
Doping Carbon Coating on Glass Fiber to Enhance Its Reinforcing Potential in a Polymer Matrix


Abstract

1. Introduction
- Affixing 2D nanomaterials such as MXene, graphene oxide (GO), or carbon nanotubes (CNTs) to GF through techniques like electrophoretic deposition, covalent grafting, or electrothermal shock, respectively. These methods introduce nanoscale hierarchies and functional groups, strengthening interactions with epoxy matrices and enhancing interfacial adhesion [33,34,35].
- Growing silane coupling sites with the general structure GF(-O-)3Si-R, in which R groups can be tailored for chemical and physical bonding with a specific polymer matrix [36,37]. R groups can also be extended into vinyl oligomers like poly(vinyl acetate), leading to entangled interfaces that improve toughness [38].
2. Materials and Methods
2.1. Material and Chemicals
2.2. Cleaning and Activating Short E-Glass Fiber
2.3. Wrapping the Prepared GF Strands with a Carbon Thin Layer
2.4. Formation of a Carbon Coating on GF Filaments
2.5. Incorporation of GF@C Filaments into ABS Matrix
2.6. Preparation of Composite Sheets for the Assessment of the GF@C Reinforced ABS Sheets
2.7. Instrumental Characterization of the Prepared GF@C-ABS Composite Sheets
2.7.1. Field-Emission Scanning Electron Microscopic and EDX Characterizations
2.7.2. Raman Spectroscopic Characterization
2.7.3. Thermogravimetric Analysis (TGA)
2.7.4. Measurement of Dielectric Property of the GF@C Reinforced ABS Matrix
2.7.5. Analysis of Mechanical Properties of the Composite Sheets
2.7.6. Dynamic Mechanical Analysis (DMA)
3. Results and Discussion
3.1. Wrapping GF Strands with a Thin Carbon Sheath
3.2. Assessment of the GF@C-ABS Composites
4. Conclusions
- The formation of a carbon coating on GF is governed by the preparation of the coating ink, which involves the selection of the solvent, the type of carbon precursor, and, critically, the molecular structure of the polyaromatic hydrocarbon (PAH) compound used as a graphitization seeding agent (dopant). These factors affect both the surface morphology and the distribution of graphite crystallites within the resulting carbon sheath.
- A doping concentration of 5 wt.% coronene in the lubricant oil produces the most effective carbon coating, characterized by a larger graphite crystallite size and a higher abundance of crystallites. This is supported by Raman spectroscopy, specifically the ID/IG ratio and the width of the G peak.
- Mechanical testing reveals that the carbon coating (CLo) on glass fiber improves the fiber–ABS interfacial resistance under tensile stress. Further enhancement is observed with coronene-doped carbon coating (CLo-Cor(5%)). However, when compared to carbon fiber (CF), both C-coated GF fibers (GF@CLo and GF@CLo-Cor(5%)) exhibit lower tensile strength (σ) and Young’s modulus (E), which is attributed to the dominant graphene structure of CF that strengthens interactions with ABS.
