FLJ32274, FLNA, FLT3LG, GALNT12, GNAQ, HCST, HOXB2, IL7R, IMAGE:5289004, INPP1, ITK, JAM, KIAA1128, KIAA1450, LEF1, LGALS2, LOC340061, NFIC, PTRF, RAB27A, RALGDS, SEMA4C, SEPW1, STAT4, TBC1D4, TEAD1, TMEPAI, TNFRSF1B, TNFRSF25, TNFSF12, and TNFSF13B [12].
The genes of the immune-response 2 signature were BLVRA, C17orf31, C1QA, C1QB, C3AR1, C4A, C6orf145, CEB1, DHRS3, DUSP3, F8, FCGR1A, GPRC5B, HOXD8, LGMN, ME1, MITF, MRVI1, NDN, OASL, PELO, SCARB2, SEPT10, and TLR5 [12].
The signatures in the survival model were named based on the biologic function of certain genes within each signature. The immune-response 1 signature includes genes encoding T-cell markers (e.g., CD7, CD8B1, ITK, LEF1, and STAT4) and genes that are highly expressed in macrophages (e.g., ACTN1 and TNFSF13B). Notably, the immune-response 1 signature is not merely a surrogate for the number of T cells in the tumor-biopsy specimen, since many other standard T-cell genes (e.g., CD2, CD4, LAT, TRIM, and SH2D1A) were not associated with survival. The immune-response 2 signature includes genes known to be preferentially expressed in macrophages, dendritic cells, or both (e.g., TLR5, FCGR1A, SEPT10, LGMN, and C3AR1). It is noteworthy that the RNA of each sample is a mixture of the RNA of different cell populations, including the tumoral B-lymphocytes, T-lymphocytes, macrophages, and dendritic cells, among others [12].
It is noteworthy that in this research, we have used a variation to define the immune response variable. Knowing that the high immune response 2 signature would associate to poor prognosis due to the high macrophage component, we created a ratio between the gene expression values of the immune response 2 and 1 as present in the series matrix file, and the best cut-off for the overall survival was searched (which corresponded to 0.97 value).
2.3. Patients and Clinicopathological Characteristic
The series was comprised of 184 cases of follicular lymphoma with diagnostic biopsies (pre-treatment). Regarding the grade, the series included 152 grades 1/2 and 32 grade 3A. In 4 cases, the follow-up was missing. The follow-up time ranged from 0.01 to 19.3 years, with a mean of 7.0 years (±4.5 STD) and a median of 6.5 years. The 1, 3, 5, and 10-year overall survival was 92% (95% CI, 88–96%), 83% (77–88%), 71% (64–78%), and 50% (42–58%), respectively. At the end of the follow-up time, 92 of 180 (51.1%) had died. The immune response scores were available for all 184 cases. The immune response score 1 (representative of T-lymphocytes) ranged from 7.3 to 10.2, with a mean of 9.3 (±0.4 STD) and a median of 9.3. The immune response score 2 (representative of macrophages) ranged from 8.1 to 9.9, with a mean of 8.7 (±0.3) and median of 8.7. An immune response ratio 2:1 high (>0.97) was found in 26.1% of the cases. This cut-off of 0.97 had been found using a receiver operating characteristic (ROC) curve and the outcome (dead/alive) of the patients. The rest of the clinicopathological characteristics of the series is shown in Table 1. In summary, the clinicopathological variables that associated with a poor prognosis of the patients were age >60-years old (Hazard risk = 2.7), number of extranodal sites >1 (HR = 1.8), increased lactate dehydrogenase levels in serum (LDH; HR = 2.0), international prognostic index (IPI; HR = 3.1), and immune response ratio 2:1 high (HR = 3.3) (Table 2). In Figure 1, Figure 2, Figure 3 and Figure 4, the distribution of the data of the different variables is shown, including the histograms as well as the overall survival plots. Follicular lymphoma is characterized by an indolent clinical course and frequent relapses, eventually leading to death or transformation to another lymphoma subtype with more aggressivity (Diffuse large b-cell lymphoma). We performed gene set enrichment analysis (GSEA) to identify biological pathways that could explain the pathological mechanism of survival outcome (dead/alive). First, the pan cancer immune oncology pathway was tested, and the result showed enrichment toward the dead phenotype, with the presence of markers related to macrophages. Of note, macrophages are associated to an immune response type 2 signature. Later, several individual pathways were tested, including ATM, BCR, CASPASE, CD8, E2F, HDAC, MAPK TRK, MYC, NFKB canonical and atypical, NOTCH, RAS, RB, RET, TP53, VEGF, and WTN canonical pathways. The most relevant, which were found to be associated to a favorable outcome, were the BCR (B-cell receptor), the MAPK TRK and atypical NFKB pathways. The relevant pathways and genes will be of interest in the neural network analysis in association to the clinical variables. In summary, follicular lymphoma is an indolent hematological neoplasia with favorable prognosis. Nevertheless, some patients have a more aggressive clinical course. To date, the most important prognostic variables are the IPI and the immune response signature. Nevertheless, the pathological mechanism is still not completely understood. Therefore, there is a need to find prognostic biomarkers that can be correlated with these parameters.

Table 1.
Clinicopathological characteristics of the follicular lymphoma series.
Table 2.
Overall survival analysis of the clinicopathological variables.