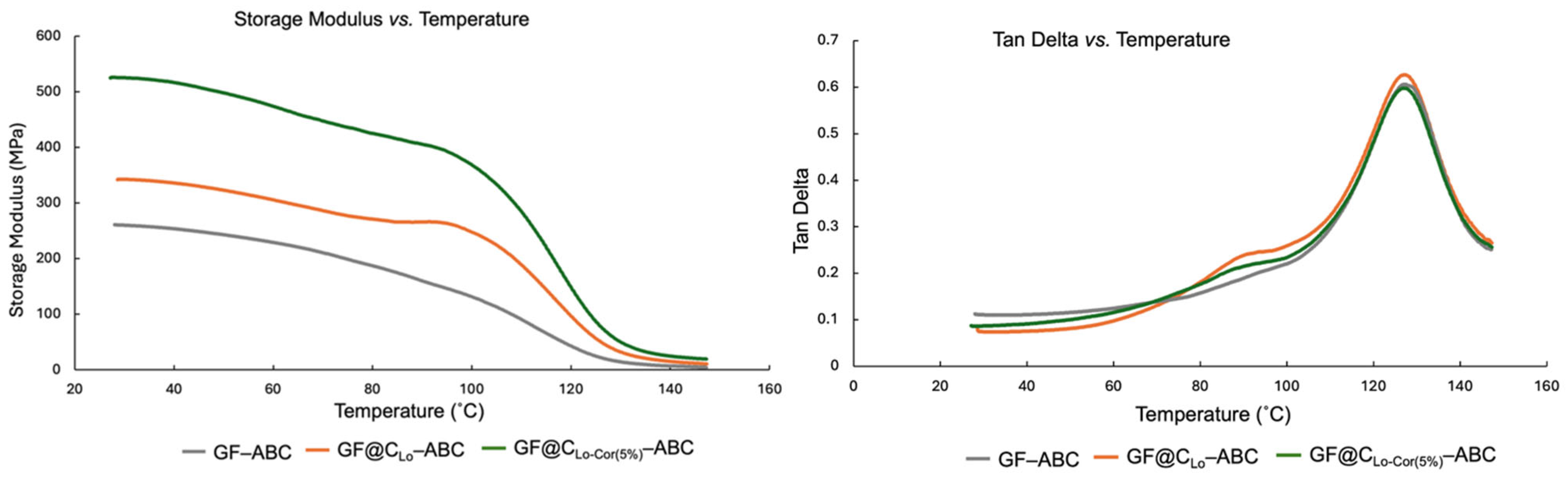
- The storage moduli obtained from dynamic mechanical analysis (DMA) follow the same trend as the Young’s moduli of the composites. However, the loss modulus data indicate that the surface heterogeneity of the C-coated fibers leads to increased chain entanglement, which influences the viscous deformation behavior of the polymer matrix.
- The carbon-coated fibers significantly enhance the dielectric constant of the composites compared to those reinforced with pristine GF or CF. This improvement is attributed to the varied adsorption states of ABS chains, primarily due to anchoring of the polar side-chain groups on the fiber surfaces.
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| GF | E-glass fiber |
| CF | Carbon fiber |
| GF@C | Glass fiber wrapped by a carbon coating |
| ABS | Acrylonitrile butadiene styrene resin |
| Lo | Lubricant oil |
| Cor | Coronene |
| Pyr | Pyrene |
| IPA | Iso-propanol |
| MEK | Methyl ethyl ketone |
| PEG | Polyethylene glycol |
| GO | Grapene oxide |
| CNTs | Carbon nanotubes |
| FRP | Fiber-reinforced polymer |
| HTC | Hydrothermal carbonization |
| 2D-MOFs | 2-Dimensional metal organic frameworks |
| PAH | Polyaromatic hydrocarbons |
| TGA | Thermogravimetric analysis |
| DMA | Dynamic mechanical analysis |
| E′ | Storage modulus |
| E″ | Loss modulus |
| EDX | Energy dispersive X-ray |
| FWHM | Full width at half maximum |
| ε′ | Dielectric constant |
References
- Mallick, P.K. Fiber-Reinforced Composites: Materials, Manufacturing, and Design, 3rd ed.; CRC Press: Boca Raton, FL, USA, 2007. [Google Scholar] [CrossRef]
- Saeedi, A.; Motavalli, M.; Shahverdi, M. Recent advancements in the applications of fiber-reinforced polymer structures in railway industry—A review. Polym Compos. 2024, 45, 77–97. [Google Scholar] [CrossRef]
- Charterer 6. “Innovating Clean Energy Technology in Advanced Manufacturing” in Quadrennial Technology Review. 2015. Available online: https://www.energy.gov/quadrennial-technology-review-2015 (accessed on 1 September 2015).
- Gao, H.; Sun, Y.; Jian, J.; Dong, Y.; Liu, H. Study on mechanical properties and application in communication pole line engineering of glass fiber reinforced polyurethane composites (GFRP). CSCM 2023, 18, e01942. [Google Scholar] [CrossRef]
- Abbood, I.S.; Odaa, S.A.; Hasan, K.F.; Jasim, M.A. Properties evaluation of fiber reinforced polymers and their constituent materials used in structures—A review. Mater. Today Proc. 2021, 43, 1003–1008. [Google Scholar] [CrossRef]
- Das, S.; Yokozeki, T. A brief review of modified conductive carbon/glass fibre reinforced composites for structural applications: Lightning strike protection, electromagnetic shielding, and strain sensing. JCOMC 2021, 5, 100162. [Google Scholar] [CrossRef]
- Zaman, A.; Gutub, S.A.; Wafa, M.A. A review on FRP composites applications and durability concerns in the construction sector. J. Reinf. Plast. Compos. 2013, 32, 1966–1988. [Google Scholar] [CrossRef]
- Hui, C.-Y.; Liu, Z.; Phoenix, S.L.; King, D.R.; Cui, W.; Huang, Y.; Gong, J.P. Mechanical behavior of unidirectional fiber reinforced soft composites. Extreme Mech. Lett. 2020, 35, 100642. [Google Scholar] [CrossRef]
- Wang, Y.; Wang, Z.; Zhu, L. A Short Review of Recent Progress in Improving the Fracture Toughness of FRP Composites Using Short Fibers. Sustainability 2022, 14, 6215. [Google Scholar] [CrossRef]
- Yang, B.J.; Ha, S.K.; Pyo, S.H.; Lee, H.K. Mechanical characteristics and strengthening effectiveness of random-chopped FRP composites containing air voids. Compos. B Eng. 2014, 62, 159–166. [Google Scholar] [CrossRef]
- Pheysey, J.; Cola, F.D.; Martinez-Hergueta, F. Short fibre/unidirectional hybrid thermoplastic composites: Experimental characterization and digital analysis. Compos. A Appl. Sci. Manuf. 2024, 181, 108121. [Google Scholar] [CrossRef]
- Fu, S.-Y.; Lauke, B.; Mäder, E.; Yue, C.-Y.; Hu, X. Tensile properties of short-glass-fiber- and short-carbon-fiber-reinforced polypropylene composites. Compos. Part A-Appl. Sci. Manu. 2000, 31, 1117–1125. [Google Scholar] [CrossRef]
- Hmeidat, N.S.; Elkins, D.S.; Peter, H.R.; Kumar, V.; Compton, B.G. Processing and mechanical characterization of short carbon fiber-reinforced epoxy composites for material extrusion additive manufacturing. Compos. B. Eng. 2021, 223, 109122. [Google Scholar] [CrossRef]
- Ogura, S. Research on strength prediction technology for fiber-reinforced plastic injection-molded products. KYB Technol. Rev. 2023, 67, 20–26. [Google Scholar]
- Rajabi, M.; Ali, A.; McConnell, M.; Cabral, J. Keratinous materials: Structures and functions in biomedical applications. Mater. Sci. Eng. C 2020, 110, 110612. [Google Scholar] [CrossRef] [PubMed]
- Callister, W.D., Jr.; Rethwisch, D.G. Materials Science and Engineering: An Introduction, 10th ed.; Wiley: Hoboken, NJ, USA, 2018; Chapter 16; ISBN 9781119405436. [Google Scholar]
- Waghmare, S.; Shelare, S.; Aglawe, K.; Khope, P. A mini review on fibre reinforced polymer composites. Mater. Today Proc. 2022, 54, 682–689. [Google Scholar] [CrossRef]
- Zhang, W.-B.; Yu, X.; Wang, C.-L.; Sun, H.-J.; Hsieh, I.-F.; Li, Y.; Dong, X.-H.; Yue, K.; Horn, R.V.; Cheng, S.Z.D. Molecular nanoparticles are unique elements for macromolecular science: From “nanoatoms” to giant molecules. Macromolecules 2014, 47, 1221–1239. [Google Scholar] [CrossRef]