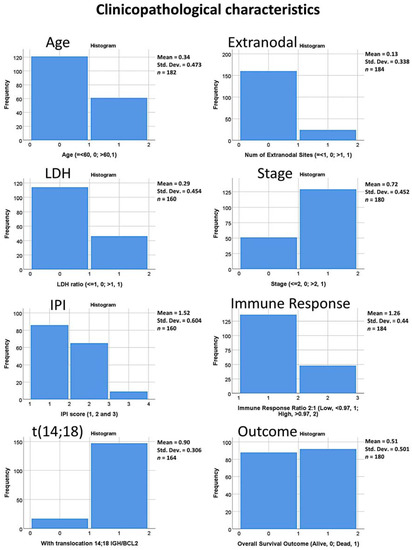
Figure 1.
Clinicopathological characteristics of the series. The histograms show the distribution of the data for each of the clinical variables including the immune response gene signature 2:1 ratio and the presence of translocation (14;18) involving the IGH/BCL2 genes.
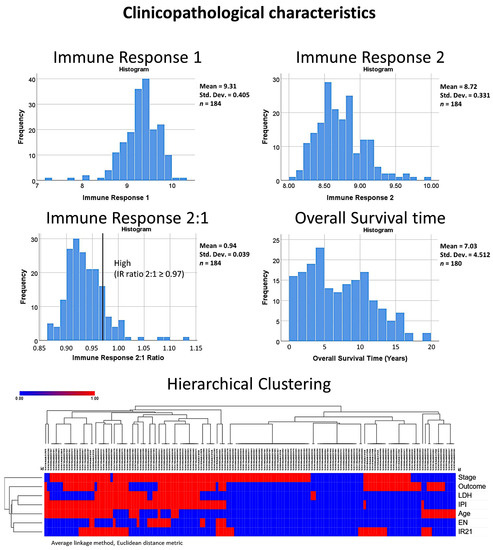
Figure 2.
Clinicopathological characteristics of the series. The histograms show the distribution of the data for the immune response variables (1, 2 and the 2:1 ratio). The immune response variables are based on the gene expression of markers that are characteristic of the tumoral immune microenvironment and host immune response (T cells, macrophages, etc.), as originally described by Dave SS et al. and published in N Engl J Med 2004 [12]. In this project, we created an immune response 2:1 ratio and the cut-off value of 0.97 was use in the overall survival analysis. The overall survival time of the series is also shown. The distribution and correlation of the different variables is also shown in the hierarchical clustering with dendrogram and heatmap.
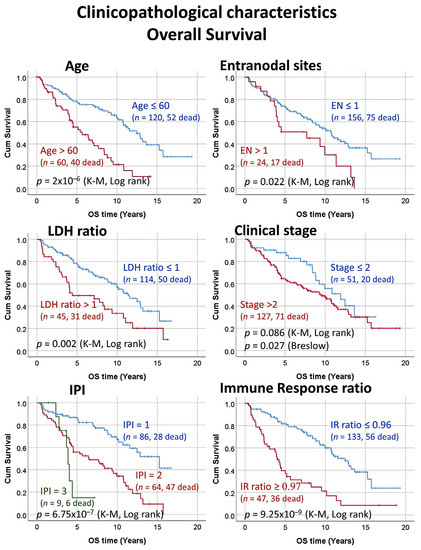
Figure 3.
Clinicopathological characteristics of the series. Correlation between the clinicopathological characteristics and the overall survival of the patients. The clinical evolution of FL is usually indolent but heterogeneous. Some clinical variables such as the IPI (FLIPI) and the immune response allows stratifying the patients according to their risk.
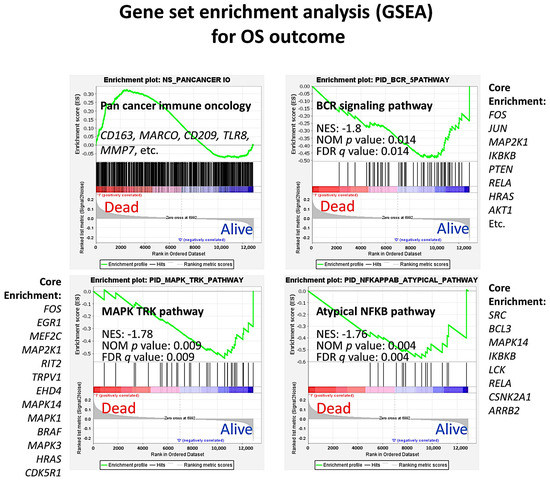
Figure 4.
Clinicopathological characteristics of the series: Gene set enrichment analysis (GSEA). GSEA was performed to identify pathways related to the overall survival outcome (dead/alive). A pan cancer immune oncology pathway was used to identify which markers were potentially responsible for a poor outcome (dead phenotype). This pathway included genes involved in the complex interplay between the tumoral B-lymphocytes, the tumor immune microenvironment, and the host immune response. The plot showed an enrichment toward the dead phenotype with the presence of genes related to macrophages. Other pathways that were highlighted were the B-cell receptor (BCR) signaling, mitogen-activated protein kinase (MAPK)—tyrosine receptor kinase (TRK), and the atypical nuclear factor-kappa B (NFKB) pathways, which were associated to a favorable outcome.
2.4. Multilayer Perceptron and Radial Basis Function Neural Network Analysis
Multilayer perceptron and radial basis function neural networks were performed as previously described [4,5]. In summary, the analysis aimed to predict a single variable or various dependent variables based on several factors (categorial variable) or covariates (continuous variable).
In the multilayer perceptron analysis setup, the rescaling of covariates was standardized. The cases were randomly assigned based on the relative numbers of cases with a training partition of 70%, test partition of 30%, and holdout partition of 0%. The architecture was comprised of hidden layers and an output layer. The hidden layers included the setup of the number of hidden layers (one or two), the activation function (hyberbolic tangent, sigmoid), and the number of units (automatically computed or custom). The output layer was defined by the activation function (identify, softmax, hyperbolic tangent and sigmoid) and by the rescaling of scale-dependent variables (standardized, normalized, adjusted normalized, or none). Of note, the activation function chosen for the output layer determined which rescaling methods were available. The type of training could be batch, online, and mini-batch, and the optimization algorithm could use the scaled conjugate gradient or the gradient descent. The network structure included the description, the diagram, and the synaptic weights. The network performance included the model summary, the classification results, the ROC curve, cumulative gains chart, lift chart, predicted by observed chart, and residual by predicted chart. The output also included a case processing summary and the analysis of the independent variable importance. The predicted value or category and the predicted pseudo-probability for each dependent variable were saved. The synaptic weight estimates were also exported to an xml file. As stopping rules, the maximum training epochs were computed automatically, the minimum relative changes in the training error was 0.0001, and the training error ratio was 0.001.
In the radial basis function setup, the parameters are similar to the multilayer perceptron but the activation function for the hidden layer can be a normalized radial basis function or an ordinary radial basis function, and the overlap among hidden units can be automatically computed or specified.
The analysis follows a series of steps. (1) The multilayer perceptron and radial basis function neural networks analysis were performed using the gene expression data of 184 patients with follicular lymphoma. The input data were the 22,215 gene probes (the predictors, covariates) and the predicted variables (the dependent variables) were the survival output (dead/alive) as well as the other relevant clinicopathological variables. The genes were ranked according to their normalized importance for prediction. The genes with more than 70% of normalized importance were selected and pooled. The genes above 1% of averaged normalized importance were also selected and pooled. (2) The final list of genes was subjected to a univariate Cox regression analysis for the prediction of survival outcome (dead/alive, enter method). Each gene was used in the univariate Cox as a quantitative variable (predictor). Subsequently, a multivariate Cox regression analysis was performed only with the significant genes of the univariate analysis. In this multivariate analysis, the method was the backward conditional. (3) The final list of significant genes from the multivariate analysis was subjected to a second round of multilayer perceptron analysis to confirm their association with the most relevant variables, the survival outcome, the international prognostic index, and the immune microenvironment immune response.
2.5. Gene Set Enrichment Analysis
The gene set enrichment analysis (GSEA) was performed using the GSEA software from the UC San Diego, Broad Institute (version 4.1.0) following the manufacturer’s instructions. In summary, three types of files were created: (1) the gene cluster text file (*.gct) that contains the gene expression data of the GSE16131 GPL96 follicular lymphoma series (n = 180); (2) the phenotype data as a categorical class (dead/alive) file format (*.cls); and (3) the gene set database as a gene matrix file format (*.gmx). When running the GSEA, the following parameters were used: number of permutations (1000), collapse to gene symbols, permutation type (phenotype), chip platform (Affymetrix HG U133), enrichment statistic (weighted), metric for ranking genes (signal2noise), gene list sorting mode (real), gene list ordering mode (descending), max size (500), and min size (15). The Java web start option (1 GB for 64-bit) was launched using Java 8 (version 261, build 1.8.O_261-b12, Oracle Corporation, 500 Oracle Parkway, Redwood Shores CA, 94065, USA).
3. Results
3.1. Multilayer Perceptron and Radial Basis Function Neural Network Analysis
In Table 3 and Table 4, all the data of the results of the MLP and RBF analyses are shown. The results include the case processing summary, the network information, the hidden layer, the output layer, the model summary, the model summary testing, the classification, and the area under the curve. The results are individualized for each of the dependent variables, as shown in the Figure 5, including survival outcome, age, extranodal sites, lactate dehydrogenase (LDH), clinical stage, international prognostic index (IPI), immune response ratio (2:1), the status of the presence of t(14;18), and the combined variable. The MLP analysis used the hyperbolic tangent activation function in the hidden layer, and the output layer used the softmax activation function and cross-entropy error function. Conversely, the RBF used the softmax activation function in the hidden layer and the identity activation function and sum of squares error function in the output layer. In both the training model and in the testing model, the error of the MLP was higher than in the RBF (p < 0.05). Conversely, the training time was higher in the RBF analysis: MLP vs. RBF, training time in seconds, 11.9 ± 20.9 vs. 384.3 ± 168.0% (p = 4.871 × 10−4), respectively. Importantly, the area under the curve of the ROC curve was higher in the MLP than in the RBF: 0.76 vs. 0.62 (p = 4.871 × 10−4). Therefore, the results indicate that the MLP is slightly more efficient than the RBF neural network. From an initial set of 22,215 gene probes, after several neural network analyses, the threshold for the 70% normalized importance and 1% averaged normalized importance, the pooling and deletion of duplicates, we finished with a set of 1005 genes. This approximately accounts for a 20-times reduction (Figure 5).
Table 3.
Multilayer perceptron analysis (MLP).
Table 4.
Radial basis function analysis (RBF).