- Fu, S.-Y.; Lauke, B. Effects of fiber length and fiber orientation distributions on the tensile strength of short-fiber-reinforced polymers. Compos. Sci. Technol. 1996, 56, 1179–1190. [Google Scholar] [CrossRef]
- Edwards, K.L. An overview of the technology of fibre-reinforced plastics for design purposes. Mater. Des. 1998, 19, 1–10. [Google Scholar] [CrossRef]
- Bernasconi, A.; Cosmi, F. Analysis of the dependence of the tensile behavior of a short fiber reinforced polyamide upon fiber volume fraction, length and orientation. Pro. Eng. 2011, 10, 2129–2134. [Google Scholar] [CrossRef]
- Zhang, R.; Shi, Z. Bi-interfacial debonding of coated fiber reinforced composites under fatigue load. Int. J. Fatigue 2008, 30, 1074–1079. [Google Scholar] [CrossRef]
- Vaidya, R.U.; Fernando, J.; Chawla, K.K.; Ferber, M.K. Effect of fiber coating on the mechanical properties of a Nextel-480-fiber-reinforced glass matrix composite. Mater. Sci. Eng. A 1992, 150, 2. [Google Scholar] [CrossRef]
- Anagnostopoulos, G.; Parthenios, J.; Galiotis, C. Thermal stress development in fibrous composites. Mater. Lett. 2008, 62, 341–345. [Google Scholar] [CrossRef]
- Chen, Y.; Prasad, V.; Yasar, M.; Murphy, N.; Ivankovic, A. Enhancing interfacial performance and fracture toughness of carbon fibre reinforced thermoplastic composites. Compos. Part A Appl. Sci. Manuf. 2024, 187, 108434. [Google Scholar] [CrossRef]
- Fu, S.Y.; Lauke, B.; Mäder, E.; Yue, C.-Y.; Hu, X.; Mai, Y.-W. Hybrid effects on tensile properties of hybrid short-glass-fiber-and short-carbon-fiber-reinforced polypropylene composites. J. Mater. Sci. 2001, 36, 1243–1251. [Google Scholar] [CrossRef]
- Ravichandran, B.; Balasubramanian, M. Joining methods for fiber reinforced polymer (FRP) composites—A critical review. Compos. A Appl. Sci. 2024, 186, 108394. [Google Scholar] [CrossRef]
- Ishtiaq, S.; Saleem, M.Q.; Naveed, R.; Harris, M.; Khan, S.A. Glass–Carbon–Kevlar fiber reinforced hybrid polymer composite (HPC): Part (A) mechanical and thermal characterization for high GSM laminates. JCOMC 2024, 14, 100468. [Google Scholar] [CrossRef]
- Yu, Y.; Yu, L.; Wang, C.; Cui, Z. Effect of polyurethane treatment on the interfacial and mechanical properties of basalt fiber-reinforced polymer composite. Fibers Polym. 2024, 25, 607–617. [Google Scholar] [CrossRef]
- De, B.; Bera, M.; Bhattacharjee, D.; Ray, B.C.; Mukherjee, S. A comprehensive review on fiber-reinforced polymer composites: Raw materials to applications, recycling, and waste management. Prog. Mater. Sci. 2024, 146, 101326. [Google Scholar] [CrossRef]
- Ari, A.; Bayram, A.; Karahan, M.; Karag, S. Comparison of the mechanical properties of chopped glass, carbon, and aramid fiber reinforced polypropylene. Polym. Polym Compos. 2022, 30, 1–13. [Google Scholar] [CrossRef]
- Morampudi, P.; Namala, K.K.; Gajjela, Y.K.; Barath, M.; Prudhvi, G. Review on glass fiber reinforced polymer composites. Mater Today Proc. 2021, 43, 314–319. [Google Scholar] [CrossRef]
- Chen, X.; Hui, Y.; Zhang, J.; Wang, Y.; Zhang, J.; Wang, X.; Cheng, S.; Wen, K.; Li, Z.; Yi, C.; et al. Single multifunctional MXene-coated glass fiber for interfacial strengthening, damage self-monitoring, and self-recovery in fiber-reinforced composites. Compos. B Eng. 2023, 259, 110713. [Google Scholar] [CrossRef]