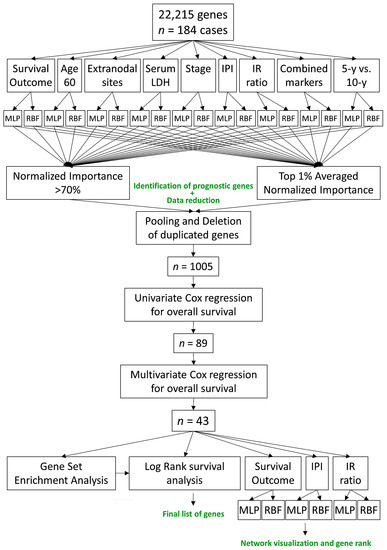
Figure 5.
Artificial Intelligence analysis of follicular lymphoma. This figure shows the method used to analyze the gene expression data, which included a multistep process. From an initial set of 22,215 gene probes, a final set of 43 genes were identified, of which 18 were associated to a poor overall survival of the patients and 25 were associated to a good overall survival of the patients.
3.2. Univariate and Multivariate Cox Survival Analysis
A univariate overall survival analysis with the Cox regression analysis was performed in each of the 1005 genes. In each regression analysis, the gene expression values were analyzed as a continuous variable. As a result, the list was reduced to 89 genes. In order to highlight the most important, a survival analysis with a multivariate Cox regression analysis (backward conditional method) was performed, and in the last step, the list of the most relevant genes was reduced to 43 genes: 18 were associated to a poor prognosis (Table 5) and 25 were associated to a good prognosis of the patients (Table 6). In the tables, the genes are ranked according to their hazard ratio for risk of survival (dead/alive).
Table 5.
Genes associated to poor prognosis in the multivariate Cox survival analysis.
Table 6.
Genes associated to good prognosis in the multivariate Cox survival analysis.
Each clinicopathological-dependent variable had a contribution of gene probes in the final set of 1005 genes, which was variable in case of MLP or RBF neural network as well as for Normalized Importance (NI) >70% or Averaged Normalized Importance. In the case of only gene probes with a normalized importance >70% and MLP analysis, the contribution was as follows: OS, 8 gene probes; Age, 47 gene probes; Extranodal, 3 gene probes; LDH, 32 gene probes; Stage, 41; IPI, 4; Immune Response, 8; OS 5-10y, 84; combined variables, 451. In case of RBF analysis, the contribution (also only gene probes with a normalized importance >70%) was as follows: OS, 56 gene probes; Age, 50 gene probes; Extranodal, 52 gene probes; LDH, 184 gene probes; Stage, 24; IPI, 66; Immune Response, 64; OS 5-10y, 85; combined variables, 451. In summary, MLP contributed in 678 gene probes into the pooling, and RBF contributed in 1032 gene probes.
This contribution may be of interest if we want to know which markers are associated to each of the dependent variables as well as to know which variables are the most relevant for predicting the overall survival of the patients in terms of gene-probes contribution.
For example, if we focus on the last step of multivariate Cox survival analysis in which a final set of 43 genes was identified, the MLP and RBF contributions are variable depending on the type of neural network and the variable tested. In general, RBF neural network contributed more frequently to the identification of the final set of genes than the MLP (35 vs. 28, respectively). Nevertheless, in the averaged normalized importance variable, the MLP identified more genes than the RBF (17 vs. 14, respectively). In summary, the combination of both MLP and RBF produces a synergic effect, the combined variable is the most efficient of the individual variables, and the averaged normalized importance is the most efficient of all. The contribution of each variable is shown in Figure 6.
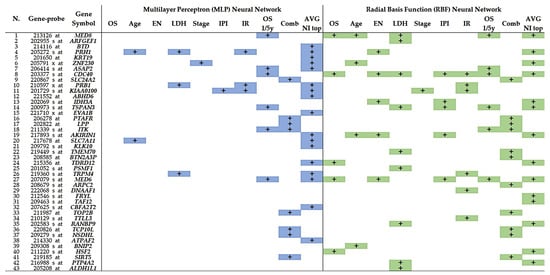
Figure 6.
Contribution of each dependent variable and neural network method in the final set of 43 predictive genes. Both multilayer perceptron (MLP) and radial basis function (RBF) contribute to the final set with a synergic effect. In general, the most efficient variables are the combined and the averaged normalized importance.
3.3. Gene Set Enrichment Analysis (GSEA)
GSEA is a computational method that determines whether an a priori defined set of genes shows statistically significant, concordant differences between two biological states (e.g., phenotypes). In our case, the two biological states were the outcome status of dead vs. alive. GSEA was performed on the set of 43 genes, and the result was a sinusoid-like plot, with some genes associated to good and other to poor prognosis. This is the same result as in the multivariate Cox regression analysis for overall survival. The GSEA association was also confirmed for the set of 18 genes (good prognosis) and the set of 25 (good). In addition, we included in the GSEA immune genes, oncogenes, and tumor-suppressor genes known to play a role in the pathogenesis of follicular lymphoma. All the results, including the genes of the core enrichment, are shown in Figure 7.
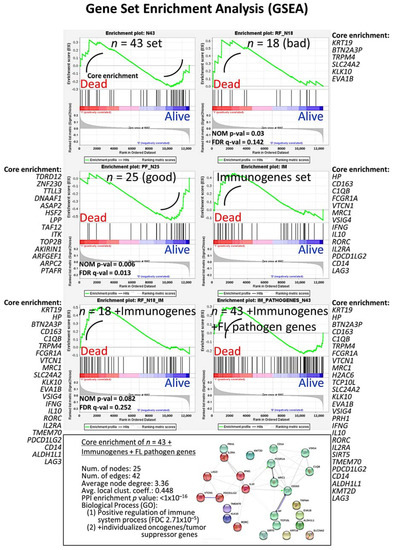
Figure 7.
Gene set enrichment analysis (GSEA) was performed to confirm the results of the multivariate Cox regression for the overall survival analysis. The set of 43 were used in addition to genes of the immune response as well as oncogenes and tumor suppressor genes related to the pathogenesis of follicular lymphoma. The genes of the core enrichment are shown as well as a functional network association analysis.
3.4. Univariate Survival Analysis with Kaplan–Meier and Log Rank Test
Based on the results of the GSEA analysis, a small group of genes was selected, and a cut-off of gene expression was searched (Figure 8). We confirmed that a high expression of EVA1B, KRT19, BTN2A3P, KLL10, and TRPM4 was associated to a poor OS of the patients, while high TDRD12 and ZNF230 was associated to a favorable OS (p < 0.05).
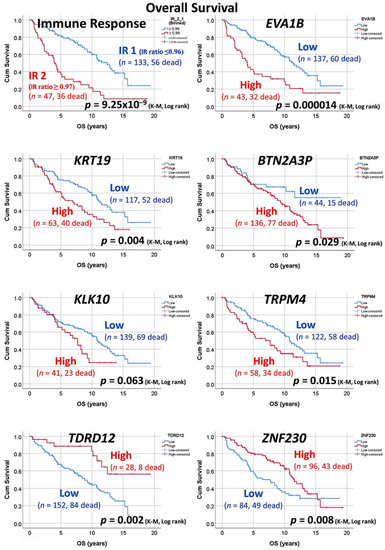
Figure 8.
Overall survival with Kaplan–Meier analysis. Based on the GSEA results, a group of genes was selected. After finding the more adequate cut-off, a group of patients with high and low expression was found with correlation to the overall survival. Bad OS genes were EVA1B, KRT19, BTN2A3P, KLL10, and TRPM4. Good OS genes were TDRD12 and ZNF230.
3.5. Final MLP and RBF Neural Network Analysis
Based only on the set of 43 genes and the 184 patients, a final MLP and RBF neural network analysis was formed. The aim was to confirm the quality control parameters of our model using the ROC curve, the cumulative gains chart, and the lift chart. In addition, the 43 were ranked according to their normalized importance to predict the OS of the patients, the immune response, and the IPI. The results are shown in Figure 9, Figure 10 and Figure 11.
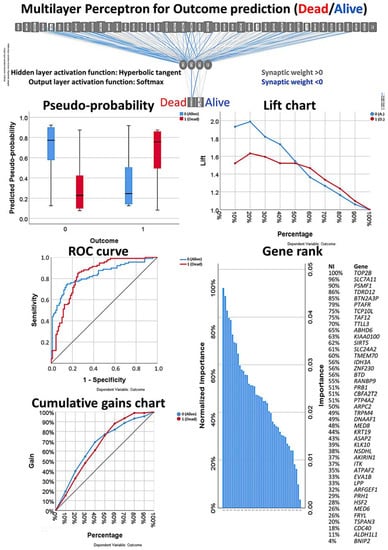
Figure 9.
Multilayer perceptron analysis based on the set of 43 genes. This MLP analysis includes only the final set of 43 genes as input variables and predicts the survival of the patients based on the survival outcome (dead/alive). The quality of the neural network analysis can be confirmed with the receiver operating characteristic (ROC) curve, and the rank of the genes is shown according to their normalized importance for prediction.
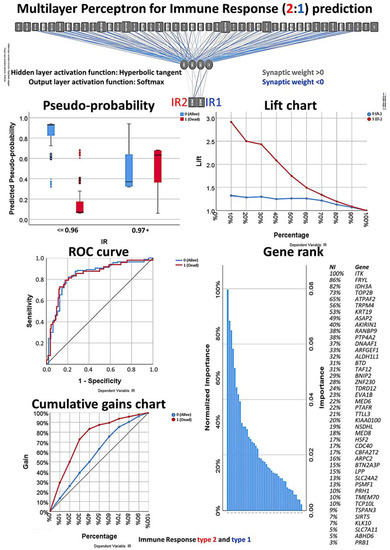
Figure 10.
Multilayer perceptron analysis based on the set of 43 genes. This MLP analysis includes only the final set of 43 genes as input variables and predicts the immune response of the patients based on the immune response 2:1 ratio. The quality of the neural network analysis can be confirmed with the ROC curve, and the rank of the genes is shown according to their normalized importance for prediction.
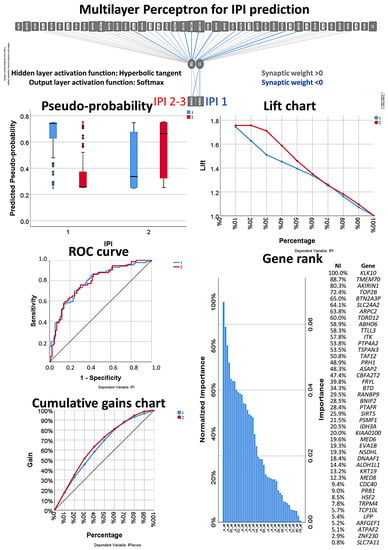
Figure 11.