- Chen, J.; Zhao, D.; Jin, X.; Wang, C.; Wang, D.; Ge, H. Modifying glass fibers with graphene oxide: Towards high-performance polymer composites. Compos. Sci. Technol. 2014, 97, 41–45. [Google Scholar] [CrossRef]
- Shang, Y.; Shi, B.; Doshi, S.M.; Chu, T.; Qiu, G.; Du, A.; Zhao, Y.; Xu, F.; Thostenson, E.T.; Fu, K.K. Rapid nanowelding of carbon coatings onto glass fibers by electrothermal shock. ACS Appl. Mater. Interfaces 2020, 12, 37722–37731. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Zhang, L.; Yu, E.; Ying, Z.; Zhang, Y.; Liu, X.; Eli, W. Modification of glass fiber surface and glass fiber reinforced polymer composites challenges and opportunities: From organic chemistry perspective. Curr. Org. Chem. 2015, 19, 991–1010. [Google Scholar] [CrossRef]
- Wu, Y.; Song, Y.; Wu, D.; Mao, X.; Yang, X.; Jiang, S.; Zhang, C.; Guo, R. Recent progress in modifications, properties, and practical applications of glass fiber. Molecules 2023, 28, 2466. [Google Scholar] [CrossRef]
- Naguib, H.M. Evaluation of filler activation for sustainable FRP composite by studying properties, mechanism, and stability. Sci. Rep. 2024, 14, 18256. [Google Scholar] [CrossRef]
- Gan, Y.X.; Yusuf, M.; Ibrahim, H. Hydrothermal carbonization coating on recycled quartz glass fiber. Mater. Lett. 2025, 382, 137877. [Google Scholar] [CrossRef]
- Abejon, R.; Garea, A.; Irabien, A. Ultrapurification of hydrogen peroxide solution from ionic metals impurities to semiconductor grade by reverse osmosis. Sep. Purif. Technol. 2010, 76, 44–51. [Google Scholar] [CrossRef]
- Jang, H.K.; Chung, Y.D.; Whangbo, S.W.; Lyo, I.W.; Whang, C.N.; Lee, S.J.; Lee, S. Effects of chemical etching with hydrochloric acid on a glass surface. J. Vac. Sci. Technol. A 2000, 18, 2563–2567. [Google Scholar] [CrossRef]
- ASTM D638-22; Standard Test Method for Tensile Properties of Plastics. ASTM: West Conshohocken, PA, USA, 2022; Volume 08.01: Plastics. [CrossRef]
- Bajdik, J.; Regdon, G.; Marek, T.; Erős, I.; Süvegh, K.; Pintye-Hódi, K. The effect of the solvent on the film-forming parameters of hydroxypropyl-cellulose. Int. J. Pharm. 2005, 301, 192–198. [Google Scholar] [CrossRef]
- Lara, J.; Zimmermann, F.; Drolet, D.; Hansen, C.M.; Chollot, A.; Monta, N. The use of the Hansen solubility parameters in the selection of protective polymeric materials resistant to chemicals. Int. J. Curr. Res. 2017, 9, 47860–47867. [Google Scholar]
- Mishra, A.; Siddiqi, H.; Kumari, U.; Behera, I.D.; Mukherjee, S.; Meikap, B.C. Pyrolysis of waste lubricating oil/waste motor oil to generate high-grade fuel oil: A comprehensive review. Renew Sustain. Energy Rev. 2021, 150, 111446. [Google Scholar] [CrossRef]
- Voorhees, K.J.; Baugh, S.F.; Stevenson, D.N. An investigation of the thermal degradation of poly(ethylene glycol). J. Anal. Appl. Pyrol. 1994, 30, 47–57. [Google Scholar] [CrossRef]
- Uematsu, H.; Yoshida, K.; Yamaguchi, A.; Fukushima, A.; Sugihara, S.; Yamane, M.; Ozaki, Y.; Tanoue, S. Enhancement of interfacial shear strength due to cooperative π-π interaction between polyphenylene sulfide and carbon fiber and molecular orientation of polyphenylene sulfide via the π-π interaction. Compos. Part A Appl. Sci. Manuf. 2023, 165, 107355. [Google Scholar] [CrossRef]
- Schwan, J.; Ulrich, S.; Batori, V.; Ehrhardt, H.; Siva, S.R.P. Raman spectroscopy on amorphous carbon films. J. Appl. Phys. 1996, 80, 440–447. [Google Scholar] [CrossRef]