Multilayer perceptron analysis based on the set of 43 genes. This MLP analysis includes only the final set of 43 genes as input variables and predicts the international prognostic index (IPI) of the patients. The quality of the neural network analysis can be confirmed with the ROC curve, and the rank of the genes is shown according to their normalized importance for prediction.
3.6. Correlation between the 7 Highlighted Genes and the Clinicopathological Characteristics of the Patients
In this project, we identified seven genes that were associated with the overall survival of the patients. In five genes (EVA1B, KRT19, BTN2A3P, KLK10, and TRPM4), high expression correlated to a poor overall survival; and in two genes (TDRD12 and ZNF230), high expression correlated to a favorable overall survival. In order to understand the reason for this association, we correlated using simple crosstabulations with the clinicopathological characteristics of the patients. The associations can be seen in Table 7.
Table 7.
Clinicopathological correlations with the final set of seven prognostic genes.
4. Discussion
Follicular lymphoma (FL) is one of the most frequent subtypes of non-Hodgkin lymphomas, and it is the second most common subtype in Western countries. According to the World Health Organization (WHO) classification of tumors of the hematopoietic and lymphoid tissues, FL is characterized by a proliferation of B lymphocytes of the germinal center of lymphoid follicles, and it almost always shows a histological follicular growth pattern under the microscope [6,7].
The molecular pathogenesis of FL is complex, and it is characterized by a multistep process in which a germinal center B-lymphocyte must acquire several genetic and epigenetic changes that eventually will lead to the malignant transformation. Most of the FL cases are characterized by the t(14;18) that conveys the overexpression of the BCL2 anti-apoptosis oncogene. Nevertheless, although the overexpression of BCL2 is necessary and seen is most of the cases even in FL “in situ”, by itself, it is not capable of malignant transformation. FL is morphologically heterogenous, comprised in morphologic terms as centrocytes (small and large cleaved cells) and centroblasts (small and large non-cleaved cells), and being in different stages of proliferation, maturation, and/or apoptosis. In addition, the tumoral immune microenvironment is rich in T-lymphocytes (including cytotoxic T-cells, follicular T helper cells, macrophages, and dendritic cells) [9,11].
Artificial Intelligence (AI) techniques make use of algorithms that can be defined as a series of instructions that a computer can execute. Many of the AI algorithms can learn from data and enhance themselves. Artificial neural networks are computer systems that try to mimic the biological neural networks. The artificial units or nodes are called neurons. The connections between neurons are called edges. Neurons and edges have a weight that adjusts in time along with the learning process. In this project, we were interested in performing AI analysis of gene expression data of FL because to the best of our knowledge, this had not been attempted before. We used two methods, two types of neural network analyses, the multilayer perceptron (MLP) and radial basis function (RBF), to analyze the gene expression of FL, and we correlated with the prognosis of the patients. In addition, due to the importance of the tumoral immune microenvironment in FL, we also queried the AI system for correlation with the two known immune responses type 1 and 2 that are associated with the prognosis of the patients [12]. Finally, the AI analysis was completed with more conventional statistical analyses including the gene set enrichment analysis (GSEA), univariate and multivariate Cox regression analyses, and the Kaplan–Meier with log rank test for overall survival analysis. Our method was successful, as we managed to simplify from 22,215 gene probes to a final list of 43 genes. Of note, we integrated both the MLP and RBF in the analysis, but we also could compare both types of neural networks, and we found that MLP was more efficient than RBF, as seen by its faster computational time and better areas under the curve of the ROC analysis. Therefore, out data show that MLP fits better the type of data of gene expression in lymphoid neoplasia.
In this project, we have highlighted several genes with prognostic relevance in FL. Some of them were associated to a poor prognosis of the patients, while others were associated to a good prognosis. Among the bad prognosis group, we found that the high gene expression of EVA1B, KRT19, BTN2A3P, KLK10, and TRPM4 was associated to an unfavorable overall survival of the FL patients. EVA1B (Protein eva-1 homolog B) is an integral component of the membrane [14]. Very little information about this marker is found in the literature (including Pubmed), but this gene was found to be highly significantly changed by altered DNA methylation [14]. According to the Human Protein Atlas [15], a high expression of EVA1B is associated to a poor prognosis of renal, breast, and liver cancers. KRT19 (Keratin, type I cytoskeletal 19) is involved in the organization of myofibers, as it is a structural constituent of the cytoskeleton. It is also associated to the Notch signaling pathway [16]. In cancer, it has been related to the pathogenesis of breast and colon cancers [15]. According to the Human Protein Atlas [15], high expression is associated to poor prognosis in renal and pancreatic cancer. BTN2A3P (Putative butyrophilin subfamily 2 member A3) regulates the cytokine production; therefore, it regulates immune responses and the T-cell receptor pathway. By genome-wide association and transcriptome studies, this gene was identified as a target gene and risk loci for breast cancer [17]. KLK10 (Kallikrein-10) is a serine-type endopeptidase involved in the cell cycle. KLK10 has a tumor-suppressor role for NES1 in breast and prostate cancer. High expression of this gene is associated to an unfavorable prognosis of the patients in pancreatic and breast cancer [15]. KLK10 is also involved in other several types of cancer. For example, the NES1/KLK10 gene represses proliferation, enhances apoptosis, and down-regulates the glucose metabolism of PC3 prostate cancer cells [15]. TRPM4 (Transient receptor potential cation channel subfamily M member 4) functions as a calcium-activated non-selective (CAN) cation channel that mediates membrane depolarization [15]. It has been involved in the adaptive immune response and in many types of cancer. For example, TRPM4 is highly expressed in human colorectal tumor buds and contributes to the proliferation, cell cycle, and invasion of colorectal cancer cells [18]. Among the good prognosis group, we found that the high gene expression of TDRD12 and ZNF230 were associated to a favorable overall survival of the FL patients. TDRD12 (Putative ATP-dependent RNA helicase TDRD12) has RNA helicase activity (nuclei acid binding) and is involved in cell differentiation. Not much information of this gene is found in the literature, but it has been related to salivary gland adenoid cystic carcinoma [19]. ZNF230 (Zinc finger protein 230) may be involved in transcriptional regulation, with DNA and metal ion binding properties. The RNA levels are detected as low levels in many cancers, and the protein level in cancer tissue has not been completed yet [15]. Therefore, using AI, we not only managed to highlight several genes with prognostic relevance in FL, but also, we identified some genes that have a role in other types of cancer. In addition, these genes were not only rated to the prognostic outcome (dead/alive) but also related to the tumoral immune microenvironment, which is a field of importance due to the development of personalized immunotherapies.
The future research directions of this research include the validation of these markers using an independent series of FL from another institution.
5. Conclusions
Using Artificial Intelligence, we have analyzed the gene expression of follicular lymphoma and managed to identify a set of 43 genes that correlated with the overall survival of the patients. Therefore, AI including the multilayer perceptron and radial basis function neural networks are useful bioinformatics tools for the identification of biomarkers in follicular lymphoma.
In this research, we used the overall survival endpoint. Nevertheless, overall survival is not the best endpoint for follicular lymphoma analysis due to the protracted and variable nature of the disease; rather, response to therapy, prediction of progression (often to large B-cell lymphoma), and quality of life are additional considerations. This aspect will be assessed in a continuation research.
Among the 43 genes, we highlighted seven genes, five associated to poor and two associated to overall survival. Those five genes also associated to M2-like macrophage markers. There is little scientific evidence about the role of these seven genes in lymphoid neoplasia, but there is some evidence of their association in other types of cancer. Therefore, these genes open a path of potential application in development of clinical trials and targeted therapies.
Author Contributions
Conceptualization, J.C.; methodology, J.C.; software, J.C.; validation, R.H.; formal analysis, J.C.; investigation, Y.Y.K., M.M., S.H., S.T., H.I., Y.K., A.I.; resources, N.N.; writing—original draft preparation, J.C.; writing—review and editing, J.C.; supervision, N.N.; project administration, J.C.; funding acquisition, J.C. and R.H. All authors have read and agreed to the published version of the manuscript.
Funding
J.C. was funded by grant KAKEN 18K15100 (JSPS and MEXT) R.H. was funded by Al-Jalila Foundation (AJF201741), the Sharjah Research Academy (Grant code: MED001) and University of Sharjah (Grant code: 1901090258).
Acknowledgments
We would like to thank all the members of the Lymphoma/Leukemia Molecular Profiling Project (LLMPP) including Louis M. Staudt, Elias Campo, WC Chan and ES Jaffe (among others) for creating and sharing publicly the GSE16131 dataset.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Aunalytics. Artificial Intelligence, Machine Learning, and Deep Learning. Available online: https://www.aunalytics.com/artificial-intelligence-machine-learning-and-deep-learning%E2%81%A0/ (accessed on 8 November 2020).
- IBM SPSS Neural Netwoks 25; IBM Corporation: Armonk, NY, USA, 2018.
- IBM Corporation. IBM SPSS Neural Networks 20. New Tools for Building Predictive Models. IBM Business Analytics. Available online: https://www.ibm.com/support/pages/node/618179 (accessed on 28 October 2020).
- Carreras, J.; Hamoudi, R.; Nakamura, N. Artificial Intelligence Analysis of Gene Expression Data Predicted the Prognosis of Patients with Diffuse Large B-Cell Lymphoma. Tokai J. Exp. Clin. Med. 2020, 45, 37–48. [Google Scholar] [PubMed]
- Carreras, J.K.; Kikuti, Y.Y.; Miyaoka, M.; Hiraiwa, S.; Tomita, S.; Ikoma, H.; Kondo, Y.; Ito, A.; Shiraiwa, S.; Hamoudi, R.; et al. Single Gene Expression Set Derived from Artificial Intelligence Predicted the Prognosis of Several Lymphoma Subtypes; and High Immunohistochemical Expression of TNFAIP8 Associated with Poor Prognosis in Diffuse Large B-Cell Lymphoma. AI 2020, 1, 342–360. [Google Scholar] [CrossRef]
- Swerdlow, S.H.; Campo, E.; Pileri, S.A.; Harris, N.L.; Stein, H.; Siebert, R.; Advani, R.; Ghielmini, M.; Salles, G.A.; Zelenetz, A.D.; et al. The 2016 revision of the World Health Organization classification of lymphoid neoplasms. Blood 2016, 127, 2375–2390. [Google Scholar] [CrossRef] [PubMed]
- Freedman, A.S.; Aster, J.C.; Lister, A.; Connor, R.F. Clinical Manifestations, Pathologic Features, Diagnosis, and Prognosis of Follicular Lymphoma. UptoDate. Available online: https://www.uptodate.com/contents/clinical-manifestations-pathologic-features-diagnosis-and-prognosis-of-follicular-lymphoma?search=lymphomas%20definition&source=search_result&selectedTitle=1~150&usage_type=default&display_rank=1 (accessed on 28 October 2020).