- Yang, F.; Hu, G.; He, H.; Yi, M.; Ge, Y.; Ran, L.; Peng, K. Effect of amorphous carbon on the tensile behavior of polyacrylonitrile (PAN)-based carbon fibers. J. Mater. Sci. 2019, 54, 8800–8813. [Google Scholar] [CrossRef]
- Mortazavian, S.; Fatemi, A. Effects of fiber orientation and anisotropy on tensile strength and elastic modulus of short fiber reinforced polymer composites. Compos. B Eng. 2015, 72, 116–129. [Google Scholar] [CrossRef]
- Ornaghi, H.L.; Almeida, J.H.S.; Monticeli, F.M.; Neves, R.M. Stress relaxation, creep, and recovery of carbon fiber non-crimp fabric composites. JCOMC 2020, 3, 100051. [Google Scholar] [CrossRef]
- Tótch, C.; Lukács, N.L.; Kovács, N.K. The role of the fiber–matrix interface in the tensile properties of short fiber–reinforced 3D-printed polylactic acid composites. Polym. Compos. 2024, 45, 13589–13602. [Google Scholar] [CrossRef]
- Chao, H.-W.; Hsu, H.-C.; Chen, Y.-R.; Chang, T.-H. Characterizing the dielectric properties of carbon fiber at different processing stages. Sci. Rep. 2021, 11, 17475. [Google Scholar] [CrossRef]
- Naka, J.; Yamamoto, S.; Komori, K.; Yamakawa, S.; Kokubo, T. Development of glass fiber with high dielectric constant. J. Non-Cryst. Solids. 1994, 177, 420–426. [Google Scholar] [CrossRef]
- Kuzmanić, I.; Vujović, I.; Petković, M.; Šoda, J. Influence of 3D printing properties on relative dielectric constant in PLA and ABS materials. Prog. Addit. Manuf. 2023, 8, 703–710. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Du, Z.; Cheng, Z.; Cao, Z.; Ye, Y.; Wang, Z.; Wei, J.; Wei, S.; Meng, X.; Song, L.; et al. Ultralow-dielectric-constant atomic layers of amorphous carbon nitride topologically derived from mxene. Adv. Mater. 2023, 35, e2301399. [Google Scholar] [CrossRef] [PubMed]
- Mekha, K.B.; Yusof, N.A.T.; Sudhakar, K.; Zainol, N.; Hasan, N.; Karim, M.S.A. Development of sustainable polymer-based dielectric composites from agricultural waste: A review. Heliyon 2024, 10, e39118. [Google Scholar] [CrossRef]
- Quader, R.; Narayanan, L.K.; Caldona, E.B. Dielectric characterization of fiber- and nanofiller-reinforced polymeric materials. J. Appl. Polym. Sci. 2024, 141, e55362. [Google Scholar] [CrossRef]
- Shakir, M.F.; Khan, A.N.; Khan, R.; Javed, S.; Tariq, A.; Azeem, M.; Riaz, A.; Shafqat, A.; Cheema, H.M.; Akram, M.A.; et al. EMI shielding properties of polymer blends with inclusion of graphene nano platelets. Results Phys. 2019, 14, 102365. [Google Scholar] [CrossRef]
- Maroulakos, G.; Wanserski, M.W.; Wanserski, M.M.; Schuler, E.J.; Egan, C.P.; Thompson, G.A. Effect of airborne-particle abrasion on 3-dimensional surface roughness and characteristic failure load of fiber-reinforced posts. J. Prosthet. Dent. 2019, 121, 461–469. [Google Scholar] [CrossRef]
- Vovchenko, L.L.; Matzui, L.Y.; Yakovenko, O.S.; Lozitsky, O.V.; Len, T.A.; Oliynyk, V.V.; Galaburda, M.V.; Borovoy, M.O.; Syvolozhskyi, O.A. Electrical and shielding properties of epoxy composites with Ni–C and Co–C core-shell nanoparticles. Phys. E Low Dimens. Syst. Nanostruct. 2022, 144, 115463. [Google Scholar] [CrossRef]












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Main components of Alfa E-Glass Fiber (wt%) | SiO2 52.0–55.0; CaO 21.2–22.2; Al2O4 14.0–15.0; B2O3 7.8–8.2 |
| Density (g/cm3) | 2.55–2.60 |
| Tensile strength (MPa) | 1950–2050 |
| Young’s modulus (GPa) | 72–85 |
| Approximate composition of Alfa ABS resin (wt%) | Butadiene 5–30; Styrene 40–60; Acrylonitrile 15–35 |
| Density (g/cm3)/Melt and glass transition temperature (°C) | 1.01–1.07/ Tm: 190–240; Tg: 88–128 |
| Tensile strength (MPa) | 27.6–55.2 |
| Young’s modulus (GPa) | 1.1–2.9 |
| Typical composition of aviation lubricant * | Main components: C5–C10 Fatty acid esters; Poly(1-olefins); Mineral oil mixtures consisting of paraffinic, naphthenic, and aromatic hydrocarbons C ≥ 15. Additives: Phosphates (RxO)3P=O as anti-wear and dispersion agent; Phenol derivatives as oxidation inhibitor; Polyisobutylene as viscosity improver; Alkyl naphthalenes as pour-point depressant; Zinc dialkyl dithiophosphate as anti-fouling and anti-corrosion compound. Solubility parameter: 15–20 MPa |
| Sample | Coating Ink | Heat Treatment | |
|---|---|---|---|
| Liquid | PAH (wt.% of Lo) | Vaporization and Curing/Carbonization | |
| GF@CLo a. | Lubricant oil (Lo) | - | 260 °C/800 °C |
| GF@CLo-Cor(x%) | Lubricant oil | Coronene (Cor, x = 3, 5, and 7) | As above |
| GF@CLo-Pyr(x%) | Lubricant oil | Pyrene (Pyr, x = 3 and 5) | As above |
| GF@CPeg | PEG-600 | - | As above |
| Solvent | Density (g/cm3) at 20 °C | δt—Solubility Parameter (MPa1/2) * | ||
|---|---|---|---|---|
| δD | δP | δH | ||
| t-Butanol | 0.775 | 21.9 | ||
| 18.1 | 5.8 | 10.9 | ||
| 2-Propanol | 0.786 | 23.5 | ||
| 15.8 | 6.2 | 16.4 | ||
| Methyl Ethyl ketone | 0.805 | 18.6 | ||
| 15.5 | 9.0 | 5.1 | ||
| Carbon Coating | ID/IG | FWHM of G-Band (cm−1) |
|---|---|---|
| CLo | 0.93 | 125 |
| CLo-Cor(3%) | 0.91 | 98 |
| CLo-Cor(5%) | 0.84 | 80 |
| CLo-Cor(7%) | 0.97 | 107 |
| CLo-Pyr(3%) | 0.79 | 148 |
| Pure Materials | ε′ | Reference |
|---|---|---|
| CF | 2.82~6.5, could also be much larger | [53] |
| E-GF | 6.5 to 6.8 | [54] |
| ABS plastic | 2.0 to 3.5 | [55] |
| Amorphous C | 2.2 | [56] |
| Processing Step/Substance | Approx. Cost (USD) * | Notes |
|---|---|---|
| Pretreatment/E-GF | ~45 | Including GF (1 kg), H2O2 (30%, Tech. grade 1.0 L), and HCl (37%, ACS grade, 1.0 L) |
| Coating-ink formulation | ~12 | Including t-Butanol (Tech. grade 10 L), Lubricant (0.25 L), and Coronene (Tech. grade 12.5 g) |
| Generation of C-coating and the above steps | ~15 | Electricity consumption ~100 KWH |
| Miscellaneous | ~10 | Filtration paper and DI water |
| Outcome | ~82 | Approx. per 1 kg C-coated GF |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tay, S.W.; Lau, I.; Hong, L. Doping Carbon Coating on Glass Fiber to Enhance Its Reinforcing Potential in a Polymer Matrix. J. Compos. Sci. 2025, 9, 348. https://doi.org/10.3390/jcs9070348
Tay SW, Lau I, Hong L. Doping Carbon Coating on Glass Fiber to Enhance Its Reinforcing Potential in a Polymer Matrix. Journal of Composites Science. 2025; 9(7):348. https://doi.org/10.3390/jcs9070348
Chicago/Turabian StyleTay, Siok Wei, Inez Lau, and Liang Hong. 2025. "Doping Carbon Coating on Glass Fiber to Enhance Its Reinforcing Potential in a Polymer Matrix" Journal of Composites Science 9, no. 7: 348. https://doi.org/10.3390/jcs9070348
APA StyleTay, S. W., Lau, I., & Hong, L. (2025). Doping Carbon Coating on Glass Fiber to Enhance Its Reinforcing Potential in a Polymer Matrix. Journal of Composites Science, 9(7), 348. https://doi.org/10.3390/jcs9070348