- Byers, R.J.; Sakhinia, E.; Joseph, P.; Glennie, C.; Hoyland, J.A.; Menasce, L.P.; Radford, J.A.; Illidge, T. Clinical quantitation of immune signature in follicular lymphoma by RT-PCR-based gene expression profiling. Blood 2008, 111, 4764–4770. [Google Scholar] [CrossRef] [PubMed]
- Carreras, J.; Lopez-Guillermo, A.; Fox, B.C.; Colomo, L.; Martinez, A.; Roncador, G.; Montserrat, E.; Campo, E.; Banham, A.H. High numbers of tumor-infiltrating FOXP3-positive regulatory T cells are associated with improved overall survival in follicular lymphoma. Blood 2006, 108, 2957–2964. [Google Scholar] [CrossRef] [PubMed]
- Carreras, J.; Lopez-Guillermo, A.; Kikuti, Y.Y.; Itoh, J.; Masashi, M.; Ikoma, H.; Tomita, S.; Hiraiwa, S.; Hamoudi, R.; Rosenwald, A.; et al. High TNFRSF14 and low BTLA are associated with poor prognosis in Follicular Lymphoma and in Diffuse Large B-cell Lymphoma transformation. J. Clin. Exp. Hematop. 2019, 59, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Carreras, J.; Lopez-Guillermo, A.; Roncador, G.; Villamor, N.; Colomo, L.; Martinez, A.; Hamoudi, R.; Howat, W.J.; Montserrat, E.; Campo, E. High numbers of tumor-infiltrating programmed cell death 1-positive regulatory lymphocytes are associated with improved overall survival in follicular lymphoma. J. Clin. Oncol. 2009, 27, 1470–1476. [Google Scholar] [CrossRef] [PubMed]
- Dave, S.S.; Wright, G.; Tan, B.; Rosenwald, A.; Gascoyne, R.D.; Chan, W.C.; Fisher, R.I.; Braziel, R.M.; Rimsza, L.M.; Grogan, T.M.; et al. Prediction of survival in follicular lymphoma based on molecular features of tumor-infiltrating immune cells. N. Engl. J. Med. 2004, 351, 2159–2169. [Google Scholar] [CrossRef] [PubMed]
- Glas, A.M.; Kersten, M.J.; Delahaye, L.J.; Witteveen, A.T.; Kibbelaar, R.E.; Velds, A.; Wessels, L.F.; Joosten, P.; Kerkhoven, R.M.; Bernards, R.; et al. Gene expression profiling in follicular lymphoma to assess clinical aggressiveness and to guide the choice of treatment. Blood 2005, 105, 301–307. [Google Scholar] [CrossRef] [PubMed]
- Lim, W.J.; Kim, K.H.; Kim, J.Y.; Jeong, S.; Kim, N. Identification of DNA-Methylated CpG Islands Associated With Gene Silencing in the Adult Body Tissues of the Ogye Chicken Using RNA-Seq and Reduced Representation Bisulfite Sequencing. Front. Genet. 2019, 10, 346. [Google Scholar] [CrossRef] [PubMed]
- Uhlen, M.; Fagerberg, L.; Hallstrom, B.M.; Lindskog, C.; Oksvold, P.; Mardinoglu, A.; Sivertsson, A.; Kampf, C.; Sjostedt, E.; Asplund, A.; et al. Proteomics. Tissue-based map of the human proteome. Science 2015, 347, 1260419. [Google Scholar] [CrossRef] [PubMed]
- Saha, S.K.; Choi, H.Y.; Kim, B.W.; Dayem, A.A.; Yang, G.M.; Kim, K.S.; Yin, Y.F.; Cho, S.G. KRT19 directly interacts with beta-catenin/RAC1 complex to regulate NUMB-dependent NOTCH signaling pathway and breast cancer properties. Oncogene 2017, 36, 332–349. [Google Scholar] [CrossRef] [PubMed]
- Ferreira, M.A.; Gamazon, E.R.; Al-Ejeh, F.; Aittomaki, K.; Andrulis, I.L.; Anton-Culver, H.; Arason, A.; Arndt, V.; Aronson, K.J.; Arun, B.K.; et al. Genome-wide association and transcriptome studies identify target genes and risk loci for breast cancer. Nat. Commun. 2019, 10, 1741. [Google Scholar] [CrossRef] [PubMed]
- Kappel, S.; Stoklosa, P.; Hauert, B.; Ross-Kaschitza, D.; Borgstrom, A.; Baur, R.; Galvan, J.A.; Zlobec, I.; Peinelt, C. TRPM4 is highly expressed in human colorectal tumor buds and contributes to proliferation, cell cycle, and invasion of colorectal cancer cells. Mol. Oncol. 2019, 13, 2393–2405. [Google Scholar] [CrossRef] [PubMed]
- Shao, C.; Sun, W.; Tan, M.; Glazer, C.A.; Bhan, S.; Zhong, X.; Fakhry, C.; Sharma, R.; Westra, W.H.; Hoque, M.O.; et al. Integrated, genome-wide screening for hypomethylated oncogenes in salivary gland adenoid cystic carcinoma. Clin. Cancer Res. 2011, 17, 4320–4330. [Google Scholar] [CrossRef] [PubMed]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).